

Abstract
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare clonal disease. To date, many reviews and series have been described. We report the experience of our center by presenting a review of 56 PNH patient cases with an average age at diagnosis of 38 yr and follow-ups beginning at approximately 40 yr; the median survival rate was 11 yr. The average clonal size upon diagnosis was 48%, presenting a variable evolution. Thrombotic episodes and cancer were five each, and the main causes of death among our patients were equal at 8.9%. Radiological study by magnetic resonance imaging is presented as a fundamental technique for estimating the deposit of iron levels in the liver and kidney, as well as in some decisive cases at the start of eculizumab therapy. Sixteen patients have been treated with eculizumab so far in our series, and being a safe drug, it provides improvement in the patients’ quality of life, and the disappearance of clinical symptoms, and avoids the emergence of new thrombosis.
Keywords: paroxysmal nocturnal hemoglobinuria, thrombosis, eculizumab, liver transplantation, secondary cancer
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare disease 1 caused by the absence of glycosylphosphatidylinositol on the membranes of blood cells and is caused by a somatic mutation in the PIG gene (most frequently in the PIG-A gene) 2–4. Because of this alteration, the blood cells become targets for the native of the complement system action causing its destruction. At the erythrocyte level, a continuous and severe intravascular hemolysis generates free hemoglobin and depletion of nitric oxide, which leads to vasoconstriction and platelet activation 5.
As a result of these phenomena, PNH manifests as a serious prothrombotic disease with severe renal insufficiency, pulmonary hypertension, and other organic damage that can lead to premature death. In a significant number of patients, it is associated with bone marrow aplasia which could constitute the pathophysiologic event triggering the disease 6,7.
Until a few years ago, the only treatment for PNH was an allogeneic hematopoietic transplant 8, reserved for selected patients. However, since the appearance of eculizumab, a monoclonal antibody directed against the C5 fraction of the complement, the situation has changed 9.
Our hospital has a long experience with PNH and is the center with the most cases in Spain. Since the first diagnoses 40 yr ago, we have had a unique opportunity to learn about the natural and clinical histories of the disease, along with the impact of a new treatment for this rare disease.
Methods
Patients
From 1970 to November 2013, 56 patients with the PNH clone have been evaluated in our hospital. Forty of them were diagnosed in our center, and the remainder were referred for possible allogeneic hemopoietic transplant evaluation or assessment prior to starting eculizumab. Thirty-six were male and 20 were female. The median age at PNH diagnosis was 32.8 yr ± 17.7 IQR (13–84). The first patient was diagnosed in 1970, and series follow-up varied between 0.1 and 41.6 yr, with a median of 11.4 ± 12.09 IQR years. Patient data were recorded retrospectively in a local database designed for the series through the compilation of historic clinical data from the digital medical records and on paper for older cases. This study has been approved by the local ethics committee. Starting from September 2010, 29 patients have been included in the International PNH Registry. Sixteen patients have been treated with the anti-C5 drug eculizumab (Soliris®; Alexion Pharmaceuticals, Inc., Cheshire, CT, USA), the first patient being treated in November 2007. All of these patients have received prophylactic vaccination with meningococcal tetravalent vaccine and revaccinated every 2 yr. Since the beginning of 2013, all patients have received ciprofloxacin 500 mg/d orally.
Diagnostics
Until 1994, patients were diagnosed using the Ham test or modifications of the test 10,11. Since the early 1990s, flow cytometry techniques have been used in our laboratory for the diagnosis and PNH patient follow-ups. For the detection of GPI-deficient granulocytes, a lyse–wash–stain technique was employed. Cells were analyzed in a FACScan® (LYSIS II) or FACScalibur® (CellQuest) instruments (Becton Dickinson, San Agustin de Guadalix, Spain). Monoclonal antibodies used were as follows:
CD55 and CD59 in an indirect immunofluorescence test 1 from 1994 to 2005 12,13.
Phycoerythrin-labeled CD55 and CD59 from 2005 to 2011.
Results were expressed as percentage of granulocytes negative for CD55/CD59 or FLAER/CD24 17,18.
The patient series was classified into three groups according to their clinical expression and percentage of PNH clone as classic PNH, PNH in the setting of another specified bone marrow disorder (SBMD), and subclinical, according to the classification of Nakakuma as amended by Parker 19,20.
Radiology by magnetic resonance imaging
In the last 4 yr, magnetic resonance imaging (MRI) studies were performed in our patients with PNH (cranioencephalic, thoracic, and/or abdominal) in acute complications (nine patients) or as a programmed protocol evaluation previous to considering and/or evaluating eculizumab treatment (14 patients).
The patients with PNH were examined with 1.5 tesla magnets for thoracic and abdominal MRI, and with 3.0 tesla magnets for cranioencephalic MRI (Achieva Magnets; Philips Healthcare, Best, the Netherlands).
Different protocols designed for the study of this pathology using morphological sequences with different empowerment, functional sequences, and angiographic studies after administration of intravenous contrast (gadobutrol) have been used. For the quantification of liver and kidney iron deposits, T2* calculations were performed.
Statistical analysis
We presented continuous data as mean and standard deviation or median and interquartile range (IQR), with extreme values. When comparing two groups, we used the Student’s t-test for normally distributed data; otherwise, the nonparametric Mann–Whitney U-test was used. For proportions, we used the Pearson chi-square test or the Fisher’s exact test for scarce data. The overall survival outcome was analyzed according to the Kaplan–Meier method and compared through the log-rank test. We considered P < 0.05 as statistically significant; all statistical tests were two-sided. We quoted 95% confidence intervals (95% CIs) whenever applicable.
Results
Clinical classification of patients
Patients were divided into three clinical groups according to the Parker classification system. This classification has been applied to each patient considering the time of maximum clinical expression of the disease and larger clonal size. This allocation has been maintained for the purposes of our analysis regardless of the clinical and clone patient evolution. According to this criterion, 29 patients have been considered as the classic form of the disease, 20 as SBMD, and seven as subclinical. The demographic and clinical characteristics of all the patients classified into Parker subgroups are contained in Table1. In the subclinical patient group, four patients were diagnosed with bone marrow failure of varying degrees, one myelodysplastic syndrome (MDS) patient with medullary blastosis (RAEB2) secondary to a bone marrow aplasia of very long evolution, and two others diagnosed MDS type RA 21.
Table 1.
Demographics and symptomatology of patients with PNH
| Parker’s classification | |||
|---|---|---|---|
| Classic | SBMD | Subclinical | |
| 29 | 20 | 7 | |
| PNH diagnosis age | 30 (13–62) | 32 (16–84) | 50 (21–78) |
| Actuarial age | 51 (25–87) | 48 (18–86) | 66 (32–85) |
| Male/Female | 17/12 | 15/5 | 4/3 |
| Bone marrow failure (BMF) | 16 (55) | 18 (90) | 4 (57) |
| Diagnosis of BMF/PNH | |||
| Simultaneous | 4 | 6 | 1 |
| Previous | 12 | 12 | 3 |
| Follow-up time (yr) | 19 (0.1–41.6) | 3.4 (0.3–30) | 7 (2.7–18) |
| Maximum PNH clone granulocytes | 80 (49–99) | 10 (2–58) | 1.8 (1–3) |
| Maximum LDH (n times over normal) | 7.8 (1.6–34) | 2 (1.2–7.4) | 1 (0.9–1.2) |
| Hemoglobinuria | 28 (96) | 6 (30) | 0 |
| Red blood transfusions | 25 (83) | 15 (75) | 6 (85) |
| Abdominal pain | 23 (79) | 4 (20) | 0 |
| Esophageal spasms | 15 (51) | 1 (0.5) | 0 |
| Erectile dysfunction [no. of male] | 9 (52.9) [17] | 1 (6) [15] | 0 [4] |
| Kidney failure3 | 13 (44.8) | 2 (10) | 1 (14) |
| Arterial hypertension | 14 (48) | 4 (20) | 2 (28) |
| Splenectomy | 2 | 1 | 0 |
| Hepatitis C | 4 | 2 | 0 |
| Allogeneic hematopoietic transplantation | 1 (Dead) | 1 (Alive) | 2 (Alive) |
| Eculizumab treatment | 16 | 0 | 0 |
| Alive/Exitus | 22/7 | 17/4 | 6/1 |
| Exitus | 7 (3 PNH: PTE, AMI, acute thrombotic stroke 4 cancer: pancreas, lung, 2 lymphomas) | 4 (1 liver disease thrombosis, 1 hepatocarcinoma, 1 hemorrhagic stroke by aplasia, 1 pneumonia) | 1 (acute thrombotic stroke) |
Data are expressed as median (interval) or median (percentage).
AMI, acute myocardial infarct; BMF, bone marrow failure; LDH, lactate dehydrogenase; PNH, paroxysmal nocturnal hemoglobinuria; PTE, pulmonary thromboembolism; SBMD, PNH in the setting of another specified bone marrow disorder.
Creatinine clearance by Cockcroft–Gault formula adjusted for sex <60 mL/min.
The most severe clinical expressivity attributable to hemolysis, including thrombotic events, is represented in the group of patients with classic disease forms in which lactate dehydrogenase (LDH) levels are significantly higher. A linear relationship is established between levels of LDH and size of clone PNH, represented in Fig.1.
Figure 1.
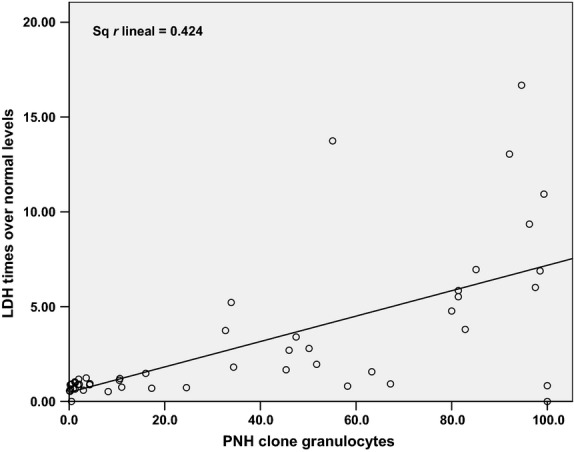
Lineal relationship between paroxysmal nocturnal hemoglobinuria (PNH) clone and lactate dehydrogenase (LDH) levels. LDH levels were correlated with the PNH clone in granulocytes in the same day in 199 samples of patients with PNH. Patients on eculizumab were excluded. LDH levels over normal could be calculated in our series in base to PNH clone in granulocytes with a mathematic equation as follows: LDH = 0.485 + 0.067 × PNH clone. r = 0.651 P < 0.001.
Renal insufficiency
Sixteen patients had various grades of kidney failure throughout its clinical evolution. Only three presented as severe renal insufficiency cases with creatinine clearance less than 15 mL/min needing dialysis: two patients who died by myocardial infarction and massive pulmonary thromboembolism, respectively, in the context of a multi-organ failure; another patient with medullary hypoplasia and chronic treatment with cyclosporine, required dialysis on one occasion, with nearly complete renal function recovery after starting treatment with eculizumab.
Relationship with bone marrow aplasia/hypoplasia
Thirty eight patients in the series had a clinical history of bone marrow aplasia of variable severity. In 11 cases, the diagnosis of aplasia/hypoplasia was simultaneous with the PNH clone and in most of them, the bone marrow deficit preceded PNH diagnosis. There was no clear relationship of the disease with a picture of bone marrow deficit in 11 patients. In cases in which the aplasia preceded the diagnosis of PNH, the interval of time between both diagnoses ranged from 4 months to 30 yr, with a median of 8.5 yr.
Most of the patients in our series showed signs of hematopoietic deficiency with cell count alteration in peripheral blood upon PNH diagnosis. Hemocytometric measurements were available on the day of PNH diagnosis for 43 patients in the series, presenting absolute neutropenia in 15 of them and thrombocytopenia in 29 patients. In all patients, characteristically, there was a MCV average increase of 100 fL ± 10.5 (IQR) with extreme values at 85 and 118. This hematologic deficit corresponds to the bone marrow biopsies findings. Characteristically, the bone marrow of the patients with classic forms of the disease showed granulocytic and megakaryocytic hypoplasia with marked erythroid hyperplasia.
Survival
Among the 56 patients, there was a loss of three follow-up patients and are not considered for survival assessments. Twelve patients have died, primarily due to cancer, and the remainder of deaths are directly attributable to causes of the underlying disease (thrombosis, mainly); see Table1. Only one of the deaths occurred of complications due to pneumonia. One patient died from cerebral hemorrhage during serious thrombocytopenia by severe aplasia. According to the Kaplan–Meier method, the survival curve is registered in Fig.2 with a median survival of 11 yr.
Figure 2.
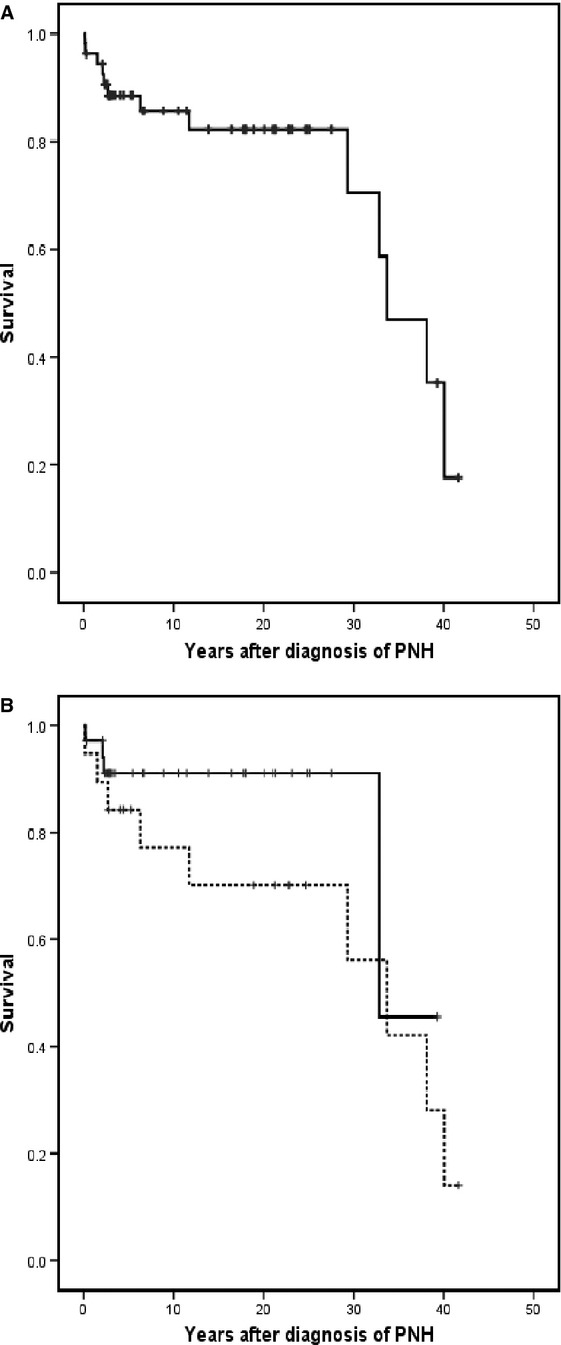
Kaplan–Meier Survival Curves: actuarial survival from the time of diagnosis of 53 patients with paroxysmal nocturnal hemoglobinuria. The monitoring of three patients from 1992, 2001, and 2005 with their respective follow-ups of 6, 3, and 14 yr has been lost and has not be included in the survival analysis. (A) The whole series. (B) Patients with thrombotic episodes (dotted line) and without it. P not significative.
A comparison was established in relation to survival among groups of patients with and without thrombosis not being statistically significant (Fig. 2), although the existence of thrombosis did seem to constitute an unfavorable factor in our series.
In our PNH series, an unexpectedly high incidence of cancer has appeared with eight patients (14.2%), displaying different hematological or non-hematological cancers in the lasts years (Table2).
Table 2.
Cancer incidence in the PNH patient series
| Sex | Age diagnosis PNH | Parker’s classification | Year diagnosis PNH | Year diagnosis cancer | Previous immunosuppression | Cancer | Year death |
|---|---|---|---|---|---|---|---|
| ♂ | 16 | Classic | 1969 | 2011 | Yes AA & liver Tx | Lymphoma | 2011 |
| ♀ | 30 | Classic | 1973 | 2003 | None | Pancreatic | 2006 |
| ♂ | 38 | Classic | 1974 | 1995 2012 | None | Gastric Lung | 2012 |
| ♂ | 26 | Classic | 1989 | 2013 | Yes, steroids | Cerebral | Alive |
| ♀ | 25 | Classic | 1994 | 2005 | Yes AA & cord blood Tx | Lymphoma | 2006 |
| ♂ | 40 | SBMD | 1995 | 1995 | Yes, steroids | Liver | 1995 |
| ♂ | 75 | SBMD | 2011 | 2009 | None | Seminoma | 20122 |
| ♂ | 56 | Subclinical | 2010 | 1999 | Yes, steroids | Prostatic | Alive |
AA, aplastic anemia; Tx, transplantation; PNH, paroxysmal nocturnal hemoglobinuria; SBMD, PNH in the setting of another specified bone marrow disorder.
Dead by bone marrow failure (hemorrhagic stroke).
Flow cytometry and clonal and clinical evolution
Cytometric analysis was available for 45 patients at the time of diagnosis and during a sufficient observation time period, to evaluate the behavior of the pathologic clone during clinical evolution.
In 16 cases, the clone increased after diagnosis. One of these patients even went from an undetectable clone to a PNH clone of greater than 60% with serious hemolysis, and with subsidiary treatment with eculizumab. This group includes 10 patients with classic forms.
In 12 patients, the PNH clone remained stable in quantity. This group includes 10 patients with classic forms of PNH.
In the majority, that is in 17 patients, the clone PNH has reduced over time, achieving complete disappearance in six patients (subclinical forms mainly), and remains detectable with variable but reduced amounts and without active hemolysis in terms of LDH elevation in the other 11 patients. Four classic types of the disease have had this favorable evolution, but in only one of these cases (after an allogeneic cord blood transplant) has the complete disappearance of the clone occurred.
In terms of the behavior of the PNH clone in patients treated with eculizumab, 13 patients were evaluated with PNH clone percentages at the beginning of treatment with an 80.14 mean ± 4.99 (45.3–99.3) and at the time of the evaluation of 87.5 ± 2.6 (73.6–98.7). Eight experienced an increase in the clone with a maximum increase of 39 points, two remained stable, and in only three patients was there a slight decrease by 8 points in the most marked case.
Allogeneic hematopoietic transplants
Four patients were subjected to allogeneic transplant treatments. Three of them were transplanted with familial identical HLA donor. At the present time, all these three cases remain alive with disappearance of the PNH clone and with recurrence in one case of MDS. One patient with a classic form of the disease received an allogeneic cord blood cell transplant with CD34+-selected cells from a haploidentical donor 22 in the era prior to the availability of eculizumab. The patient remained in complete remission of their disease with disappearance of the PNH clone for more than 1 yr, but dying eventually by post-transplant lymphoma of the CNS.
Eculizumab
Sixteen patients in our series have received eculizumab. The follow-up treatment period ranges from 3 months to 6 yr. All of them are patients with classic forms of the disease according to the Parker classification, with severe hemolysis and PNH clone granulocytes percentage of >45%, and symptoms attributable to the disease in all cases. All patients except three met the requirement of transfusion dependence due to hemolysis by PNH. Of the three without transfusion dependency, two had been transfused at some time during their illness due to extreme anemia, and all of them had serious impairments in their quality of life attributable to PNH. Five patients were also attributable to PNH thrombosis. In all cases, transfusion independence was achieved at the beginning of treatment with normalization of LDH figures and disappearance of the symptomatology attributable to the disease.
All patients presented maintenance at 900 mg of the drug every 14 d. Only one patient, due to breakthrough hemolysis 6 months after starting treatment, has been receiving 1200 mg every 2 wk since then. Four patients presented a positive Coombs test after beginning eculizumab with positive C3d complement fraction. Only one of the patients (the one receiving 1200 mg doses of eculizumab biweekly) has presented analytical changes, which could be compatible with the presence of an extravascular hemolysis.
Three patients have received concomitant treatment with eculizumab and cyclosporine as an immunosuppressive for its marrow deficit:
The first began treatment with eculizumab in aplasia recovery phase after appearance of hemolysis with hemoglobinuria and a distinct increase in the PNH clone. The cyclosporine was withdrawn 2 yr later.
The second received cyclosporine continuously and continues with treatment with the two medications.
The third has started treatment with cyclosporine 4 yr after starting eculizumab because of the worsening of their bone marrow failure.
Only one of the patients treated with eculizumab died from lymphoma after liver transplantation.
None of the patients treated with eculizumab has presented relevant infections without hospital admissions for this group of patients. Only one patient, undergoing liver transplantation, presented an abscess during the splenectomy procedure and is attributable to the same procedure.
Four of the patients underwent surgical treatment while on eculizumab treatment. In the first case (a liver transplant with splenectomy), there were complications (breakthrough hemolysis). In the following three cases, levels of LDH were monitored anticipating eculizumab doses where necessary, without presenting hemolysis.
Pregnancy
Two patients in our series had pregnancies to term in the pre-eculizumab period. One of them, a classic form, had to undergo a Cesarean section for fetal distress in a term pregnancy. With a worsening of the postpartum hemolysis, she required multiple transfusions and a hysterectomy was performed. The other patient with a minor clone and bone marrow hypoplasia had two pregnancies to term and childbirths without complications. No patient in our series in treatment with eculizumab had pregnancies.
Liver transplantation
A male with the classic form of the disease underwent a cadaveric liver transplant due to hepatitis C (HCV) terminal liver disease 2 yr after starting treatment with eculizumab. During the procedure, he also underwent a splenectomy. After the procedure, the patient presented two episodes of breakthrough hemolysis, with low levels of eculizumab because of extraordinary plasma losses by drainages. These episodes were resolved through administration of extra doses of eculizumab. After the transplantation, he also presented with immediate hemoperitoneal complications associated with hemodynamic compromise with acute renal insufficiency of prerenal origin, perihepatic Escherichia coli abscesses, lobular hepatitis by recurrent HCV, and CMV infection; all were resolved favorably. The patient survived the procedure for a year with excellent quality of life. After this period, he presented a post-transplant hepatic lymphoma, dying as a result.
Radiology with MRI
Thrombosis was found in four cases: one in the inferior cava vein and three in arteries (two cerebral and one in descending aorta). In three patients, these findings implied initiating eculizumab therapy. Minor ischemic brain changes were displayed by three patients. None of the 18 patients explored with thoracic MRI displayed pulmonary hypertension signs despite the elevation of pro-BNP in eight of them. Iron overload in the liver and/or kidneys was very frequent. The finding of a reversal of the normal cortical and medullary intensities on T1- and T2-weighted images of both kidneys 23,24 was evident in the majority of patients with severe PNH types on eculizumab treatment. This finding was also evident in patients with active hemolysis in the past but with very low PNH clones and clinical remission of the disease. In livers, we have found low T2* echo times (4–29 ms) that correlate with high liver iron deposits, of unknown cause. In blood, large serum ferritin values were found (121–709 ng/mL) after eculizumab treatment with normal or diminished values before the treatment. Many other incidental discoveries include cholelithiasis, splenomegaly, kidney arterial vessel constriction, vascular anomalies, kidney and vesical stones, adrenal adenoma, atheromatosis at different levels, Tornwaldt cyst, hamartomas, hemangiomas, and abnormal bone marrow signals.
Thrombosis
Five patients with PNH previously anticoagulated with coumarins because of thrombosis, continued with therapy after the addition of eculizumab. Also, patients with more than 50% PNH clone (established by cytometry with FLAER on granulocytes) and platelets >50 × 109/L received anticoagulation from the year 2009.
The major thrombotic events in our series of patients prior to the use of the eculizumab are shown in Table3. After introduction of eculizumab, active thrombosis resolved in all cases, as was the case of a patient with a large persistent thrombosis in the inferior cava vein despite the isolated anticoagulation therapy. Only one patient on eculizumab therapy experienced a thrombotic event, which was a transient ischemic attack with aphasia after a prolonged catheter ablation procedure for atrial fibrillation. This patient previously had signs of small vessel disease in MRI techniques. The episode occurred despite heparin anticoagulation and anticipated additional eculizumab doses and resolved thereafter.
Table 3.
PNH and thrombosis: The incidence and localization of thrombotic events in our series prior Eculizumab
| Parker’s classification | Classic | SBMD | Subclinical |
|---|---|---|---|
| Number of patients | 29 | 20 | 7 |
| Thrombosis cases (%) | 12 (41) | 4 (20) | 4 (57) |
| Thrombotic episodes | 25 | 6 | 6 |
| Deep calf | 5 | 3 | 2 |
| Cerebral ischemic infarcts | 3 | 1 | |
| Large cerebrovascular | 2 (1 death) | 3 (1 death) | |
| Budd–Chiari | 1 | 1 | 1 |
| Portal | 2 | 1 (1 death) | |
| Retinal | 2 | ||
| Cava | 1 | ||
| PTE | 2 (1 death) | ||
| Myocardial infarct | 3 (1 death) | ||
| Arterial ischemia | 2 (1 amputation) | ||
| Skin ischemic | 2 (vasculitis, livedo reticularis) |
PNH, paroxysmal nocturnal hemoglobinuria; SBMD, PNH in the setting of another specified bone marrow disorder; PTE, pulmonary thromboembolism.
A multivariate analysis was performed to establish a relationship between different parameters involved in the increased risk of thrombosis to see whether any of them had a predictive character. Only one statistical significance was found between the existence of esophageal spasms and abdominal pain (P = 0.04 and 0.012, respectively), and the existence of thrombosis. Two patients with arterial thrombosis (myocardial and skin necrosis) have been reported previously 25,26.
Discussion
In our series of patients, the majority of the patients were diagnosed before age 33 with a slight predominance in males, especially in the SBMD Parker group. In the series published with the largest number of cases 27–31 (Table4), a majority of the patients were diagnosed before age 40, with a slight predominance of males in the Asian series 28,31. In our series, the presence of hemoglobinuria and other serious signs of hemolysis, such as esophageal spasms, abdominal pain, and erectile dysfunction in males, were much more frequent in the patients group with the classic form and non-existent in subclinicals, clearly in relation to the size of the PNH clone. With our patients, a linear relationship between the size of the PNH clone in granulocytes and elevation of above normal LDH has been established, and thus, we can anticipate complications from cytometrics in the laboratory examination.
Table 4.
Comparison of Puerta de Hierro PNH series with previous published series
| First author (yr) (reference) | Number of patients Period of study | Population site | Age of PNH diagnosis Sex | Survival & complications Spontaneous remission | Patients with aplastic anemia, % | Adverse prognostic factors | Number of deaths by cancer |
|---|---|---|---|---|---|---|---|
| Dacie (1972) & Hillmen (1995) 27,29 | 80 >30 yr | London (UK) Hammersmith Hospital | 42 (15–75) 33♂/47♀ | 60 dead Median survival 10 yr Hemoglobinuria 26% Thrombosis 39% 12 survivors with spontaneous remission | 29 | NR | 1 (lymphoma) |
| Kruatrachue (1978) 28 | 85 25 yr | Bangkok (Thailand) Siriraj Hospital | 30 (10–80) 52♂/23♀ | 7 dead Hemoglobinuria 32% Thrombosis 1% Abdominal pain <1% | 10 | NR | None |
| Socié (1996) 30 | 220 46 yr | France Multicenter | 33 (6–82) 100♂/120♀ | 71 dead Median survival 15 yr Thrombosis 6% Abdominal pain 13% | 30 | Age >55 Thrombosis MDS/AML Thrombocytopenia | 4 (3 MDS/AML) |
| Nishimura (2004) 31 | 385 36 yr (USA) 28 yr (Japan) | 176 USA (Duke University) 209 Japan (Multicenter) | 30 (4–80) 77♂/99♀ 45 (10–86) 118♂/91♀ | Median survival 19 and 32 yr Hemoglobinuria 41% >Thrombosis in USA 19% vs. 6% | 33 | Age >50 Leukopenia infection thrombosis (USA) Kidney failure (Japan) | 13 (9 MDS/AML) |
| Muñoz-Linares (2013) | 56 40 yr | Spain Puerta de Hierro Majadahonda Hospital | 32 (13–84) 36♂/20♀ | 12 dead Median survival 11 yr Hemoglobinuria 60% Thrombosis 35% Abdominal pain 48% 17 survivors with spontaneous remission | 67 | Thrombosis Esophageal spasms Abdominal pain | 5 (Pancreas, lung, hepatocarcinoma, and 2 lymphomas) |
AML, acute myeloid leukemia; MDS, myelodysplastic syndrome; NR, not reported; PNH, paroxysmal nocturnal hemoglobinuria.
Paroxysmal nocturnal hemoglobinuria should be considered a variable disease. Flow cytometry is an important tool for tracking it. In our experience, the behavior of clonal size is highly variable, stressing that subclinical forms could reach complete remission. On the contrary, in the classic forms, only patients who are undergoing allogeneic hemopoietic transplant reach an undetectable remission at the clonal level 32. Eculizumab did not seem to have, at least in our series, an effect on decrease in clonal size.
According to Kelly et al. 33, the size of the clone at the diagnosis has a clear prognostic implication because patients with very small clones (around 1%) tend to spontaneous remission and never evolve into classical forms. Although this fact is corroborated in our series of patients, there may be some exceptions to this rule: In our series, a young patient diagnosed with aplasia with several determinations of negative PNH cytometry, presented an emerging PNH clone a year after diagnosis with a progressive increase, starting with a severe symptomatic hemolysis, and within 2 yr requiring treatment with eculizumab.
The association with bone marrow failure is extremely common in our series of patients, preceding the time of PNH diagnosis in a large number of cases, which is an association corroborated in other series 28,31. However, aplasia/PNH chronology is not entirely clear, because in our series, and with more refined cytometry techniques, the detection of small clones with high frequency in patients with bone marrow deficit is currently possible at the time of diagnosis of the aplasia. This bone marrow deficiency persists in varying degrees throughout the clinical and clonal evolution in the patients with PNH and evolves in some cases, regardless of the treatment with eculizumab.
Thrombosis, mainly venous, but also arterial, constitutes the first cause of death in all reported Western patient series 27–31. Of emphasis in our series, is the particularly high presence of thrombosis in patients with low PNH clone, demonstrating the relevance of the PNH clone presence as an isolated prothrombotic factor 34,8. The widespread use of thrombotic prophylaxis in patients with PNH clones demonstrating great utility in other series 35 did not prevent the appearance of serious cases of thrombosis in all our patients. In this sense, the case is especially revealing of a young male with hemoglobinuria and a high PNH clone that displayed an inferior cava vein thrombosis despite anticoagulation therapy. Three months later, the thrombosis was resolved after treatment with eculizumab. This drug had a clearly favorable impact of preventing thrombosis in our series of patients with PNH, as in other series published elsewhere 36,37. Careful monitoring of LDH levels and shortening of the interval between doses of the drug are indicated in any surgical or invasive procedures in these patients. To accentuate the relationship of the thrombosis with the existence of abdominal pain and esophageal spasms which reflect an intense hemolysis corroborates the findings obtained in the Korean PNH Registry 38.
Magnetic resonance imaging is the best imaging technique to diagnose thrombosis in patients with PNH and to control evolution, especially in the cerebrovascular setting. MRI is the only imaging technique that permits to evaluate the iron overload that in some PNH cases could be underestimated, thus needing chelation therapy. In our opinion, all new patients with classic PNH must be evaluated prospectively with MRI.
The high incidence of cancer in our series is striking. The incidence of cancer described in other series is relatively low, ranging between 1% and 4% 28–31. Some of the cancers could be attributed to the therapy applied in particular patients: The two secondary lymphomas after organ transplantation could be explained by the immunosuppression employed in these procedures. This result, never reported before in PNH, merits an investigational survey of cancer incidence in patients with PNH in the PNH International Registry.
Regarding the treatment of the patients with eculizumab, the drug is presented as safe and one that improves the quality of life of our patients with complete disappearance of clinical signs of hemolysis and improvement in analytical parameters. In one case, it has allowed the patient to safely undergo a liver transplant without thrombotic recurrence after the procedure 39. The extravascular hemolysis attributable to treatment with eculizumab and described by other authors 40–42 was not a particularly relevant issue in our series: Of four patients with positive direct Coombs test after eculizumab, only one had signs of occasional extravascular hemolysis. After a splenectomy, this patient has achieved figures for normal peripheral blood cell counts without hemolysis, similar to the case described by Risitano et al. 43, as an effect attributable to eculizumab, highlighting increasing levels of ferritin, as well as the deposition of hepatic iron observed in half of the patients treated with eculizumab in our series, and which is presented as a phenomenon already reported by other groups 41–44. In two of our patients, it has motivated treatment with iron chelators.
In conclusion, we must emphasize the importance of following up patients with PNH in the long term, because it allows us to learn more about its different aspects, as well as its management. For this reason, there is a need to centralize its study at experienced centers to learn more about this disease.
Acknowledgments
The authors would like to thank Isabel Millán for statistical analysis and Inés Moreno for cytometric analysis.
Authorship and disclosures
CML, EO, and RF were the principal investigators and take primary responsibility for the paper. CML, EO, MC, and DM collected the data. RF performed the cytometric analysis. MP performed the radiology studies. IB collaborated in liver transplantation case. EO, RF, GB, and JRC were the clinical responsible for hematopoietic transplants. CM, PB, MJG, TC, JLS, and AV submitted patients for evaluation in our unit. All authors contributed to the data analysis and writing of the manuscript. EO, RF, MP, and AV have served as speakers for Alexion Pharma.
References
- Rosse WF. Epidemiology of PNH. Lancet. 1996;348:560. doi: 10.1016/S0140-6736(05)64792-7. [DOI] [PubMed] [Google Scholar]
- Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, Takahashi M, Kitani T, Kinoshita T. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–11. doi: 10.1016/0092-8674(93)90250-t. [DOI] [PubMed] [Google Scholar]
- Bessler M, Mason PJ, Hillmen P, Miyata T, Yamada N, Takeda N, Luzzatto L, Kinoshita T. Paroxysmal nocturnal hemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J. 1994;13:110–7. doi: 10.1002/j.1460-2075.1994.tb06240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawitz PM, Höchsmann B, Murakami Y, et al. A case of paroxysmal nocturnal hemoglobinuria caused by a germline mutation and a somatic mutation in PIGT. Blood. 2013;122:1312–5. doi: 10.1182/blood-2013-01-481499. [DOI] [PubMed] [Google Scholar]
- Hill A, Kelly RJ, Hillmen P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood. 2013;121:4985–96. doi: 10.1182/blood-2012-09-311381. [DOI] [PubMed] [Google Scholar]
- Luzzatto L, Bessler M, Rotoli B. Somatic mutations in paroxysmal nocturnal hemoglobinuria: a blessing in disguise? Cell. 1997;88:1–4. doi: 10.1016/s0092-8674(00)81850-4. [DOI] [PubMed] [Google Scholar]
- Young NS, Maciejewski JP, Sloand E, Chen G, Zeng W, Risitano A, Miyazato A. The relationship of aplastic anemia and PNH. Int J Hematol. 2002;76(Suppl 2):168–72. doi: 10.1007/BF03165111. [DOI] [PubMed] [Google Scholar]
- de Latour RP, Schrezenmeier H, Bacigalupo A, et al. Allogeneic stem cell transplantation in paroxysmal nocturnal hemoglobinuria. Hematologica. 2012;97:1666–73. doi: 10.3324/haematol.2012.062828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355:1233–43. doi: 10.1056/NEJMoa061648. [DOI] [PubMed] [Google Scholar]
- Ham T. Chronic hemolytic anemia with paroxysmal nocturnal hemoglobinuria. A study of the mechanism of hemolysis relation to acid-base equilibrium. N Engl J Med. 1937;217:915–7. [Google Scholar]
- Hartmann RC, Jenkins DE. The “sugar-water” test for paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1966;275:155–7. doi: 10.1056/NEJM196607212750308. [DOI] [PubMed] [Google Scholar]
- Forés R, Alcocer M, Diez-Martin JL, Fernandez MN. Flow cytometric analysis of decay-accelerating factor (CD55) on neutrophils from aplastic anaemia patients. Br J Haematol. 1995;90:728–30. doi: 10.1111/j.1365-2141.1995.tb05611.x. [DOI] [PubMed] [Google Scholar]
- Forés R, Alcocer M, Cabrera R, Sanjuán I, Briz M, Lago C, Fernández MN. Detection of PNH clones using flow cytometry in aplastic anemia and paroxysmal nocturnal hemoglobinuria. Sangre (Barc) 1999;44:199–203. [PubMed] [Google Scholar]
- Forés R, Bautista G, Steegmann JL, Javier Peñalver F, Cabrera R, Fernández MN. De novo smoldering paroxysmal nocturnal hemoglobinuria: a flow cytometric diagnosis. Haematologica. 1997;82:695–7. [PubMed] [Google Scholar]
- Brodsky RA, Mukhina GL, Li S, Nelson KL, Chiurazzi PL, Buckley JT, Borowitz MJ. Improved detection and characterization of paroxysmal nocturnal hemoglobinuria using fluorescent aerolysin. Am J Clin Pathol. 2000;114:459–66. doi: 10.1093/ajcp/114.3.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peghini PE, Fehr J. Clinical Evaluation of an aerolysin-based screening test for paroxysmal nocturnal haemoglobinuria. Cytometry B Clin Cytom. 2005;67B:13–8. doi: 10.1002/cyto.b.20059. [DOI] [PubMed] [Google Scholar]
- Sutherland DR, Kuek N, Davidson J, Barth D, Chang H, Yeo E, Bamford S, Chin-Yee I, Keeney M. Diagnosing PNH with FLAER and multiparameter flow cytometry. Cytometry B Clin Cytom. 2007;72B:167–77. doi: 10.1002/cyto.b.20151. [DOI] [PubMed] [Google Scholar]
- Borowitz MJ, Craig FE, Digiuseppe JA, Illingworth AJ, Rosse W, Sutherland DR, Wittwer CT, Richards SJ Clinical Cytometry Society. Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry B Clin Cytom. 2010;78:211–30. doi: 10.1002/cyto.b.20525. [DOI] [PubMed] [Google Scholar]
- Nakakuma H, Kawaguchi T, Horikawa K, Hidaka M, Nagakura S, Iwamoto N, Kagimoto T, Takatsuki K. Proposal for a classification of the clinical stages of paroxysmal nocturnal hemoglobinuria. Blood. 1995;86:2051–2. [PubMed] [Google Scholar]
- Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106:3699–709. doi: 10.1182/blood-2005-04-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117:5019–32. doi: 10.1182/blood-2011-01-293050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista G, Cabrera JR, Regidor C, et al. Cord blood transplants supported by co-infusion of mobilized hematopoietic stem cells from a third-party donor. Bone Marrow Transplant. 2009;43:365–73. doi: 10.1038/bmt.2008.329. [DOI] [PubMed] [Google Scholar]
- Pastrana M, Muñoz-Linares C, Ojeda E, et al. Paroxysmal Nocturnal Hemoglobinuria and magnetic resonance imaging (abstract) Blood. 2013;122:4873. [Google Scholar]
- Rodríguez Jornet A, Martín Martínez J, Roig I. Diagnostic role of magnetic resonance in the renal involvement in nocturnal paroxysmal hemoglobinuria. Nefrologia. 2000;20:375–8. [PubMed] [Google Scholar]
- del Rincon I, Yebra M, Gea JC, Diego FJ, Lacoma F. Improvement with corticotherapy of skin lesions of paroxysmal nocturnal hemoglobinuria. Presse Med. 1990;19:870. [PubMed] [Google Scholar]
- Antorrena I, Castro A, Alonso A, Oteo JF, Fores R, de Artaza M. Acute myocardial infarction in nocturnal paroxysmal hemoglobinuria. Rev Esp Cardiol. 2001;54:117–9. doi: 10.1016/s0300-8932(01)76273-2. [DOI] [PubMed] [Google Scholar]
- Dacie JV, Lewis SM. Paroxysmal nocturnal haemoglobinuria: clinical manifestations, haematology, and nature of the disease. Ser Haematol. 1972;5:3–23. [PubMed] [Google Scholar]
- Kruatrachue M, Wasi P, Na-Nakorn S. Paroxysmal nocturnal haemoglobinuria in Thailand with special reference to as association with aplastic anaemia. Br J Haematol. 1978;39:267–76. doi: 10.1111/j.1365-2141.1978.tb01097.x. [DOI] [PubMed] [Google Scholar]
- Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333:1253–8. doi: 10.1056/NEJM199511093331904. [DOI] [PubMed] [Google Scholar]
- Socié G, Mary JY, de Gramont A, Rio B, Leporrier M, Rose C, Heudier P, Rochant H, Cahn JY, Gluckman E. Paroxysmal nocturnal haemoglobinuria: long-term followup and prognostic factors. Lancet. 1996;348:573–7. doi: 10.1016/s0140-6736(95)12360-1. [DOI] [PubMed] [Google Scholar]
- Nishimura JI, Kanakura Y, Ware RE, et al. Clinical course and flow cytometric analysis of paroxysmal nocturnal hemoglobinuria in the United States and Japan. Medicine (Baltimore) 2004;83:193–207. doi: 10.1097/01.md.0000126763.68170.46. [DOI] [PubMed] [Google Scholar]
- Perez-Oteyza J, Roldan E, Brieva JA, Cancelas JA, Garcia-Larana J, Odriozola J, Hernandez-Jodra M, Navarro JL. Expression of phosphatidylinositol anchored membrane proteins in paroxysmal nocturnal hemoglobinuria after bone marrow transplantation. Bone Marrow Transplant. 1992;10:297–9. [PubMed] [Google Scholar]
- Kelly R, Hill A, Dickinson A, Cullen F, Shingles J, Cullen M, Hillmen P. Insights into the natural history of Paroxysmal Nocturnal Hemoglobinuria (PNH): analysis of the presenting clinical, haematological and flow cytometric features of 705 patients leads to improved classification and prediction of clinical course (abstract) Blood. 2013;122:3718. [Google Scholar]
- Movalia MK, Weitz IC, Lim SH, Illingworth A. Incidence of PNH clones by diagnostic code utilizing high sensitivity flow cytometry (abstract) Blood. 2011;118:1033. [Google Scholar]
- Hall C, Richards S, Hillmen P. Primary prophylaxis with warfarin prevents thrombosis in paroxysmal nocturnal hemoglobinuria (PNH) Blood. 2003;102:3587–91. doi: 10.1182/blood-2003-01-0009. [DOI] [PubMed] [Google Scholar]
- Hillmen P, Muus P, Dürhsen U, et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;110:4123–8. doi: 10.1182/blood-2007-06-095646. [DOI] [PubMed] [Google Scholar]
- Hillmen P, Muus P, Röth A, Elebute MO, Risitano AM, Schrezenmeier H. Long-term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2013;162:62–73. doi: 10.1111/bjh.12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Jang JH, Kim JS, Yoon SS, Lee JH, Kim YK, Jo DY, Chung J, Sohn SK. Clinical signs and symptoms associated with increased risk for thrombosis in patients with paroxysmal nocturnal hemoglobinuria from a Korean Registry. Int J Hematol. 2013;97:749–57. doi: 10.1007/s12185-013-1346-4. [DOI] [PubMed] [Google Scholar]
- Singer AL, Locke JE, Stewart ZA, Lonze BE, Hamilton JP, Scudiere JR, Anders RA, Rother RP, Brodsky RA, Cameron AM. Successful liver transplantation for Budd-Chiari syndrome in a patient with paroxysmal nocturnal hemoglobinuria treated with the anti-complement eculizumab. Liver Transpl. 2009;15:540–3. doi: 10.1002/lt.21714. [DOI] [PubMed] [Google Scholar]
- Hill A, Rother RP, Arnold L, Kelly R, Cullen MJ, Richards SJ, Hillmen P. Eculizumab prevents intravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria and unmasks low-level extravascular hemolysis occurring through C3 opsonization. Haematologica. 2010;95:567–73. doi: 10.3324/haematol.2009.007229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risitano AM, Notaro R, Marando L, et al. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by Eculizumab. Blood. 2009;113:4094–100. doi: 10.1182/blood-2008-11-189944. [DOI] [PubMed] [Google Scholar]
- Höchsmann B, Leichtle R, von Zabern I, Kaiser S, Flegel WA, Schrezenmeier H. Paroxysmal nocturnal haemoglobinuria treatment with eculizumab is associated with a positive direct antiglobulin test. Vox Sang. 2012;102:159–66. doi: 10.1111/j.1423-0410.2011.01530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risitano AM, Marando L, Seneca E, Rotoli B. Hemoglobin normalization after splenectomy in a paroxysmal nocturnal hemoglobinuria patient treated by eculizumab. Blood. 2008;112:449–51. doi: 10.1182/blood-2008-04-151613. [DOI] [PubMed] [Google Scholar]
- Risitano AM, Imbriaco M, Marando L, Seneca E, Soscia E, Malcovati L, Iori AP, Mano F, Notaro R, Matarazzo M. From perpetual haemosiderinuria to possible iron overload: iron redistribution in paroxysmal nocturnal haemoglobinuria patients on eculizumab by magnetic resonance imaging. Br J Haematol. 2012;158:415–8. doi: 10.1111/j.1365-2141.2012.09145.x. [DOI] [PubMed] [Google Scholar]