

Abstract
The HIV-1 Vif protein suppresses the inhibition of viral replication caused by the human antiretroviral factor APOBEC3G. As a result, HIV-1 mutants that do not express the Vif protein are replication incompetent in ‘nonpermissive' cells, such as primary T cells and the T-cell line CEM, that express APOBEC3G. In contrast, Vif-defective HIV-1 replicates effectively in ‘permissive' cell lines, such as a derivative of CEM termed CEM-SS, that do not express APOBEC3G. Here, we show that a second human protein, APOBEC3F, is also specifically packaged into HIV-1 virions and inhibits their infectivity. APOBEC3F binds the HIV-1 Vif protein specifically and Vif suppresses both the inhibition of virus infectivity caused by APOBEC3F and virion incorporation of APOBEC3F. Surprisingly, APOBEC3F and APOBEC3G are extensively coexpressed in nonpermissive human cells, including primary lymphocytes and the cell line CEM, where they form heterodimers. In contrast, both genes are quiescent in the permissive CEM derivative CEM-SS. Together, these data argue that HIV-1 Vif has evolved to suppress at least two distinct but related human antiretroviral DNA-editing enzymes.
Keywords: APOBEC3F, DNA editing, HIV-1, innate resistance, Vif
Introduction
The human immunodeficiency virus type 1 (HIV-1) Vif protein is essential for virus replication in primary lymphoid and myeloid cells, but is dispensable for efficient replication in several transformed T-cell lines as well as in nonlymphoid cell lines such as HeLa and 293T (Gabuzda et al, 1992; Sakai et al, 1993; von Schwedler et al, 1993). Cells that are unable to support the replication of Vif-defective HIV-1 (HIV-1ΔVif) have been termed ‘nonpermissive', while cells that can sustain HIV-1ΔVif replication are termed ‘permissive'. The observation that heterokaryons formed by fusion of nonpermissive and permissive cells exhibit the nonpermissive phenotype (Madani and Kabat, 1998; Simon et al, 1998a) led to the hypothesis that nonpermissive cells express an inhibitor of HIV-1 replication, lacking in permissive cells, that is blocked by the viral Vif protein.
Based on this hypothesis, Sheehy et al (2002) sought to identify a gene expressed in the nonpermissive human T-cell line CEM, which was not expressed in a permissive clone of CEM termed CEM-SS that, when expressed in CEM-SS or other permissive cells, would be sufficient to confer the nonpermissive phenotype. Sheehy et al (2002) were able to identify a single human gene product, termed CEM15 or APOBEC3G(h3G), that fully satisfied these criteria. Moreover, analysis of a small number of other nonpermissive and permissive cells demonstrated that h3G is expressed in the former but lacking in the latter (Sheehy et al, 2002; Stopak et al, 2003). Recent research has shown that h3G is specifically packaged into HIV-1 virions and then blocks productive infection by massively editing dC residues to dU on the DNA minus strand during reverse transcription (Harris et al, 2003; Mangeat et al, 2003; Mariani et al, 2003; Zhang et al, 2003). Vif in turn inhibits h3G function by binding h3G directly and sequestering h3G away from progeny virion particles and/or activating h3G degradation via the proteasome (Conticello et al, 2003; Kao et al, 2003; Marin et al, 2003; Sheehy et al, 2003; Yu et al, 2003; Mehle et al, 2004).
While h3G is clearly sufficient to confer a nonpermissive phenotype, it has been less certain that it is invariably necessary. In this context, it is interesting to note that h3G is actually encoded by one of seven related human genes, termed APOBEC3A–3G, that which are tandemly arrayed along one arm of chromosome 22 (Jarmuz et al, 2002). This array likely appeared fairly recently in vertebrate evolution as, nonprimate species, such as mice, contain only a single APOBEC3 gene at the equivalent location. In humans, APOBEC3E appears to be a pseudogene, while APOBEC3D expression has not yet been observed (Jarmuz et al, 2002). The remaining five genes, here termed h3A, h3B, h3C, h3F and h3G, can be divided into two groups based on whether they contain one or two copies of the cytidine deaminase active site also present in human APOBEC1 (hAPO1), the prototype of this family of nucleic acid-editing enzymes. Thus, h3A (at 199 aa) and h3C (at 190 aa) are similar in size to hAPO1 (229 aa), while h3B (382 aa), h3F (373 aa) and h3G (384 aa) are predicted to be considerably larger (Jarmuz et al, 2002).
Given the extensive predicted sequence identity between h3G and h3F (Supplementary Figure 1), we were intrigued by the possibility that h3F might also display an antiretroviral phenotype. Here, we report that h3F is indeed incorporated into HIV-1 virions and can inhibit their infectivity by editing reverse transcripts. The h3F protein, like h3G, binds the HIV-1 Vif protein specifically, and both HIV-1 and HIV-2 Vif are able to suppress the virion incorporation of h3F and the inhibition of HIV-1 infectivity caused by h3F. Finally, we report that h3G and h3F are extensively coexpressed in human cells in vivo, as well as in the prototypic nonpermissive T-cell line CEM, but not its permissive clonal derivative CEM-SS.
Results
Human APOBEC3F inhibits HIV-1 virion infectivity
To test whether other members of the APOBEC3 family, in addition to h3G, would reduce the infectivity of HIV-1 virions produced in their presence, we first cloned h3A, h3C, h3F and h3G, as well as hAPO1 and mouse APOBEC3 (m3) as controls, into an expression plasmid that attaches a carboxy-terminal influenza hemaggluttinin (HA) epitope tag, as previously described (Bogerd et al, 2004). Western analysis of APOBEC protein expression in transfected cells, using a monoclonal antibody specific for the tag, showed comparable levels of expression of all the six proteins (Figure 1A). All the APOBEC proteins displayed the predicted electrophoretic mobility. Each APOBEC expression plasmid was then transfected into human 293T cells, which are normally permissive for HIV-1ΔVif (Sheehy et al, 2002), along with a previously described HIV-1 proviral indicator construct termed pNL-HXB-LUCΔVif (Bogerd et al, 2004). This plasmid encodes a full-length HIV-1 provirus bearing a defective, truncated vif gene, and also contains the luciferase (luc) indicator gene substituted in place of the dispensable viral nef gene. At ∼44 h post-transfection, the virus-containing supernatant media were collected and used to infect 293T cells engineered to express CD4 and CXCR4, and also for analysis of secreted p24 Gag protein levels by ELISA. A further ∼28 h later, the infected cells were lysed and the level of virus-encoded luciferase was determined.
Figure 1.
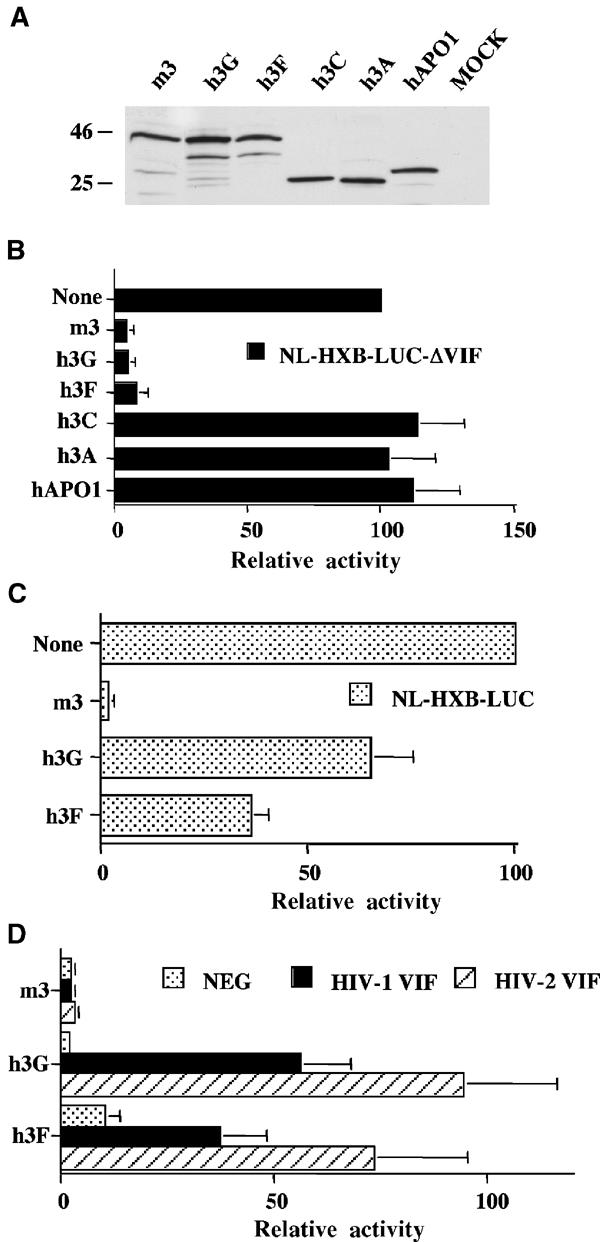
Inhibition of productive HIV-1 infection by h3F is suppressed by both HIV-1 and HIV-2 Vif. (A) Western analysis of HA epitope-tagged forms of the indicated APOBEC proteins expressed in transfected 293T cells. (B) 293T cells were transfected with pNL-HXB-LUCΔVIF (1.5 μg) and the indicated APOBEC expression plasmid (125 ng). At 44 h after transfection, supernatant media were collected and used to infect CD4+ CXCR4+ 293T cells. A further 28 h later, these cells were lysed and induced luciferase activities were quantified. The average of three independent experiments with standard deviation is indicated. Activities are given relative to the virus obtained from the culture transfected with pNL-HXB-LUCΔVIF and the parental pcDNA3 plasmid, which was set at 100. (C) Similar to panel B, except that the Vif+ pNL-HXB-LUC indicator virus was used. (D) Similar to panel B, except that the cells were additionally transfected with 250 ng of a plasmid expressing HIV-1 Vif or HIV-2 Vif, or the pgΔVif plasmid as a negative control.
As shown in Figure 1B, both h3G and m3, which have previously been shown to effectively block HIV-1ΔVif infectivity (Sheehy et al, 2002; Mariani et al, 2003), reduced production of the virus-encoded luciferase enzyme ∼20-fold. In contrast, h3A, h3C and hAPO1 all had no significant effect on the infectivity of the NL-HXB-LUCΔVIF virus. Finally, and most importantly, h3F was also observed to dramatically reduce the infectivity of this HIV-1 indicator virus. As expected, none of the APOBEC proteins had a significant effect on the level of supernatant Gag protein secreted by the transfected 293T cells (data not shown).
In parallel, we also asked whether the inhibition of viral infectivity caused by h3F would be observable in the presence of the viral vif gene product. Initially, we asked whether m3, h3G and h3F would affect the infectivity of an indicator virus, NL-HXB-LUC, that is identical to NL-HXB-LUCΔVif, except that the vif gene is intact. As shown in Figure 1C, the presence of an intact vif gene almost entirely restored the infectivity of HIV-1 virions produced in the presence of h3G, but had no effect on the minimal infectivity of virions produced in the presence of m3, as previously reported (Mariani et al, 2003). Expression of Vif from the HIV-1 provirus also largely, but not fully, rescued the infectivity of HIV-1 virions produced in the presence of h3F (Figure 1C).
As an alternative approach to the rescue of HIV-1 infectivity in the presence of m3, h3G or h3F, we also co-transfected 293T cells with the Vif-defective pNL-HXB-LUCΔVif indicator virus expression plasmid, together with an APOBEC expression plasmid and a plasmid expressing either HIV-1 Vif or HIV-2 Vif. The data obtained in these experiments (Figure 1D) again demonstrated substantial rescue by HIV-1 Vif of virion infectivity repressed by h3G or h3F, but no rescue in the presence of m3. The data obtained using the HIV-2 Vif expression plasmid proved comparable, except that a somewhat more effective rescue of viral infectivity repressed by either h3G or h3F was observed (Figure 1D).
HIV-1 and HIV-2 Vif inhibit virion incorporation of h3F
It is now well established that h3G inhibits HIV-1 infectivity by being packaged into progeny virions and then interfering with the production of a functional provirus in infected cells. HIV-1 Vif suppresses this effect by binding h3G and blocking h3G incorporation into virions (Goff, 2003). It has remained controversial whether this block is due entirely to degradation of h3G subsequent to Vif binding or whether sequestration of h3G away from budding virions also plays a significant role (Kao et al, 2003; Marin et al, 2003; Sheehy et al, 2003).
To analyze the effect of HIV-1 and HIV-2 Vif on the incorporation of APOBEC proteins into HIV-1 virions, we transfected 293T cells with expression plasmids encoding HA-tagged forms of h3G, h3F or m3, or an HA-tagged version of an irrelevant cytoplasmic protein, β arrestin 2 (βarr2) (Oakley et al, 2000). The cells were also co-transfected with the proviral expression plasmid pNL4-3ΔVifΔEnv, which bears defective forms of both the env and vif genes but is otherwise fully intact (Bogerd et al, 2004). Finally, cells were also co-transfected with a plasmid encoding HIV-2 or HIV-1 Vif, or a negative control plasmid. At ∼44 h post-transfection, released virions were collected by ultracentrifugation, as previously described (Bogerd et al, 2004), and the level of each APOBEC protein present in the virion and producer cell lysates was analyzed by Western blot (Figure 2). We also quantified p24 Gag expression by Western analysis in each virion and cell lysate (Figure 2 and data not shown).
Figure 2.

Specific packaging of h3F into HIV-1 virions is inhibited by both HIV-1 and HIV-2 Vif. 293T cells were transfected with 1.5 μg of the pNL4-3ΔVifΔEnv proviral expression plasmid, together with plasmids expressing HA-tagged forms of the indicated APOBEC proteins or the βarr2 control protein. Cells were also transfected with plasmids expressing HIV-1 or HIV-2 Vif, or a negative control plasmid (pgΔVif). At 44 h after transfection, supernatant media were harvested and released virus collected by ultracentrifugation, while the producer cells were collected and lysed. The cell and virion lysates were then subjected to gel electrophoresis, followed by Western analysis using a rabbit polyclonal antiserum specific for the HA tag or a monoclonal antibody specific for p24 Gag. While only the Gag Western performed with the disrupted virions is presented, closely similar results were also obtained using the cell lysates (data not shown). The level of expression of each APOBEC protein in the cell or virion lysates was quantified by scanning, and is presented for each panel normalized to the sample obtained in the absence of any Vif protein, which was set at 100. NA, not applicable.
Analysis of virions produced in the presence of the HA-tagged βarr2 protein, which is expressed at high levels in the cytoplasm of transfected cells (Figure 2) (Oakley et al, 2000), revealed that βarr2 is not incorporated into HIV-1 virions and that Vif expression has no effect on βarr2 expression in co-transfected cells (Figure 2). In contrast, analysis of the m3 protein (Figure 2) showed that m3 is efficiently incorporated into HIV-1 virions, as previously reported (Mariani et al, 2003). However, m3 virion incorporation, as well as the level of m3 expression in transfected cells, was also not affected by expression of either the HIV-1 or HIV-2 Vif protein in trans.
Like the m3 protein, h3G was also incorporated into HIV-1 virions at readily detectable levels. However, the level of h3G observed in virions was reduced by ∼90% in the presence of HIV-1 Vif and ∼96% in the presence of HIV-2 Vif. Analysis of h3G expression in the producer cell lysate showed a significantly less profound drop in h3G expression in the presence of HIV-1 Vif (∼58% reduction), although the effect of HIV-2 Vif on h3G expression appeared similar in both producer cells and released virions.
Finally, analysis of the effect of HIV-1 and HIV-2 Vif on h3F incorporation showed a pattern closely similar to h3G. Specifically, we observed readily detectable levels of h3F in HIV-1 virions produced in the presence of h3F but absence of Vif. This incorporation was profoundly inhibited (∼83%) upon expression of HIV-1 Vif, and became undetectable upon coexpression of HIV-2 Vif. Analysis of producer cell lysates showed a comparable, but again attenuated, effect on the level of h3F expression in this compartment. We therefore conclude that h3F, like h3G and m3, is specifically incorporated into HIV-1 virions. Like h3G, but unlike m3, this incorporation is strongly inhibited upon coexpression of either HIV-1 or HIV-2 Vif.
HIV-1 Vif binds h3F specifically
Vif is believed to prevent h3G incorporation into HIV-1 virions by binding h3G directly and then sequestering h3G from progeny virions and/or targeting h3G for degradation by the proteasome (Goff, 2003). We therefore asked whether we would be able to detect a specific interaction between h3F and the HIV-1 Vif protein by immunoprecipitation of h3F from coexpressing cells using an HA-tag-specific monoclonal antibody, followed by Western analysis of the immunoprecipitate using a rabbit polyclonal anti-Vif antiserum.
As shown in Figure 3, we were readily able to detect co-immunoprecipitation of both h3G and h3F with the HIV-1 Vif protein in lysates derived from coexpressing cells. Although the level of Vif co-immunoprecipitated with h3F was consistently somewhat lower than that seen with h3G (compare lanes 4 and 6), we note that the level of h3F expressed in the co-transfected cells, and recovered in the immunoprecipitate, was also consistently somewhat lower (Figure 3, see also Figure 1A). By quantifying the relative recovery of the HIV-1 Vif protein by co-immunoprecipitation with h3G or h3F, and controlling for the somewhat lower level of h3F expression, we calculate that the recovery of Vif protein in the presence of h3F was 0.7±0.3-fold as efficient as the recovery seen using h3G; that is, this difference, if real, is quite minor. We were unable to detect any immunoprecipitation of the HIV-1 Vif protein by the HA-monoclonal antibody in the absence of an APOBEC protein (Figure 3, lane 5) or in the presence of h3A, h3C or hAPO1 (data not shown). We therefore conclude that HIV-1 Vif interacts specifically with not only h3G but also h3F, and that this interaction is likely key to the ability of HIV-1 Vif to suppress both the inhibition of virion infectivity induced by h3F (Figure 1) and h3F virion incorporation (Figure 2).
Figure 3.
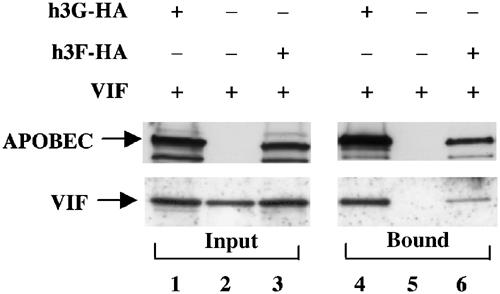
The HIV-1 Vif protein specifically binds both h3G and h3F. 293T cells were transfected with an h3G or h3F expression plasmid, together with the HIV-1 Vif expression plasmid pgVif. The parental pcDNA3 plasmid served as a negative control. After 48 h, cells were lysed and a portion subjected to immunoprecipitation using an anti-HA monoclonal antibody. In all, 10% of the input lysate (lanes 1–3) and 25% of the bound fraction (lanes 4–6) were then subjected to electrophoresis, followed by Western analysis using rabbit polyclonal antisera specific for the HA epitope tag (upper panels) or the Vif protein (lower panels).
Both h3G and h3F inhibit HIV-1 by editing retroviral reverse transcripts
As noted above, it is now well established that the h3G protein inhibits the replication of Vif-deficient HIV-1 by extensively editing dC residues to dU on the DNA minus strand during reverse transcription (Goff, 2003). This editing results in the misincorporation of A in place of G in the plus strand. Analysis of editing sites has revealed that h3G prefers to edit C residues flanked by C residues at the −1 and −2 positions (Harris et al, 2003; Beale et al, 2004).
To test whether h3F also induces editing of HIV-1 reverse transcripts, we used PCR amplification to recover proviral DNA from cells infected with Vif-deficient HIV-1 virions produced in the presence of either h3F or h3G. Sequence analysis of the plus strand of a limited sample of HIV-1 reverse transcripts produced in the presence of h3G revealed 19 independent G to A mutations out of 4232 bases sequenced, of which 619 were expected to be G residues. In addition, we noted three T to C mutations, six A to G mutations and five C to T mutations. These latter mutations could have arisen due to errors during reverse transcription or during PCR or, in the case of the C to T mutations, could be meaningful despite being in the plus strand.
Tabulation of the 19 G to A mutations observed (Table I) showed that these occurred in the context of a consensus sequence essentially identical to that previously reported for editing by h3G (Harris et al, 2003), that is, the mutated C residues were generally flanked at the −1 position by a C and were invariably flanked at the −2 position by a C or T. While this sample of mutations is too small to give rise to a reliable consensus, we did note that the +1 position was frequently an A residue while G residues appeared to be discriminated against, including at the −3 and +1 positions relative to the edited C residue.
Table 1.
Consensus sequences of h3G and h3F editing
| −3 | −2 | −1 | 0 | +1 | |
|---|---|---|---|---|---|
| h3G | |||||
| A | 47 | 0 | 16 | 0 | 74 |
| C | 21 | 72 | 74 | 100 | 5 |
| G | 0 | 0 | 5 | 0 | 0 |
| T | 32 | 28 | 5 | 0 | 21 |
| Consensus | ? | C | C | C | A |
| h3F | |||||
| A | 33 | 17 | 3 | 0 | 35 |
| C | 22 | 11 | 6 | 100 | 9 |
| G | 11 | 6 | 6 | 0 | 0 |
| T | 33 | 67 | 85 | 0 | 56 |
| Consensus |
? |
T |
T |
C |
T |
| This table presents the flanking sequences surrounding 19 distinct h3G-induced and 41 distinct h3F-induced dC to dU editing events. Mutations were detected by PCR amplification and sequencing of HIV-1 proviral sequences derived from cells infected with Vif-deficient HIV-1 virions produced in the presence of h3G or h3F. The data presented in the h3G panel, while limited, are nevertheless closely comparable to a more complete analysis presented previously by Harris et al (2003). | |||||
A more extensive analysis of the sequence of HIV-1 reverse transcripts recovered from cells infected by Vif-deficient HIV-1 virions produced in the presence of h3F revealed 41 independent G to A mutations out of 6160 bases sequenced, of which 770 were expected to be G residues. In addition, we noted six A to G mutations, two C to T mutations and two T to C mutations. Tabulation of the 41 G to A mutations revealed a consensus sequence that was distinct from the consensus editing site previously reported for h3G (Harris et al, 2003) and confirmed here (Table I). Specifically, we detected a very strong selection for a T residue at the −1 position relative to the edited C, and strong selection for a T in the −2 position. We noted a fairly weak positive selection at the −3 and +1 positions, although G residues again seemed to be selected against. These data therefore confirm that h3F, like h3G, is indeed able to edit the minus strand of HIV-1 reverse transcripts and identify a distinct consensus DNA-editing site for h3F.
h3G and h3F are widely coexpressed in vivo
Although the evidence presented thus far clearly demonstrates that h3F can inhibit HIV-1 replication, it was important to examine whether h3F is expressed in cell types that are subject to HIV-1 infection. We therefore used RT–PCR to analyze the in vivo expression pattern of h3F when compared to h3G. Owing to the extensive sequence similarity between h3F and h3G (Supplementary Figure 1), we first confirmed that the two sets of DNA primers used were able to effectively distinguish between these two related genes (Supplementary Figure 2A). Using a cDNA preparation derived from a human chronic myelogenous leukemia cell line, K562, that has previously been reported to express h3F (Jarmuz et al, 2002), we next established amplification conditions that gave rise to levels of the amplified DNA fragment that were linearly related to the level of the input cDNA preparation used (Supplementary Figure 2B).
Using these same conditions, we analyzed the relative level of h3F and h3G mRNA expression using cDNA preparations, derived from a range of human tissues, that had been previously normalized based on the expression level of four housekeeping genes (β-actin, α-tubulin, GAPDH and phospholipase A2). cDNA preparations derived from K562 cells and from pooled, Epstein–Barr virus-transformed human B cells served as positive controls. As shown in Figure 4A, we in fact observed h3F mRNA expression in a wide range of human tissues. More strikingly, we noted that the h3F gene, which is located less than 30 kb away from the h3G gene on chromosome 22 (Jarmuz et al, 2002), displays a pattern of tissue expression that is closely similar to h3G, with, for example, very low expression of both h3G and h3F mRNA in human brain and muscle, and readily detectable expression in pancreas and liver. Although we are unaware of any published description of the tissue expression pattern of h3F mRNA in vivo, these data are in good agreement with the limited expression data for h3F and h3G reported in the SOURCE database (source.stanford.edu).
Figure 4.
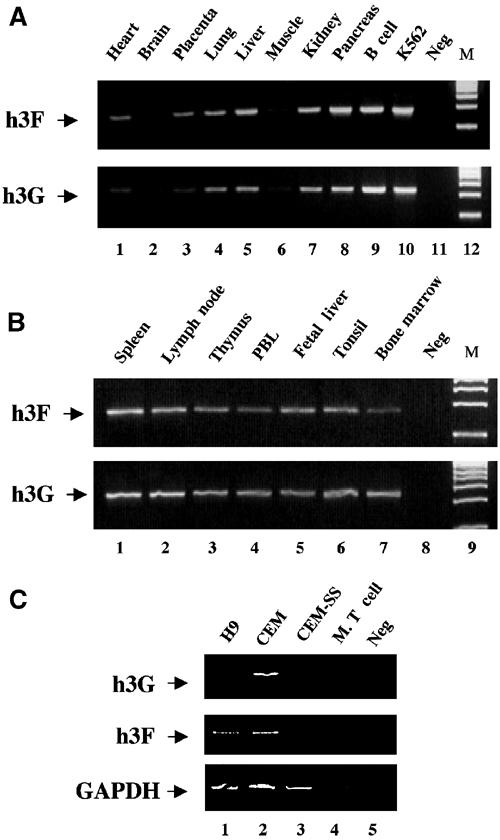
Analysis of the expression pattern of h3G and h3F mRNA by semiquantitative RT–PCR. (A) This experiment utilized a commercially obtained pre-normalized human multiple tissue cDNA panel. Positive controls represent non-normalized cDNA preparations derived from EBV-transformed B cells (lane 9) or human K562 cells (lane 10). Neg: no added cDNA. (B) Similar to panel A, except that a pre-normalized panel of human lymphoid tissue cDNAs was used. (C) This RT–PCR experiment utilized total RNA preparations derived from human H9, CEM or CEM-SS cells or mouse T cells. The GAPDH gene was used as an internal control. Neg: no added mRNA.
The critical question is, of course, not the general pattern of h3F mRNA expression in vivo, but rather whether h3F is expressed in human tissues that are susceptible to HIV-1 infection. In fact, as shown in Figure 4B, we observed readily detectable levels of both h3F and h3G mRNA expression in a wide range of human lymphoid tissues, including thymus and PBL. Therefore, these data strongly suggest that h3F is indeed expressed in cells that are normally susceptible to HIV-1 infection.
The h3G gene was originally identified based on the fact that it is expressed at readily detectable levels in the nonpermissive human T-cell line CEM, but not in a permissive clone of CEM termed CEM-SS (Sheehy et al, 2002). We therefore performed an RT–PCR analysis of the level of expression of both h3G and h3F mRNA in RNA preparations derived from CEM and CEM-SS cells, as well as from a second nonpermissive T-cell line called H9. These preparations were normalized using RT–PCR of the housekeeping gene GAPDH. As shown in Figure 4C, we observed readily detectable levels of both h3G and h3F mRNA expression in H9 and CEM cells, but little or no expression of either human gene in CEM-SS.
h3G and h3F form heteromultimers
The data reported in Figure 4 suggest that h3F and h3G are coexpressed in a wide range of human tissues. APOBEC1 has previously been reported to form homodimers and data have also been presented showing specific multimerization, presumably dimerization, of h3G in the yeast two-hybrid assay (Jarmuz et al, 2002). We were therefore intrigued by the possibility that the similar h3G and h3F proteins might also form heterodimers.
To test this idea, we tagged the h3G protein with a second, distinct epitope tag, the 20 aa N-peptide tag derived from phage P22 (Wiegand et al, 2003), and coexpressed the N-tagged h3G protein with HA-tagged forms of h3G, h3F and h3C, as described in Figure 1. After co-transfection into 293T cells, the cells were lysed and a fraction was retained for direct analysis, while the remainder was immunoprecipitated using a mouse monoclonal anti-HA antibody. These cell lysates and immunoprecipitates were then subjected to Western analysis using rabbit polyclonal antibodies specific for the HA tag (Figure 5A) or the N-peptide tag (Figure 5B). This experiment revealed that the N-h3G protein was able to effectively co-immunoprecipitate both h3G-HA and h3F-HA, but failed to interact with h3C-HA. We therefore conclude that h3G and h3F form specific heteromultimers when coexpressed in human cells.
Figure 5.
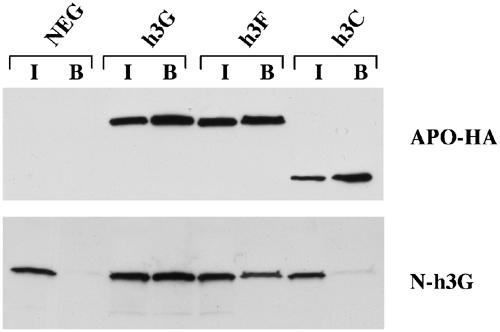
h3F and h3G form heteromultimers. This experiment was performed essentially as described in Figure 2A, except that cells were transfected with 500 ng each of N-tagged h3G and HA-tagged h3G, h3C or h3F (or pcDNA3 as a negative control). After immunoprecipitation using an HA-specific monoclonal, the recovered proteins were subjected to Western analysis using rabbit polyclonal antisera specific for the HA tag (panel A) or the N tag (panel B). Each panel shows both the input lysate (I, 10% of total) and the bound fraction (B, 25% of total).
Discussion
Analysis of heterokaryons formed between cells that are permissive and those that are nonpermissive for replication of HIV-1ΔVif has revealed the the nonpermissive phenotype is dominant, that is, that nonpermissive cells express a protein(s) that blocks replication of HIV-1 in the absence of Vif (Madani and Kabat, 1998; Simon et al, 1998a). Efforts to identify this host antiretroviral defense factor(s) led to the demonstration that h3G is selectively expressed in nonpermissive cells and, most importantly, sufficient to confer the nonpermissive phenotype when expressed in permissive cell types (Sheehy et al, 2002; Goff, 2003).
While h3G expression is therefore sufficient to confer the nonpermissive phenotype, we now present evidence that it may not invariably be necessary. In particular, we here demonstrate that the h3F gene, a second member of the human APOBEC3 family that is similar to h3G in sequence (Supplementary Figure 1), also encodes an antiretroviral activity. We show that the h3F protein, like h3G, is selectively packaged into HIV-1 virions (Figure 2) and profoundly inhibits HIV-1 virion infectivity when expressed in trans (Figure 1). Moreover, the ability of h3F to inhibit HIV-1 virion infectivity, and package into virions, is effectively blocked by expression of either the HIV-1 or HIV-2 Vif protein (Figures 1 and 2). We also demonstrate that HIV-1 Vif can specifically bind h3F in co-transfected cells, an interaction that is likely critical to the ability of HIV-1 Vif to suppress the inhibition of HIV-1 infectivity caused by h3F (Figure 3). Finally, we report that h3F, like h3G, can also edit dC residues to dU on the DNA minus strand during HIV-1 reverse transcription. The antiviral mechanisms of action of h3F are therefore very close to those previously described for h3G, except that these two editing enzymes appear to have evolved distinct consensus editing sites, with h3G preferring to edit CCC* (where the asterisk indicates the edited base) while h3F prefers to edit TTC* (Table I).
An interesting aspect of the distinct editing preferences of h3G and h3F reported in this article is that it may help to explain the pattern of hypermutation seen in some HIV-1 sequences (Vartanian et al, 1991; Fitzgibbon et al, 1993; Borman et al, 1995). While this hypermutation consists almost entirely of G to A transitions, it has been reported that the mutated sites are almost invariably G*A or G*G dinucleotides, with G*A being, if anything, more prevalent. The action of h3G can clearly explain hypermutation of G*G to AG, but it has been unclear how hypermutation of G*A to AA could occur, given the strong preference of h3G for the target sequence CC* on the DNA minus strand (Harris et al, 2003). Our observation that h3F selectively edits TC* on the DNA minus strand (Table I) may explain this phenomenon, and suggests that HIV-1 hypermutation may in fact reflect the combined action of h3G and h3F. We note that Beale et al (2004) recently hypothesized the existence of an APOBEC family member able to edit TC* sequences as a way to explain exactly this conundrum.
The different DNA target preferences of h3G and h3F reported in this article may also explain the evolutionary duplication of the original APOBEC3 gene into a small gene family in humans, and possibly other primates; however mice retain only a single APOBEC3 gene (Jarmuz et al, 2002). Alternately, it is possible that this duplication permitted the selection of APOBEC3 variants that retain the ability to interfere with retroviral reverse transcription, yet had acquired the ability to escape from inhibition by specific viral Vif orthologs. In this context, it is interesting to note that h3G is not subject to inhibition by the Vif protein encoded by the African green monkey simian immunodeficiency virus, due to a minor sequence difference with the African green monkey APOBEC3G (Bogerd et al, 2004; Schröfelbauer et al, 2004).
A striking result reported in Figure 4 is that h3G and h3F are extensively coexpressed in human tissues. Moreover, h3G and h3F are also coexpressed in the nonpermissive T-cell lines H9 and CEM, but not in CEM-SS, a permissive clonal derivative of CEM (Figure 4C). It therefore seems possible that expression of the h3G and h3F genes, which are located immediately proximal to one another on chromosome 22 (Jarmuz et al, 2002), is coordinately regulated, possibly by recruitment of transcription factors to the same adjacent enhancer element(s). Of note, the fact that h3G is expressed in nonpermissive CEM cells, but not its permissive derivative CEM-SS, together with the observation that expression of h3G in permissive cells is sufficient to confer the nonpermissive phenotype, represented the key observations reported by Sheehy et al (2002) in support of the identification of h3G as the cellular target for inhibition by HIV-1 Vif. In this article, we present essentially the same data in support of the identification of h3F as a physiological target for both HIV-1 and HIV-2 Vif, and moreover extend these results by demonstrating that h3F is incorporated in HIV-1 virions (Figure 2), that HIV-1 and HIV-2 Vif inhibit this incorporation (Figure 2), that HIV-1 Vif binds h3F specifically (Figure 3) and that h3F induces editing of HIV-1 reverse transcripts (Table I).
An interesting observation reported in Figure 5 is that h3G not only forms homodimers, as previously reported (Jarmuz et al, 2002), but also some form of heteromultimer with h3F. This suggests that the HIV-1 Vif protein has likely evolved to bind and inactivate at least three distinct but related cellular targets, that is, h3G homodimers, h3F homodimers and h3G:h3F heterodimers (Figure 2). In contrast, closely related h3G orthologs expressed in several heterologous species are unable to bind HIV-1 Vif, and are therefore refractory to HIV-1 Vif-induced degradation (Mariani et al, 2003; Bogerd et al, 2004; Schröfelbauer et al, 2004) (Figure 2). Based on these observations, we hypothesize that HIV-1 Vif has been selected to target the two proteins that pose a threat to efficient HIV-1 replication in human cells, that is, h3G and h3F, but has not been under selective pressure to target closely related antiretroviral factors found in nontarget species. We propose that efforts to understand the molecular basis for the specific interaction between Vif and its cellular protein targets should take account of this flexibility.
Materials and methods
Construction of molecular clones
The HIV-1 proviral expression plasmids pNL-HXB-LUC, pNL-HXB-LUC-ΔVIF and pNL4-3ΔVifΔEnv have been previously described (Bogerd et al, 2004). The HIV-1 Vif expression plasmid pgVif, the control plasmid pgΔVif and the h3G expression plasmid pcDNA3-h3G/HA have also been described (Simon et al, 1998b; Bogerd et al, 2004). A plasmid expressing an HA-tagged form of βarr2 was obtained from Dr R Lefkowitz (Oakley et al, 2000). The h3A cDNA was PCR amplified from a plasmid obtained from Dr P Madsen (Madsen et al, 1999). h3C and hAPO1 were both PCR amplified from a directional HeLa cDNA library. h3F was PCR amplified from a cDNA preparation derived from the human cell line K562. A mouse APOBEC3 cDNA was PCR amplified from a murine spleen cDNA preparation (BD-Clontech). All PCR products (KpnI/EcoRI or KpnI/MfeI fragments) were subcloned in frame into ph3G-HA in place of the h3G sequence (Bogerd et al, 2004). The N-h3G construct was created by PCR amplification of h3G (EcoRI/XhoI), followed by ligation into pCMV/N (Wiegand et al, 2003). pgVif2 was created by replacing the XbaI/SalI fragment from pgVIF, containing the entire HIV-1 vif gene, with a similar PCR-generated HIV-2 vif gene fragment derived from the ROD HIV-2 proviral clone (Guyader et al, 1987). All constructs were fully verified by DNA sequence analysis.
Western blot analysis
Cell lysates, virion lysates and immunoprecipitates were subjected to gel electrophoresis and then transferred to a nitrocellulose membrane. The membranes were probed with a mouse monoclonal antibody specific for the HIV-1 capsid protein (Chesebro et al, 1992) or rabbit polyclonal antisera specific for the HA epitope tag (Covance), HIV-1 Vif (Goncalves et al, 1996) or the N-peptide tag (Wiegand et al, 2003). Reactive proteins were detected using the Lumi-Light Western Blotting Substrate (Roche), as previously described (Bogerd et al, 2004).
Cell culture and analysis
293T cells were cultured in 5% fetal bovine serum in DMEM and transfected using the calcium phosphate method. HIV-1 infectivity assays were performed as previously described (Bogerd et al, 2004) using supernatant media from 293T cells transfected with pNL-HXB-LUCΔVIF or pNL-HXB-LUC, together with an APOBEC expression plasmid and/or a Vif expression plasmid.
Experiments analyzing virion packaging of APOBEC proteins were performed as previously described (Bogerd et al, 2004). Briefly, 293T cells were transfected using pNL4-3ΔVifΔEnv, together with an APOBEC and/or a Vif expression plasmid. At 44 h post-transfection, virus-containing supernatants were collected and 9 ml was layered on a 2 ml 20% w/v sucrose cushion (in PBS). The virus was pelleted by centrifugation at 35 000 rpm for 2 h at 4°C in an SW41 rotor. Pellets were resuspended and normalized to HIV-1 p24 values determined from the original supernatant, and then the levels of p24, h3G, m3, h3B and h3F were analyzed by Western, as described above.
Editing of HIV-1 proviral DNA by h3G and h3F
VSV-G pseudotyped ΔVif virus was produced by co-transfecting 293T cells with pNL-Luc-HXBΔVif, with inhibiting amounts of either h3G or h3F as described in Figure 1A. The supernatant media were passed through a 0.45 μm pore size filter and applied to fresh 293T cells and allowed to infect for 4 h. The cells were then washed and fresh media added, and infection was allowed to proceed for an additional 12 h. At this point, the cells were harvested and total DNA isolated using the DNeasy tissue kit (Qiagen). Isolated DNA was digested with DpnI (New England Biolabs) to remove any contaminating plasmid DNA. Regions of the envelope gene containing DpnI sites were amplified using TAQ Plus® precision polymerase (Stratagene), using primers with introduced EcoRI and XbaI restriction sites. Amplified DNA fragments were then digested with these enzymes, cloned into pGEM(3+) (Promega) and sequenced.
mRNA expression analysis
Analysis of the tissue expression pattern of h3G and h3F mRNA (Figure 4A and B) was performed by PCR analysis using pre-normalized human multiple tissue cDNA panels (BD/Clontech). Reactions were set up largely according to the manufacturer's instructions. Briefly, gene-specific primers for h3G or h3F (see Supplementary Figure 2C) were used to amplify a small fragment of either gene. In all, 35 cycles of PCR were performed at the following temperatures and for the indicated lengths of time: 98°C, 45 s; 51°C, 45 s; 72°C, 45 s.
After isolation of total RNA from H9, CEM, CEM-SS, K562 or mouse T cells, 10 μg was subjected to reverse transcription using the StrataScript First Strand Synthesis System (Stratagene) and the included oligo(dT) primers, according to the manufacturer's instructions. Prior to the reverse transcription step, all RNA preparations were treated with RQ1 RNase-free DNase (Promega) to ensure removal of any genomic DNA contamination. PCR reactions were performed as described above for the h3G and h3F mRNA. Linear amplification of the GAPDH mRNA internal control required a reduction to 30 cycles of PCR, and used standard primers obtained from BD/Clontech.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Acknowledgments
We thank Michael Malim, Robert Lefkowitz and Peter Madsen for reagents used in this research. The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: rabbit polyclonal anti-Vif antiserum from Dr Dana Gabuzda, and a mouse monoclonal antibody specific for the HIV-1 p24 protein from Dr Bruce Chesebro and Dr Hardy Chen. This work was supported by the Howard Hughes Medical Institute and by grant 1R01 AI057099 from the National Institute of Allergy and Infectious Diseases.
References
- Beale RCL, Petersen-Mahrt SK, Watt IN, Harris RS, Rada C, Neuberger MS (2004) Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J Mol Biol 337: 585–596 [DOI] [PubMed] [Google Scholar]
- Bogerd HP, Doehle BP, Wiegand HL, Cullen BR (2004) A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV-1 Vif. Proc Natl Acad Sci USA 101: 3770–3774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borman AM, Quillent C, Charneau P, Kean KM, Clavel F (1995) A highly defective HIV-1 group O provirus: evidence for the role of local sequence determinants in G → A hypermutation during negative-strand viral DNA synthesis. Virology 208: 601–609 [DOI] [PubMed] [Google Scholar]
- Chesebro B, Wehrly K, Nishio J, Perryman S (1992) Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J Virol 66: 6547–6554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conticello SG, Harris RS, Neuberger MS (2003) The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol 13: 2009–2013 [DOI] [PubMed] [Google Scholar]
- Fitzgibbon JE, Mazar S, Dubin DT (1993) A new type of G → A hypermutation affecting human immunodeficiency virus. AIDS Res Hum Retroviruses 9: 833–839 [DOI] [PubMed] [Google Scholar]
- Gabuzda DH, Lawrence K, Langhoff E, Terwilliger E, Dorfman T, Haseltine WA, Sodroski J (1992) Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J Virol 66: 6489–6495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff SP (2003) Death by deamination: a novel host restriction system for HIV-1. Cell 114: 281–283 [DOI] [PubMed] [Google Scholar]
- Goncalves J, Korin Y, Zack J, Gabuzda D (1996) Role of Vif in human immunodeficiency virus type 1 reverse transcription. J Virol 70: 8701–8709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyader M, Emerman M, Sonigo P, Clavel F, Montagnier L, Alizon M (1987) Genome organization and transactivation of the human immunodeficiency virus type 2. Nature 326: 662–669 [DOI] [PubMed] [Google Scholar]
- Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH (2003) DNA deamination mediates innate immunity to retroviral infection. Cell 113: 803–809 [DOI] [PubMed] [Google Scholar]
- Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, Navaratnam N (2002) An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 79: 285–296 [DOI] [PubMed] [Google Scholar]
- Kao S, Kahn MA, Miyagi E, Plishka R, Buckler-White A, Strebel K (2003) The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J Virol 77: 11398–11407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani N, Kabat D (1998) An endogenous inhibitor of human immunodeficiency virus in human lymphocytes is overcome by the viral Vif protein. J Virol 72: 10251–10255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen P, Anant S, Rasmussen HH, Gromov P, Vorum H, Dumanski JP, Tommerup N, Collins JE, Wright CL, Dunham I, MacGinnitie AJ, Davidson NO, Celis JE (1999) Psoriasis upregulated phorbolin-1 shares structural but not functional similarity to the mRNA-editing protein apobec-1. J Invest Dermatol 113: 162–169 [DOI] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D (2003) Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424: 99–103 [DOI] [PubMed] [Google Scholar]
- Mariani R, Chen D, Schröfelbauer B, Navarro F, König R, Bollman B, Münk C, Nymark-McMahon H, Landau NR (2003) Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114: 21–31 [DOI] [PubMed] [Google Scholar]
- Marin M, Rose KM, Kozak SL, Kabat D (2003) HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med 9: 1398–1403 [DOI] [PubMed] [Google Scholar]
- Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D (2004) Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem 279: 7792–7798 [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS (2000) Differential affinities of visual arrestin, βarrestin1, and βarrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem 275: 17201–17210 [DOI] [PubMed] [Google Scholar]
- Sakai H, Shibata R, Sakuragi J, Sakuragi S, Kawamura M, Adachi A (1993) Cell-dependent requirement of human immunodeficiency virus type 1 Vif protein for maturation of virus particles. J Virol 67: 1663–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröfelbauer B, Chen D, Landau NR (2004) A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc Natl Acad Sci USA 101: 3927–3932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH (2002) Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418: 646–650 [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Malim MH (2003) The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med 9: 1404–1407 [DOI] [PubMed] [Google Scholar]
- Simon JHM, Gaddis NC, Fouchier RAM, Malim MH (1998a) Evidence for a newly discovered cellular anti-HIV-1 phenotype. Nat Med 4: 1397–1400 [DOI] [PubMed] [Google Scholar]
- Simon JHM, Miller DL, Fouchier RAM, Soares MA, Peden KWC, Malim MH (1998b) The regulation of primate immunodeficiency virus infectivity by Vif is cell species restricted: a role for Vif in determining virus host range and cross-species transmission. EMBO J 17: 1259–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopak K, de Noronha C, Yonemoto W, Greene WC (2003) HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell 12: 591–601 [DOI] [PubMed] [Google Scholar]
- Vartanian JP, Meyerhans A, Asjo B, Wain-Hobson S (1991) Selection, recombination, and G–A hypermutation of human immunodeficiency virus type 1 genomes. J Virol 65: 1779–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Schwedler U, Song J, Aiken C, Trono D (1993) vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J Virol 67: 4945–4955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand HL, Lu S, Cullen BR (2003) Exon junction complexes mediate the enhancing effect of splicing on mRNA expression. Proc Natl Acad Sci USA 100: 11327–11332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu X-F (2003) Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302: 1056–1060 [DOI] [PubMed] [Google Scholar]
- Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L (2003) The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424: 94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2