

Abstract
Objectives
To identify switch modalities used when initiating second- or third-line anagrelide for essential thrombocythemia (ET), assess whether anagrelide is initiated consistently with Summary of Product Characteristics (SPC) recommendations, and determine whether different observed switch regimens have any relationship with maintenance, platelet response, or tolerability.
Methods
This observational study was conducted across 43 centers in France. High-risk patients (>60 yr of age and/or history of thrombosis and/or platelet count >1000 × 109/L) with ET starting second- or third-line anagrelide therapy were identified and monitored for 6 months.
Results
A total of 177 patients were enrolled. The SPC-recommended starting dose (1 mg/d) was used in 52.6% of patients; 0.5 mg/d was used in 41.1%. 77.1% of patients underwent an anagrelide dose increase during the study. At 6-month follow-up, 84.7% of patients (n = 144/170) were still receiving anagrelide; 70.6% (n = 120/170) achieved a platelet response. A higher proportion of patients who discontinued previous cytoreductive therapy (CRT) after initiating anagrelide achieved a platelet response (n = 34/39, 87.2%) vs. patients who discontinued their previous CRT before anagrelide initiation (n = 77/115, 67.0%). Platelet response rates were higher in patients whose anagrelide initiation was consistent (n = 100/133, 75.2%) vs. inconsistent (n = 20/37, 54.1%) with the SPC. The incidence of adverse drug reactions was lower in patients whose anagrelide treatment was consistent (n = 52/133, 39.1%) vs. inconsistent (n = 25/37, 67.6%) with the SPC.
Conclusions
To our knowledge, the FOX study provides the first comprehensive real-world data on the modalities used when switching from previous CRT to anagrelide. Highest platelet responses were observed when previous CRT was discontinued after anagrelide initiation or when anagrelide was initiated consistently with the SPC. Safety data corresponded with the SPC.
Keywords: myeloproliferative disorders, essential thrombocythemia, anagrelide, hydroxycarbamide, switch, resistance, intolerance, blood platelets, platelet count
The World Health Organization classifies essential thrombocythemia (ET) as a chronic myeloproliferative neoplasm characterized by an elevated platelet count and an increased risk of developing thrombohemorrhagic complications 1. The prevalence of ET is approximately 30/100 000 in Western populations 2.
The primary objective of ET treatment is to decrease the incidence of thrombohemorrhagic events 3. According to European LeukemiaNet (ELN) guidelines, cytoreductive therapy (CRT) is indicated in patients with ET who are at high risk of developing thrombohemorrhagic events (>60 yr of age and/or history of thrombosis and/or platelet count >1500 × 109/L) 4.
There are two licensed treatments available for the management of high-risk patients with ET in Europe. Hydroxycarbamide (HC) is licensed for first-line therapy 4 and, as of 2004, anagrelide is licensed by the European Medicines Agency for patients with ET who are intolerant or refractory to their current CRT 5.
The ELN guidelines recommend HC as first-line treatment and suggest that physicians consider non-leukemogenic drugs such as anagrelide or interferon (IFN; off label) in high-risk patients who are intolerant or resistant to therapy with HC 4. Anagrelide is the preferred choice, with IFN being reserved for young females or those with anagrelide contraindications 4. Results from previous studies have demonstrated that anagrelide is effective at reducing platelet counts by producing response rates of between 77% and 93% 6–9. In addition, anagrelide has been shown to reduce symptoms associated with ET as well as major and minor thromboembolic complications 7,9,10.
The anagrelide Summary of Product Characteristics (SPC) defines a lower platelet count than the ELN guidelines as high risk (>1000 × 109/L vs. >1500 × 109/L) 4,5. The SPC recommends starting treatment at 1 mg/d, in two divided doses (0.5 mg/dose), and maintaining this dose for ≥1 wk 5. The dose may then be titrated to a patient-specific optimal dosage required to reduce and/or maintain a platelet count <600 × 109/L and ideally between 150 × 109/L and 400 × 109/L. The dose increment must not exceed 0.5 mg/d per week, and 2.5 mg is the maximum single dose. Starting doses >1 mg/d may be used, but platelet counts must be monitored regularly until a stable maintenance dose is achieved 5. However, neither the SPC nor the ELN provide any guidance on how to transition from another CRT to anagrelide. Therefore, it would be useful to identify the optimal modality for transitioning patients onto anagrelide from their current CRT in order to achieve prolonged maintenance, efficacy, and tolerability.
The aim of the France Observatoire Xagrid® (FOX) observational study was to identify the switch modalities used when initiating anagrelide and to describe any possible relationship with platelet response, tolerability, and maintenance of anagrelide at 6 months.
Methods
Study design and patients
The FOX study (SPD422-702; ClinicalTrials.gov identifier: NCT01192347) was a non-interventional (observational) study conducted in France to investigate the switch modalities used when initiating anagrelide in adult patients with ET. The study included two periods: a case screening period in which eligible patients who had initiated anagrelide treatment in the previous month or were about to start anagrelide treatment (those patients with a documented physician decision to prescribe) were identified and enrolled in the study, and a prospective follow-up period that was 5–6 months (depending on whether a patient initiated anagrelide in the previous month) in length to allow each patient to be observed for their first 6 months of anagrelide treatment. Before the study started, approvals were obtained from le Comité Consultatif sur le Traitement de l’Information en matière de Recherche dans le domaine de la Santé (CCTIRS) and la Commission Nationale de l’Information et des Libertés (CNIL).
Eligible patients were aged ≥18 yr with a diagnosis of ET uncontrolled by first-line (or previous) CRT (for efficacy or tolerability reasons) and categorized as high risk according to the SPC. Patients must have been on second- or further-line anagrelide treatment for ≤1 month or had a decision documented to commence second- or further-line anagrelide treatment. Patients were excluded from the study if they were participating in another clinical trial where their treatment was defined by that study protocol. Written informed consent was obtained for each of the participants before they entered the study.
Anagrelide therapy
The decision to prescribe anagrelide or continue with an existing treatment was made at the investigator’s discretion and was not influenced by the study measures. No treatment or medication was contraindicated during the study; however, patients who were intended to be treated with prolonged combinations of CRT at anagrelide initiation could not be included in the study. Dosing and timing of the doses was determined by the treating physician. Patients were categorized at the end of the study according to the treatment protocol they had received.
Study objectives
The objectives of this study were to observe how different treatment regimens for initiation of anagrelide therapy affect continuation with treatment in the first 6 months and to investigate whether the different treatment regimens have a relationship with platelet response (full response, platelet count of ≤400 × 109/L; partial response, platelet count between 400 and 600 × 109/L or a platelet count reduction of at least 200 × 109/L), patient characteristics, or adverse drug reactions (ADRs).
In addition, the study aimed to collect information on the incidence of various starting doses; the titration increments used in clinical practice when initiating anagrelide; the various down titration and withdrawal strategies used to stop previous ET therapy during initiation of anagrelide; and hematological parameters (platelet counts, white blood cell counts, neutrophils, red blood cell counts, hematocrit, and hemoglobin).
Sample size and statistical methods
Sixty centers were required to enroll approximately 180 patients so that full analyses could be carried out on at least 160 patients. Statistical analyses were carried out using SAS® Version 9.1.3 (SAS Institute, Cary, NC, USA).
Results
Patients
Between September 2010 and April 2012, 177 patients were enrolled into the study across 43 centers in France. Of these, 175 patients received one or more doses of anagrelide and had one or more postbaseline safety assessment (NB: two patients did not meet the criteria and were not included in the safety set). The full analysis set (FAS) comprised the 170 patients who received previous CRT prior to initiating anagrelide (the other five patients received first-line anagrelide).
Patients who had been treated with a previous CRT were divided into defined subgroups based upon the treatment regimens used when anagrelide treatment was initiated. These are described in Fig.1.
Figure 1.
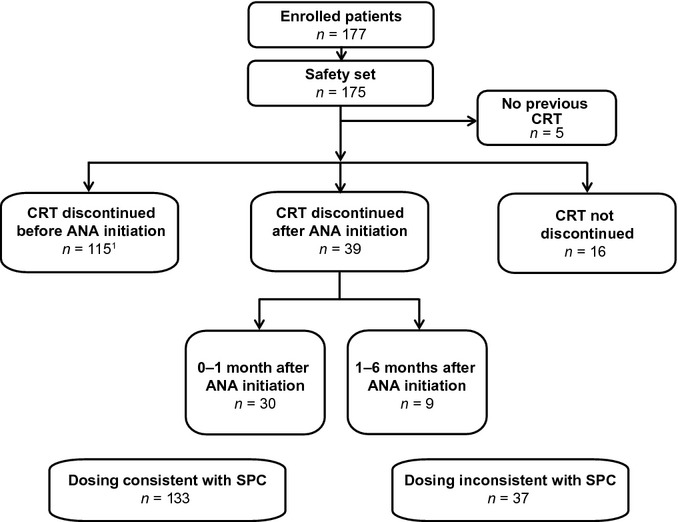
Patient disposition. ANA, anagrelide; CRT, cytoreductive therapy; SPC, Summary of Product Characteristics. 0–1 month: CRT was discontinued within the first 30 d of anagrelide treatment (median 10 d). 1–6 months: CRT was continued for ≥30 d (median 65 d), but was discontinued before anagrelide was discontinued or the patient completed the study period. Not discontinued: all other cases (i.e., patients received both their current CRT and anagrelide throughout the 6-month follow-up period). 1Includes two patients who restarted CRT after anagrelide initiation that was discontinued again in the 6-month follow-up period.
Patients were also categorized based upon whether anagrelide treatment was initiated consistently or inconsistently with the SPC dosing recommendations—consistent dosing: anagrelide starting dose was ≤1 mg/d, any increase in dose was ≤0.5 mg/d, any increase in dose was made ≥7 d after first initiation or ≥7 d after any previous modification (up or down), and the maximum dose did not exceed 10 mg/d at any stage; inconsistent dosing: any deviations from the above anagrelide starting dose and titration schedule were considered to be inconsistent with the SPC. Although the SPC recommends an anagrelide starting dose of 1 mg/d, this study also considered patients who received a starting dose of 0.5 mg/d to have initiated anagrelide consistently with the SPC dosing recommendations.
The disposition of patients across the main and switch subgroups is displayed in Fig.1. The majority of patients discontinued their previous CRT prior to initiating anagrelide (n = 115/175, 65.7%), and most patients’ treatment with anagrelide was consistent with the SPC dosing recommendations (n = 133/175, 76.0%). Although patients whose previous CRT was not planned to be discontinued were ineligible for the study, 16 patients (9.1%) did not discontinue their previous CRT at any time point throughout the 6-month follow-up period. Patient baseline and demographic characteristics are displayed in Table1. Of note, one patient received anagrelide as fourth-line therapy. Some slight differences were observed between the main subgroups (data not shown), but as none were thought to influence the efficacy and safety results, these were not presented.
Table 1.
Patient baseline and demographic characteristics: safety set
| Characteristic | n = 175 | |||
|---|---|---|---|---|
| Age, years | ||||
| Median (range) | 70.0 (23.0–89.0) | |||
| Classes of age, n (%) | ||||
| ≤60 yr | 42 (24.0) | |||
| >60 yr | 133 (76.0) | |||
| Gender, n (%) | ||||
| Male | 67 (38.3) | |||
| Female | 108 (61.7) | |||
| Duration since diagnosis, months | ||||
| Median (range) | 58.8 (0.0–366.8) | |||
| Baseline platelet count (109/L) | ||||
| Median (range) | 553.0 (179.0–1549.0) | |||
| Reasons for anagrelide initiation, n (%) | ||||
| Lack of efficacy of previous CRT | 71 (40.6) | |||
| Intolerance to previous CRT | 114 (65.1) | |||
| Other | 31 (17.7) | |||
| Symptoms of disease at inclusion,2 n (%) | 49 (28.0) | |||
| History of thrombohemorrhagic events, n (%) | ||||
| Arterial thrombosis | 30 (17.6) | |||
| Venous thrombosis | 29 (17.1) | |||
| Hemorrhage | 13 (7.6) | |||
| Main cardiovascular risk factors, n (%) | ||||
| Hypertension | 75 (42.9) | |||
| Hypercholesterolemia | 46 (26.3) | |||
| Overweight | 32 (18.3) | |||
| Other | 108 (61.7) | |||
| JAK2 V617F positive,3 n (%) | 77 (44.0) | |||
| Cardiovascular check-up performed, n (%) | ||||
| Before anagrelide initiation | 78 (44.6) | |||
| Since anagrelide initiation | 31 (17.7) | |||
| Prior cytoreductive treatments, n (%) | ||||
| Any treatment | 170 (97.1) | |||
| First line | Second line | Third line | ||
| Hydroxycarbamide | 158 (92.9) | 5 (2.9) | 0 | |
| Pipobroman | 9 (5.3) | 19 (11.2) | 0 | |
| Peginterferon alfa-2a | 2 (1.2) | 3 (1.8) | 1 (0.6) | |
| Busulfan | 1 (0.6) | 0 | 0 | |
| First line | Second line | Third line | Fourth line | |
| Anagrelide initiation | 5 (2.9) | 143 (81.1) | 26 (14.9) | 1 (0.6) |
CRT, cytoreductive therapy.
Symptoms of disease at inclusion included hemorrhagic or ischemic manifestations, erythromelalgia, fatigue, paresthesia, headache, or other symptoms considered related by the investigator.
JAK2 V617F testing was not undertaken in 20 patients.
Anagrelide therapy (safety set)
Lack of efficacy (n = 71, 40.6%) and intolerance (n = 114, 65.1%) to patients’ previous CRT were the most frequently reported reasons for anagrelide initiation. Approximately half of patients (n = 92, 52.6%) commenced anagrelide treatment at the 1 mg/d starting dose recommended in the SPC 5 and 41.1% started at 0.5 mg/d (Figure S1). A greater proportion of patients who discontinued their previous CRT after anagrelide initiation (i.e., had a period of combination therapy) received an anagrelide starting dose of 0.5 mg/d (n = 22/39, 56.4%) compared with those who discontinued their previous CRT prior to anagrelide initiation (n = 42/115, 36.5%). All nine patients who received an anagrelide starting dose of 1.5 mg/d had stopped their previous CRT prior to commencing anagrelide treatment. None of these patients withdrew from the study, and only two experienced an ADR, with one leading to a dose reduction. The median maximum daily dose and the median last daily dose of anagrelide were 1.5 mg/d in the total population, with a slightly higher dose of 2.0 mg/d in patients who underwent prolonged transition and in patients whose anagrelide dosing schedule was inconsistent with the SPC (Table2).
Table 2.
Anagrelide dosing and modification: safety set
| ANA therapy | Subgroup totals | Total (n = 175) | ||||||
|---|---|---|---|---|---|---|---|---|
| Discontinuation of previous CRT2 | CRT discontinued after ANA | Dosing with SPC recommendations2 | ||||||
| Before ANA (n = 115) | After ANA (n = 39) | Not discontinued (n = 16) | 0–1 month (n = 30) | 1–6 months (n = 9) | Consistent (n = 133) | Inconsistent (n = 37) | ||
| Starting daily dose (mg/d) Median (range) | 1.0 (0.3–1.5) | 0.5 (0.5–1.3) | 1.0 (0.5–1.0) | 0.5 (0.5–1.3) | 0.5 (0.5–1.0) | 1.0 (0.5–1.0) | 1.0 (0.3–1.5) | 1.0 (0.3–1.5) |
| Maximum daily dose (mg/d) Median (range) | 1.5 (0.5–4.0) | 1.5 (0.5–3.0) | 1.3 (1.0–2.5) | 1.5 (0.5–3.0) | 2.0 (1.0–3.0) | 1.5 (0.5–4.0) | 2.0 (1.0–3.5) | 1.5 (0.5–4.0) |
| Last daily dose (mg/d) Median (range) | 1.5 (0.3–4.0) | 1.5 (0.5–3.0) | 1.0 (1.0–2.0) | 1.4 (0.5–3.0) | 1.5 (0.8–3.0) | 1.0 (0.3–4.0) | 2.0 (0.5–3.5) | 1.5 (0.3–4.0) |
| Dose modifications, n (%) | ||||||||
| Total dose increases | 83 (72.2) | 35 (89.7) | 13 (81.3) | 27 (90.0) | 8 (88.9) | 98 (73.7) | 33 (89.2) | 135 (77.1) |
| Reasons | ||||||||
| ≥1 platelet objective not reached | 64 (77.1) | 24 (68.6) | 8 (61.5) | 19 (70.4) | 5 (62.5) | 71 (72.4) | 25 (75.8) | 99 (73.3)3 |
| ≥1 preplanned dose titration | 30 (36.1) | 21 (41.4) | 6 (41.2) | 17 (63.0) | 4 (50.0) | 43 (43.9) | 14 (42.4) | 58 (43.0)3 |
| Total dose decreases | 24 (20.9) | 19 (48.7) | 5 (31.3) | 15 (50.0) | 4 (44.4) | 35 (26.3) | 13 (35.1) | 50 (28.6) |
| Reasons | ||||||||
| ≥1 ADR | 10 (41.7) | 6 (31.6) | 4 (80.0) | 6 (40.0) | 0 | 13 (37.1) | 7 (53.8) | 21 (42.0)3 |
| ≥1 other reason | 9 (37.5) | 8 (42.1) | 0 | 6 (40.0) | 2 (50.0) | 13 (37.1) | 4 (30.8) | 18 (36.0)3 |
| ≥1 platelet objective not reached | 4 (16.7) | 2 (10.5) | 1 (20.0) | 1 (6.7) | 1 (25.0) | 4 (11.4) | 3 (20.1) | 7 (14.0)3 |
| ≥1 preplanned dose titration | 5 (20.8) | 3 (15.8) | 0 | 2 (13.3) | 1 (25.0) | 6 (17.1) | 2 (15.4) | 8 (16.0)3 |
| Total dose interruptions | 10 (8.7) | 5 (12.8) | 1 (6.3) | 3 (10.0) | 2 (22.2) | 11 (8.3) | 5 (13.5) | 16 (9.1) |
| Reasons | ||||||||
| ≥1 ADR | 6 (60.0) | 3 (60.0) | 0 | 2 (66.7) | 1 (50.0) | 6 (54.5) | 3 (60.0) | 9 (56.3)3 |
| ≥1 other reason | 4 (40.0) | 2 (40.0) | 1 (100.0) | 1 (33.3) | 1 (50.0) | 5 (45.5) | 2 (40.0) | 7 (43.8)3 |
| Total anagrelide discontinuations | 20 (17.4) | 5 (12.8) | 3 (18.8) | 4 (13.3) | 1 (11.1) | 24 (18.0) | 4 (10.8) | 30 (17.1) |
| Reasons | ||||||||
| ADR | 16 (80.0) | 4 (80.0) | 2 (66.7) | 4 (100.0) | 0 | 19 (79.2) | 3 (75.0) | 24 (80.0)3 |
| Death | 2 (10.0) | 1 (20.0) | 0 | 0 | 1 (100.0) | 2 (8.3) | 1 (25.0) | 3 (10.0)3 |
| Disease progression | 1 (5.0) | 0 | 0 | 0 | 0 | 1 (4.2) | 0 | 1 (3.3)3 |
| Intercurrent disease | 1 (5.0) | 0 | 0 | 0 | 0 | 1 (4.2) | 0 | 1 (3.3)3 |
| Lack of efficacy | 0 | 0 | 1 (33.3) | 0 | 0 | 1 (4.2) | 0 | 1 (3.3)3 |
| No modification since starting dose | 21 (18.3) | 2 (5.1) | 3 (18.8) | 2 (6.7) | 0 | 22 (16.5) | 4 (10.8) | 26 (14.9) |
ADR, adverse drug reaction; ANA, anagrelide; CRT, cytoreductive therapy; SPC, Summary of Product Characteristics.
Data not shown for five additional patients included in the study who had no prior cytoreductive therapy and therefore received anagrelide as a first-line treatment.
Percentage calculated from the total of each dose modification.
In total, 135 patients (77.1%) underwent an anagrelide dose increase at some point during the 6-month follow-up period; 99/135 patients (73.3%) because they did not achieve their platelet target and 58/135 patients (43.0%) had one or more preplanned dose increase. Nearly all patients who received an anagrelide starting dose of 0.5 mg/d underwent a dose increase (n = 67/72, 93.1%) compared with two-thirds of those who received an anagrelide starting dose of 1 mg/d (n = 61/92, 66.3%). Overall, 50 patients (28.6%) underwent a dose reduction and 16 patients (9.1%) underwent a dose interruption during the 6-month follow-up period, with an ADR the most frequent reason (Table2). ADRs were also the most frequent reason why anagrelide was discontinued in 24/30 patients (80.0%) during the 6-month follow-up period. Twenty-six patients (14.9%) did not undergo any dose modification during the 6-month follow-up period (Table2).
Continuation of anagrelide therapy (full analysis set)
A total of 144 patients (84.7%) were still receiving anagrelide therapy at the end of the 6-month follow-up period, with >80% in each main subgroup (Fig.2). A greater proportion of patients who discontinued their previous CRT after anagrelide initiation were still receiving anagrelide therapy at the end of the 6-month follow-up period (n =37/39, 94.9%) compared with patients who discontinued their previous CRT prior to anagrelide initiation (n = 94/115, 81.7%). Notably, 100% of patients (n = 9) who underwent prolonged transition were still receiving anagrelide therapy at the end of the 6-month follow-up period.
Figure 2.
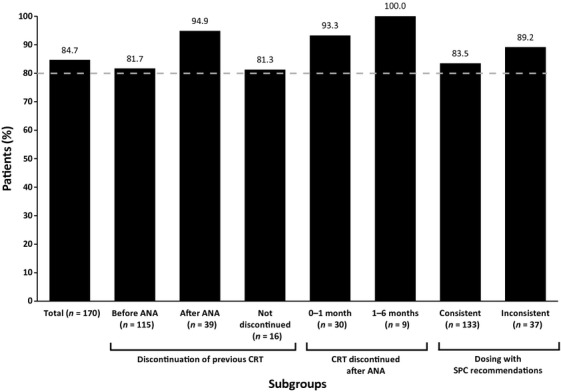
Proportion of patients continuing anagrelide treatment at 6 months: full analysis set. ANA, anagrelide; CRT, cytoreductive therapy; SPC, Summary of Product Characteristics.
Platelet response (full analysis set)
In the study population, the median platelet count recorded at diagnosis and baseline was 816 × 109/L (range 413–1862) and 553 × 109/L (range 179–1549), respectively. The last median platelet count, calculated from the last value recorded during the 6-month follow-up, was 411.5 × 109/L (range 167–1265) (Table3).
Table 3.
Change from baseline in platelet count, red blood cells, leukocytes, neutrophils, hemoglobin, and hematocrit: safety set
| Baseline | Last value during follow-up | Absolute change from baseline to last value during follow-up | |
|---|---|---|---|
| Platelet count, 109/L; median (range) | 553.0 (179–1549) | 411.5 (167–1265) | −94.5 (−1344 to 837) |
| Red blood cells, 1012/L; median (range) | 3.6 (1.9–5.9) | 4.2 (2.4–6.2) | 0.5 (−1.2 to 2.3) |
| Leukocytes, 109/L; median (range) | 6.3 (1.6–15.1) | 7.8 (2.8–22.3) | 1.7 (−6.2 to 17.3) |
| Neutrophils, 109/L; median (range) | 3.9 (0.7–11.2) | 4.8 (1.2–20.4) | 1.3 (−4.8 to 15.6) |
| Hemoglobin, g/dL; median (range) | 12.7 (8.1–17.9) | 12.8 (8.4–17.8) | 0.0 (−3.9 to 5.2) |
| Hematocrit, %; median (range) | 38.0 (26.0–53.0) | 38.0 (25.0–52.0) | 0.0 (−11.0 to 14.0) |
A total of 120 patients (70.6%) achieved a platelet response, of whom 71 (41.8%) achieved a full response and 49 (28.8%) achieved a partial response (Fig.3). The platelet response rate (full and partial) was slightly lower in patients who received anagrelide as second-line therapy (n = 98/143, 68.5%) compared with third-line therapy (n = 21/26, 80.8%). The platelet response rate was similar in patients who started anagrelide at 0.5 mg/d (n = 51/69, 73.9%) and 1 mg/d (n = 61/90, 67.8%), but highest in those that started at 1.5 mg/d (n = 8/9, 88.9%).
Figure 3.
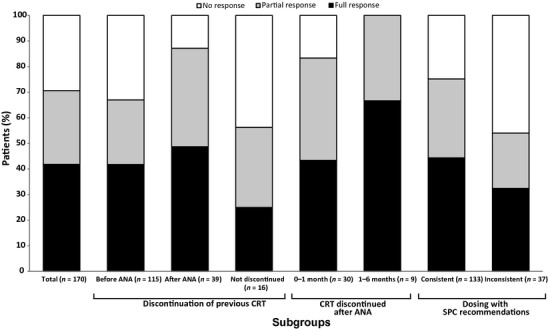
Platelet responses by main subgroups: full analysis set. ANA, anagrelide; CRT, cytoreductive therapy; SPC, Summary of Product Characteristics.
A greater proportion of patients who discontinued their previous CRT after anagrelide initiation achieved a platelet response (n = 34/39, 87.2%) compared with those who discontinued their previous CRT before anagrelide initiation (n = 77/115, 67.0%) and compared with patients who did not discontinue their CRT at any time point throughout the 6-month follow-up period (n = 9/16, 56.3%). Notably, 100% of patients (n = 9) who discontinued their previous CRT 1–6 months after anagrelide initiation achieved a platelet response (Fig.3). More patients whose anagrelide treatment was consistent with the SPC dosing recommendations experienced a platelet response (n = 100/133, 75.2%) than those whose treatment was inconsistent (n =20/37, 54.1%). Platelet response rates, as stratified by the reasons for inconsistency with the SPC dosing recommendations, were as follows: anagrelide starting dose >1 mg/d (n = 8/10, 80.0%); anagrelide dose titrated <7 d (n = 5/12, 41.7%); anagrelide dose increased >0.5 mg/d per week (n = 8/16, 50.0%).
Most patients’ treatment followed one of the following three modalities: discontinuation of previous CRT before anagrelide initiation that was consistent with the SPC dosing recommendations (n = 85/170, 50.0%); discontinuation of previous CRT before angrelide initiation that was inconsistent with the SPC dosing recommendations (n = 30/170, 17.6%); and discontinuation of previous CRT 0–1 month after anagrelide initiation (less prolonged transition; i.e., combination therapy) that was consistent with the SPC dosing recommendations (n = 28/170, 16.5%). The proportion of patients experiencing platelet responses in these groups were 71.8%, 53.3%, and 85.7%, respectively. The group with less prolonged transition to anagrelide initiation that was consistent with the SPC dosing recommendations experienced greater response rates (n = 24/28, 85.7%) than the group that discontinued prior CRT before the initiation of anagrelide that was inconsistent with the SPC dosing recommendations (n = 16/30, 53.3%).
The median change from baseline platelet count to the last platelet count during the 6-month follow-up period was −94.5 × 109/L (range −1344.0 to 837.0). No notable changes were observed from baseline in any of the other laboratory parameters (Table3), and no differences were observed between subgroups. No thrombosis was reported during the course of the study.
Safety (safety set)
A total of 81/175 patients (46.3%) experienced one or more ADRs during the 6-month follow-up period (Table4), with a median time to first ADR of 28.0 d. The incidence of ADRs was similar in patients who started anagrelide at 0.5 mg/d (n = 35/72, 48.6%) and 1 mg (n = 42/92, 45.7%), but the median time to first ADR was 14.0 d in patients who started at 0.5 mg/d and 37.0 d in patients who started at 1 mg/d. The proportion of patients who experienced an ADR was highest in the first 7 d of anagrelide initiation in both the anagrelide 0.5 mg/d (n = 13/35, 37.1%) and 1 mg/d starting group (n = 10/42, 23.8%).
Table 4.
Number of patients who had adverse drug reactions during the follow-up period by main subgroup: safety set
| ADR type | Subgroup totals | Total (n = 175) | ||||||
|---|---|---|---|---|---|---|---|---|
| Discontinuation of previous CRT2 | CRT discontinued after ANA | Dosing with SPC recommendations2 | ||||||
| Before ANA (n = 115) | After ANA (n = 39) | Not discontinued (n = 16) | 0–1 month (n = 30) | 1–6 months (n = 9) | Consistent (n = 133) | Inconsistent (n = 37) | ||
| Summary of ADRs | ||||||||
| ADRs | 51 (44.3) | 20 (51.3) | 6 (37.5) | 16 (53.3) | 4 (44.4) | 52 (39.1) | 25 (67.6) | 81 (46.3)3 |
| Serious ADRs | 6 (5.2) | 1 (2.6) | 1 (6.3) | 1 (3.3) | 0 | 6 (4.5) | 2 (5.4) | 8 (4.6) |
| ADRs leading to discontinuation | 19 (16.5) | 6 (15.4) | 2 (12.5) | 6 (20.0) | 0 | 25 (18.8) | 2 (5.4) | 29 (16.6)3 |
| ADRs leading to death | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
ADR, adverse drug reaction; ANA, anagrelide; CRT, cytoreductive therapy; SPC, Summary of Product Characteristics.
Data not shown for five additional patients included in the study who had no prior cytoreductive therapy and therefore received anagrelide as a first-line treatment.
These total values also include the patients who did not receive prior cytoreductive therapy (these were not further subdivided into the different subgroups).
The incidence of ADRs was lower in patients whose anagrelide treatment was consistent with the SPC dosing recommendations (n = 52/133, 39.1%) than in the group of patients whose anagrelide treatment was inconsistent with the SPC dosing recommendations (n = 25/37, 67.6%). The incidence of ADRs, as stratified by the reasons for inconsistency with the SPC dosing recommendations, was as follows: anagrelide starting dose >1 mg/d (n = 3/10, 30.0%); anagrelide dose titrated <7 d (n = 11/12, 91.7%); anagrelide dose increased >0.5 mg/d per week (n = 12/16, 75.0%). The proportion of patients who experienced an ADR was fairly similar across the study, irrespective of when (or if) patients had discontinued their previous CRT prior to anagrelide initiation. The most frequent ADRs by preferred term were palpitations (13.1%), headache (10.9%), diarrhea (5.7%), and asthenia (5.7%) (Table5). The incidences of diarrhea (21.6% vs. 1.5%) and palpitations (18.9% vs. 11.3%) were higher in patients whose anagrelide treatment was inconsistent with the SPC dosing recommendations than in patients whose anagrelide treatment was consistent with the SPC dosing recommendations. No clear differences were observed between the main subgroups in patients who experienced headache. Of the 81 patients, 49 experienced a mild ADR and 36 a moderate ADR. Seventeen patients experienced a severe ADR, the most frequent being anemia (n = 3, 1.7%). Severe cases of asthenia, headache, and hypertensive crisis were each reported in two patients (1.1%). In total, 10 serious ADRs were reported in eight patients (4.6%), of whom two had hypertensive crisis (Table5).
Table 5.
Summary of adverse drug reactions in three or more patients and serious adverse drug reactions in all patients reported during the 6-month follow-up period: safety set
| ADR type | Total (n = 175) |
|---|---|
| ADRs in three or more patients by preferred term | |
| Palpitations | 23 (13.1) |
| Headache | 19 (10.9) |
| Asthenia | 10 (5.7) |
| Diarrhea | 10 (5.7) |
| Abdominal pain | 5 (2.9) |
| Anemia | 5 (2.9) |
| Nausea | 5 (2.9) |
| Tachycardia | 5 (2.9) |
| Edema peripheral | 4 (2.3) |
| Tinnitus | 4 (2.3) |
| Myalgia | 3 (1.7) |
| Pruritus | 3 (1.7) |
| Rash | 3 (1.7) |
| Vertigo | 3 (1.7) |
| Serious ADRs in all patients by preferred term | |
| Hypertensive crisis | 2 (1.1)2 |
| Anemia | 1 (0.6) |
| Angina pectoris | 1 (0.6) |
| Cardiac failure | 1 (0.6) |
| Cerebrovascular accident | 1 (0.6)3 |
| Headache | 1 (0.6)2 |
| Palpitations | 1 (0.6) |
| Pulmonary arterial hypertension | 1 (0.6) |
ADR, adverse drug reaction.
One patient experienced both serious headache and serious hypertensive crisis.
One patient had two incidences of cerebrovascular accident.
Anagrelide was discontinued because of an ADR in 29 patients (16.6%) (Table4). The most frequent ADRs by preferred term that led to anagrelide discontinuation were palpitations (n = 7, 4.0%), headache (n = 5, 2.9%), and peripheral edema (n = 3, 1.7%). Three deaths occurred during the 6-month follow-up period, but all were considered unrelated to anagrelide.
Discussion
Since the discovery of the JAK2 V617F mutation in myeloproliferative neoplasms, targeted agents have been developed demonstrating clinical efficacy in myelofibrosis 11,12. However, limited data are available for agents targeting this mutation in ET and polycythemia vera; thus, optimal management of patients using ‘classic’ drugs is required. Currently, the anagrelide SPC 5 and the ELN 4 do not provide specific guidance on switching methods from one ET treatment to another, and so the question of how to initiate anagrelide most effectively remains unanswered. To our knowledge, the FOX study provides the first comprehensive real-world data on the modalities used when switching from previous CRT to anagrelide, based on a representative population of patients with ET requiring second- or third-line treatment. In this study, the transition methods identified for initiating second- or third-line anagrelide were as follows: discontinuing previous CRT prior to anagrelide initiation; discontinuing previous CRT after anagrelide initiation (either less prolonged transition 0–1 months or prolonged transition 1–6 months); or not discontinuing previous CRT; and whether anagrelide initiation was consistent or inconsistent with the SPC dosing recommendations. The SPC recommends that patients should start anagrelide at 1 mg/d and be monitored carefully on a regular basis. However, a proportion of patients were observed to initiate anagrelide as combination therapy in this study. Therefore, as the SPC dosing recommendations do not specify how to initiate anagrelide as combination therapy, the observed approach of starting anagrelide at ≤1.0 mg/d was considered consistent with the SPC dosing recommendations. The various approaches observed in this study reflect that patients with ET are treated on an individualized basis.
At the end of the 6-month follow-up period, most patients were still being treated with anagrelide and had achieved a platelet response (full or partial). However, some differences were observed according to the switching modalities.
Our results showed that more patients who discontinued their previous CRT after initiating anagrelide achieved a platelet response and remained on anagrelide therapy at the end of the 6 months compared with patients who discontinued their previous CRT before starting anagrelide. A period of treatment overlap may avoid a platelet rebound as continuation of prior therapy supports gradual increase in anagrelide dosage. This overlap may improve short-term tolerance and platelet response. In addition, patients whose anagrelide treatment was consistent with the SPC dosing recommendations were more likely to achieve a platelet response than those whose anagrelide treatment was inconsistent with the SPC dosing recommendations. Of the latter group of patients, the lowest responses were observed in those who had their anagrelide dose titrated <7 d or increased >0.5 mg/d per week. This could indicate that patients were not achieving a sufficient platelet response on the current dose and so had their dose increased earlier than recommended in an attempt to achieve a platelet response. This approach may have been employed more frequently in the presence of resistant thrombocytosis and refractory disease. The highest response rates (full and partial) were seen in patients who discontinued previous CRT after anagrelide initiation (n = 34/39) or those receiving anagrelide that was consistent with the SPC dosing recommendations (n = 111/133). The highest median maximum daily dose of anagrelide was observed in patients undergoing prolonged transition to anagrelide or those receiving anagrelide inconsistent with the SPC dosing recommendations. The prolonged transition period may have allowed the gradual increase in anagrelide to a higher daily dose with improved tolerance resulting in better response rates.
The lowest platelet responses were observed in patients who did not discontinue their previous therapy (i.e., had a period of combination therapy throughout the duration of the study) or in those whose anagrelide treatment was inconsistent with the SPC dosing recommendations. The presence of refractory disease could be a reason why previous CRT was not discontinued in the former group, and the dose was increased sooner than recommended to a higher dose in the latter group. A proportion of high-risk patients with ET are difficult to treat and present with refractory thrombocytosis. Guidance is available to help identify these patients 13. Moreover, in patients whose anagrelide treatment was inconsistent with the SPC dosing recommendations, the higher incidence of ADRs may be related to the rapid increases in anagrelide dose. Thus, further dose titrations may not have been possible, and a platelet response was not achieved. In addition, low anagrelide doses were used in patients who did not discontinue their previous therapy, perhaps with the aim of preventing ADRs, but this may have in turn reduced efficacy.
Approximately half of patients started anagrelide therapy at the recommended dose of 1 mg/d and received a maximum daily dose consistent with the SPC (1–3 mg/d) 5 and findings of the Europe-wide EXELS study 14. In this study, it was observed that 41.1% of patients received an anagrelide starting dose of 0.5 mg/d. More patients who received an anagrelide starting dose of 0.5 mg/d discontinued their previous CRT after anagrelide initiation (i.e., had a period of combination therapy) than patients who received an anagrelide starting dose of 1 mg/d (56.4% vs. 36.5%, respectively). Physicians may opt to use a more conservative approach when initiating anagrelide during a planned period of combination therapy to minimize side effects, especially as intolerance to previous CRT was the most frequently reported reason for initiating anagrelide. Moreover, patients may have been started on a low dose because the platelet counts already appeared to be controlled at baseline. The majority of patients had their anagrelide dose increased during the study, irrespective of starting dose, mainly for insufficient platelet response at the lower dose. This was expected, as anagrelide is usually titrated to achieve the lowest effective dose required to reduce and/or maintain a platelet count <600 × 109/L and ideally normalized while minimizing potential side effects.
In this study, anagrelide was well tolerated and the most frequent ADRs were consistent with the SPC 5 and results from previous studies 8,9,15. The incidence of ADRs, including diarrhea and palpitations, was lower in patients whose anagrelide treatment was consistent with the SPC dosing recommendations than in the group of patients whose anagrelide treatment was inconsistent with the SPC dosing recommendations. Of the latter group, the highest incidence of ADRs was observed in those whose anagrelide dose was titrated <7 d or whose dose was increased >0.5 mg/d per week. This supports the belief that a rapid dose increase in anagrelide and/or increase of dose >0.5 mg/d per week may not be an appropriate management strategy in terms of safety. The serious ADRs of palpitations, cardiac failure, angina pectoris, anemia, and headache that were reported in this study are all listed in the SPC 5. None of the serious ADRs were fatal. The average time to first ADR was shorter in patients who received an anagrelide starting dose of 0.5 mg/d than those who received a starting dose of 1 mg/d. This was an unexpected finding, but could be because approximately half of the patients who started anagrelide at 0.5 mg/d initiated anagrelide as combination therapy, thereby increasing the incidence of side effects.
As this was a non-randomized, observational study, subgroup numbers were not well balanced and some groups had very low patient numbers. These added an element of potential bias to the results. Formal significance testing was not presented, as P-values would also be potentially biased and misleading. In addition, although French hematologists may use World Health Organization criteria as mandatory, bone marrow biopsies were not collected in this study to confirm ET diagnosis as this study was non-interventional and focused on ET management in a population being treated under the assumption that they had ET. It was also deemed inappropriate to use bone marrow biopsies over 1 yr old. Furthermore, as the median time from diagnosis to trial inclusion was approximately 60 months, this delay would usually be sufficient to detect myelofibrotic symptoms if patients had initial prefibrotic myelofibrosis. Patients were still considered to have ET by the investigators if secondary myelofibrosis symptoms were absent after the follow-up.
In conclusion, data from the FOX study provide novel information on the switch modalities used when initiating anagrelide and their potential impact on patient outcomes in clinical practice. Highest platelet responses were observed when previous CRT was discontinued after anagrelide initiation or when anagrelide was initiated consistently with the SPC dosing recommendations. The safety data corresponded with the SPC, and the incidence of ADRs was lowest in patients whose anagrelide treatment was consistent with the SPC dosing recommendations. Limitations of the study are mainly related to the observational design, and so future randomized controlled trials would be required to confirm these findings.
Acknowledgments
The study was funded by Shire Development LLC. The authors thank the patients and investigators who participated in this study. Under the direction of the authors, Emma Burke and Louise Prince, employees of iMed Comms, Macclesfield, UK, provided medical writing assistance for this publication. Editorial assistance in formatting, proofreading, copy editing, and fact checking was also provided by iMed Comms. iMed Comms was funded by Shire for support in writing and editing this manuscript. JR, J-FV, and J-JK received funding for consultancy and advisory boards (J-FV and J-JK only) from Shire. KK and PW are employees of Shire and hold stock/stock options. JS is a statistical contractor employed with Shire.
FOX investigators: Abgrall JF (Brest), Adiko D (Libourne), Allangba O (Saint-Brieuc), Bakir R (Aulnay-sous-Bois), Besson C (Le Kremlin-Bicétre), Bons JM (Montluçon), Bordessoule D (Limoges), Boyer Perrard F (Angers), Cambier N (Lille), Camo JM (Perpignan), Casadevall N (Paris), Christian B (Metz), Cony Makhoul P (Annecy), De Faucal P (Nantes), De Jaureguiberry JP (Toulon), Demory JL (Lille), Dine G (Troyes), Espinouse D (Pierre-Bénite), Feugier P (Vandœuvre-les-Nancy), Fezoui H (Toulon), Fitoussi O (Bordeaux), Frenkiel N (Poissy), Genereau T (Nantes), Guyotat D (Saint-Etienne), Hacini M (Chambéry), Hutin P (Quimper), Ianotto JC (Brest), Kiladjian JJ (Paris), Lionne P (Arras), Liu V (Strasbourg), Maloisel F (Strasbourg), Ojeda Uribe M (Mulhouse), Orfeuvre H (Bourg-en-Bresse), Pautas Chambon C (Créteil), Pignon B (Reims), Rey J (Marseille), Rodon P (Blois), Rosenthal E (Nice), Sanhes L (Perpignan), Sebahoun G (Marseille), Thioliere B (Paris), Viallard JF (Pessac), Wattel E (Pierre-Bénite).
Supporting Information
Proportion of patients on different starting, maximum, and last daily doses of anagrelide therapy: safety set.
References
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. WHO Classification of Tumours of Haemopoietic and Lymphoid Tissues. 4th edn. Lyon: IARC Press; 2008. [Google Scholar]
- Johansson P. Epidemiology of the myeloproliferative disorders polycythemia vera and essential thrombocythemia. Semin Thromb Hemost. 2006;32:171–3. doi: 10.1055/s-2006-939430. [DOI] [PubMed] [Google Scholar]
- Tefferi A, Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J Clin Oncol. 2011;29:573–82. doi: 10.1200/JCO.2010.29.8711. [DOI] [PubMed] [Google Scholar]
- Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29:761–70. doi: 10.1200/JCO.2010.31.8436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Medicines Agency. Xagrid Summary of Product Characteristics. Last update 2013. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000480/WC500056557.pdf. Accessed May 7, 2013.
- Anagrelide Study Group. Anagrelide, a therapy for thrombocythemic states: experience in 577 patients. Am J Med. 1992;92:69–76. doi: 10.1016/0002-9343(92)90017-6. [DOI] [PubMed] [Google Scholar]
- Mills AK, Taylor KM, Wright SJ, et al. Efficacy, safety and tolerability of anagrelide in the treatment of essential thrombocythaemia. Aust N Z J Med. 1999;29:29–35. doi: 10.1111/j.1445-5994.1999.tb01585.x. [DOI] [PubMed] [Google Scholar]
- Penninga E, Jensen BA, Hansen PB, Clausen NT, Mourits-Andersen T, Nielsen OJ, Hasselbalch HC. Anagrelide treatment in 52 patients with chronic myeloproliferative diseases. Clin Lab Haematol. 2004;26:335–40. doi: 10.1111/j.1365-2257.2004.00637.x. [DOI] [PubMed] [Google Scholar]
- Steurer M, Gastl G, Jedrzejczak WW, Pytlik R, Lin W, Schlogl E, Gisslinger H. Anagrelide for thrombocytosis in myeloproliferative disorders: a prospective study to assess efficacy and adverse event profile. Cancer. 2004;101:2239–46. doi: 10.1002/cncr.20646. [DOI] [PubMed] [Google Scholar]
- Gisslinger H, Gotic M, Holowiecki J, Penka M, Thiele J, Kvasnicka HM, Kralovics R, Petrides PE. Anagrelide compared to hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood. 2013;121:1720–8. doi: 10.1182/blood-2012-07-443770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–98. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363:1117–27. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barosi G, Birgegard G, Finazzi G, et al. Response criteria for essential thrombocythemia and polycythemia vera: result of a European LeukemiaNet consensus conference. Blood. 2009;113:4829–33. doi: 10.1182/blood-2008-09-176818. [DOI] [PubMed] [Google Scholar]
- Besses C, Kiladjian JJ, Griesshammer M, Gugliotta L, Harrison C, Coll R, Smith J, Abhyankar B, Birgegard G. Cytoreductive treatment patterns for essential thrombocythemia in Europe. Analysis of 3643 patients in the EXELS study. Leuk Res. 2013;37:162–8. doi: 10.1016/j.leukres.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Fruchtman SM, Petitt RM, Gilbert HS, Fiddler G, Lyne A. Anagrelide: analysis of long-term efficacy, safety and leukemogenic potential in myeloproliferative disorders. Leuk Res. 2005;29:481–91. doi: 10.1016/j.leukres.2004.10.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Proportion of patients on different starting, maximum, and last daily doses of anagrelide therapy: safety set.