

Abstract
Objective
To determine whether copy number variations (CNVs) in FCGR3A and FCGR3B are associated with systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) in Taiwanese individuals.
Methods
FCGR3A and FCGR3B CNV genotypes were determined in 846 patients with SLE, 948 patients with RA, and 1,420 healthy control subjects, using custom TaqMan CNV assays. The FCGR3A and FCGR3B CNV genotypes were compared between healthy control subjects and patients and among patients stratified according to clinical characteristics.
Results
A low (<2) FCGR3A copy number was significantly associated with SLE (for <2 copies versus 2 copies, P = 5.06 × 10−4, false discovery rate–corrected P [PFDR] = 0.001, odds ratio [OR] 3.26, 95% confidence interval [95% CI] 1.68−6.35) and RA (for <2 copies versus 2 copies, P = 5.83 × 10−4, PFDR = 0.0012, OR 2.82, 95% CI 1.56−5.1). A low FCGR3B copy number was also significantly associated with SLE (for <2 copies versus 2 copies, P = 0.0032, PFDR = 0.0032, OR 1.59, 95% CI 1.17−2.18). Notably, a high (>2) FCGR3A copy number was also associated with SLE (for >2 copies versus 2 copies, P = 0.003, PFDR = 0.0061, OR 1.6, 95% CI 1.17−2.18). Additionally, the FCGR3A low copy number genotype was significantly enriched in subsets of patients with SLE (those with ulcer, arthritis, rash, discoid rash, photosensitivity, nephritis, leukopenia, thrombocytopenia, depressed complement levels, and autoantibody positivity) and patients with RA (those positive for rheumatoid factor) compared with healthy control subjects. The FCGR3B low copy number genotype was also significantly enriched in SLE patients with ulcer, rash, discoid rash, photosensitivity, ascites, nephritis, complement level depression, and anti–double-stranded DNA antibody positivity compared with control subjects. However, FCGR3B CNVs were not associated with RA susceptibility (for <2 copy numbers versus 2 copy numbers, P = 0.3584, OR 1.15, 95% CI 0.85–1.55) and clinical characteristics.
Conclusion
In Taiwanese individuals, a low FCGR3A copy number is a common risk factor for SLE and RA, while a low FCGR3B copy number confers a risk of SLE but not RA.
IgG Fcγ receptors (FcγRs) mediate a variety of immune functions that are critical in immune responses. In humans, 5 classic low-affinity FcγRs (FcγRIIA, FcγRIIB, FcγRIIC, FcγRIIIA, and FcγRIIIB) are coded by 5 genes (FCGR2A, FCGR2B, FCGR2C, FCGR3A, and FCGR3B, respectively) in the FCGR cluster on chromosome 1. Activating FcγRs (FcγRIIA, FcγRIIC, FcγRIIIA, and FcγRIIIB) mediate immune cell activation promoting inflammation, while the classic inhibitory FcγRIIB dampens immune responses and restricts inflammation (1,2). Activating FcγRs mediate functions including immune complex clearance, phagocytosis, antigen presentation, antibody-dependent cellular cytotoxicity, and cytokine production (3). In contrast, the inhibitory FcγRIIB abrogates immune cell activation. FcγRIIB also plays a role in the maintenance of peripheral B cell tolerance and the prevention of autoimmunity (2).
The variations in FcγR expression significantly affect IgG immune complex–mediated signal thresholds (3,4). Notably, proinflammatory and antiinflammatory cytokines could modulate the expression levels of activating and inhibitory FcγRs (5) that affect the threshold immune cell response to IgG immune complexes (6). FcγR-knockout mouse models indicate that both activating and inhibitory FcγRs influence the development of autoimmune diseases (7–9). The contributions of FcγR genes to autoimmune diseases have attracted substantial attention, and functional FcγR polymorphisms have been reported to play important roles in the pathogenesis of autoimmune diseases (4,10,11).
Gene copy number variation (CNV) is a rich source of genetic heterogeneity (12,13). The FCGR cluster on chromosome 1q23 shows a complex pattern of CNVs. Among 5 FcγR genes in the cluster, 3 genes (FCGR3A, FCGR2C, and FCGR3B) have CNVs, while FCGR2A and FCGR2B do not have CNVs (14,15). CNVs play important roles in human disease pathogenesis (16). FCGR3B copy number deficiency is associated with autoimmune disease, including systemic lupus erythematosus (SLE) (17–19), Sjögren's syndrome (20), and systemic sclerosis (21). Although FCGR3B CNVs were reported to associate with rheumatoid arthritis (RA) (22–24), no association between RA and FCGR3B CNVs was observed in other studies (25,26). Moreover, no association between FCGR3A CNVs and RA was observed (24,27). In the current study, we investigated whether FCGR3A and FCGR3B CNVs are associated with susceptibility to SLE and RA in Taiwanese individuals. The results provide new insights into the role of FcγRIII family members in the pathogenesis of SLE and RA in an Asian population.
PATIENTS AND METHODS
Study subjects
Taiwanese healthy control donors (512 men and 908 women) were recruited locally. The mean ± SD age of the healthy control donors was 40.2 ± 11.6 years (range 18–64 years). Taiwanese patients with SLE (72 men and 774 women) who fulfilled the 1982 and/or 1997 revised American College of Rheumatology criteria for SLE (28,29) and patients with RA (141 men and 807 women) who fulfilled the 1987 American College of Rheumatology criteria for the classification of RA (30) were recruited at Chang Gung Memorial Hospital, Tao-Yuan, Taiwan. Stratification of the clinical characteristics of the patients with RA was previously described (31). The ethics committee of Chang Gung Memorial Hospital approved the human study, and all donors provided written consent.
Nucleic acid isolation
Anticoagulated peripheral blood was obtained from healthy control donors, patients with SLE, and patients with RA. All genomic DNA samples were isolated from anticoagulated peripheral blood using a Puregene DNA isolation kit (Gentra Systems) in the same laboratory as previously described (32).
Determination of FCGR3 CNVs
FCGR3A and FCGR3B CNVs were genotyped using custom TaqMan CNV real-time quantitative polymerase chain reaction (PCR) assays with FAM/minor groove binder dual-labeled probes that were produced by Applied Biosystems (see Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38813/abstract). A TaqMan Copy Number Reference Assay (RNase P) with a VIC dye–labeled TAMRA probe (catalog no. 4403328; Applied Biosystems) was used as the internal control for copy number targets.
Duplex quantitative real-time PCRs were carried out on an Applied Biosystems ViiA 7 Real-Time PCR System (Life Technologies), according to the manufacturer's instructions. All samples were tested in duplicate, and fluorescence signals were normalized to ROX. The quantitative PCR amplification curves were analyzed using ViiA 7 software (Applied Biosystems) on a plate-by-plate basis, and the copy number was assigned from the raw quantification cycle (Cq) values using CopyCaller software version 2.0 (Applied Biosystems). This software uses a clustering algorithm and assigns a copy number value of 2 to the cluster with the most samples. CopyCaller software also provides extensive diagnostics for the validity of the results, which were set to accept the copy number assignment only when confidence was >95%, the SD of the sample replicate ΔCq estimates was <0.20, and a reference gene Cq was <32. More than 85% of samples had >99% confidence level in the copy number assignment. In addition, a repeat copy number assay was carried out on all samples with a copy number of <2, 10% of healthy control samples with a copy number of 2, and 20% of all samples with a copy number of >2, to confirm the copy number calls. Copy number assignment with >95% confidence levels completely matched (100% reproducibility) for all samples in repeat copy number assays. Overall, our methodology resulted in clear assignment of FCGR3 copy numbers for 99% of the samples (histograms of FCGR3A and FCGR3B CNV analyses are shown in Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38813/abstract). The accuracy of the assays was further confirmed by determining CD16 expression on natural killer (NK) cells and neutrophils.
Evaluation of FcγRIII (CD16) expression levels
To determine the expression of CD16 on NK cells and neutrophils, 100-μl samples of fresh whole blood were stained with fluorescein isothiocyanate (FITC)–conjugated anti-human CD16 monoclonal antibodies (clone 16B; eBiosciences) and phycoerythrin (PE)–conjugated anti-human CD56 (Beckman Coulter). Whole blood samples stained with FITC-conjugated murine IgG1 and PE-conjugated anti-human CD56 (in separate tubes) were used as isotype controls. After incubation at room temperature for 30 minutes, blood samples were treated with 1× BD FACS Lysing Solution (BD Biosciences) to lyse red blood cells, followed by analysis on a Beckman Coulter FC500 flow cytometer. NK cells within the lymphocyte population were identified as CD56+ cells. Characteristic light scatter properties were used to identify neutrophils in flow cytometric analyses. The expression of FcγRIII (CD16) was analyzed using FlowJo software (Tree Star).
Autoantibody assays
Autoantibody titers were determined by enzyme-linked immunosorbent assay (ELISA). The autoantibody status of the patients was assessed at the time of diagnosis of SLE or RA. Antinuclear antibody (ANA) positivity was defined as a serum titer of ≥1:80 in a Hep-G2 cell assay. Anti–extractable nuclear antigen antibodies (Ro/SSA, La/SSB, Sm, and RNP) and anticardiolipin antibodies were assessed by commercial ELISA according to the manufacturer's instructions (Pharmacia Diagnostics).
Statistical analysis
The distribution of FCGR3 copy numbers between patients with SLE or patients with RA and healthy control subjects was compared using chi-square and Fisher's exact tests. P values, odds ratios (ORs), and 95% confidence intervals (95% CIs) were calculated based on the identified risk copy number. To investigate the association of copy number with clinical manifestations of SLE and RA, we assigned those SLE and RA patients with a clinical phenotype as “1” cases and those without it as “0” cases. The clinical phenotypes of SLE patients were stratified according to the SLE diagnostic criteria. The phenotypes of RA patients were stratified based on the presence or absence of rheumatoid factor (RF), anti–cyclic citrullinated peptide (anti-CCP) antibodies, and radiographic erosions. We compared the copy number distributions between “1” cases and “0” cases and between “1” cases and healthy controls using chi-square and Fisher's exact tests. In addition, logistic regression models adjusted for sex and age were used to investigate the relationship between each clinical manifestation and copy number among patients. The logistic regression models adjusted for sex and age were also used to calculate the ORs of high copy number (>2) and low copy number (<2) in disease susceptibility.
To correct for multiple comparisons, an SAS MULTTEST procedure was performed, in which false discovery rate (FDR)–adjusted P values (PFDR) (33) were defined in a step-up manner but with less conservative multipliers. Mann-Whitney U tests were used to evaluate the effect of FCGR3 copy numbers on FcγRIII (CD16) expression levels. P values less than 0.05 were considered significant.
RESULTS
Characteristics of the SLE patients and the RA patients
The SLE cohort comprised 846 patients ranging in age from 8 years to 77 years (mean ± SD age 30.88 ± 11.87 years). Seventy-two (8.51%) of the patients were male, and 774 (91.49%) were female. The mean ± SD age of the male patients was 31.46 ± 12.52 years, and that of the female patients was 30.64 ± 11.56 years. The clinical characteristics of the patients with SLE are shown in Table 1. The RA cohort comprised 948 patients (141 males and 807 females). The mean ± SD age of these patients was 46.49 ± 13.9 years; 76.4% (670 of 887) were anti-CCP antibody positive, 78.81% (744 of 944) were RF positive, 50.9% (424 of 833) were ANA positive, and 71.04% (601 of 846) had destructive disease.
Table 1.
Clinical characteristics of the patients with SLE (n = 846)*
| Oral ulcer | 224/846 (26.48) |
| Arthritis | 532/846 (62.88) |
| Malar rash | 466/846 (55.08) |
| Discoid rash | 161/846 (19.03) |
| Photosensitivity | 189/846 (22.34) |
| Pleural effusion | 162/846 (19.15) |
| Pericardial effusion | 102/846 (12.06) |
| Ascites | 44/846 (5.2) |
| Nephritis | 471/846 (55.67) |
| Neuropsychiatric manifestations | 135/846 (15.96) |
| Leukopenia† | 473/846 (55.91) |
| Anemia‡ | 257/846 (30.38) |
| Thrombocytopenia§ | 220/846 (26) |
| Anti-dsDNA | 628/828 (75.85) |
| Complement level depression | 643/833 (77.19) |
| Anti-RNP | 295/685 (43.07) |
| Anti-Sm | 260/686 (37.9) |
| Anti-SSA | 365/568 (64.26) |
| Anti-SSB | 150/568 (26.41) |
| Anticardiolipin IgG | 187/664 (28.16) |
| Anticardiolipin IgM | 55/607 (9.06) |
Values are the no. of positive cases/no. of total cases (%). SLE = systemic lupus erythematosus; anti-dsDNA = anti–double-stranded DNA.
White blood cell count <3,500 units/liter.
Hemoglobin concentration <9 gm/dl.
Platelet count <105/μl.
Association of both FCGR3A and FCGR3B CNVs with SLE susceptibility
We examined the single-locus associations between FCGR3 CNVs and susceptibility to SLE. As shown in Table 2, a low (<2) FCGR3A copy number was significantly associated with SLE disease susceptibility (for <2 copies versus 2 copies, P = 5.06 × 10−4, PFDR = 0.001, OR 3.26, 95% CI 1.68–6.35). Notably, the high (>2) FCGR3A copy number was also a risk factor for SLE susceptibility (for >2 copies versus 2 copies, P = 0.003, PFDR = 0.0061, OR 1.6, 95% CI 1.17–2.18), suggesting that an FCGR3A abnormality has a role in the development of SLE. Similarly, a low FCGR3B copy number was significantly associated with SLE disease susceptibility (for <2 copies versus 2 copies, P = 0.0032, PFDR = 0.0032, OR 1.59, 95% CI 1.17–2.18). Moreover, a high FCGR3B copy number tended to have a protective role against SLE disease development (adjusted P [Padj] = 0.0574, PFDR = 0.0574, OR 0.77, 95% CI 0.59–1.01). Our data suggested that the FcγRIIIB deficiency is also a risk factor for SLE in Taiwanese individuals.
Table 2.
Association of FCGR3A and FCGR3B CNVs with SLE susceptibility*
| FCGR3A | FCGR3B | |||
|---|---|---|---|---|
| SLE | Controls | SLE | Controls | |
| Unadjusted | ||||
| CN <2 | 31 (3.68) | 19 (1.34) | 121 (14.34) | 126 (8.97) |
| CN = 2 | 700 (83.04) | 1,280 (90.14) | 605 (71.68) | 1,026 (73.02) |
| CN >2 | 112 (13.29) | 121 (8.52) | 118 (13.98) | 253 (18.01) |
| χ2 | 27.8166 | 19.1486 | ||
| P | 9.11 × 10−7 | 6.95 × 10−5 | ||
| Fisher's exact P | 1.23 × 10−6 | 7.95 × 10−5 | ||
| Adjusted for age and sex | ||||
| CN >2 vs. CN = 2 | ||||
| P | 0.0030 | 0.0574 | ||
| PFDR | 0.0061 | 0.0574 | ||
| OR (95% CI) | 1.6 (1.17−2.18) | 0.77 (0.59−1.01) | ||
| CN <2 vs. CN = 2 | ||||
| P | 5.06 × 10−4 | 0.0032 | ||
| PFDR | 0.001 | 0.0032 | ||
| OR (95% CI) | 3.26 (1.68−6.35) | 1.59 (1.17−2.18) | ||
Except where indicated otherwise, values are the number (%). CNVs = copy number variations; SLE = systemic lupus erythematosus; PFDR = false discovery rate–adjusted P value; OR = odds ratio; 95% CI = 95% confidence interval.
Association of RA susceptibility with FCGR3A CNVs but not with FCGR3B CNVs
As shown in Table 3, a low (<2) FCGR3A copy number was significantly associated with RA disease susceptibility (for <2 copies versus 2 copies, P = 5.83 × 10−4, PFDR = 0.0012, OR 2.82, 95% CI 1.56–5.10). In contrast to the findings in SLE patients, the high (>2) FCGR3A copy number had no effect on RA susceptibility (P = 0.3335). In addition, neither low FCGR3B copy number nor high FCGR3B copy number was associated with RA susceptibility. Our data indicated that FCGR3A deficiency is a susceptibility factor for RA, while FCGR3B CNVs appear not to have a role in the development of RA in Taiwanese individuals.
Table 3.
Association of FCGR3A and FCGR3B CNVs with RA susceptibility*
|
FCGR3A |
FCGR3B |
|||
|---|---|---|---|---|
| RA | Controls | RA | Controls | |
| Unadjusted | ||||
| CN <2 | 36 (3.81) | 19 (1.34) | 98 (10.35) | 126 (8.97) |
| CN = 2 | 836 (88.47) | 1,280 (90.14) | 685 (72.33) | 1,026 (73.02) |
| CN >2 | 73 (7.72) | 121 (8.52) | 164 (17.32) | 253 (18.01) |
| χ2 | 15.5197 | 1.3208 | ||
| P | 4.27 × 10−4 | 0.5167 | ||
| Fisher's exact P | 5.00 × 10−4 | 0.5160 | ||
| Adjusted for age and sex | ||||
| CN >2 vs. CN = 2 | ||||
| P | 0.3335 | 0.4320 | ||
| PFDR | 0.432 | 0.432 | ||
| OR (95% CI) | 0.86 (0.62−1.17) | 0.91 (0.72−1.15) | ||
| CN <2 vs. CN = 2 | ||||
| P | 5.83 × 10−4 | 0.3584 | ||
| PFDR | 0.0012 | 0.3584 | ||
| OR (95% CI) | 2.82 (1.56−5.1) | 1.15 (0.85−1.55) | ||
Except where indicated otherwise, values are the number (%). CNVs = copy number variations; RA = rheumatoid arthritis; PFDR = false discovery rate–adjusted P value; OR = odds ratio; 95% CI = 95% confidence interval.
Effects of FCGR3A and FCGR3B CNVs on SLE phenotypes and autoantibody production
Patients with SLE exhibit heterogeneous disease manifestations and variations in the severity, nature, and spectrum of clinical involvement. We subsequently examined the effects of FCGR3A and FCGR3B CNVs on SLE clinical phenotypes. Copy number frequencies were compared between the SLE patients with each disease characteristic and the healthy control subjects (positive versus healthy controls) and among SLE patients stratified by each clinical characteristic (positive versus negative).
The FCGR3A low copy number genotype was significantly enriched in SLE patients positive for ulcer, arthritis, rash, discoid rash, photosensitivity, nephritis, leukopenia, thrombocytopenia, complement level depression, anti–double-stranded DNA (anti-dsDNA), anti-RNP, anti-Sm, anti-SSA, anti-SSB, and anticardiolipin IgG compared with healthy control subjects (see Supplementary Table 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38813/abstract). However, a low (<2) FCGR3A copy number was only marginally associated with nephritis among subsets of SLE patients stratified according to clinical manifestations (for patients with nephritis versus patients without nephritis, Padj = 0.0457, OR 2.32, 95% CI 1.02–5.28) (see Supplementary Table 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38813/abstract). Our data suggested that FCGR3A deficiency may play a role in the development of lupus nephritis.
FCGR3B low copy number genotypes were significantly enriched in SLE patients with ulcer, rash, discoid rash, photosensitivity, ascites, nephritis, complement level depression, and anti-dsDNA antibody positivity compared with healthy controls (see Supplementary Table 4, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38813/abstract). Nevertheless, only the phenotypes of oral ulcer and nephritis were associated with a low (<2) FCGR3B copy number (PFDR < 0.05) among SLE patients stratified according to clinical manifestations (see Supplementary Table 5, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38813/abstract).
FCGR3 CNVs and clinical characteristics of patients with RA. We also examined whether FCGR3A CNVs are associated with RA disease characteristics. The FCGR3A low copy number genotype was significantly increased among RF-positive patients with RA and patients with destructive RA compared with healthy control subjects (see Supplementary Table 6, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38813/abstract), but the increased enrichment was not significant when the association was compared among the subsets of patients with RA (see Supplementary Table 7, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38813/abstract). In contrast, a low (<2) FCGR3A copy number was protective against anti-CCP antibody production (PFDR = 0.008, OR 0.35, 95% CI 0.17–0.72) in RA patients stratified according to anti-CCP positivity (see Supplementary Table 7). These data suggested that the functions of FCGR3A play a role in RA. However, FCGR3B was not associated with any clinical characteristics of RA (see Supplementary Tables 8 and 9, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38813/abstract).
Correlation between FCGR3A CNVs and CD16A expression on NK cells
To evaluate whether the FCGR3A CNVs affect FcγRIIIA (CD16A) expression on NK cells, peripheral blood samples from individuals carrying 1 (n = 4), 2 (n = 17), and 3 (n = 8) copies of FCGR3A were used in flow cytometric assays. As shown in Figure 1A, NK cells carrying 1 copy of FCGR3A expressed significantly less CD16A than those carrying 2 copies of FCGR3A (P = 0.028), and the NK cells carrying 3 copies of FCGR3A also expressed significantly more CD16A than those carrying 1 copy of FCGR3A (P = 0.040), suggesting that FCGR3A CNVs may affect NK cell functions.
Figure 1.
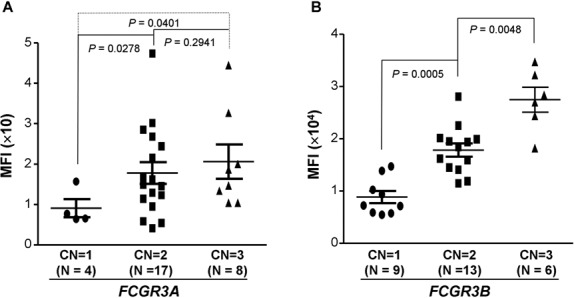
FCGR3A and FCGR3B copy numbers (CNs) affect Fcγ receptor expression. A, Effect of FCGR3A copy number on CD16A expression on natural killer (NK) cells. NK cells carrying 1 copy of FCGR3A expressed significantly less CD16A than those carrying 2 copies (P = 0.0278). NK cells carrying 3 copies of FCGR3A expressed significantly more CD16A than those carrying 1 copy (P = 0.0401). B, Effect of FCGR3B copy number on CD16B expression on neutrophils. Neutrophils from donors carrying 1 copy of FCGR3B expressed significantly less CD16B than those from donors carrying 2 copies (P = 0.0005). Neutrophils from donors carrying 3 copies of FCGR3B expressed significantly more CD16B than those from donors carrying 2 copies (P = 0.0048). Each symbol represents an individual subject; bars show the mean ± SD. MFI = mean fluorescence intensity.
Significant effect of FCGR3B CNVs and CD16B expression on neutrophils
To assess the effect of FCGR3B copy number on neutrophil FcγRIIIB (CD16B) expression, we determined the expression of CD16B in individuals carrying 1 (n = 9), 2 (n = 13), or 3 (n = 6) copies of FCGR3B. As shown in Figure 1B, neutrophils from individuals carrying 1 copy of FCGR3B expressed significantly less CD16B compared with neutrophils from individuals carrying 2 copies of FCGR3B (P = 0.0005). Neutrophils from individuals carrying 3 copies of FCGR3B expressed significantly more CD16 compared with neutrophils from individuals carrying 2 copies of FCGR3B (P = 0.0048). These data confirmed the previous observation that FcγRIIIB gene doses affect the expression of CD16B on neutrophils (19).
DISCUSSION
Although FCGR3A CNVs were previously detected (14,34,35), the role of FCGR3A CNVs in SLE is unknown. Moreover, no association between FCGR3A CNVs and RA was observed in European Caucasians (24,27). In this study, we observed that a low (<2) FCGR3A copy number was significantly associated with both SLE and RA in Taiwanese individuals, suggesting that FcγRIIIA deficiency may be a common risk factor for SLE and RA in Taiwanese. Surprisingly, a high (>2) FCGR3A copy number was also associated with susceptibility to SLE (Table 2), suggesting that the unbalanced functions of FCGR3A (both a deficiency and an overload) play a role in the pathogenesis of SLE.
FCGR3A copy number frequencies are similar between healthy European individuals (24,27) and healthy Taiwanese individuals. However, FCGR3A deficiency is a risk factor for RA in Taiwanese, while no association between FCGR3A CNVs and RA susceptibility was observed in European Caucasians (24,27). There are 2 explanations for this discrepancy.
First, because the frequency of FCGR3A deficiency (copy number <2) is very low in all populations (<2%), large numbers of patients and control subjects are required to detect an effect of FCGR3A CNVs. Compared with previous studies (24,27), the current study included more healthy control subjects and patients with RA. To detect associations of FCGR3A CNVs with SLE and RA by setting the alpha level at 0.05, the current study had >95.8% power to detect an OR of 1.55 in 846 patients with SLE and 1,420 healthy control subjects based on a 13.29% frequency of a high (>2) copy number. The study had >99.5% power to detect an OR of 2.86 in 945 patients with RA and 1,420 healthy control subjects based on 3.81% frequency of a low (<2) copy number. Therefore, the current study had much better power to detect associations of FCGR3A CNVs with SLE and RA.
Second, epistatic interactions between FCGR3A CNVs and other genes may be required for development of the RA phenotype. In European and African American populations, the composition of epistatic modifiers may differ from that in the Taiwanese population. Therefore, the effect of FCGR3A CNVs on RA may not be the same in different races. Similarly, the functional FcγRIIB single-nucleotide polymorphism (SNP) is associated with SLE in Asians (32,36–38), while the association between the FcγRIIB SNP and SLE in Caucasians requires studies with a large sample size because of epistatic interactions (39). Notably, a low FCGR3A copy number showed a negative association with anti-CCP antibody positivity, which highlighted a complicated role that FCGR3A CNVs play in autoimmune disease phenotypes and suggested that the genetic backgrounds may be different between anti-CCP antibody–positive and anti-CCP antibody–negative patients with RA. Most importantly, FCGR3A deficiency is associated with 2 distinct autoimmune diseases (SLE and RA), suggesting that defective FcγRIIIA functions may represent a common risk factor for various autoimmune diseases.
The functional FCGR3A SNP (rs396991) has been associated with lupus nephritis in several different ethnic groups in multiethnic meta-analyses (40,41). In the current study, we observed that a low FCGR3A copy number is a strong risk factor for nephritis in patients with SLE compared with healthy control subjects (OR 4.51, P < 0.0001) (Supplementary Table 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38813/abstract). However, the association with nephritis was marginal among patients with SLE (OR 2.32, P = 0.0457) stratified according to the presence of nephritis (see Supplementary Table 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38813/abstract). Because a low FCGR3A copy number is also a predisposing factor for SLE, the overall enrichment of the FCGR3A low copy number genotype in the entire SLE patient population may somehow decrease the significance of the association between lupus nephritis and a low FCGR3A copy number among patients with SLE. The association of a low FCGR3A copy number with nephritis in Taiwanese individuals reaffirms the notion that less-efficient immune complex handling by FcγRIIIA is an important contributor to lupus nephritis pathogenesis (11).
Surprisingly, we observed that a high FCGR3A copy number is also a risk factor for SLE. Higher frequencies of cytokine-producing FcγRIIIA-positive dendritic cells (DCs) were observed in SLE patients, particularly in those with active disease, suggesting that FcγRIIIA-mediated inflammatory cytokine production in DCs might contribute to disease pathogenesis (42). The higher density of activating FcγRIIIA on the surface of immune cells (NK cells, monocytes, DCs, macrophages, and subsets of T cells) could tip the delicate balance of immune responses toward intense inflammation, which may result in the development of SLE.
Our in vitro data demonstrated a correlation between a low FCGR3A copy number and low CD16A expression on NK cells, suggesting that FCGR3A copy number has physiologic implications in NK cell functions. The disease associations suggest that modulation of FcγRIIIA function may be an important therapeutic target for lupus nephritis. Because the sample size of subjects with a low (<2) FCGR3A copy number in the current study was not very large, further study using a much larger sample size is required to confirm our findings.
Consistent with previous studies in SLE (17–19,25,43), we observed that an FCGR3B deficiency was significantly associated with SLE susceptibility in Taiwanese individuals, with an OR of 1.59 (99.4% statistical power based on 14.34% frequency of a low [<2] copy number and an alpha level of 0.05), which is within the range of a meta-analysis (OR 1.32–1.92 for SLE) (44). Moreover, a high (>2) FCGR3B copy number seems to have a protective role in SLE. Our findings highlight the growing evidence that FcγRIIIB functions play important roles in the pathogenesis of SLE across multiple ethnicities. Additionally, our data demonstrate a clear correlation between FCGR3B copy number and CD16B cell surface expression, suggesting that FCGR3B copy number has physiologic implications in neutrophil functions. FcγRIIIB plays a critical role in the adherence of neutrophils to immune complexes and their subsequent clearance (19,45). The high density of FcγRIIIB on neutrophils with weak signaling capacity may suit FcγRIIIB for efficient capture and internalization of immune complexes with minimal neutrophil activation (45,46). Thus, FcγRIIIB expression on neutrophils may have an important role in autoimmunity, and the expression levels of FcγRIIIB might have a significant impact on the pathogenesis of autoimmune diseases. Insufficient FcγRIIIB-mediated immune complex clearance may be the underlying mechanism of a low FCGR3B copy number predisposing to SLE and a high FCGR3B copy number having a protective role in SLE.
Our study failed to show an association between FCGR3B CNVs and RA susceptibility in Taiwanese individuals, suggesting that FCGR3B CNVs may not have a significant role in the development of RA, which is consistent with the results of a previous meta-analysis (44). The association between FCGR3B CNVs and RA observed in different studies was not consistent (22–26). If the lowest estimate (OR 1.36) of an association in the meta-analysis (44) is used, our current study would only have 55.8% power to detect a significant difference between 945 RA patients and 1,420 healthy control subjects based on the low (<2) copy number frequency of 10.35%, which may be the reason that we failed to detect a significant association of FCGR3B CNVs with RA in Taiwanese individuals. Nevertheless, the lack of an association between a low FCGR3B copy number and RA in Taiwanese individuals suggests that CD16B function may not play a critical role in the development of RA in an Asian population.
Two recent studies demonstrated that FcγR gene CNVs in chromosomal region 1q23 are the result of independent and recurrent non-allelic homologous recombination (NAHR) events between the 2 segments that carry FCGR3A or FCGR3B (15,47). Deletions of either FCGR3A or FCGR3B differ only in the position of the NAHR breakpoint. The FCGR CNV mutation rate per generation is estimated to be ∼1.008 × 10−3 (15). No linkage disequilibrium exists between FCGR3A CNVs and FCGR3B CNVs (15,47) or between FCGR3 CNVs and functional SNPs in FcγRIII genes (34). Therefore, the association of individual FCGR3 CNVs with SLE and RA most likely reveals independent FcγRIII gene effects in autoimmune diseases.
In conclusion, the present study demonstrates a role of FCGR3 CNVs in autoimmunity. The deficiency of FCGR3 family members is one of mechanisms involved in the development of SLE and RA in Taiwanese individuals.
Acknowledgments
We greatly appreciate Miss Wen-Chi Chen and Hui-Ting Chan for providing technical support and personnel at the Hsin Chu Blood Donor Center for collecting blood samples.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Drs. Chen and Wu had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Chen, Wang, J. Wu.
Acquisition of data. Chen, Chang, Cheng, Y.-J. Wu, Lin, Yang, Ho, J. Wu.
Analysis and interpretation of data. Chen, Wang, Chang, J. Wu.
REFERENCES
- 1.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–90. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 2.Ravetch JV, Lanier LL. Immune inhibitory receptors. Science. 2000;290:84–9. doi: 10.1126/science.290.5489.84. [DOI] [PubMed] [Google Scholar]
- 3.Dijstelbloem HM, van de Winkel JG, Kallenberg CG. Inflammation in autoimmunity: receptors for IgG revisited. Trends Immunol. 2001;22:510–6. doi: 10.1016/s1471-4906(01)02014-2. [DOI] [PubMed] [Google Scholar]
- 4.Salmon JE, Pricop L. Human receptors for immunoglobulin G: key elements in the pathogenesis of rheumatic disease. Arthritis Rheum. 2001;44:739–50. doi: 10.1002/1529-0131(200104)44:4<739::AID-ANR129>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 5.Pricop L, Redecha P, Teillaud JL, Frey J, Fridman WH, Sautes-Fridman C, et al. Differential modulation of stimulatory and inhibitory Fc γ receptors on human monocytes by Th1 and Th2 cytokines. J Immunol. 2001;166:531–7. doi: 10.4049/jimmunol.166.1.531. [DOI] [PubMed] [Google Scholar]
- 6.Boruchov AM, Heller G, Veri MC, Bonvini E, Ravetch JV, Young JW. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J Clin Invest. 2005;115:2914–23. doi: 10.1172/JCI24772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolland S, Ravetch JV. Spontaneous autoimmune disease in FcγRIIB-deficient mice: results from strain-specific epistasis. Immunity. 2000;13:277–85. doi: 10.1016/s1074-7613(00)00027-3. [DOI] [PubMed] [Google Scholar]
- 8.Kleinau S, Martinsson P, Heyman B. Induction and suppression of collagen-induced arthritis is dependent on distinct fcγ receptors. J Exp Med. 2000;191:1611–6. doi: 10.1084/jem.191.9.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nabbe KC, Blom AB, Holthuysen AE, Boross P, Roth J, Verbeek S, et al. Coordinate expression of activating Fcγ receptors I and III and inhibiting Fcγ receptor type II in the determination of joint inflammation and cartilage destruction during immune complex–mediated arthritis. Arthritis Rheum. 2003;48:255–65. doi: 10.1002/art.10721. [DOI] [PubMed] [Google Scholar]
- 10.Morgan AW, Keyte VH, Babbage SJ, Robinson JI, Ponchel F, Barrett JH, et al. FcγRIIIA-158V and rheumatoid arthritis: a confirmation study. Rheumatology (Oxford) 2003;42:528–33. doi: 10.1093/rheumatology/keg169. [DOI] [PubMed] [Google Scholar]
- 11.Wu J, Edberg JC, Redecha PB, Bansal V, Guyre PM, Coleman K, et al. A novel polymorphism of FcγRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest. 1997;100:1059–70. doi: 10.1172/JCI119616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–51. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 13.Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–8. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 14.Breunis WB, van Mirre E, Geissler J, Laddach N, Wolbink G, van der Schoot E, et al. Copy number variation at the FCGR locus includes FCGR3A, FCGR2C and FCGR3B but not FCGR2A and FCGR2B. Hum Mutat. 2009;30:E640–50. doi: 10.1002/humu.20997. [DOI] [PubMed] [Google Scholar]
- 15.Machado LR, Hardwick RJ, Bowdrey J, Bogle H, Knowles TJ, Sironi M, et al. Evolutionary history of copy-number-variable locus for the low-affinity Fcγ receptor: mutation rate, autoimmune disease, and the legacy of helminth infection. Am J Hum Genet. 2012;90:973–85. doi: 10.1016/j.ajhg.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fanciulli M, Petretto E, Aitman TJ. Gene copy number variation and common human disease. Clin Genet. 2010;77:201–13. doi: 10.1111/j.1399-0004.2009.01342.x. [DOI] [PubMed] [Google Scholar]
- 17.Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–5. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 18.Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, Kamesh L, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. 2007;39:721–3. doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willcocks LC, Lyons PA, Clatworthy MR, Robinson JI, Yang W, Newland SA, et al. Copy number of FCGR3B, which is associated with systemic lupus erythematosus, correlates with protein expression and immune complex uptake. J Exp Med. 2008;205:1573–82. doi: 10.1084/jem.20072413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nossent JC, Rischmueller M, Lester S. Low copy number of the Fc-γ receptor 3B gene FCGR3B is a risk factor for primary Sjogren's syndrome. J Rheumatol. 2012;39:2142–7. doi: 10.3899/jrheum.120294. [DOI] [PubMed] [Google Scholar]
- 21.McKinney C, Broen JC, Vonk MC, Beretta L, Hesselstrand R, Hunzelmann N, et al. Evidence that deletion at FCGR3B is a risk factor for systemic sclerosis. Genes Immun. 2012;13:458–60. doi: 10.1038/gene.2012.15. [DOI] [PubMed] [Google Scholar]
- 22.Graf SW, Lester S, Nossent JC, Hill CL, Proudman SM, Lee A, et al. Low copy number of the FCGR3B gene and rheumatoid arthritis: a case-control study and meta-analysis. Arthritis Res Ther. 2012;14:R28. doi: 10.1186/ar3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKinney C, Fanciulli M, Merriman ME, Phipps-Green A, Alizadeh BZ, Koeleman BP, et al. Association of variation in Fcγ receptor 3B gene copy number with rheumatoid arthritis in Caucasian samples. Ann Rheum Dis. 2010;69:1711–6. doi: 10.1136/ard.2009.123588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson JI, Carr IM, Cooper DL, Rashid LH, Martin SG, Emery P, et al. Confirmation of association of FCGR3B but not FCGR3A copy number with susceptibility to autoantibody positive rheumatoid arthritis. Hum Mutat. 2012;33:741–9. doi: 10.1002/humu.22031. [DOI] [PubMed] [Google Scholar]
- 25.Mamtani M, Anaya JM, He W, Ahuja SK. Association of copy number variation in the FCGR3B gene with risk of autoimmune diseases. Genes Immun. 2010;11:155–60. doi: 10.1038/gene.2009.71. [DOI] [PubMed] [Google Scholar]
- 26.Marques RB, Thabet MM, White SJ, Houwing-Duistermaat JJ, Bakker AM, Hendriks GJ, et al. Genetic variation of the Fcγ receptor 3B gene and association with rheumatoid arthritis. PLoS One. 2010:5. doi: 10.1371/journal.pone.0013173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thabet MM, Huizinga TW, Marques RB, Stoeken-Rijsbergen G, Bakker AM, Kurreeman FA, et al. Contribution of Fcγ receptor IIIA gene 158V/F polymorphism and copy number variation to the risk of ACPA-positive rheumatoid arthritis. Ann Rheum Dis. 2009;68:1775–80. doi: 10.1136/ard.2008.099309. [DOI] [PubMed] [Google Scholar]
- 28.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 29.Hochberg MC Diagnostic and Therapeutic Criteria Committee of the American College of Rheumatology. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. for the [letter] [DOI] [PubMed] [Google Scholar]
- 30.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 31.Chen JY, Wang CM, Wu YJ, Kuo SN, Shiu CF, Chang SW, et al. Disease phenotypes and gender association of FCRL3 single-nucleotide polymorphism -169T/C in Taiwanese patients with systemic lupus erythematosus and rheumatoid arthritis. J Rheumatol. 2011;38:264–70. doi: 10.3899/jrheum.100437. [DOI] [PubMed] [Google Scholar]
- 32.Chen JY, Wang CM, Ma CC, Luo SF, Edberg JC, Kimberly RP, et al. Association of a transmembrane polymorphism of Fcγ receptor IIb (FCGR2B) with systemic lupus erythematosus in Taiwanese patients. Arthritis Rheum. 2006;54:3908–17. doi: 10.1002/art.22220. [DOI] [PubMed] [Google Scholar]
- 33.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- 34.Hollox EJ, Detering JC, Dehnugara T. An integrated approach for measuring copy number variation at the FCGR3 (CD16) locus. Hum Mutat. 2009;30:477–84. doi: 10.1002/humu.20911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niederer HA, Willcocks LC, Rayner TF, Yang W, Lau YL, Williams TN, et al. Copy number, linkage disequilibrium and disease association in the FCGR locus. Hum Mol Genet. 2010;19:3282–94. doi: 10.1093/hmg/ddq216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu ZT, Tsuchiya N, Kyogoku C, Ohashi J, Qian YP, Xu SB, et al. Association of Fcγ receptor IIb polymorphism with susceptibility to systemic lupus erythematosus in Chinese: a common susceptibility gene in the Asian populations. Tissue Antigens. 2004;63:21–7. doi: 10.1111/j.1399-0039.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- 37.Kyogoku C, Dijstelbloem HM, Tsuchiya N, Hatta Y, Kato H, Yamaguchi A, et al. Fcγ receptor gene polymorphisms in Japanese patients with systemic lupus erythematosus: contribution of FCGR2B to genetic susceptibility. Arthritis Rheum. 2002;46:1242–54. doi: 10.1002/art.10257. [DOI] [PubMed] [Google Scholar]
- 38.Siriboonrit U, Tsuchiya N, Sirikong M, Kyogoku C, Bejrachandra S, Suthipinittharm P, et al. Association of Fcγ receptor IIb and IIIb polymorphisms with susceptibility to systemic lupus erythematosus in Thais. Tissue Antigens. 2003;61:374–83. doi: 10.1034/j.1399-0039.2003.00047.x. [DOI] [PubMed] [Google Scholar]
- 39.Willcocks LC, Carr EJ, Niederer HA, Rayner TF, Williams TN, Yang W, et al. A defunctioning polymorphism in FCGR2B is associated with protection against malaria but susceptibility to systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2010;107:7881–5. doi: 10.1073/pnas.0915133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karassa FB, Trikalinos TA, Ioannidis JP. The FcγRIIIA-F158 allele is a risk factor for the development of lupus nephritis: a meta-analysis. Kidney Int. 2003;63:1475–82. doi: 10.1046/j.1523-1755.2003.00873.x. [DOI] [PubMed] [Google Scholar]
- 41.Li LH, Yuan H, Pan HF, Li WX, Li XP, Ye DQ. Role of the Fcγ receptor IIIA-V/F158 polymorphism in susceptibility to systemic lupus erythematosus and lupus nephritis: a meta-analysis. Scand J Rheumatol. 2010;39:148–54. doi: 10.3109/03009740903292304. [DOI] [PubMed] [Google Scholar]
- 42.Henriques A, Ines L, Carvalheiro T, Couto M, Andrade A, Pedreiro S, et al. Functional characterization of peripheral blood dendritic cells and monocytes in systemic lupus erythematosus. Rheumatol Int. 2012;32:863–9. doi: 10.1007/s00296-010-1709-6. [DOI] [PubMed] [Google Scholar]
- 43.Morris DL, Roberts AL, Witherden AS, Tarzi R, Barros P, Whittaker JC, et al. Evidence for both copy number and allelic (NA1/NA2) risk at the FCGR3B locus in systemic lupus erythematosus. Eur J Hum Genet. 2010;18:1027–31. doi: 10.1038/ejhg.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKinney C, Merriman TR. Meta-analysis confirms a role for deletion in FCGR3B in autoimmune phenotypes. Hum Mol Genet. 2012;21:2370–6. doi: 10.1093/hmg/dds039. [DOI] [PubMed] [Google Scholar]
- 45.Tsuboi N, Asano K, Lauterbach M, Mayadas TN. Human neutrophil Fcγ receptors initiate and play specialized nonredundant roles in antibody-mediated inflammatory diseases. Immunity. 2008;28:833–46. doi: 10.1016/j.immuni.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coxon A, Cullere X, Knight S, Sethi S, Wakelin MW, Stavrakis G, et al. FcγRIII mediates neutrophil recruitment to immune complexes: a mechanism for neutrophil accumulation in immune-mediated inflammation. Immunity. 2001;14:693–704. doi: 10.1016/s1074-7613(01)00150-9. [DOI] [PubMed] [Google Scholar]
- 47.Mueller M, Barros P, Witherden AS, Roberts AL, Zhang Z, Schaschl H, et al. Genomic pathology of SLE-associated copy-number variation at the FCGR2C/FCGR3B/FCGR2B locus. Am J Hum Genet. 2013;92:28–40. doi: 10.1016/j.ajhg.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]