

Abstract
Background and Purpose
Activation of hepatic stellate cells (HSCs) is a crucial step in the pathogenesis of hepatic fibrosis. Histone deacetylase (HDAC) is an attractive target in liver fibrosis because it plays a key role in gene expression and cell differentiation. We have developed a HDAC inhibitor, N-hydroxy-7-(2-naphthylthio)heptanomide (HNHA), and investigated the anti-fibrotic activity of HNHA in vitro and in vivo.
Experimental Approach
We investigated the anti-fibrotic effect of HNHA on mouse and human HSC activation in vitro and in the liver of bile duct-ligated (BDL) rats in vivo using cell proliferation assays, cell cycle analysis, biochemical assay, immunohistochemistry and Western blots. Liver pathology was assessed with histochemical techniques.
Key Results
HNHA inhibited proliferation and arrested the cell cycle via p21 induction in HSCs. In addition, HNHA induced apoptosis of HSCs, which was correlated with reduced COX-2 expression, NF-κB activation and cell death signals. HNHA restored liver function and decreased the accumulation of extracellular matrix in the liver via suppression of HSC activation in BDL rats in vivo. HNHA administration also increased survival in BDL rats.
Conclusions and Implications
HNHA improved liver function, suppressed liver fibrosis and increased survival of BDL rats, accompanied by reduction of cell growth, activation and survival of HSCs. These findings show that HNHA may be a potent anti-fibrosis agent against hepatic fibrosis because of its multi-targeted inhibition of HSC activity in vivo and in vitro.
Links to online information in IUPHAR/DB and BPS Concise Guide to PHARMACOLOGY
| Targets | Ligands |
|---|---|
| Caspases | Bilirubin |
| Collagen | Ibuprofen |
| COX-2, cyclooxygenase 2 | Meloxicam |
| HDAC, histone deacetylase | SAHA (vorinostat) |
| MMP, matrix metallopeptidase | TGF-β1 |
This table lists key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Liver fibrosis is an overactive wound healing process in which excessive connective tissue builds up in the liver and results in the distortion of hepatic architecture and liver failure (Branton and Kopp, 1999). Chronic liver injury activates and transforms quiescent hepatic stellate cells (HSCs), which transdifferentiate into activated HSCs, called myofibroblasts, acquiring contractile, proliferative and fibrogenic properties as the primary cellular source of extracellular matrix (ECM) components in the liver (Moreira, 2007). As a consequence, the excessive accumulation of ECM initiates fibrosis by releasing inflammatory mediators such as PDGF, TGF and connective tissue growth factor (Pellicoro et al., 2012). Therefore, suppression of HSC activation and proliferation as well as induction of apoptosis in activated HSCs have been proposed as therapeutic strategies for the treatment and prevention of hepatic fibrosis (Elsharkawy et al., 2005).
Anti-fibrotic drugs are not efficiently taken up by activated HSCs and are often toxic to hepatic cells, producing unwanted side effects, and there are no proven drugs to treat liver fibrosis in patients (Bataller and Brenner, 2005). Recent studies suggest that liver fibrosis can be blocked by induction of histone hyperacetylation by histone deacetylase (HDAC) inhibitors (Kaimori et al., 2010; Liu et al., 2013). We have already described the HDAC inhibitor, N-hydroxy-7-(2-naphthylthio)heptanomide (HNHA), as inhibiting nuclear HDAC enzyme activity in human umbilical endothelial cells and breast cancer cells, an effect that was accompanied by histone hyperacetylation and cell cycle arrest, as well as inhibition of ECM regulators such as MMPs (Kim et al., 2007; Park et al., 2011). However, the effects of HNHA on the regulation of fibrogenic responses in HSCs and liver fibrosis have not been studied.
Models of liver injury following bile duct ligation (BDL) in rats have been widely used to dissect the molecular mechanisms underlying acute and chronic liver injury. Here, we evaluated the biological effects of HNHA and other known treatments on the fibrogenic responses, including the proliferation, apoptosis and activation of mouse and human HSCs, and have also evaluated the anti-fibrotic effect of HNHA in liver fibrosis induced by BDL in rats.
Methods
Isolation and culture of HSCs
The livers of 6-week-old male C57BL/6 mice (Orient Bio Inc., Seong-nam, South Korea) were sequentially digested with pronase (Roche Applied Science, Mannheim, Germany) and type IV collagenase (Sigma-Aldrich, St Louis, MO, USA) by in situ perfusion for primary HSC isolation. Hepatocytes were removed by centrifuging the digested liver at 50× g for 3 min. The non-parenchymal cells present in the supernatant were laid on top of a gradient of arabinogalactan (Sigma-Aldrich; with densities of 1.035, 1.045, 1.058 and 1.085), followed by centrifugation at 74 000× g for 40 min at 25°C. The fraction containing HSCs was recovered from the interface between the medium (1.045) and the lowest density (1.035). The purity of HSCs was evaluated by examining the characteristic stellate shape of the cells with phase contrast microscopy and by the presence of lipid droplets by autofluorescence using an excitation light of 320 nm. The viability of HSCs assessed by Trypan blue staining always exceeded 95%. Primary mouse HSCs were cultured in DMEM (Life Technologies, Inc., Grand Island, NY, USA) with 10% FBS and 100 units·mL−1 penicillin-streptomycin and were maintained at 37°C in an atmosphere of 5% CO2. We grew human primary HSCs from human resection specimens (Sciencell, San Diego, CA, USA) and used them for up to five passages. For experiments, we plated cells at a density of 3 × 104 cells·cm−2. When the cultures reached 70–80% confluence, they were trypsinized and passaged.
Cell proliferation assay, cell viability assay and cell cycle phase distribution by flow cytometric DNA analysis
Mouse or human primary HSCs were pre-cultured for 24 h at a density of 2.3 × 105 cells·mL−1 and the growth medium was replaced with an experimental medium containing ibuprofen, meloxicam, suberoylanilide hydroxamic acid (SAHA) or HNHA at final concentrations of 10 μM in cell proliferation assay and cell cycle analysis, or ranging from 1 to 160 μM in the cell viability assay. Growth medium containing 0.1% (v/v) saline was used as a vehicle control. These assays were performed as previously described (Park et al., 2011).
Western blot analysis
Western blots were carried out as previously reported by Park et al. (2011) using primary antibodies against Bcl-2, Bcl-w, Bcl-xL, HDAC1, HDAC4, Bad, Bax, Bak, COX-2, cyclin D1 (all from Abcam, Cambridge, UK); phospho-IκB-α (Cell Signaling Technology, Danvers, MA, USA); phospho-NF-κB p65, IκB-α (from Santa Cruz Biotechnology, Santa Cruz, CA, USA); p21 and p53 (Abcam); pro-caspase-3 and cleaved-caspase-3, pro-caspase-7 and cleaved-caspase-7, pro-caspase-8 and caspase-8, pro-caspase-9 and cleaved-caspase-9 (all from Santa Cruz Biotechnology), collagen type I (COL-I) (R&D Systems, Minneapolis, MN, USA), α-smooth muscle actin (α-SMA) (Abcam), TGF-β1 (R&D Systems), MMP-2 and MMP-9 (Abcam), and β-actin (Santa Cruz Biotechnology).
RNA extraction and RT-PCR
Total RNA prepared from HSCs was extracted with RNeasy Mini Kit (Qiagen, Valencia, CA, USA), including treatment with DNase I to prevent genomic DNA contamination using RURBO DNA-free Kit (Ambion, Foster City, CA, USA) according to the manufacturer's instructions. RT-PCR was carried out with total RNA (1 μg) using the one-step RT-PCR kit (Qiagen) according to the manufacturer's protocol. The resultant cDNA was used to carry out 40 PCR cycles consisting of 15 s at 95°C, 30 s at 57°C and 60 s at 72°C on an ABI Prism 7000 sequence detection system (Applied Biosystems, Foster City, CA, USA). The reactions were performed using SYBR green 1 (Qiagen), using primers for p21 (forward 5′-tccacagcgatatccagaca-3′, reverse 5′-ggacatcaccaggattggac-3′) and Gapdh (forward 5′-atcaagaaggtggtgaagcggaa-3′, reverse 5′-tggaagagtgggagttgctgttga-3′). Each PCR was set up in triplicate wells in a total volume of 25 μL. The reaction mixture contained the cDNA equivalent of 20 ng total RNA. The quantitative values of the genes of interest were normalized using Gapdh as the endogenous reference, and fold-increase over control values was calculated using the relative quantification method of 2−ΔΔCt.
Serum biochemistry
For testing the liver function, the activities of aspartate transaminase (AST), alanine transaminase (ALT) and the contents of total bilirubin (Tbil) in the serum were determined after 3 weeks of experimental treatment using an automated analyser (RA-XT; Technicon, Tarrytown, NY, USA).
Animals and experimental design
All animal care and experimental procedures complied with local guidelines and were approved by the Animal Experiments Committee of Yonsei University. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 111 animals (106 rats, 5 mice) were used in the experiments described here.
Male Sprague Dawley rats (Orient Bio Inc.) weighing 250–300 g (7 weeks old) were used. All animals were housed in cages (five mice or two rats per cage) for 7 days. The light cycle was controlled to provide 12 h light and 12 h darkness; temperature was 22°C and humidity was 40–60%. A standard diet of rodent pellets and tap water (membrane-filter purified and autoclaved) were provided ad libitum. Rats were anaesthetized with an i.m. injection of Zoletil (30 mg·kg−1; Virbac, Carros, France). After a midline abdominal incision, the common bile duct (CBD) was doubly ligated with 4-0 silk sutures and cut with #11 blade. Sham operation was just exposure of the CBD by passing the cotton applicator below the foramen of Winslow. After saline irrigation, abdominal closure was performed with continuous 3-0 silk suture. After the surgical procedure, animals were divided into five groups in efficacy experiments and survival experiments: sham operation group (SHAM, n = 10); BDL (+Veh) group (n = 20), in which saline was i.p. injected beginning at 24 h after BDL; BDL + meloxicam group (n = 20), in which meloxicam was i.m. injected at 1.6 mg·kg−1 beginning at 24 h after BDL; BDL + SAHA group (n = 20); and BDL + HNHA group (n = 20), in which the treatments (30 mg·kg−1) were given i.p. 24 h after BDL. All drugs were administered once every 3 days for 3 weeks, in efficacy experiments or until death, in survival experiments. For in vivo toxicity experiments, meloxicam was i.m. injected (1.6 mg·kg−1) or HNHA (30 mg·kg−1), beginning at 24 h after the sham operation (n = 16).
Tissue preparation
After 3 weeks, all animals were killed, and the blood, spleens and livers were collected and the organ weights were determined. Plasma was separated by centrifugation at 2300× g for 5 min at room temperature within 15 min of sample collection. Livers were sliced into several parts. Some were immediately snap-frozen in liquid nitrogen for further analysis and kept at −80°C. Other samples of liver were immersed into 10% neutral buffered formalin solution for histopathological examinations.
Haematoxylin and eosin (H&E) staining, Masson trichrome staining and immunohistochemistry
Liver biopsy specimens fixed in 10% buffered formalin solution were embedded with paraffin. Sections (5 μm) were stained with H&E. The degree of fibrosis was assessed with Masson trichrome staining according to the protocol provided by the manufacturer (Sigma-Aldrich). This procedure stains nuclei dark red, cytoplasm and muscle fibres red, and ECM components blue.
Immunohistochemical analysis was conducted as previously reported (Park et al., 2011) using primary antibodies against acetyl histone H3 (1:200; Cell Signaling Technology, Beverly, MA, USA), HDAC-1 (1:200), COL-I (1:200; Cell Signaling Technology), α-SMA (1:20), TGF-β1 (1:100; Abcam) and COX-2 (1:500; Santa Cruz Biotechnology). The images were quantified by image analysis using Metamorph 6.0 software (Universal Imaging Corp., Downingtown, PA, USA) according to the software manual. Total area of collagen-stained area and its optical density were calculated from three fields of every slide and compared between groups by non-parametric statistical methods.
Data analysis
Data are expressed as means ± SEM, unless otherwise stated. Statistical analysis was performed with SPSS software, version 11.5 (SPSS Inc., Chicago, IL, USA). Differences between means were assessed by two-tailed Student's t-test or by one-way anova followed by Bonferroni's multiple-comparison post hoc test. P values lower than 0.05 were considered statistically significant.
Materials
HNHA and SAHA (vorinostat) were provided by Dr H-J. Kwon. Meloxicam and PDGF-B were purchased from R&D Systems.
Results
HNHA inhibits the proliferation and activation of HSCs and induces the cell cycle arrest and apoptosis of HSCs
HNHA treatment for 1 week showed greater inhibition of mouse and human primary HSC proliferation, a feature of HSC activation, compared with ibuprofen (a non-selective COX inhibitor), meloxicam (a COX-2 selective inhibitor) and SAHA (another HDAC inhibitor) (Figure 1A and B).
Figure 1.

Effects of HNHA on proliferation, apoptosis and activation of HSCs. (A, B) Cell growth assay of mouse (A) or human (B) primary HSCs using cell counting. (C) Western blot analysis of HSC activation markers in mouse HSCs. (D) Cell cycle phase distribution by flow cytometric DNA in mouse HSCs. (E) Western blot analysis of cell proliferation protein levels in activated mouse HSCs. (F) Quantitative RT-PCR analysis of p21 in activated mouse HSCs. Mouse primary HSCs were cultured in complete media with increasing concentrations of drugs for 48 h. (G) Cell viability assay. Mouse primary HSCs were cultured in complete media with increasing concentrations of drugs for 96 h (n = 3). (H) Western blot analysis of apoptosis-related proteins and HDAC levels in mouse HSCs. In the experiments summarized in (C)–(F) and in (H), mouse HSCs were incubated with ibuprofen, meloxicam, SAHA or HNHA at a final concentration of 10 μM. Data shown are means ± SD (n = 4 except for G). *P < 0.05, **P < 0.01, ***P < 0.005 versus control or ibuprofen.
To assess further the effects of HNHA on HSC activation, the expression of COL-I, α-SMA and TGF-β1 in PDGF-stimulated HSCs was examined by Western blot analysis. Stimulation of mouse HSCs with PDGF (20 ng·mL−1) for 24 h significantly induced the expression of α-SMA, TGF-β1 and COL-I expression, activation markers of myofibroblasts. HNHA treatment suppressed this increased expression of α-SMA, TGF-β1 and COL-I in activated HSCs (Figure 1C).
Incubation with HNHA (10 μM) arrested mouse HSCs in the sub-G0/G1 and G0/G1 phase (about 90%) more effectively than the other compounds used (about 70%; Figure 1D). Consistent with the observed effects on cell proliferation, this concentration of HNHA also reduced the expression of COX-2, the phosphorylation of IκB-α and NF-κB p65, and the degradation of IκB-α in HSCs, unstimulated or stimulated with PDGF, a key molecule in the sustained activation and proliferation of HSC survival (Figure 1E). Furthermore, HNHA induced p21 and p53, a well-known arrestor of the cell cycle, but reduced cyclin D1, a positive regulator of the cell cycle (Figure 1E, F, H). These results were compatible with the concentration-dependent cytotoxicity of HNHA, which induced greater loss of viability at low concentrations (1–10 μM) than the other inhibitors used (Figure 1G and Table 1).
Table 1.
Inhibition of HSC proliferation by drug treatments
| Meloxicam (μM) | SAHA (μM) | HNHA (μM) | |
|---|---|---|---|
| Mouse primary HSCs | 54 (±1.0) | 23 (±1.7) | 15 (±1.0) |
| Human primary HSCs | 50 (±1.1) | 20 (±1.9) | 12 (±1.1) |
Each data point represents the mean IC50 value (±SD) from three independent experiments performed in triplicate using sulforhodamine B to assay proliferation.
To investigate pro-apoptotic signalling pathways activated following the incubation with these inhibitors, the expression of pro- (Bad, Bax and Bak) and anti-apoptotic (Bcl-2, Bcl-w and Bcl-xL) members of the Bcl-2 family and the activation of caspase-3, caspase-7, caspase-8 and caspase-9, major effectors in apoptosis, in HSCs as well as the expression of HDACs (HDAC1 and HDAC4) were investigated by Western blotting. HNHA led to increased expression of pro-apoptotic members of the Bcl-2 family and cleavage of pro-caspase in HSCs (Figure 1H), which are barely detectable in PDGF-treated HSCs. Moreover, HNHA and SAHA almost abolished PDGF-induced expression of anti-apoptotic members of the Bcl-2 family and decreased HDAC protein levels, key molecules in the signalling pathways of HSC survival, but the COX inhibitors (ibuprofen and meloxicam) were less effective.
HNHA restores liver function in BDL rats
BDL, the most commonly used animal model of obstructive cholestasis, causes obstructive jaundice and cholestasis, which over time leads to histological changes in the liver, including the development of cirrhosis. After 3 weeks of BDL, livers were grossly enlarged and had lost compliance, compared with those from sham-operated rats. BDL caused a significant increase in liver/body weight ratio, serum AST, ALT and Tbil levels, compared with sham-operated rats. In the BDL + HNHA group, the changes in body weights and liver/body weight ratios were significantly less than in the other groups (Table 2). The increased serum activities of AST, ALT and the contents of Tbil were attenuated in the BDL + HNHA group compared with the BDL, BDL + meloxicam or BDL + SAHA groups (Table 3). In addition, there was no evidence of systemic toxicity attributable to the doses of meloxicam, SAHA and HNHA used here. These data suggest that HNHA was the most effective agent for decreasing hepatic damage after BDL in this liver fibrosis model.
Table 2.
Effects of HNHA on body and organ weight changes in BDL rats
| Groups | Body weight (g) | Organ weight | ||||
|---|---|---|---|---|---|---|
| Liver (g) | Spleen (g) | Liver/body (%) | Spleen/body (%) | |||
| SHAM | Veh | 346 ± 7.9 | 14.00 ± 0.29 | 1.07 ± 0.07 | 3.86 ± 0.11 | 0.30 ± 0.05 |
| Meloxicam | 368 ± 9.0 | 14.35 ± 0.33 | 1.12 ± 0.05 | 3.89 ± 0.10 | 0.30 ± 0.02 | |
| SAHA | 360 ± 4.2 | 13.56 ± 0.19 | 1.08 ± 0.02 | 3.76 ± 0.05 | 0.29 ± 0.03 | |
| HNHA | 353 ± 5.9 | 13.65 ± 0.17 | 1.03 ± 0.09 | 3.86 ± 0.07 | 0.29 ± 0.05 | |
| BDL | Veh | 298 ± 4.7 | 21.13 ± 1.02 | 2.09 ± 0.12 | 7.08 ± 0.95 | 0.70 ± 0.11 |
| Meloxicam | 308 ± 5.5*;##; | 18.00 ± 0.97*;##; | 1.80 ± 0.55*;##; | 5.84 ± 0.45*;##; | 0.58 ± 0.05*;#; | |
| SAHA | 311 ± 6.4*;#; | 15.74 ± 0.51**;#; | 1.52 ± 0.43**;#; | 5.06 ± 0.41**;#; | 0.49 ± 0.52**;#; | |
| HNHA | 324 ± 5.1**; | 14.05 ± 2.06***; | 1.32 ± 0.55***; | 4.33 ± 0.54**; | 0.40 ± 0.05**; | |
Meloxicam was injected i.m. at a daily dose of 1.6 mg, and SAHA and HNHA were injected i.p. in rats at a dose of 30 mg·kg−1 once every 3 days for 3 weeks. Data shown are means ± SEM.
P < 0.05,
P < 0.01,
P < 0.005 versus BDL;
P < 0.05,
P < 0.01 versus BDL + HNHA.
Table 3.
Serum biochemistry
| AST (IU·L−1) | ALT (IU·L−1) | Tbil (mg·L−1) | |
|---|---|---|---|
| SHAM | 69.73 ± 7.98 | 27.17 ± 4.84 | 1.1 ± 0.2 |
| BDL | 342.11 ± 81.92 | 104.84 ± 38.12 | 81.4 ± 29.7 |
| BDL + meloxicam | 189.45 ± 47.12*;## | 79.48 ± 29.59*;## | 68.4 ± 14.5*;## |
| BDL + SAHA | 113.17 ± 19.75**# | 59.08 ± 18.22**# | 50.7 ± 11.1**# |
| BDL + HNHA | 98.35 ± 20.44**; | 46.35 ± 17.24***; | 34.2 ± 9.5***; |
| SHAM + meloxicam | 77.35 ± 10.01 | 30.27 ± 4.01 | 1.8 ± 0.4 |
| SHAM + SAHA | 76.98 ± 9.24 | 29.59 ± 3.87 | 1.7 ± 0.4 |
| SHAM + HNHA | 76.14 ± 9.97 | 29.87 ± 3.99 | 1.6 ± 0.3 |
Meloxicam was injected i.m. at a daily dose of 1.6 mg, and SAHA and HNHA were injected i.p. in rats at a dose of 30 mg·kg−1 once every 3 days for 3 weeks. Serum levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT) and total bilirubin (Tbil) of sham-operated (SHAM) and bile duct ligation (BDL) rats were determined. Data shown are means ± SEM (n = 25).
P < 0.05,
P < 0.01,
P < 0.005 versus BDL;
P < 0.05,
P < 0.01 versus BDL + HNHA.
HNHA ameliorates histopathological deterioration and ECM accumulation in BDL-liver
Each lobe of the BDL-liver was equally affected grossly by BDL upon examination by light microscopy of liver sections stained with H&E. There was marked architectural distortion of lobular structure and extensive bile duct proliferation infiltrating into hepatocytes with periportal fibrogenesis and occasional fibrogenesis in the parenchymal area, suggesting activation of HSCs. The groups treated with meloxicam, SAHA or HNHA showed an overall reduction of pathological changes compared with the BDL group. The BDL + HNHA group showed a greater suppression of lower bile duct proliferation and periportal fibrosis compared with the meloxicam or SAHA-treated groups (Figure 2A). Because liver fibrosis is characterized by an increased accumulation of ECM, mainly collagens, we assessed the collagen contents based upon Masson's trichrome staining and Western blots for type I collagen. Consistent with the histopathology, HNHA markedly suppressed collagen accumulation in BDL rat livers (Figure 2B) and COL-I expression (Figure 2C). This suggests that HNHA improved the histological changes and reduced ECM accumulation in liver fibrosis.
Figure 2.
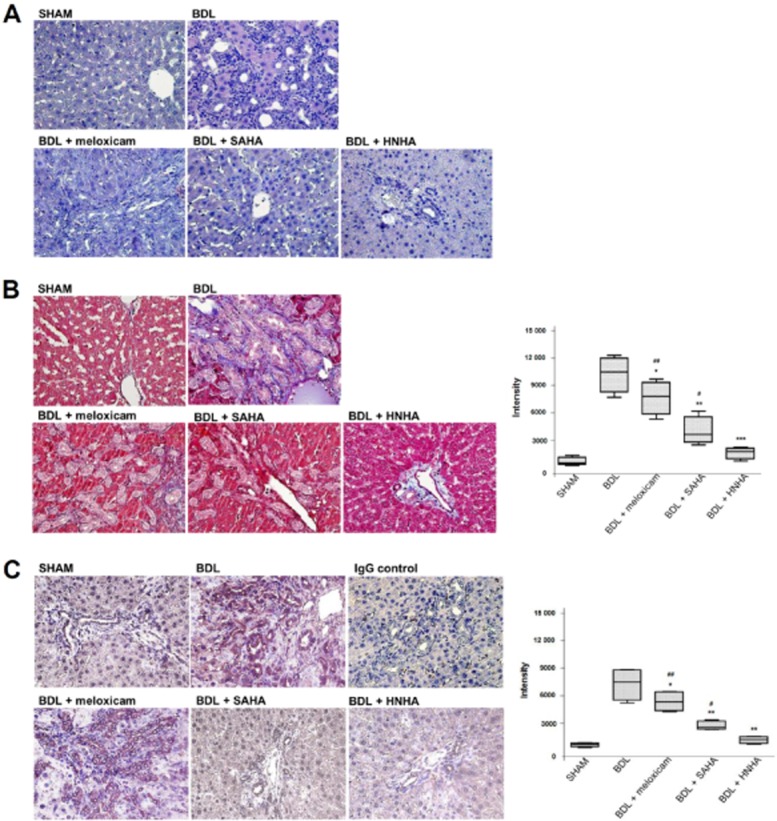
Effects of HNHA on histopathology and ECM accumulation of BDL-livers. (A) The histological changes in the livers of bile duct ligated (BDL) rats as shown by haematoxylin and eosin (H&E) staining. (B, C) Representative staining (left) and densitometric analysis (right) of Masson's trichrome staining (B) and collagen staining (C) of liver tissues of BDL rats. Masson's trichrome staining stains nuclei black, cytoplasm and muscle fibres red, and ECM components blue. Negative control slides (IgG control) were treated with isotype-matched IgG. Quantitative data are shown as box and whisker plots with the median indicated as a horizontal bar, first and third quartiles as the box and the range shown by the whiskers (n = 15). *P < 0.05, **P < 0.01, ***P < 0.005 versus BDL; #P < 0.05, ##P < 0.01 versus BDL+HNHA.
HNHA suppresses activation of myofibroblasts induced by BDL
Because the appearance of α-SMA-positive myofibroblasts is considered as a key event in the progression of liver fibrosis and TGF-β1, predominantly expressed by HSCs, is a key mediator for transdifferentiation, we assessed α-SMA or TGF-β1-positive cells by morphometric quantification to ascertain the accumulation of fibrogenic myofibroblasts. Activated myofibroblasts, as shown by positive staining for α-SMA, were predominant in the peri-ductular zone 1 area and, to a lesser degree, in the peri-sinusoidal area in zone 2 and zone 3. Immunohistochemical staining for TGF-β1 showed that the positively stained brown-coloured cells were mainly proliferating bile ductules and limiting plate hepatocytes. However, liver samples from rats treated with meloxicam, SAHA or HNHA for 3 weeks after BDL showed less bile duct proliferation and weak or no cytoplasmic immunostaining for α-SMA (Figure 3A) compared with BDL rats. Liver sections from meloxicam, SAHA and HNHA group rats showed weak or no cytoplasmic TGF-β1 staining in bile duct epithelial cells and limiting plate hepatocytes (Figure 3B). Treatment with HNHA was more effective in suppressing α-SMA and TGF-β1 in BDL-livers than treatment with meloxicam or SAHA. We then compared COX-2 expression in liver tissue samples. With vehicle treatment after BDL, COX-2 was expressed mainly in the proliferating bile ductules and, to a lesser degree, in hepatocytes. In livers from BDL rats receiving meloxicam, SAHA or HNHA, there was decreased hepatic COX-2 expression (Figure 3C) with the lowest expression in the HNHA-treated group. These results were confirmed by Western blot analysis of total lysates of livers, which showed that production of COL-I, α-SMA, TGF-β1, COX-2, MMP-2 and MMP-9 appeared to be most reduced in the BDL + HNHA group compared with other groups (Figure 3D). The acetylation of histone H3 in liver tissue was much stronger in the HNHA-treated group than in the BDL group (Supporting Information Fig. S2), whereas the level of HDAC1 was decreased in the HNHA-treated group compared with the BDL group (Supporting Information Fig. S3).
Figure 3.

Effects of HNHA on myofibroblast activation in BDL-liver. Representative staining (left) and densitometric analysis (right) of α-SMA (A), TGF-β1 (B) and COX-2 (C) staining of liver tissues of BDL rats. (D) Representative Western blot of COL-1, α-SMA, TGF-β1, COX-2, MMP-2 and MMP-9 in liver tissues of BDL rats (n = 4). Quantitative data are shown as box and whisker plots with the median indicated as a horizontal bar, first and third quartiles as the box and the range shown by the whiskers (n = 15 except for D). *P < 0.05, **P < 0.01 ***P < 0.005 versus BDL; #P < 0.05, ##P < 0.01 versus BDL + HNHA.
HNHA increases survival of BDL rats
Meloxicam, SAHA and HNHA were injected i.p. to evaluate their effect on the survival of BDL rat, over 63 days. The survival rate of the HNHA-treated group was higher than that of the other groups (Figure 4). Administration of the drug treatments to rats without BDL did not affect survival, which was the same as that for sham-operated animals, indicating no systemic toxicity attributable to meloxicam, SAHA and HNHA. These findings were consistent with the results of histopathology or liver function analysis. Taken together, these results showed that HNHA improved liver function and overall survival of rats with BDL.
Figure 4.
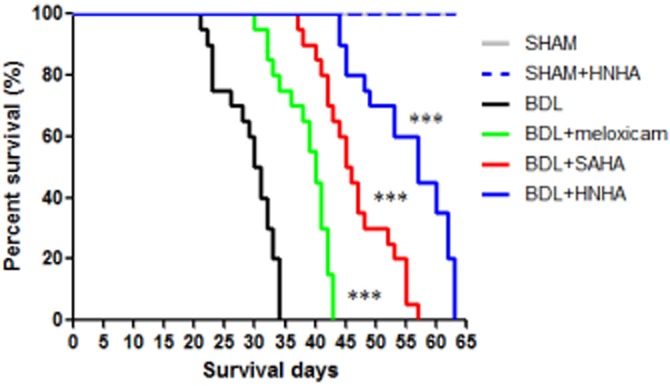
Effects of HNHA on survival in BDL rats. Kaplan–Meier plot for overall survivals. Differences between survival curves for 63 days in groups (SHAM group, n = 10; SHAM + HNHA, n = 16; BDL group, n = 20; BDL + meloxicam group, n = 20; BDL + SAHA group, n = 20; BDL + HNHA group, n = 20) were compared using a log-rank test. ***P < 0.005 versus BDL.
Discussion
The induction of apoptosis in activated HSCs has emerged as a treatment strategy for liver fibrosis (Schuppan and Kim, 2013). HDAC inhibitors can modulate the expression of apoptosis-related genes (Cao et al., 2001) and recent studies have shown that HDAC4 is accumulated during HSC transdifferentiation (Mann et al., 2007; Qin and Han, 2010). For this reason, the inhibition of HDAC has been considered as a promising suppressor of HSC proliferation and activation in vitro and in vivo (Rombouts et al., 2002; Watanabe et al., 2011). HNHA is a new synthetic HDAC inhibitor with better pharmacological properties than other HDAC inhibitors including SAHA and was more effective in inducing G1/S cell cycle arrest in breast cancer cells, compared with SAHA (Kim et al., 2007). The anti-proliferative effect of HNHA on HSCs appeared also to be mediated through cell cycle arrest. In concordance with the earlier data, we found here that HNHA suppressed cell proliferation and increased, very effectively, cell cycle arrest at the G0/G1 phase in mouse and human primary HSC proliferation than ibuprofen, meloxicam or SAHA. These results were related to an increase in the endogenous cyclin-dependent kinase inhibitor, p21, and p53 and accompanied by a decrease in cyclin D1 expression by HNHA, which led to profound inhibition of HSC growth. It is well established that histone hyperacetylation induces apoptosis via p21, whose promoter is amplified from acetylated histone H3- or H4-associated chromatin by HDAC inhibition (Blagosklonny et al., 2002). HNHA also strongly rescued histone acetylation in FM3A and MCF-7 cells (Kim et al., 2007; Park et al., 2011), a function possibly linked to its ability to arrest the cell cycle in HSCs.
Several cytokines are central to the pathogenesis of fibrosis and the process of HSC activation, particularly PDGF and TGF-β1. PDGF induces the activation of the downstream signalling kinase ERK in activated HSCs, which is associated with myofibroblast differentiation, cell proliferation and stimulation of ECM production, the most important event in tissue fibrogenic progression (Marra et al., 1999; Friedman, 2000; Reif et al., 2003). Simultaneously, inhibition of PDGF signalling pathways have also been shown to induce HSC apoptosis and attenuate liver fibrosis (Borkham-Kamphorst et al., 2004). Furthermore, Bcl-2 enhances the resistance to apoptosis in activated HSCs, which may contribute to the development of fibrosis, while increased HSC apoptosis was associated with reduced anti-apoptotic Bcl-2 expression in the liver (Novo et al., 2006). HNHA showed marked inhibition of HSC activation and Bcl-2 expression and led to activation of caspases in PDGF-stimulated HSCs. Therefore, our results suggested that HNHA not only significantly inhibited the proliferation and activation of HSCs through suppression of NF-κB activation but also had a marked apoptotic effect on HSCs through inhibition of Bcl-2 activation, which is important for fibrogenesis.
Quiescent HSCs do not express COX-2 but activated HSCs do express COX-2, suggesting that the COX-2/PG pathway is involved in hepatic fibrogenesis (Rahman et al., 2000; Cheng et al., 2002). Because the effects of non-selective COX inhibitors such as ibuprofen on the fibrogenic response in HSCs and in the liver still remain controversial (Rahman et al., 2000; Cheng et al., 2002), specific inhibition of COX-2 would appear to be safer. Recent studies reported that selective COX-2 inhibitors, including meloxicam, suppressed proliferation of human HSCs (Furst, 1997). Interestingly, inhibitors of HDAC suppress COX-2 expression by blocking the induction of c-Jun and lead to the inhibition of viability and the induction of apoptosis (Yamaguchi et al., 2005). In our experiment, HNHA mainly acted on portal myofibroblasts in cholestasis, by suppressing COX-2 in bile duct epithelium, resulting in diminished collagen formation in activated myofibroblasts. As shown by histology and image analysis of collagen formation in the present study, HNHA showed powerful repression of bile duct proliferation and periportal fibrosis. Cholestatic liver fibrosis led to hepatocyte apoptosis/necrosis during acute liver damage, transformation of portal fibroblast or HSCs to activated forms of myofibroblasts producing ECM, proliferation of bile duct and increase in secretion of cytokines such as PDGF and TGF-β (Ramm et al., 2009). In our experiments, TGF-β1 was strongly stained in hepatocyte and HSCs and, to a lesser degree, in cholestatic liver, and this staining was diminished by HNHA administration. Consequently, HNHA suppressed α-SMA expression and collagen formation in cholestasis induced by BDL as well as PDGF and TGF-β, effects confirmed in primary HSCs. In addition to expressing type I collagen, activated HSCs also express MMPs, which are important in the ECM remodelling that accompanies most liver diseases. Our previous study demonstrated that treatment of tumour cell lines with the HDAC inhibitor resulted in modulation of secreted MMP-2 and MMP-9, accompanied by global histone H3 and H4 hyperacetylation (Rodriguez-Salvador et al., 2005). As shown in our present experiments, HNHA inhibited MMP-2 and MMP-9 production in activated HSCs and BDL livers. Because of their role in fibrosis, the inhibition of TGF-β, α-SMA, MMP and COX-2 expression in the present study showed that HNHA could be an effective treatment for fibrotic diseases.
HNHA showed no toxic effect on hepatocytes at the concentrations used in this study (data not shown) and restored the body weight of BDL rats. In addition, a hepatoprotective activity of HNHA was expressed through the reduction in ALT, AST and TBil, which offers an additional therapeutic advantage of HNHA, compared with the other treatments, as it could both improve hepatocyte integrity and reduce stellate cell activation, as two independent but complementary activities. Furthermore, HNHA increased survival in BDL rats. As HNHA itself did not affect liver function and improved survival of BDL rats, this HDAC inhibitor may serve both for the chemoprevention and for the treatment of biliary fibrosis.
In conclusion, we have demonstrated that treatment with HNHA, a new HDAC inhibitor, markedly attenuated the process of liver fibrosis both in vitro and in vivo, showing a marked decrease of HSC proliferation and increased apoptosis, as well as suppression of profibrogenic factors such as PDGF and TGF-β1. Our data also provided the first in vivo evidence that HNHA increased survival in the BDL-induced liver fibrosis model. Although further studies to determine drug doses from long-term studies are clearly required, our present results encourage the development of HNHA as a possible treatment for hepatic fibrosis.
Acknowledgments
The authors acknowledge the support for every experiment; special thanks to Ryoo Sun Ha, Chief Executive Officer, DAWIN bio corp. This work was supported by a faculty research grant from Yonsei University College of Medicine for 6-2012-0172 and the Brain Korea 21 Project for Medical Science, Yonsei University. H. J. K. was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (NRF-2009-0092964, 2010-0017984).
Glossary
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BDL
bile duct ligation
- COL-I
collagen type I
- ECM
extracellular matrix
- H&E
haematoxylin and eosin
- HNHA
N-hydroxy-7-(2-naphthylthio)heptanomide
- HSC
hepatic stellate cell
- SAHA
suberoylanilide hydroxamic acid
- Tbil
total bilirubin
- α-SMA
α-smooth muscle actin
Conflict of interest
The authors disclose no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12590
Figure S1 Quantification of Western blots. The box and whisker plots show median, with first and third quartiles (as box) and range (as whiskers). Data are from n = 4 experiments. *P < 0.05, **P < 0.01, ***P < 0.005 versus PDGF, control, or BDL.
Figure S2 Effects of HNHA on acetylation of histone H3 in BDL-liver. Representative staining (A) and densitometric analysis (B) of acetyl histone H3. The box and whisker plots show median, with first and third quartiles (as box) and range (as whiskers). Data are from n = 5 experiments. *P < 0.05, **P < 0.01, ***P < 0.005 versus BDL.
Figure S3 Effects of HNHA on levels of HDAC1 in BDL-liver. Representative staining (A) and densitometric analysis (B) of HDAC1 levels. The box and whisker plots show median, with first and third quartiles (as box) and range (as whiskers). Data are from n = 5 experiments. **P < 0.01, ***P < 0.005 versus BDL.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny MV, Robey R, Sackett DL, Du L, Traganos F, et al. Histone deacetylase inhibitors all induce p21 but differentially cause tubulin acetylation, mitotic arrest, and cytotoxicity. Mol Cancer Ther. 2002;1:937–941. [PubMed] [Google Scholar]
- Borkham-Kamphorst E, Stoll D, Gressner AM, Weiskirchen R. Antisense strategy against PDGF B-chain proves effective in preventing experimental liver fibrogenesis. Biochem Biophys Res Commun. 2004;321:413–423. doi: 10.1016/j.bbrc.2004.06.153. [DOI] [PubMed] [Google Scholar]
- Branton MH, Kopp JB. TGF-beta and fibrosis. Microbes Infect. 1999;1:1349–1365. doi: 10.1016/s1286-4579(99)00250-6. [DOI] [PubMed] [Google Scholar]
- Cao XX, Mohuiddin I, Ece F, McConkey DJ, Smythe WR. Histone deacetylase inhibitor downregulation of bcl-xl gene expression leads to apoptotic cell death in mesothelioma. Am J Respir Cell Mol Biol. 2001;25:562–568. doi: 10.1165/ajrcmb.25.5.4539. [DOI] [PubMed] [Google Scholar]
- Cheng J, Imanishi H, Amuro Y, Hada T. NS-398, a selective cyclooxygenase 2 inhibitor, inhibited cell growth and induced cell cycle arrest in human hepatocellular carcinoma cell lines. Int J Cancer. 2002;99:755–761. doi: 10.1002/ijc.10409. [DOI] [PubMed] [Google Scholar]
- Elsharkawy AM, Oakley F, Mann DA. The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis. 2005;10:927–939. doi: 10.1007/s10495-005-1055-4. [DOI] [PubMed] [Google Scholar]
- Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- Furst DE. Meloxicam: selective COX-2 inhibition in clinical practice. Semin Arthritis Rheum. 1997;26(6 Suppl. 1):21–27. doi: 10.1016/s0049-0172(97)80049-2. [DOI] [PubMed] [Google Scholar]
- Kaimori A, Potter JJ, Choti M, Ding Z, Mezey E, Koteish AA. Histone deacetylase inhibition suppresses the transforming growth factor beta1-induced epithelial-to-mesenchymal transition in hepatocytes. Hepatology. 2010;52:1033–1045. doi: 10.1002/hep.23765. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Lee J, Kim KN, Kim HJ, Jeung HC, Chung HC, et al. Anti-tumor activity of N-hydroxy-7-(2-naphthylthio) heptanomide, a novel histone deacetylase inhibitor. Biochem Biophys Res Commun. 2007;356:233–238. doi: 10.1016/j.bbrc.2007.02.126. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wang Z, Wang J, Lam W, Kwong S, Li F, et al. A histone deacetylase inhibitor, largazole, decreases liver fibrosis and angiogenesis by inhibiting transforming growth factor-beta and vascular endothelial growth factor signalling. Liver Int. 2013;33:504–515. doi: 10.1111/liv.12034. [DOI] [PubMed] [Google Scholar]
- Mann J, Oakley F, Akiboye F, Elsharkawy A, Thorne AW, Mann DA. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: implications for wound healing and fibrogenesis. Cell Death Differ. 2007;14:275–285. doi: 10.1038/sj.cdd.4401979. [DOI] [PubMed] [Google Scholar]
- Marra F, Arrighi MC, Fazi M, Caligiuri A, Pinzani M, Romanelli RG, et al. Extracellular signal-regulated kinase activation differentially regulates platelet-derived growth factor's actions in hepatic stellate cells, and is induced by in vivo liver injury in the rat. Hepatology. 1999;30:951–958. doi: 10.1002/hep.510300406. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira RK. Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med. 2007;131:1728–1734. doi: 10.5858/2007-131-1728-HSCALF. [DOI] [PubMed] [Google Scholar]
- Novo E, Marra F, Zamara E, Valfre di Bonzo L, Monitillo L, Cannito S, et al. Overexpression of Bcl-2 by activated human hepatic stellate cells: resistance to apoptosis as a mechanism of progressive hepatic fibrogenesis in humans. Gut. 2006;55:1174–1182. doi: 10.1136/gut.2005.082701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KC, Kim SW, Park JH, Song EH, Yang JW, Chung HJ, et al. Potential anti-cancer activity of N-hydroxy-7-(2-naphthylthio) heptanomide (HNHA), a histone deacetylase inhibitor, against breast cancer both in vitro and in vivo. Cancer Sci. 2011;102:343–350. doi: 10.1111/j.1349-7006.2010.01798.x. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucleic Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicoro A, Ramachandran P, Iredale JP. Reversibility of liver fibrosis. Fibrogenesis Tissue Repair. 2012;5(Suppl. 1):S26. doi: 10.1186/1755-1536-5-S1-S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Han YP. Epigenetic repression of matrix metalloproteinases in myofibroblastic hepatic stellate cells through histone deacetylases 4: implication in tissue fibrosis. Am J Pathol. 2010;177:1915–1928. doi: 10.2353/ajpath.2010.100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MA, Dhar DK, Masunaga R, Yamanoi A, Kohno H, Nagasue N. Sulindac and exisulind exhibit a significant antiproliferative effect and induce apoptosis in human hepatocellular carcinoma cell lines. Cancer Res. 2000;60:2085–2089. [PubMed] [Google Scholar]
- Ramm GA, Shepherd RW, Hoskins AC, Greco SA, Ney AD, Pereira TN, et al. Fibrogenesis in pediatric cholestatic liver disease: role of taurocholate and hepatocyte-derived monocyte chemotaxis protein-1 in hepatic stellate cell recruitment. Hepatology. 2009;49:533–544. doi: 10.1002/hep.22637. [DOI] [PubMed] [Google Scholar]
- Reif S, Lang A, Lindquist JN, Yata Y, Gabele E, Scanga A, et al. The role of focal adhesion kinase-phosphatidylinositol 3-kinase-akt signaling in hepatic stellate cell proliferation and type I collagen expression. J Biol Chem. 2003;278:8083–8090. doi: 10.1074/jbc.M212927200. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Salvador J, Armas-Pineda C, Perezpena-Diazconti M, Chico-Ponce de Leon F, Sosa-Sainz G, Lezama P, et al. Effect of sodium butyrate on pro-matrix metalloproteinase-9 and -2 differential secretion in pediatric tumors and cell lines. J Exp Clin Cancer Res. 2005;24:463–473. [PubMed] [Google Scholar]
- Rombouts K, Knittel T, Machesky L, Braet F, Wielant A, Hellemans K, et al. Actin filament formation, reorganization and migration are impaired in hepatic stellate cells under influence of trichostatin A, a histone deacetylase inhibitor. J Hepatol. 2002;37:788–796. doi: 10.1016/s0168-8278(02)00275-1. [DOI] [PubMed] [Google Scholar]
- Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123:1887–1901. doi: 10.1172/JCI66028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Tajima H, Hironori H, Nakagawara H, Ohnishi I, Takamura H, et al. Sodium valproate blocks the transforming growth factor (TGF)-beta1 autocrine loop and attenuates the TGF-beta1-induced collagen synthesis in a human hepatic stellate cell line. Int J Mol Med. 2011;28:919–925. doi: 10.3892/ijmm.2011.768. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Lantowski A, Dannenberg AJ, Subbaramaiah K. Histone deacetylase inhibitors suppress the induction of c-Jun and its target genes including COX-2. J Biol Chem. 2005;280:32569–32577. doi: 10.1074/jbc.M503201200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Quantification of Western blots. The box and whisker plots show median, with first and third quartiles (as box) and range (as whiskers). Data are from n = 4 experiments. *P < 0.05, **P < 0.01, ***P < 0.005 versus PDGF, control, or BDL.
Figure S2 Effects of HNHA on acetylation of histone H3 in BDL-liver. Representative staining (A) and densitometric analysis (B) of acetyl histone H3. The box and whisker plots show median, with first and third quartiles (as box) and range (as whiskers). Data are from n = 5 experiments. *P < 0.05, **P < 0.01, ***P < 0.005 versus BDL.
Figure S3 Effects of HNHA on levels of HDAC1 in BDL-liver. Representative staining (A) and densitometric analysis (B) of HDAC1 levels. The box and whisker plots show median, with first and third quartiles (as box) and range (as whiskers). Data are from n = 5 experiments. **P < 0.01, ***P < 0.005 versus BDL.