

Abstract
A major question in cell biology is how molecular specificity is achieved by different growth factor receptors that activate apparently identical signaling events. For the neurotrophin family, a distinguishing feature is the ability to maintain a prolonged duration of signal transduction. However, the mechanisms by which neurotrophin receptors assemble such a sustained signaling complex are not understood. Here we report that an unusual ankyrin-rich transmembrane protein (ARMS+kidins220) is closely associated with Trk receptor tyrosine kinases, and not the EGF receptor. This association requires interactions between transmembrane domains of Trk and ARMS. ARMS is rapidly tyrosine phosphorylated after binding of neurotrophins to Trk receptors and provides a docking site for the CrkL–C3G complex, resulting in Rap1-dependent sustained ERK activation. Accordingly, disruption of Trk–ARMS or the ARMS–CrkL interaction with dominant-negative ARMS mutants, or treatment with small interference RNA against ARMS substantially reduce neurotrophin-elicited signaling to ERK, but without any effect upon Ras or Akt activation. These findings suggest that ARMS acts as a major and neuronal-specific platform for prolonged MAP kinase signaling by neurotrophins.
Keywords: BDNF, Crk, NGF, Rap1, signal transduction
Introduction
Growth factor signaling is characterized by a common set of events, including receptor dimerization, protein phosphorylation, recruitment of adaptor proteins and stimulation of downstream signaling cascades. Many signal transduction events are shared between different cellular receptor systems, raising the issue of how molecular specificity is achieved to give a unique biological response (Chao, 1992). This is particularly relevant to neurotrophin signaling, which is responsible for a wide variety of neuronal functions including survival, differentiation, axonal and dendritic growth, cell death, neurotransmitter secretion and neuronal activity (Bibel and Barde, 2000; Poo, 2001; Chao, 2003; Huang and Reichardt, 2003).
The actions of neurotrophins depend upon two different transmembrane receptor types, the Trk receptor tyrosine kinase and the p75 neurotrophin receptor that belongs to the TNF receptor family (Huang and Reichardt, 2003). Nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3) and NT-4 all recognize the p75 receptor, but NGF binds preferentially to TrkA, BDNF and NT-4 to TrkB, and NT-3 to TrkC receptors. Trk and p75 receptors can engage distinct signaling modules when expressed independent of each other (Carter et al, 1996; Yoon et al, 1998). In the nervous system where both receptors are frequently co-expressed, however, p75 receptor also acts as a co-receptor for Trk to increase neurotrophin's binding affinity and to enhance Trk specificity for cognate ligands (Bibel and Barde, 2000; Chao, 2003).
What are the critical signaling events that emanate from Trk receptors? After neurotrophin binding, activated Trk receptors recruit adaptor proteins, such as Shc and FRS-2, and effectors, such as phosphatidylinositol 3-kinase (PI3-kinase) and phospholipase-C-γ (PLC-γ) (Chao, 2003; Huang and Reichardt, 2003). The key docking sites on the Trk receptor are tyrosine residue 490 in the juxtamembrane region and tyrosine residue 785 at the end of the cytoplasmic domain, which binds to PLC-γ. Activation of PI3-kinase and MAP kinase activities are both mediated by Shc protein binding to the Trk receptor at tyrosine 490 (Huang and Reichardt, 2003).
Despite our understanding of neurotrophin signaling, many of these pathways, such as ERK, Akt, PLC, PKC and Ras, are not unique to neurotrophin signaling. Each signaling component is universally used in many different contexts and by other growth factors and cytokines. This problem is accentuated by the findings that, in addition to cell differentiation, neurotrophins have effects upon many aspects of neuronal activity that result in synaptic responses that are extremely rapid (Blum et al, 2002) or that can be long lasting (Sherwood and Lo, 1999).
A distinguishing feature of neurotrophin signaling has been the sustained activation of MAP kinase activity (Marshall, 1995). Continuous ERK activity occurs for hours after NGF treatment of PC12 cells, whereas EGF treatment provides a transient activation (Qui and Green, 1992; Cowley et al, 1994). The small G protein Rap1 accounts at least in part for the ability of neurotrophins to signal for long time periods, by activating the B-Raf and the MEK/ERK pathway (York et al, 1998). Recent studies further implicate a critical role of the Rap1–B-Raf pathway in retrograde endosomal signaling (Kao et al, 2001; Wu et al, 2001; Stork, 2003).
A still unanswered question is how prolonged signaling through the Rap1/ERK pathway is connected to the Trk receptor. A resolution to this issue would provide an insight into how neurotrophins are able to signal in a manner that selectively triggers a sustained MAP kinase response, as well as explain how trophic responses unique to neurotrophins are generated. Here we report that a novel transmembrane protein substrate of Trk receptors, Ankyrin-Rich Membrane Spanning (ARMS) protein, is directly involved in these downstream signaling events for neurotrophins.
Results
Kinetics of neurotrophin signal transduction
The ARMS gene was originally identified as an interacting protein for the p75 neurotrophin receptor (Kong et al, 2001), as well as a target for protein kinase D, Kidins220, or a kinase D-interacting substrate of 220 kDa (Iglesias et al, 2000). ARMS is a highly conserved 220 kDa protein possessing 11 ankryin repeats, four closely spaced transmembrane domains and a large C-terminal cytoplasmic tail. The ARMS protein is tyrosine phosphorylated by Trk receptors in response to NGF and BDNF (Kong et al, 2001), but it lacks the usual binding motifs associated with tyrosine kinase substrates, such as pleckstrin or SH2 domains. Since propagation of Trk signaling is associated with rapid tyrosine phosphorylation, we assessed how rapidly the ARMS protein is phosphorylated compared to more well-known Trk substrates, such as Shc and PLC-γ.
In PC12 cells treated with NGF for brief intervals, tyrosine phosphorylation of ARMS occurred simultaneously with the detection of phosphorylated TrkA receptors (Figure 1A). The phosphorylation of ARMS also appeared at the same time as Shc and PLC-γ, indicating that ARMS was among the first proteins to be activated by NGF. This rapid phosphorylation was also observed in primary cultures of cortical neurons treated with BDNF (Figure 1B and Supplementary Figure S1). By contrast, other downstream events, such as activation of ERK and Akt, proceeded after the phosphorylation of ARMS, Shc and PLC-γ (Figure 1A), consistent with their known roles in the Trk signal transduction cascade (Huang and Reichardt, 2003).
Figure 1.
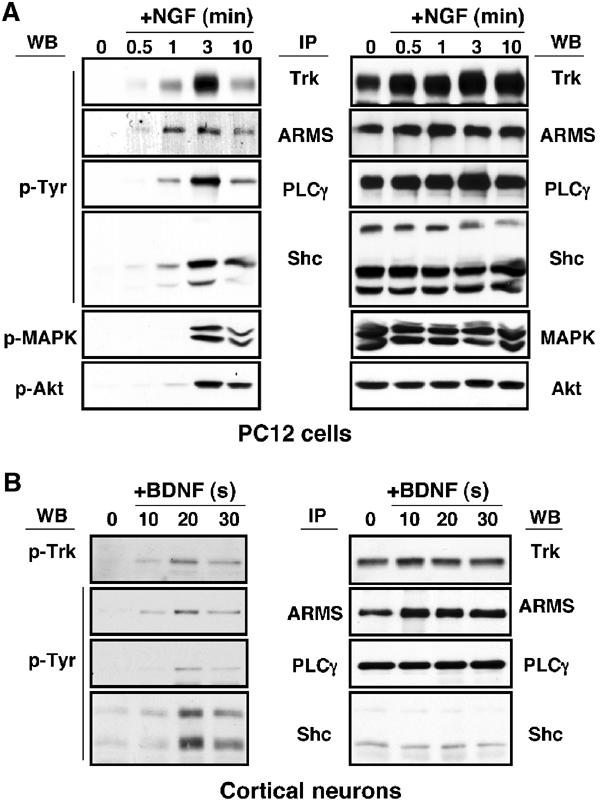
ARMS is rapidly tyrosine phosphorylated upon neurotrophin treatment. (A) Kinetics of TrkA signaling. PC12 cells were treated with NGF (100 ng/ml) for the indicated time (minutes). Immunoprecipitation was performed for Trk, ARMS, PLC-γ and Shc, followed by Western blot with 4G10, an anti-phosphotyrosine antibody, whereas 50 μg of whole lysates was used to detect the phosphorylation status of MAPK and Akt using phosphospecific antibodies (left panel). The same blot was re-probed with Trk, ARMS, PLC-γ, Shc, MAPK and Akt antibodies to verify the equality of protein loading (right panel). (B) Kinetics of TrkB signaling. Cortical neurons (8–10 DIV) treated with BDNF (50 ng/ml) were added for the indicated time (s). Immunoprecipitation was carried out as described in (A). The left blot was probed with 4G10 and the total amount of proteins is shown in the right panel.
Transmembrane interaction of ARMS with Trk receptors
The rapid phosphorylation of ARMS by Trk suggests that, like Shc and PLC-γ, ARMS may be directly associated with Trk receptors. To test whether an interaction exists between these two proteins, TrkA, TrkB and TrkC receptors were coexpressed with FLAG-tagged ARMS protein in HEK293 cells. The associations were assessed by co-immunoprecipitation. Indeed, all the three Trk receptors associate with ARMS (Figure 2A). In contrast, ARMS did not interact with the EGF receptor (Figure 2B), consistent with our previous finding that EGF does not stimulate ARMS tyrosine phosphorylation (Kong et al, 2001).
Figure 2.
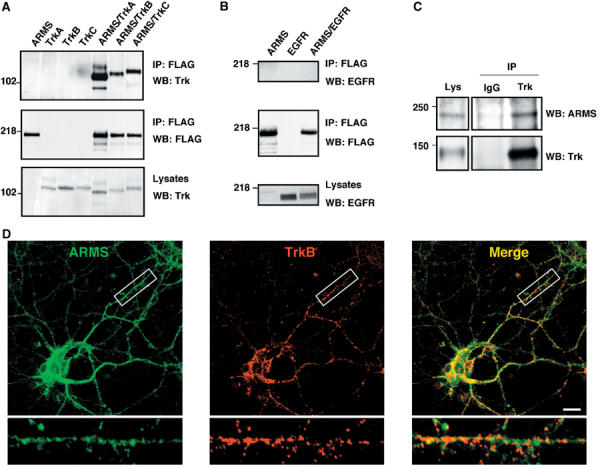
ARMS and Trk receptors interact. (A) ARMS interacts with Trk receptors. Lysates from HEK293 cells transfected with FLAG-ARMS and/or TrkA, B, or C receptor were immunoprecipitated with FLAG antibodies, followed by Western blotting by anti-Trk antisera. (B) Immunoprecipitated ARMS does not bind EGF receptors. Experiments were carried out in the same way as described in panel A, but EGF receptor was analyzed. (C) ARMS interacts with TrkB in primary cortical neurons. Cells were cultured for 8–12 DIV and crosslinked with the lipid-soluble compound DSP (Pierce), a thiol-cleavable reagent. Control rabbit IgG was used instead of anti-Trk to demonstrate the specificity of the Trk–ARMS interaction. (D) Co-localization of TrkB and ARMS in primary cortical neurons. Staining was carried out with affinity-purified rabbit polyclonal antibody 892 against ARMS (green; Kong et al, 2001) and the mouse monoclonal antibody B3 (Santa Cruz) against TrkB (red). Co-localization of both proteins is shown in yellow. Images were collected on a confocal microscope. An area within the cortical neuron was enlarged to highlight TrkB and ARMS colocalization. Scale bar=10 μm.
To verify that the Trk–ARMS interaction also occurs in vivo, detergent lysates of E18 cortical neurons were immunoprecipitated with an antiserum raised against Trk, followed by Western blotting with anti-ARMS antibody. As shown in Figure 2C, stable complex formation between TrkB and ARMS was clearly detected. Furthermore, punctate staining of ARMS protein was found to co-localize significantly with TrkB in E18 cortical neurons (Figure 2D), thus supporting the immunoprecipitation analysis of an endogenous Trk–ARMS interaction.
To identify the domains responsible for the interaction between ARMS and the Trk receptor, a series of truncations were made in the Trk receptor and the ARMS protein (Figure 3). Each truncated mutant was tested in cotransfection and immunoprecipitation experiments. Trk receptors contain two extracellular IgG domains responsible for ligand binding, a single transmembrane sequence and a consensus tyrosine kinase domain in the intracellular domain. Notably, neither the cytoplasmic domain nor the extracellular domains of Trk were sufficient to maintain an interaction with ARMS (Figure 3B; Supplementary Figure S2), suggesting that transmembrane or nearby sequences of Trk were responsible. Indeed, this interpretation was supported by an analysis of a chimeric receptor of TrkA that contained the transmembrane domain of the EGF receptor flanked by the extracellular and intracellular domains of TrkA (Figure 3A). Unlike other Trk mutants that were tested, TrkA-TM EGFR did not interact with ARMS in immunoprecipitation experiments (Figure 3A and B).
Figure 3.
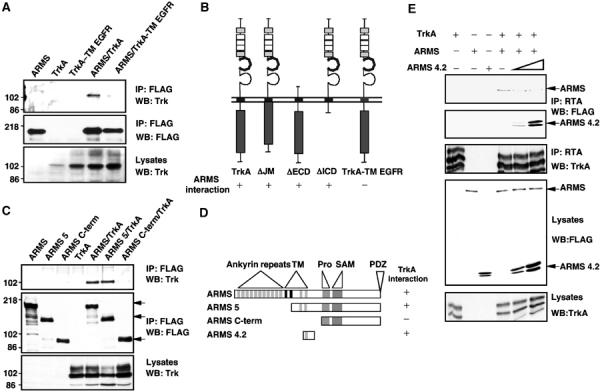
Transmembrane domains are responsible for the interaction between Trk receptors and ARMS. (A) Transmembrane region of TrkA is necessary for the interaction with ARMS. Anti-FLAG immunoprecipitation was performed with extracts from HEK293 cells transfected with FLAG-ARMS and wild-type TrkA or a chimera of TrkA/EGFR that contains the transmembrane region of the EGF receptor. The presence of Trk was verified by Western blotting with an anti-Trk antisera. (B) Summary of Trk/ARMS interaction using different Trk mutants. The regions deleted were the juxtamembrane (ΔJM), extracellular (ΔECD) or intracellular (ΔICD) domain (Arévalo et al, 2000; Yano et al, 2000, 2001). The chimera TrkA-TM EGF receptor was generated by swapping the transmembrane region of TrkA for the corresponding domain from the EGF receptor. See Supplementary data for ARMS binding to Trk receptor mutants (Supplementary Figure S2). (C) Lack of transmembrane regions in ARMS abolished the interaction with TrkA. Anti-FLAG immunoprecipitation experiments were performed with extracts from HEK293 cells transfected with wild-type TrkA and FLAG-tagged full-length ARMS or truncated ARMS, followed by Western blotting with an anti-Trk antisera. Arrows indicate the migration of different truncated ARMS proteins. (D) Summary of TrkA/ARMS interaction using different ARMS mutants. Similar experiments as in Figure 4C were carried out to identify the ARMS regions involved in the TrkA interaction. TM=transmembrane domain; Pro=polyproline-rich region; SAM=sterile α-motif; PDZ=binding motif (SIL). See Supplementary data for analysis of TrkA and ARMS mutants (Supplementary Figure S3). (E) A truncated form of ARMS impairs the interaction of TrkA and full-length ARMS. Extracts from HEK293 cells transfected with full-length FLAG-ARMS, TrkA and increasing concentrations of FLAG-ARMS4.2 were immunoprecipitated with anti-TrkA antibodies (Clary et al, 1994) and Western blot were performed with anti-FLAG antibody to detect ARMS and ARMS4.2 expression. Note that increasing levels of ARMS4.2 diminish TrkA interaction with full-length ARMS.
The domain of ARMS necessary for Trk association was also determined. A large deletion of the N-terminal ankyrin repeats as well as a deletion of the C-terminal tail of ARMS did not interfere with its association with the Trk receptor (Figure 3C and D; Supplementary Figure S3). Consistent with the Trk mapping analysis (Figure 3A and B), the transmembrane segments of ARMS (i.e. ARMS3 and ARMS4.2) were found to interact with Trk receptors and can do so without the majority of intracellular sequences (Supplementary Figure S3). These results were corroborated by testing whether the ARMS transmembrane domains alone exerted any effect upon Trk association with ARMS. We therefore expressed the fourth transmembrane segment of ARMS (ARMS4.2) in the presence of full-length epitope-tagged ARMS and TrkA receptors in HEK293 cells. The association of FLAG-tagged ARMS and Trk was assessed as a function of increasing levels of ARMS4.2. A noticeable decrease in the association of ARMS and TrkA was observed in the presence of ARMS4.2 protein (Figure 3E). Taken together, we conclude that the transmembrane domains are major sites of interaction between ARMS and Trk, which can be disrupted by expressing the membrane-spanning segments of ARMS.
ARMS-dependent neurotrophin signaling
The interaction between Trk receptors and ARMS requires transmembrane domains of each protein, suggesting that the association is constitutive and that ARMS is uniquely positioned to coordinate rapid Trk signal transduction via its multiple protein interaction motifs. We therefore sought to characterize the role of ARMS in neurotrophin action. Since ectopic expression of the transmembrane domains of ARMS (ARMS4.2) disengages endogenous TrkA–ARMS complexes (Figure 3E), the effect of ARMS4.2 on NGF signaling was first examined. PC12 cells transfected with full-length ARMS or ARMS4.2 were compared for neuritogenic responsiveness to NGF. While the majority of control (GFP-transfected) PC12 cells displayed pronounced neurite outgrowth after 2 days of NGF treatment, expression of wild-type ARMS augmented NGF-induced neuronal differentiation (Figure 4A). Significantly, PC12 transfectants expressing the ARMS4.2 mutant protein displayed a 50% decrease in neurite-bearing population (Figure 4A) after comparable duration of NGF treatment. These data suggest that ARMS functionally interacts with TrkA and that disruption of this interaction results in aborted NGF-dependent signal transduction critical for neurite outgrowth.
Figure 4.
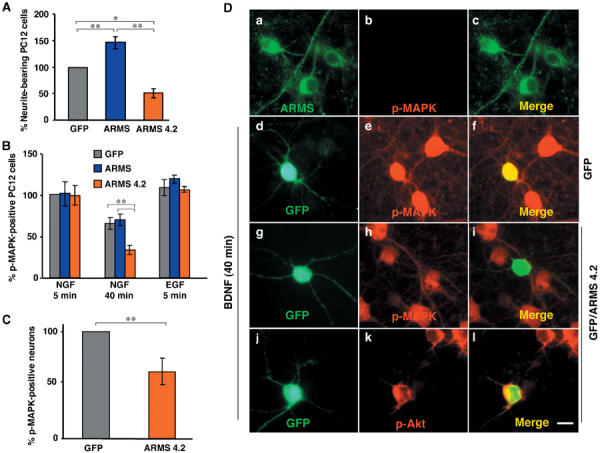
ARMS4.2 interferes with sustained MAPK phosphorylation. (A) Neurite outgrowth of PC12 cells transfected with GFP, FLAG-ARMS or FLAG-ARMS4.2. Cells were stimulated with NGF for 24 h after transfection and neurite outgrowth was quantified 3 days later by assessing the percentage of fluorescence-positive cells bearing neurites at least twice the length of the cell body. At least 50 cells were counted for each experiment and condition. Results were normalized to control GFP-transfected cells (set as 100%). Values are calculated from at least four independent experiments. Mean and standard deviation (s.d.) are shown. Where applicable, statistical analyses were carried out by Student's t-test (*P<0.02; **P<0.001). (B) Activation of p-MAPK in PC12 cells transfected with GFP, FLAG-ARMS or FLAG-ARMS4.2. Respective cultures were stimulated with NGF for 5 or 40 min or EGF for 5 min. Quantification was carried out as described in panel A (**P<0.001). (C) Immunofluorescence analysis of primary cortical neurons transfected with ARMS4.2 constructs. Cells were transfected with GFP or GFP/FLAG-ARMS4.2 and stimulated 2 days later with BDNF for 40 min. Panel b shows the absence of p-MAPK staining in untreated cells. Panels d–l show transfected neurons with GFP and GFP/FLAG-ARMS4.2. Panels d–f and g–l show GFP and GFP/FLAG-ARMS4.2-transfected neurons that are positive (yellow, panel f) and negative (green, panel i) for p-MAPK, respectively. Panels j–l show GFP/FLAG-ARMS4.2-transfected neurons stained for p-Akt (yellow, panel l). Scale bar=5 μm. (D) Activation of p-MAPK in cortical neurons transfected with GFP and GFP/ARMS4.2 stimulated with BDNF for 40 min (**P<0.001).
Neuronal differentiation of PC12 cells is closely associated with sustained MAP kinase activation elicited by NGF, whereas mitogenic growth factors, such as EGF, induce a relatively transient activation of MAP kinase (Marshall, 1995). To assess if ARMS modulates these signaling events, MAP kinase activation in ARMS4.2-transfected PC12 cells was monitored by immunostaining with phospho-MAP kinase (p-MAPK)-specific antibodies. As shown in Figure 4B, PC12 cells expressing FLAG-ARMS or FLAG-ARMS4.2 were stimulated with NGF for 5 or 40 min, two time points that can discriminate between transient and sustained MAP kinase activation (Qui and Green, 1992; York et al, 1998). The total numbers of p-MAPK-positive cells expressing GFP, FLAG-ARMS, or FLAG-ARMS4.2 were similar after NGF treatment for 5 min (Figure 4B). However, PC12 cells transfected with FLAG-ARMS4.2 construct showed a significant reduction in the number of p-MAPK-positive cells at the later time point (Figure 4B). Importantly, ARMS4.2 mutant did not perturb the transient MAP kinase activation by EGF, consistent with earlier data that EGF treatment normally evokes a transient MAP kinase cascade in PC12 cells (Qiu and Green, 1991; Qui and Green, 1992). The results suggest that ARMS is a specific substrate for Trk receptor signaling (Figures 2 and 3; Kong et al, 2001). In addition, the lack of an effect of ARMS4.2 on transient MAP kinase activation evoked by either NGF or EGF suggests that the previously identified pathway for Ras-dependent MAP kinase activation via FRS2 (Kouhara et al, 1997; Stork, 2003) is not affected by ARMS4.2 expression.
Since in vitro binding studies indicated that TrkB also interacts with ARMS (Figure 2A), the role of ARMS in BDNF signaling was evaluated in cortical neurons, which express endogenous TrkB receptor and ARMS (Barbacid, 1994; Kong et al, 2001). Accordingly, transfected cortical neurons were treated with BDNF (50 ng/ml) for 40 min (Figure 4D, panels d–l) and the proportion of p-MAPK-positive cells was determined (Figure 4C). The results indicated a clear decrease in the number of p-MAPK-positive neurons after ARMS4.2 expression (Figures 4C and D, panels g–i), when compared to control cultures (Figures 4C and D, panels d–f). Interestingly, we found no effect of ARMS4.2 on BDNF-dependent activation of Akt (Figure 4D, panels j–l), suggesting that functional TrkB–ARMS interaction is specifically required for modulating the kinetics of MAP kinase activation. These observations lend support to the hypothesis that ARMS is involved in the sustained activation of MAP kinase mediated by Trk receptors upon neurotrophin treatment.
NGF induces complex formation between Trk, ARMS and CrkL
The ability of neurotrophins to activate the prolonged MAP cascade requires the integration of Ras- and Rap1-dependent pathways. In PC12 cells, Rap1 is responsible for the sustained phase of MAP kinase activation via B-Raf, whereas Ras activity is associated with the earliest initial response (Vossler et al, 1997; York et al, 1998). Our findings with the ARMS4.2 mutant suggest a specific role for ARMS in maintaining late-phase MAP kinase activation (Figure 4). Since the Crk adaptor protein family is involved in neurotrophin-dependent differentiation and signaling to Rap1 (Teng et al, 1995, 1996; York et al, 1998; Feller, 2001), we asked whether NGF regulates the interactions between Trk and Crk proteins.
To address this question, Crk proteins and TrkA were co-immunoprecipitated in PC12nnr cells (Green et al, 1986), which stably expressed either exogenous wild-type or mutant TrkA receptors (ΔShc) that do not interact with the adaptor protein FRS2 (Kouhara et al, 1997). An inducible interaction was clearly detectable with wild-type Trk and a Trk receptor mutant (ΔShc) missing the consensus Shc-binding site ENPQY (Figure 5A). It had been proposed that association of Crk proteins with Trk was facilitated by FRS2 (Meakin et al, 1999; Kao et al, 2001). However, the interaction of Crk with Trk receptors did not depend upon the Shc site or FRS2 (Figure 5A). Therefore, the association of Crk with TrkA did not require Shc phosphorylation or FRS2 activation.
Figure 5.
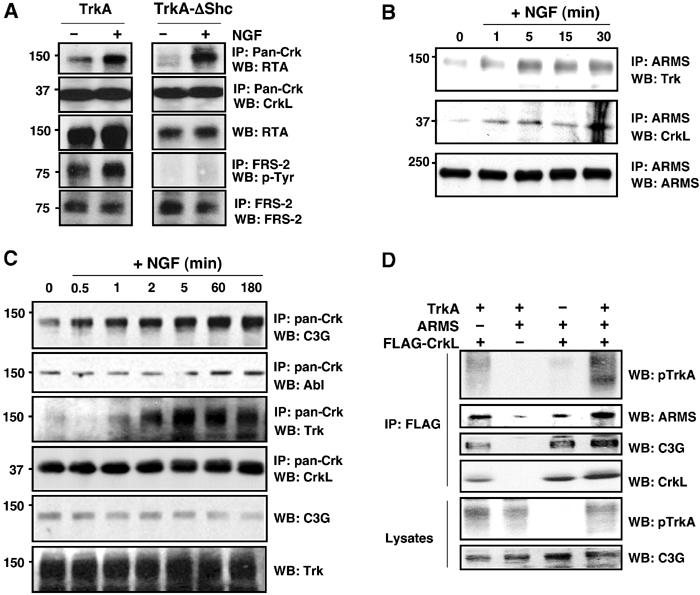
TrkA, ARMS and CrkL association is enhanced by NGF. (A) Deletion of the Shc-binding site abolished NGF-induced FRS-2 interaction, but did not affect complex formation between TrkA and CrkL/c-Crk II. PC12nnr5 cells stably expressing wild-type TrkA or a mutant Trk (ΔShc) carrying a deletion (ENPQY490F) that does not bind Shc or FRS2 were treated with NGF for 10 min. Lysates were immunoprecipitated either with an anti-Crk antibody, followed by blotting with an anti-Trk antiserum (RTA), or with anti-FRS2 antiserum and blotting with antiphosphotyrosine (p-Tyr). Reprobing the blots with anti-CrkL and anti-FRS2 verified sample loading. (B) TrkA, ARMS and CrkL association in PC12 cells is enhanced by NGF treatment. PC12-615 cells were treated for the indicated times with 100 ng/ml NGF. Detergent lysates were immunoprecipitated with anti-ARMS polyclonal antibody. Western blotting was performed with Trk (C-14) and CrkL polyclonal antibodies. (C) CrkL increases the binding of C3G and Trk upon NGF treatment. PC12-615 cells were stimulated for the indicated time with NGF and extracts were immunoprecipitated with pan-Crk antibodies. Western blotting analysis was performed with C3G, Abl, CrkL and TrkA to detect these proteins. Note that the increasing levels of C3G and activated TrkA were pulled down together with CrkL in response to NGF treatment. (D) Association of Trk, ARMS and CrkL. HEK293T cells were transfected with the indicated plasmids. Anti-FLAG immunoprecipitates were blotted with the indicated antibodies. The association of TrkA with CrkL was enhanced by ARMS expression (lane 1 versus lane 4).
In endogenous co-immunoprecipitation experiments, we also found that CrkL is specifically associated with ARMS and TrkA in an NGF-inducible manner in PC12 cells (Figure 5B). This interaction increased minutes after NGF treatment. Consistent with the mapping results from transfected cells, these findings suggest that ARMS is involved in the association between TrkA and CrkL. This complex was observed constitutively, but increased with NGF treatment (Figure 5B).
Tyrosine phosphorylation events may change the specificity of Crk binding, leading to differential association with downstream proteins. This may be manifested by a change in the binding specificities of Crk to different signaling proteins, such as C3G or Abl (Feller, 2001). To evaluate this possibility, we carried out a time course of association of CrkL, activated TrkA and the guanine nucleotide exchange factor (GEF) C3G, in response to NGF in PC12 cells. After NGF treatment, there was a noticeable increase in the amount of endogenous C3G and TrkA associated with CrkL over a time period of 3 h (Figure 5C). This increase was specific to C3G, since Abl, another interactor of CrkL, did not show a corresponding change. The increased association of C3G with CrkL paralleled the activation of the TrkA receptor. NGF, therefore, induced an association of CrkL with C3G, which can lead to increases in Rap1 and MAPK activities associated with prolonged responses by neurotrophins. Taken together with the association of ARMS and CrkL, these results indicate that a complex of proteins forms downstream of the Trk receptor, which is enhanced upon neurotrophin treatment.
ARMS may therefore serve as a molecular scaffold for Trk-dependent signaling via CrkL. If so, ARMS overexpression would be expected to enhance complex formation between CrkL and TrkA. To address this possibility, we carried out co-immunoprecipitation analysis of FLAG-tagged CrkL with TrkA in the presence or absence of exogenous ARMS. HEK293 cells harbor a low level of endogenous ARMS protein that can facilitate TrkA–CrkL binding. As shown in Figure 5D, complex formation between TrkA and CrkL was augmented by additional ARMS protein expression, suggesting that one function of ARMS is to link activated Trk to CrkL. Consistent with the role of Crk adaptors in Rap1 signaling (Feller, 2001), the Rap1 GEF C3G was also found to complex with CrkL in this assay (Figure 5D). These results suggest that endogenous ARMS–CrkL and CrkL–C3G complexes occur constitutively, but also that these associations can be significantly augmented by NGF stimulation.
Specificity of ARMS–CrkL interaction
Analysis of the ARMS protein sequence reveals a region rich in polyprolines (Pro) in the cytoplasmic domain (Figure 6A). In particular, P1081PLP, P1089PRPP and P1131QHP on ARMS conform to the consensus SH3-binding site for the Crk family of adaptor proteins, which modulate Rap1 activity via C3G (Feller, 2001). To determine which of these polyproline sequences of ARMS might be involved in binding to Crk family members, we carried out in vitro GST-binding experiments with the polyproline segment of ARMS. Utilizing cellular lysates from HEK293 cells that express endogenous c-Crk II and the related CrkL, a specific interaction of CrkL, but not c-Crk II, with GST–ARMS7.2 was detected (Figure 6B). No binding was detected with the GST–ARMS7.3 or GST–ARMS7.6 proteins. Binding of CrkL was therefore localized to the second of these three polyproline sequences of ARMS (P1089PRPP). To confirm these results, a corresponding GST–ARMS7.2 fusion protein lacking the P1089PRPP sequence (ARMS 7.2ΔP) was next tested for CrkL binding. No interaction of GST–ARMS7.2ΔP proteins with CrkL was observed (Figure 6B). These findings verify that there is direct binding of ARMS and CrkL, and identify the requirement of the P1089PRPP sequence for this interaction.
Figure 6.
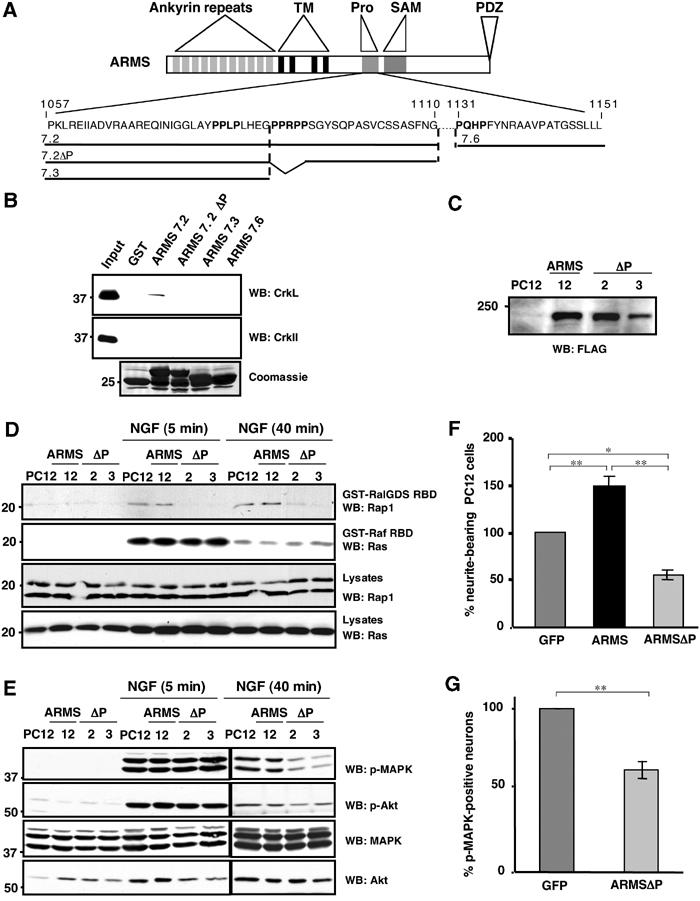
Ligand dependency of ARMS, CrkL and C3G interactions. (A) The polyproline-rich region of ARMS contains consensus sequences (PXXP) for binding to SH3 domains. Sequences within the polyproline-rich region of ARMS (P1057–L1151) present three PXXP motifs (bold) that are consensus for binding of SH3 domain-containing molecules (Feller, 2001) and are predicted by website sequence analysis: http://scansite.mit.edu/. (B) In vitro interaction between ARMS and CrkL, but not c-CrkII. GST–ARMS recombinant proteins were incubated with HEK293 cell extracts and subjected to Western blotting analysis with anti-c-Crk II or anti-CrkL antisera. A Coomassie-stained gel of the input GST fusion proteins is shown (bottom panel). (C) PC12 cells were stably transfected with FLAG-ARMS and FLAG-ARMSΔP. Individual clones were analyzed for the ectopic expression of ARMS or ARMSΔP proteins using anti-FLAG antibodies. Reprobing the blot with anti-ARMS antibody revealed that the overexpression levels for Flag-ARMS and Flag-ARMSΔP are approximately three-fold. (D) Expression of ARMSΔP abolishes Rap1 but not Ras activation elicited by NGF. PC12 clones stably expressing FLAG-ARMS or FLAG-ARMSΔP were serum starved for 16 h, lysates were prepared and subjected to incubation with 10 μg of GST-RalGDS RBD or the Raf-1 Ras-binding domain protein (GST-Raf RBD) in pull-down assays (Hermann et al, 1996) to detect active Rap1 and Ras, respectively. Immunoblots using anti-Rap1 and anti-Ras were carried out as indicated. (E) Expression of FLAG-ARMSΔP in PC12 cells impairs sustained MAP kinase activation elicited by NGF. Cell extracts from PC12 cells or PC12 clones described above (C), nontreated or treated with NGF for 5 or 40 min, were obtained and Western blots were performed with p-MAPK and p-Akt antibodies. A reduction in MAPK activation in PC12 clones expressing FLAG-ARMSΔP was seen at 40 min of NGF treatment, but no differences were observed in the activation of Akt. (F) Neurite outgrowth of PC12 cells transiently transfected with GFP, FLAG-ARMS or FLAG-ARMSΔP. At 24 h after transfection with the indicated plasmids, cells were treated with NGF for 2 additional days. PC12 transfectants were scored as positive as described in Figure 4A (*P<0.02; **P<0.001). (G) Cortical neurons transfected with GFP/FLAG-ARMSΔP showed a reduced response to BDNF. Cells were transfected with GFP or GFP/FLAG-ARMSΔP. After 2 days, cultures were stimulated with BDNF for 40 min and staining for p-MAPK was performed. The statistical significance by Student's t-test was demonstrated for (F) and (G) (*P<0.02; **P<0.001).
ARMS mediates Rap1-dependent MAP kinase activation
The binding interactions between ARMS and CrkL suggest that downstream signaling to Rap1 may be directly affected by disrupting this complex. ARMS interacts with Trk receptors via transmembrane domains, but with CrkL via its polyproline region (Figures 2, 3 and 6B). Therefore, ARMS mutants with a deleted P1089PRPP polyproline sequence (ARMSΔP) might be expected to disengage Trk signaling to CrkL. Given that activation of Rap1 is necessary for prolonged MAP kinase activity in PC12 cells (York et al, 1998), we considered whether expression of ARMSΔP specifically affects Rap1 and MAP kinase activation by neurotrophins.
Toward this end, we generated stable PC12 cell clones expressing FLAG-ARMS or FLAG-ARMSΔP (Figure 6C). The state of Rap1 activity in these cells was evaluated with a GST protein fused to the Rap1-binding domain of RalGDS (GST-RalGDS RBD), which specifically binds only to the active, GTP-bound form of Rap1 (Hermann et al, 1996). Cells were treated with NGF for 5 and 40 min and cell lysates were subjected to GST-RalGDS RBD pull-down assays. Consistent with the above model, NGF-dependent Rap1 activation was detected in control as well as FLAG-ARMS-expressing PC12 cells, but is completely abolished in cells expressing the ARMSΔP protein (Figure 6D). In contrast, we observed no differences in the amount of active Ras between PC12 cells that express FLAG-ARMS and FLAG-ARMSΔP (Figure 6D). These results therefore verify the specificity of the ARMSΔP mutant on NGF-dependent Rap1 signaling.
Next, we sought to correlate the effects of ARMSΔP on Rap1 activation to downstream MAP kinase events. In contrast to wild-type ARMS-expressing cells, expression of ARMSΔP resulted in a marked decrease in p-MAPK protein at longer but not short time points (Figure 6E). Consistent with the NGF role in maintaining MAP kinase activity (York et al, 1998), ARMSΔP did not affect the initial activation of MAP kinase, but the longer duration of this activity. Interestingly, this was not accompanied by a decrease in pAkt by immunoblot analysis (Figure 6E), a result that was previously observed by staining primary cultures treated with the ARMS deletion mutant 4.2 (Figure 4D). Similar to the previous experiments using ARMS4.2, NGF-dependent neurite outgrowth in PC12 cells is also significantly reduced by ARMSΔP expression (Figure 6F). Finally, transient expression of ARMSΔP in cultured cortical neurons was found to perturb sustained MAPK activation by BDNF (Figure 6G), suggesting a conservation of ARMS function among the neurotrophins.
Loss of function of ARMS
The function of ARMS in mediating prolonged MAPK responses has been gleaned largely from transfection of mutant constructs into cells. As another means of investigating the effects of ARMS in neurotrophin signaling, we attempted to decrease the expression of the endogenous ARMS proteins in PC12 cells. Small interference RNA (siRNA) was generated against the rat ARMS cDNA sequence. ARMS expression was specifically reduced by at least 80% in cells transfected with siRNA (Figure 7). This reduction impaired the ability of the PC12 cells to activate Rap1 (Figure 7A) and the sustained MAPK pathway (40-min treatment; Figure 7B), but without appreciable effect on the Ras activation or on early MAP kinase activation (5-min treatment) in response to NGF. By contrast, Akt activation was not affected at any time point (Figure 7B), consistent with the specificity of ARMS signaling to the prolonged MAP kinase pathway (Figures 4D and 6E). The lack of effect upon Akt separates this signaling pathway from the prolonged MAPK pathway influenced by ARMS. Therefore, the siRNA experiments indicated that levels of ARMS are instrumental in the sustained Rap1-dependent MAPK activation elicited by NGF. Finally, the observations that ARMS siRNA abolished both early and delayed Rap1 activation but only interfered with late-phase MAP kinase activity are consistent with a bimodal mechanism of Ras and Rap1 in early- and late-phase MAP kinase induction (York et al, 1998).
Figure 7.
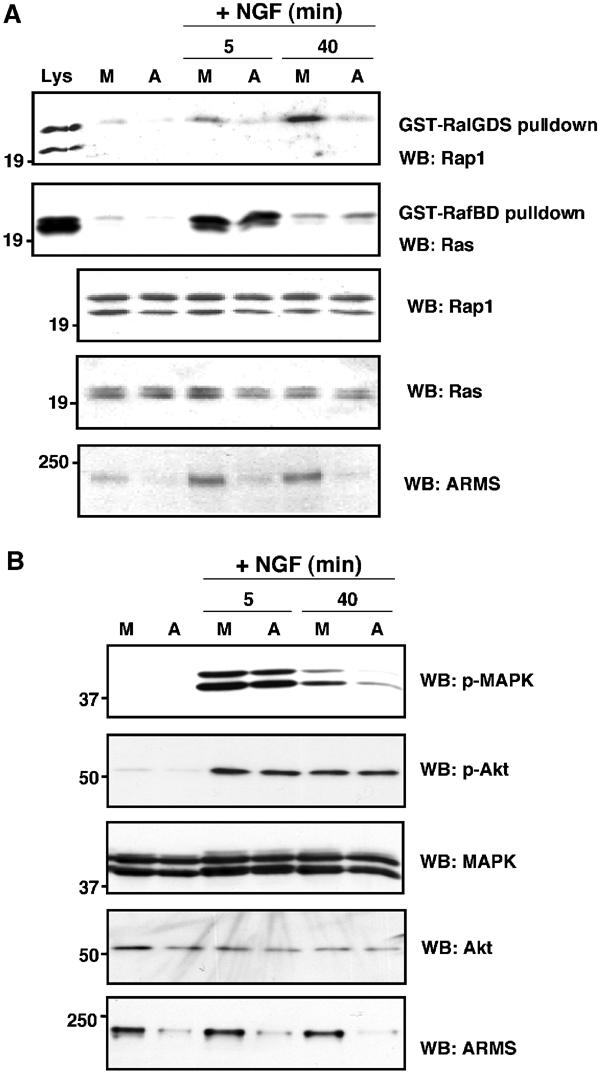
Downregulation of ARMS expression by siRNA impairs Rap1 and prolonged MAPK activation in PC12 cells upon NGF treatment. (A) ARMS siRNA impairs Rap1 but not Ras activation upon NGF treatment of PC12 cells. ARMS siRNA was produced as described in Materials and methods. PC12 cells were either mock transfected or transfected with ARMS siRNA. NGF treatment was applied for 5 or 40 min. Equal amounts of cellular lysates were subjected to Ras or Rap1 pulldown, as described in Figure 6D. Total lysates under each treatment condition were subjected to Western blotting with the indicated antibodies. (B) siRNA downregulates ARMS in PC12 cells and specifically decreases the Rap1-dependent prolonged MAPK activation elicited by NGF. Cellular extracts from control or ARMS siRNA-transfected PC12 cells were Western blotted with p-MAPK or pAkt antibody. The levels of ARMS protein in siRNA-transfected cells were assessed by blotting with anti-ARMS (892). A decrease of p-MAPK signal was detected in the cells with a reduced amount of ARMS at 40 min of NGF treatment, but not at 5 min. M, mock; A, ARMS siRNA.
Discussion
Many signaling pathways are not unique to neurotrophins but are utilized by other growth factors and cytokines in different cell contexts. Here we have found that the ARMS protein serves as an important downstream platform for the Trk neurotrophin receptor in neurons. There are several unique features of the ARMS protein. First, it is closely associated with Trk receptors through interactions between transmembrane domains. Second, ARMS becomes tyrosine phosphorylated by neurotrophins for many hours and days (Kong et al, 2001), consistent with the proposed role of maintaining neurotrophin signaling. Third, although ARMS lacks conventional motifs such as SH2, SH3 and PH domains, it contains a C-terminal PDZ-binding motif (SIL), a SAM domain, 11 ankyrin repeats and a polyproline region. In this study, we have found that the P1089RPPP sequence in the polyproline region of ARMS is responsible for sustaining MAP kinase activity and that this polyproline sequence interacts specifically with CrkL. Our data support a model in which the interactions of ARMS and CrkL lead to activation of the small G protein Rap1 that accounts for the ability of neurotrophins to signal for sustained duration.
ARMS-specific neurotrophin signaling
This report has identified a functional interaction between ARMS and CrkL in maintaining late-phase ERK activity via the Rap1 GTPase. The ARMS–CrkL interaction is specific, as the related c-CrkII proein does not bind to ARMS (Figure 6B). Consistent with nonoverlapping functions for CrkL and c-Crk II in Trk signaling, inactivation of the CrkL gene resulted in neural crest defects (Guris et al, 2001), similar to defects when the neurotrophins and Trk genes are inactivated (Huang and Reichardt, 2003).
Although MAP kinase activities are influenced by multiple neurotrophin-elicited signal transduction mechanisms (Kouhara et al, 1997; Qian et al, 1998; Atwal et al, 2000), our findings identify a unique pathway that accounts for the prolonged MAP kinase activity. This conclusion is supported by Trk receptor mutant knockin experiments, in which the Shc-binding site in the TrkB receptor was mutated by changing the consensus tyrosine residue to phenylalanine (Minichiello et al, 1998). This mutation resulted in a complete lack of association of Shc with TrkB. When BDNF was administered to cortical neurons isolated from TrkBShc/Shc mutant mice, MAP kinase activation was reduced, but not eliminated completely (Minichiello et al, 1998). Likewise, TrkCShc/Shc mutation also yielded a similar pattern of response to NT-3 (Postigo et al, 2002). In the latter study, MAP kinase activity still persisted after 60 min of neurotrophin treatment, indicating that sustained MAP kinase activation does not rely solely upon Shc binding.
Our findings with the ARMS protein suggest a new mechanism to account for the in vitro and in vivo findings that have been published. Association of ARMS with Trk receptors generates a scaffold, by which the CrkL–C3G–Rap1 proteins are assembled. Prolonged MAP kinase activation is facilitated by the formation of this complex. Disruption of the Trk–ARMS–CrkL interaction with two different mutant ARMS proteins (ARMS4.2 and ARMSΔP) perturbs this pathway, but still allows transient MAP kinase signaling events to occur.
Role of tyrosine phosphorylation
Since ARMS consists of multiple consensus motifs for serine/threonine and tyrosine kinases (Iglesias et al, 2000), an outstanding issue is the role of tyrosine phosphorylation in modulating the ARMS–CrkL interaction. The available data indicate that the constitutive interactions between Trk and ARMS and between ARMS and CrkL are critical for the initiation of Rap1-dependent signaling (Figures 4 and 6). One potential mechanism is that a Trk–ARMS–CrkL complex changes its recruitment for C3G upon NGF-induced phosphorylation. This is supported by the increase in C3G recruitment with CrkL (Figure 5C). We therefore propose that a preformed Trk–ARMS–CrkL complex (via the CrkL SH3 domain) facilitates CrkL SH2 domain binding to tyrosine-phosphorylated ARMS upon neurotrophin stimulation, thereby releasing the CrkL SH3 domain for recruitment of C3G. Previous studies indicated that conformational changes occur intramolecularly in Crk proteins that modify the binding specificities of Crk to other signaling proteins (Escalente et al, 1999; Kurokawa et al, 2001; Ting et al, 2001). Resolution of this issue will require the future identification and mutational analysis of ARMS phosphorylation sites.
Implications of ARMS signaling
The rapid and sustained kinetics of ARMS tyrosine phosphorylation by Trk is reminiscent of other receptor systems that utilize scaffolds for signal transduction. This includes the insulin receptor, which mediates the bulk of its signaling through IRS proteins (Shpakov and Pertseva, 2000) and by T-cell antigen receptors that use LAT, a transmembrane protein which serves as a scaffold for a variety of SH2 domain-containing proteins. The specificity of ligand–receptor signaling appears to involve the coupling of each receptor to unique multidomain adaptors.
Presynaptic and postsynaptic functions are dramatically influenced by neurotrophins (Poo, 2001). It is attractive to consider that neurons have evolved distinctive mechanisms to regulate synaptic function over different time courses. The recent finding that BDNF can exert synaptic responses over millisecond time intervals (Blum et al, 2002), coupled with the ability of neurotrophins to produce physiological effects of extended duration, suggests that there are specialized mechanisms for neurotrophin receptors. This is further supported by recent observations that AMPA receptor distribution is determined by the opposing actions of Ras and Rap (Zhu et al, 2002), which can display specific consequences upon LTP (Morozov et al, 2003). Due to the close proximity of the ARMS protein with Trk receptors and its time course of phosphorylation, it is tempting to speculate that ARMS may serve as a bridge between neurotrophins and changes in short-term and long-term synaptic strength, in addition to the process of neuronal differentiation.
Materials and methods
Materials
NGF was obtained from Harlan (Indianapolis, IN) and BDNF and EGF were from Peprotech (Rocky Hill, NJ). The following antibodies were used: 4G10, CrkL were obtained from Upstate Biotech (Lake Placid, NY), Trk (C-14 and B-3), PLC-γ, EGFR, Crk II, C3G and Rap1 were from Santa Cruz Biotechnology (Santa Cruz, CA), p-MAPK, p-Akt, MAPK and Akt were from Cell Signaling Technology (Beverly, MA), anti-FLAG was from Sigma (St Louis, MO), polyclonal ARMS antibody has been previously described (Kong et al, 2001). Anti-TrkA antiserum (RTA) that recognizes the extracellular domain of TrkA was generously provided by L Reichardt (UCSF).
Plasmids
Constructs of different ARMS domains were amplified by PCR using the rat ARMS cDNA (Kong et al, 2001) as a template. Deletion of amino acids P1089–P1093 in ARMS was performed by site-directed mutagenesis to generate ARMSΔP. Deletion constructs of ARMS were cloned in pFLAG-CMV1 or pFLAG-CMV2 vectors, or in pGEX6P to generate recombinant proteins in Escherichia coli. The TrkAΔJM construct was made in pCMV5, by ligating PCR-generated cDNA fragments of rat TrkA (Yano et al, 2000). All PCR fragments were verified by DNA sequencing.
DNA transfections, preparation of cell lysates and immunoblotting
Plasmid DNA was transiently transfected into HEK293 cells (2 × 106 cells/plate) using the calcium phosphate method. PC12 cells and cortical neurons were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. For PC12 cell immunoprecipitations, 1–2 × 107 cells were used. Western blotting was performed according to Arévalo et al (2000).
Preparation of GST fusion proteins and in vitro binding assays
ARMS7, ARMS7.2 (amino acids P1057–G1110), ARMS7.2ΔP (ARMS 7.2 with amino acids P1089–P1093 deleted), ARMS7.3 (amino acids P1057–G1088) and ARMS-7.6 (amino acids P1132–L1151) were amplified by PCR and subcloned in pGEX6P-1 vector. GST–ARMS proteins were purified according to Yano et al (2001).
Stably transfected PC12 cells
PC12 cells were cotransfected with 20 μg of pFLAG-ARMS or pFLAG-ARMSP and 2 μg of pCDNA3 with Lipofectamine 2000. To obtain stable transfectants, cells were treated with 500 μg/ml G418. After 15–20 days of G418 selection, resistant clones were subjected to Western blot analysis using FLAG antibodies to verify ARMS or ARMSΔP protein expression. Positive clones were maintained in 200 μg/ml G418 thereafter.
siRNA against ARMS
ARMS siRNA was obtained using the Silencer™ siRNA Cocktail Kit (RNase III) (Ambion). As a template for the in vitro transcription, a fragment of 400 bp corresponding to the nucleotides 3952–4357 of the ARMS rat sequence was subcloned in pcDNA3 in both orientations. PC12 cells in 24-well plates were transfected with ARMS siRNA (0.5 μg/well) or mock transfected using Lipofectamine 2000 in two consecutive days. After 24 h, cells were starved overnight and treated with NGF (100 ng/ml) for different time points before immunoblotting.
Preparation and crosslinking of cortical neuron lysates
Cortical neurons were obtained from E17–19 rats and cultured in Neurobasal medium (Gibco) supplemented with B-27 and 0.4 mM glutamine in poly-L-lysine-coated plates. For co-immunoprecipitation, primary cultured neurons DIV 8–11 were rinsed with PBS and proteins were crosslinked with 1 mM dithiobis-succinimidylpropionate (DSP, Pierce Biotechnology, Rockford, IL) in PBS for 30 min. Immunoprecipitated complexes were boiled for 5 min to cleave the crosslinked proteins, and resolved by SDS–PAGE and Western blotting with antibodies against Trk and ARMS.
Ras and Rap1 activation assays
PC12 cells and stable PC12 transformants expressing FLAG-ARMS or FLAG-ARMSΔP were used for these experiments. Recombinant GST-RalGDS Rap-binding domain (RBD) and GST-Raf-Ras-binding domain were produced in bacteria as described above. Pull-down assays were used to detect endogenous levels of GTP-bound Rap1 or Ras proteins (Wu et al, 2001).
Supplementary Material
Supplementary data
Acknowledgments
We thank Belén Domínguez, Pilar Pérez, Dionisio Martin-Zanca, Barbara Hempstead and the members of the lab for comments and support, Louis Reichardt for generously providing RTA antibody, and Lani Mutstacchi for primary cultures. This work was supported by the NIH (NS21072 and HD023315) and by a Postdoctoral Fellowship from the Spanish Education Ministry (JCA).
References
- Arévalo JC, Conde B, Hempstead BL, Chao MV, Martin-Zanca D, Perez P (2000) TrkA immunoglobulin-like ligand binding domains inhibit spontaneous activation of the receptor. Mol Cell Biol 20: 5908–5916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwal J, Massie B, Miller F, Kaplan D (2000) The TrkB-Shc site signals neuornal survival and local axon growth via MEK and PI3-kinase. Neuron 27: 265–277 [DOI] [PubMed] [Google Scholar]
- Barbacid M (1994) The trk family of neurotrophin receptors. J Neurobiol 25: 1386–1403 [DOI] [PubMed] [Google Scholar]
- Bibel M, Barde Y (2000) Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev 14: 2919–2937 [DOI] [PubMed] [Google Scholar]
- Blum R, Kafitz K, Konnerth A (2002) Neurotrophin-evoked depolarization requires the sodium channel NaV1.9. Nature 419: 687–693 [DOI] [PubMed] [Google Scholar]
- Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhauser N, Bohm-Matthaei R, Baeuerle PA, Barde Y-A (1996) Selective activation of NK-kB by nerve growth factor through the neurotrophin receptor p75. Science 272: 542–545 [DOI] [PubMed] [Google Scholar]
- Chao MV (1992) Growth factor signaling: where is the specificity? Cell 68: 995–997 [DOI] [PubMed] [Google Scholar]
- Chao MV (2003) Neurotrophins and their receptors: a convergence point for many signaling pathways. Nat Rev Neurosci 4: 299–309 [DOI] [PubMed] [Google Scholar]
- Clary DO, Weskamp G, Austin LR, Reichardt LF (1994) TrkA cross-linking mimics neuronal responses to nerve growth factor. Mol Biol Cell 5: 549–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley S, Paterson H, Kemp P, Marshall CJ (1994) Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell 77: 841–852 [DOI] [PubMed] [Google Scholar]
- Escalente M, Courtney J, Chin W, Teng K, Kim J, Fajardo J, Mayer B, Hempstead B, Birge R (1999) Phosphorylation of c-Crk II on the negative regulatory Tyr222 mediates nerve growth factor-induced cell spreading and morphogenesis. J Biol Chem 275: 24787–24797 [DOI] [PubMed] [Google Scholar]
- Feller S (2001) Crk family adaptors-signalling complex formation and biological roles. Oncogene 20: 6348–6371 [DOI] [PubMed] [Google Scholar]
- Green SH, Rydel RE, Connolly JL, Greene LA (1986) PC12 mutant that possesses low- but not high-affinity NGF receptors neither respond to nor internalize NGF. J Cell Biol 102: 830–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guris D, Fantes J, Tara D, Druker B, Imamoto A (2001) Mice lacking the homologue of the human 22q11.2 gene CrkL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet 27: 293–298 [DOI] [PubMed] [Google Scholar]
- Hermann C, Horn G, Spaargaren M, Wittinghofer A (1996) Differential interaction of the ras family GTP-binding proteins H-Ras, Rap1A, and R-Ras with the putative effector molecules Raf kinase and Ral-guanine nucleotide exchange factor. J Biol Chem 271: 6794. [DOI] [PubMed] [Google Scholar]
- Huang E, Reichardt L (2003) Trk receptors: roles in neuronal signal transduction. Ann Rev Biochem 72: 609–642 [DOI] [PubMed] [Google Scholar]
- Iglesias T, Cabrera-Poch N, Mitchell M, Naven T, Rozengurt E, Schiavo G (2000) Identification and cloning of Kidins220, a novel neuronal substrate of protein kinase D. J Biol Chem 275: 40048–40056 [DOI] [PubMed] [Google Scholar]
- Kao S, Jaiswal R, Kolch W, Landreth G (2001) Identification of the mechanisms regulating the differential activation of the Mapk cascade by epidermal growth factor and nerve growth factor in PC12 cells. J Biol Chem 276: 18169–18177 [DOI] [PubMed] [Google Scholar]
- Kong H, Boulter J, Weber J, Lai C, Chao M (2001) An evolutionarily conserved transmembrane protein that is a novel downstream target of neurotrophin and ephrin receptors. J Neurosci 21: 176–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J (1997) A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 89: 693–702 [DOI] [PubMed] [Google Scholar]
- Kurokawa K, Mochizuki N, Ohba Y, Mizuno H, Miyawaki A, Matsuda M (2001) A pair of fluorescent resonance energy transfer-based probes for tyrosine phosphorylation of the CrkII adaptor protein in vivo. J Biol Chem 276: 31305–31310 [DOI] [PubMed] [Google Scholar]
- Marshall C (1995) Specificity of receptor tyrosine kinase signalling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80: 179–185 [DOI] [PubMed] [Google Scholar]
- Meakin S, MacDonald J, Gryz E, Kubu C, Verdi J (1999) The signaling adapter FRS-2 competes for Shc for binding to the nerve growth factor receptor TrkA. J Biol Chem 274: 9861–9870 [DOI] [PubMed] [Google Scholar]
- Minichiello L, Casagranda F, Tatche RS, Stucky CL, Postigo A, Lewin GR, Davies AM, Klein R (1998) Point mutation in trkB causes loss of NT4-dependent neurons without major effects on diverse BDNF responses. Neuron 21: 335–345 [DOI] [PubMed] [Google Scholar]
- Morozov A, Muzzio I, Bourtchouladze R, Van-Strien N, Lapidus K, Yin D, Winder D, Admas J, Sweatt J, Kandel E (2003) Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning and memory. Neuron 39: 309–325 [DOI] [PubMed] [Google Scholar]
- Poo M-m (2001) Neurotrophins as synaptic modulators. Nat Rev Neurosci 2: 24–31 [DOI] [PubMed] [Google Scholar]
- Postigo A, Calella A, Fritzsch B, Knipper M, Katz D, Eilers A, Schimmang T, Lewin G, Klein R, Minichiello L (2002) Distinct requirements for TrkB and TrkC signaling in target innervation by sensory neurons. Genes Dev 16: 633–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Riccio A, Zhang Y, Ginty D (1998) Identification and characterization of novel substrates of Trk receptors in developing neurons. Neuron 21: 1017–1029 [DOI] [PubMed] [Google Scholar]
- Qiu M-S, Green SH (1991) NGF and EGF rapidly activate p21ras in PC12 cells by distinct, convergent pathways involving tyrosine phosphorylation. Neuron 7: 937–946 [DOI] [PubMed] [Google Scholar]
- Qui M-S, Green SH (1992) PC12 cell neuronal differentiation is associated with prolonged p21ras activity and consequent prolonged ERK activity. Neuron 9: 705–717 [DOI] [PubMed] [Google Scholar]
- Sherwood N, Lo D (1999) Long-term enhancement of central synaptic transmission by chronic brain-derived neurotrophic factor treatment. J Neurosci 19: 7025–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpakov A, Pertseva M (2000) Structural and functional characterization of insulin receptor substrate proteins and the molecular mechanisms of their interaction with insulin superfamily tyrosine kinase receptors and effector proteins. Membr Cell Biol 13: 455–484 [PubMed] [Google Scholar]
- Stork P (2003) Does Rap1 deserve a bad Rap? Trends Biochem Sci 28: 267–275 [DOI] [PubMed] [Google Scholar]
- Teng KK, Courtney J, Henegouwen P, Birge R, Hempstead B (1996) Dissociation of NGF induced signal transduction from neurite elongation by expression of a mutant adaptor protein v-Crk in PC12 cells. Mol Cell Neurosci 8: 157–170 [DOI] [PubMed] [Google Scholar]
- Teng KK, Landers H, Fajardo JE, Hanafusa H, Hempstead BL, Birge RB (1995) v-Crk modulation of growth factor-induced PC12 cell differentiation involves the Src homology 2 domain of v-crk and sustained activation of the ras/mitogen activated protein kinase pathway. J Biol Chem 270: 20677–20685 [DOI] [PubMed] [Google Scholar]
- Ting A, Kain K, Klemke R, Tsien R (2001) Genetically encoded fluorescent reporters of protein tyrosine kinase activities in living cells. Proc Natl Acad Sci USA 98: 15003–15008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossler M, Yao H, York R, Pan M-G, Rim C, Stork P (1997) cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell 89: 73–82 [DOI] [PubMed] [Google Scholar]
- Wu C, Lai C, Mobley W (2001) Nerve growth factor activates persistent Rap1 signaling in endosomes. J Neuroscience 21: 5406–5416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano H, Cong F, Birge RB, Goff SP, Chao MV (2000) Association of the Abl tyrosine kinase with the Trk nerve growth factor receptor. J Neurosci Res 59: 356–364 [DOI] [PubMed] [Google Scholar]
- Yano H, Lee F, Kong H, Chuang J-Z, Arévalo J, Perez P, Sung C-H, Chao M (2001) Association of tyrosine kinase neurotrophin receptors with components of the cytoplasmic dynein motor. J Neurosci 21: RC125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SO, Carter BD, Casaccia-Bonnefil P, Chao MV (1998) Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci 18: 3273–3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- York R, Yao H, Dillon T, Ellig C, Eckert S, McCleskey E, Stork P (1998) Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature 392: 622–626 [DOI] [PubMed] [Google Scholar]
- Zhu J, Qin Y, Zhao M, Van Aelst L, Malinow R (2002) Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 110: 443–455 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data