

Abstract
Turoctocog alfa (NovoEight®) is a recombinant factor VIII (rFVIII) with a truncated B-domain made from the sequence coding for 10 amino acids from the N-terminus and 11 amino acids from the C-terminus of the naturally occurring B-domain. Turoctocog alfa is produced in Chinese hamster ovary (CHO) cells without addition of any human- or animal-derived materials. During secretion, some rFVIII molecules are cleaved at the C-terminal of the heavy chain (HC) at amino acid 720, and a monoclonal antibody binding C-terminal to this position is used in the purification process allowing isolation of the intact rFVIII. Viral inactivation is ensured by a detergent inactivation step as well as a 20-nm nano-filtration step. Characterisation of the purified protein demonstrated that turoctocog alfa was fully sulphated at Tyr346 and Tyr1664, which is required for optimal proteolytic activation by thrombin. Kinetic assessments confirmed that turoctocog alfa was activated by thrombin at a similar rate as seen for other rFVIII products fully sulphated at these positions. Tyr1680 was also fully sulphated in turoctocog alfa resulting in strong affinity (low nm Kd) for binding to von Willebrand factor (VWF). Half-lives of 7.2 ± 0.9 h in F8-KO mice and 8.9 ± 1.8 h haemophilia A dogs supported that turoctocog alfa bound to VWF after infusion. Functional studies including thromboelastography analysis of human haemophilia A whole blood with added turoctocog alfa and effect studies in mice bleeding models demonstrated a dose-dependent effect of turoctocog alfa. The non-clinical data thus confirm the haemostatic effect of turoctocog alfa and, together with the comprehensive clinical evaluation, support the use as FVIII replacement therapy in patients with haemophilia A.
Keywords: haemophilia A, N8, rFVIII, recombinant factor VIII, turoctocog alfa
The human gene for factor VIII (FVIII) was cloned and expressed in 1984 1–5, making it possible to produce recombinant FVIII (rFVIII) for prevention and treatment of bleeds in patients with haemophilia A. The FVIII gene encodes a single chain of 2332 amino acid residues with the domain structure A1-A2-B-A3-C1-C2 (Fig.1) 1. During cellular processing, the molecule undergoes posttranslational modifications, including sulphation of specific tyrosine residues and glycosylation. Furthermore, the protein is processed into a heterodimer consisting of a heavy chain (HC) with the A1-A2-B domains and a light chain (LC) with the A3-C1-C2 domains. The two chains are held together by metal ions 6. While the B-domain plays a role in restricting the expression of endogenous FVIII, it is apparently not needed for the function of FVIII 7–9. Acidic regions C-terminal to the A1 and the A2 domains and N-terminal to the A3 domain (a1, a2 and a3, respectively) are important for interaction with thrombin and von Willebrand factor (VWF). Sulphation of Tyr1680 is essential for high affinity binding of FVIII to VWF 10,11, which is required for protection of FVIII from degradation and rapid clearance. During haemostasis, FVIII is activated by specific thrombin cleavages, thereby producing the A1, A2 and A3-C1-C2 fragments of activated FVIII (FVIIIa). This results in dissociation of VWF and assembly of the tenase complex (FVIIIa/FIXa) on the surface of activated platelets. FXa is generated, resulting in a thrombin burst, and ultimately leading to the formation of a stable haemostatic plug.
Figure 1.
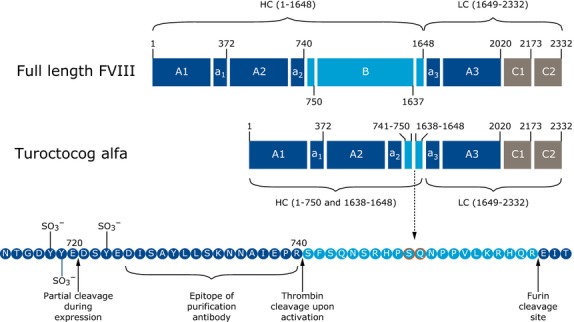
Molecular structure of full-length FVIII and turoctocog alfa. The upper part of the figure illustrates full-length FVIII coding for 2332 amino acid residues. The middle part of the figure shows the structure of turoctocog alfa. The molecule consists of the heavy chain containing the A1-A2 domains, a truncated B-domain coding for 21 amino acids of the naturally occurring B-domain (amino acid 741–750 fused with 1638–1648) and the light chain (A3-C1-C2 domains). The lower part of the figure shows the sequence coding for the 21 amino acid residue truncated B-domain (light blue), which represents 10 amino residues from the N-terminal of the B-domain linked to 11 amino acid residues from the C-terminal of the B-domain. Cleavage sites, tyrosine sulphation sites and the epitope detected by F25 during purification are indicated in this region 13. Reproduced from Thim et al. 13 with permission from John Wiley and Sons.
The design of turoctocog alfa
Turoctocog alfa (NovoEight®) is a new rFVIII molecule with a truncated B-domain (Fig.1). The molecule consists of a heavy chain containing the A1-A2 domains, a truncated B-domain coding for 21 amino acids of the naturally occurring B-domain (amino acid 741–750 fused with 1638–1648) and the light chain (A3-C1-C2 domains). While the B-domain is dispensable for procoagulant activity of FVIII 8, cellular expression and secretion is regulated by the B-domain 12. The truncated B-domain was selected to achieve a well-defined product when produced in Chinese hamster ovary (CHO) cells, thereby providing a solid basis for establishing a robust purification process. In addition, an O-glycosylation site is present in the B-domain allowing for future glyco-engineering 13,14. Upon thrombin activation, the truncated B-domain is removed resulting in rFVIIIa with a structure similar to endogenous FVIIIa 13,15.
Although the truncated B-domain is derived from the N-and C-terminal sequences of the B-domain from the native full-length FVIII molecule, the junction between these sequences is engineered and might therefore in theory pose an immunological risk. In silico methods, based on in vitro affinity of peptides towards major histocompatibility complex (MHC) class II molecules, are used to evaluate potential risks of immunogenicity of non-native sequences. Kimchi-Sarfaty et al. 16 used this method to compare the predicted immunogenicity of turoctocog alfa with that of a B-domain deleted rFVIII product [ReFacto AF®, Swedish Orphan Biovitrum AB (publ), Stockholm, Sweden]. While the junction between the N- and C-terminal parts of the B-domain is the same for the two proteins, the N-terminal part of the B-domain for the comparator molecule comprises only three amino acids in contrast to 10 in turoctocog alfa, thereby creating a different immunological environment. The in silico analysis predicted that the B-domain of turoctocog alfa bound to fewer MHC class II molecules and with lower affinity than the comparator FVIII product 16, suggesting that the B-domain of turoctocog alfa is not associated with an additional risk of inducing an immune response. As the predictive value of in silico methods is not established, clinical data are required to assess the immunogenicity of turoctocog alfa. No inhibitors were observed in the pivotal clinical development programme for turoctocog alfa, which included 213 previously treated patients with severe haemophilia A 17,18.
Manufacturing of turoctocog alfa
Turoctocog alfa is produced in CHO cells grown in serum-free conditions and formulated without animal- or human-derived materials. The CHO cell is a well-known production cell line, which has been used to produce therapeutic proteins within many different therapeutic fields 19. The FVIII molecule is intracellularly processed by furin, resulting in cleavage between the B-domain and the LC (Fig.1). The purification process (Fig.2) includes the following steps: concentration (using hydrophobic interaction/cation exchange), anti-rFVIII immunoaffinity chromatography (using a recombinant monoclonal antibody), anion exchange chromatography, and size exclusion 13. Viral inactivation is ensured by detergent treatment (included in the initial purification step) and a 20-nm filtration step prior to the size exclusion step.
Figure 2.
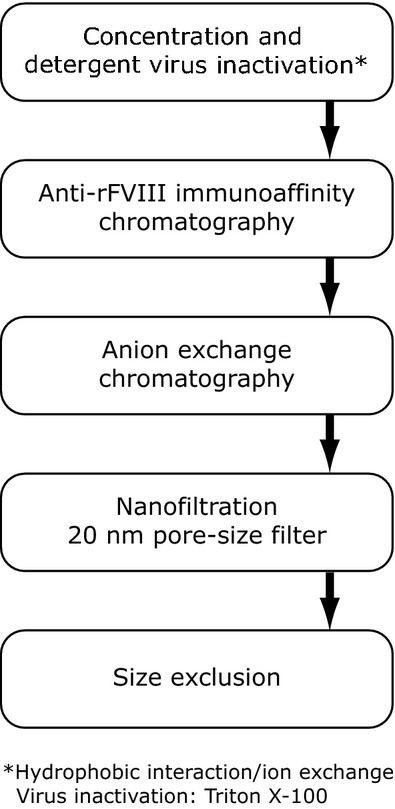
Purification of turoctocog alfa. From Thim et al. 13.
During secretion, some rFVIII molecules are cleaved in the A2 domain leaving part of the FVIII molecules with a heavy chain with C-terminus at amino acid 720 or 729 20,21. As the C-terminal end of the A2 domain (amino acids 720–740) contains residues required for optimal interaction with thrombin 22, the purification process for turoctocog alfa was optimised in order to secure isolation of molecules with intact A2 domain. This was obtained by selecting an antibody (F25) for immunoaffinity chromatography that binds selectively to molecules with intact A2 domain. The antibody binds to the C-terminus of the A2 domain (Fig.1), enabling removal of FVIII degraded within the A2 domain 13. The gene encoding the antibody was transferred to CHO cells for production and therefore no additional murine antigens or host cell proteins are introduced 23. The removal of degraded heavy chain is clearly seen by SDS–PAGE analysis after thrombin cleavage, where more than one band represents the A2 domain for ReFacto AF® (Fig.3, lane 8). For turoctocog alfa (Fig.3, lane 2) or rFVIII derived from the full-length genes (Fig.3, lanes 4 and 6), no C-terminal degradation in the A2 domain was observed.
Figure 3.
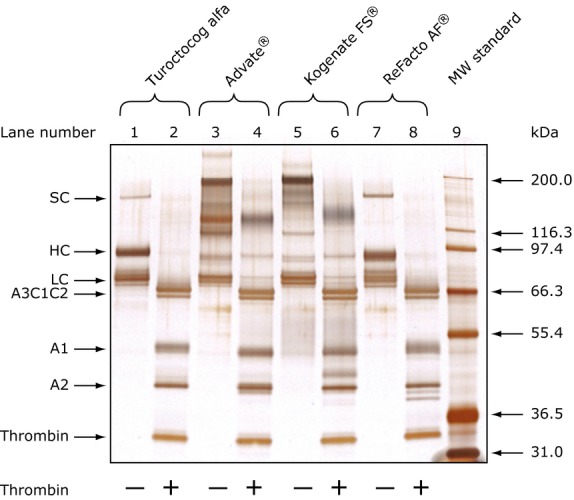
SDS-PAGE of rFVIII molecules before and after thrombin cleavage. From Kristensen et al. 26.
The SDS-PAGE analysis also shows that the rFVIII proteins produced by the full-length gene, due to multiple cleavage sites within the B-domain, are a heterogeneous mixture of heavy chains with different lengths of the B-domain included (Fig.3, lanes 3 and 5). For Advate® (lane 3), one of these bands contains both B-domain sequences and LC sequences, suggesting incomplete processing between the B-domain and the LC 13,24–26. For turoctocog alfa and ReFacto AF®, no such heterogeneity of HC chain fragments was observed and mainly one band at 90 kDa and a minor band representing the single chain protein were detected (lanes 1 and 7). The single chain (representing approximately 3–4% in turoctocog alfa 13) can be converted into the A1 and A2 domains after thrombin cleavage (lane 2 and 8). The SDS-PAGE analysis also illustrates that the purification process for turoctocog alfa results in a homogeneous rFVIII product, which after thrombin cleavage is identical to native FVIIIa.
Structural characterisation of turoctocog alfa
The purity and homogeneity of turoctocog alfa allowed crystallisation of the protein. The resulting X-ray crystallographic structure of turoctocog alfa (Fig.4) confirms that the protein has a structure similar to those previously reported for other rFVIII molecules 27,28. The X-ray fluorescence wavelength scan of the turoctocog alfa crystals identified two significant peaks, indicating copper (Cu+) and zinc (Zn2+), respectively 29. The presence of both copper and zinc was supported by colorimetric data, indicating that the ion-binding site in the A1 domain is predominantly populated by zinc, while that in the A3 domain is predominantly populated by copper 29.
Figure 4.
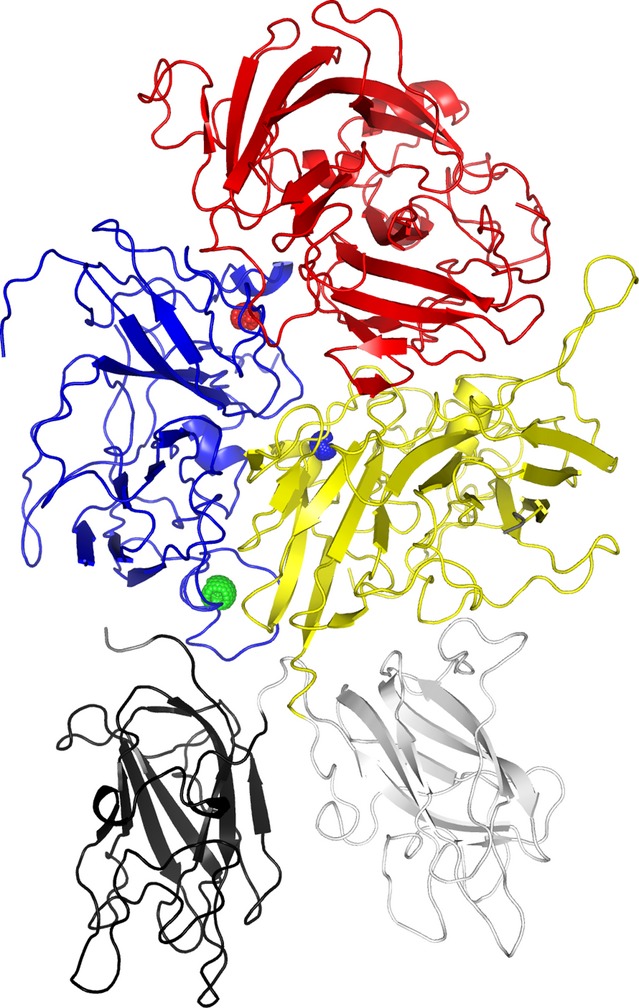
Crystallographic structure of turoctocog alfa. The heavy chain consists of the A1 (blue) and A2 (red) domain while the light chain consists of the A3 (yellow), C1 (grey) and C1 (black) domains. The three ions, calcium (green), copper (blue) and zink (red), observed in the structure are shown as spheres. The structure is available at protein database with the entry code 4bdv (http://www.rcsb.org/pdb/explore/explore.do?structureId=4BDV) 29.
Posttranslational modifications
Known posttranslational modifications of plasma-derived FVIII include N- and O-linked glycosylations 30. As for the plasma-derived FVIII, turoctocog alfa contains four N-linked glycosylations (Asn41 and Asn239 in the A1 domain, Asn1810 in the A3 domain and Asn2118 in the C1 domain) and one O-linked glycosylation (Ser750) in the B-domain 13. Structure analyses have shown that the N-linked glycosylation sites are fully glycosylated, while the degree of occupancy of the O-linked glycan at Ser750 is approximately 65%. The majority of the complex N-linked glycans at Asn41 in the A1 domain and Asn1810 in A3 are di-sialylated core fucosylated bi-antennary structures, while high mannose glycans are present at Asn239 in A1 and Asn2118 in C1 13.
Recombinant proteins derived from non-human cell lines can contain detectable amount of non-human glycoforms 31. As observed for Advate® 31, N-glycolyl neuraminic acid is also detected in low amounts in turoctocog alfa on di-sialylated core fucosylated bi-antennary structures 13. Furthermore, the O-linked glycosylation at Ser750 contains a doubly sialylated GalNAc-Gal structure, which has also been described to be present in other rFVIII molecules 32. The clinical relevance of these observations is unknown. In agreement with the lack of capacity of producing the antigenic epitope in Galα(1,3)Gal structures in CHO cell lines 33, these structures have not been detected in turoctocog alfa 13.
An additional posttranslational modification of FVIII is sulphation of specific tyrosines. A total of six tyrosine sulphation sites (Tyr346 in a1, Tyr718, 719, 723 in a2 and Tyr1664 and Tyr1680 in a3) have been described for plasma-derived FVIII 30. The tyrosine sulphation in turoctocog alfa was analysed, and Tyr346, Tyr1664 and Tyr1680 were found to be fully sulphated (Table1) 34. The high negative charge of the peptide containing Tyr718, Tyr719 and Tyr723 resulted in weak response in the mass spectrometry analysis thereby preventing reliable quantification. However, only fully sulphated peptides containing Tyr718, Tyr719 and Tyr723 were detected. Similar data were found for a plasma-derived FVIII concentrate Octanate®, whereas Tyr1680 was not fully sulphated in Advate® 34. The existence of a small amount (2.6-16.7%) of non-sulphated Tyr1680 in Advate® was also observed in other studies 31,35. In these studies, other rFVIII products (Kogenate FS® and ReFacto AF®) also contained similar amounts of non-sulphated Tyr1680, while FVIII produced in human embryonic kidney (HEK) cells 31 or plasma-derived FVIII 35 did not have detectable amounts of non-sulphated Tyr1680 (Table1). The full sulphation of Tyr1680 in turoctocog alfa demonstrates that CHO cells are capable of providing full tyrosine sulphation.
Table 1.
Tyrosine sulphation in turoctocog alfa and other FVIII products
| Product | Origin | Non-sulphated Tyr1680 (%) |
|---|---|---|
| Turoctocog alfa | CHO | Below detection limit (0.5%) |
| Octanate® | Plasma-derived | Below detection limit (0.5%) |
| Advate® | CHO | 2.6–16.7 |
| ReFacto AF® | CHO | 4.5–13.9 |
| Kogenate FS® | BHK | 1.0–6.5 |
| Human-cl rhFVIII | HEK | Below detection limit |
In agreement with the existence of a small amount of non-sulphated Tyr1680 in Advate®, the affinity of turoctocog alfa to immobilised VWF was slightly higher than observed for Advate®, both when measured by ELISA and by surface plasmon resonance (SPR) (Table2) 15. However, for both FVIII proteins, the Kd values are in the sub-nanomolar range 15 and the excess VWF in the circulation (30–50 nm VWF monomer) means that essentially all FVIII will be bound to VWF after infusion. In agreement with this, comparable pharmacokinetics of turoctocog alfa and Advate® were observed in F8-knockout (F8-KO) mice 36, in haemophilia A dogs 37 and in human clinical trials 38 (see below).
Table 2.
VWF binding in turoctocog alfa and Advate®
| VWF binding | Turoctocog alfa | Advate® |
|---|---|---|
| VWF binding (ELISA) Kd (nm) (n = 4)† | 0.24 ± 0.04* | 0.48 ± 0.13 |
| VWF binding (SPR) (turoctocog alfa n = 9, Advate® n = 2)1 | ||
| Kd (nm) | 0.23 ± 0.13* | 0.45 ± 0.07 |
| kon (×106/m/s) | 4.5 ± 2.1 | 1.2 ± 0.6 |
| koff (×10−4/s) | 9.3 ± 4.7 | 5.5 ± 1.9 |
From Christiansen et al. 2010 15.
VWF, von Willebrand factor; SPR, surface plasmon resonance.
Significantly different (P < 0.05) from the value obtained for Advate® using one-tailed Student’s t-test.
Values are mean and SD of the numbers of experiments indicated.
Functional characteristics – in vitro and in vivo
Sulphation of Tyr346 and Tyr1664 are required for optimal interaction with thrombin 11. In line with the observed full tyrosine sulphation at Tyr346 and Tyr1664 for both turoctocog alfa and Advate®, similar rates of activation by thrombin were observed for turoctocog alfa (13.5 ± 6.7 ×10−3 per minute) and Advate® (10.6 ± 3.5 × 10−3 per minute) 15. The kinetic assessment furthermore revealed similar affinity towards FIXa and similar Michaelis constants (Km) of FX activation for turoctocog alfa and Advate®, suggesting the same cofactor activity of the two FVIII compounds. Inactivation of activated turoctocog alfa and Advate® by activated protein C (APC) was also similar for the two products 15. In standard FVIII:C assays, such as one-stage clotting assay and chromogenic assay, similar activities were obtained showing in essence no assay discrepancies 15. The specific activity was 9300 ± 400 U/mg (mean and SD of seven batches) 15. In addition, a field study using spiked haemophilia A plasma samples mimicking post-infusion samples showed that turoctocog alfa can be reliably measured using routine FVIII:C assays in clinical laboratories without use of a product-specific standard. The field study also showed that the variability between Advate® and turoctocog was similar across all the laboratories irrespectively of the assay conditions used 39.
In additional functional studies, the thrombin generation assay and thromboelastography (TEG®) were used to characterise the activity of turoctocog alfa in human blood or plasma 15. In the thrombin generation assay, haemophilia A was mimicked either by using severe haemophilia A plasma and adding normal human platelets and lipidated tissue factor 40, or by a reconstituted cell-based model 41 where coagulation factors V, VIII, VIIa, IX, X XI, prothrombin, tissue factor pathway inhibitor (TFPI) and antithrombin were added to normal human platelets and monocytes stimulated to express tissue factor. In both systems, a delayed and lowered thrombin generation was seen when no FVIII was present, while addition of turoctocog alfa improved the thrombin generation in a concentration-dependent manner. When turoctocog alfa was added at 1 IU/mL, the rate of thrombin generation in the plasma-based model system was 3.7 ± 0.8 nm/min similar to the value obtained for Advate® (3.7 ± 0.7 nm/min) 15. Thromboelastography using whole blood from patients with haemophilia A showed as expected that the clot formation time was delayed and the rate of clot formation decreased in this population. Addition of turoctocog alfa resulted in a dose-dependent improvement and normalisation of these parameters.
The in vivo effect of turoctocog alfa was evaluated in F8-KO mice. These mice do not have spontaneous bleeds, but bleed significantly after injury. In a tail bleeding model using a 4 mm tail cut, turoctocog alfa induced a dose-dependent shortening of the bleeding time and a reduction of the blood loss 36. The turoctocog alfa dose causing 50% reduction (ED50) of these parameters were 29 and 27 IU/kg, respectively (Fig.5). These ED50 values were not significantly different from the values obtained for Advate® (39 and 28 IU/kg, respectively). At a sufficiently high dose, both FVIII compounds normalised the bleeding time and blood loss. Lower FVIII doses were required for normalisation when more subtle injuries were made in the mice. In a saphenous vein bleeding model, the ED50 of turoctocog alfa was 5.7 ± 1.3 IU/kg 42, while an ED50 of 1.1 IU/kg [95% confidence intervals 0.09–2.0 IU/kg] was observed in a tail vein transection model (Peter Johansen and Tom Knudsen, Novo Nordisk, manuscript submitted). A single high dose of turoctocog alfa (280 IU/kg) was evaluated in a needle-induced joint bleeding model in F8-KO mice 36. While untreated mice showed bleeds in the joint and concomitant swelling 1 and 3 d after injury, turoctocog alfa-treated animals showed no signs of or only minor bleeds in the joints. In conclusion, all mice studies demonstrated haemostatic efficacy of turoctocog alfa, and turoctocog alfa was capable of normalising the bleeding phenotype.
Figure 5.
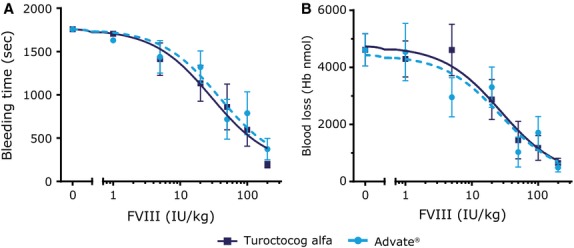
Effect of turoctocog alfa on bleeding in F8-KO mice. Turoctocog alfa or Advate® was administered intravenously at the doses noted to F8-KO mice 5 min prior to a 4-mm tail cut. Bleeding time (A) and blood loss (B) were measured for 30 min. Data are means and SEM of n = 8. The left data point (0 IU/kg) reflects the bleeding observed in F8-KO mice receiving vehicle only. Reproduced from Elm et al. 38 with permission from John Wiley and Sons.
Pharmacokinetics
The pharmacokinetics of turoctocog alfa were assessed in F8-KO mice and in haemophilia A dogs before initiating the human clinical trials 36,37. In the human phase 1 clinical trial, where the pharmacokinetic profile of a single i.v. dose of turoctocog alfa was comparable to the profile of a single i.v. dose of Advate® (Table3) 38.
Table 3.
Pharmacokinetic parameters for turoctocog alfa in patients with haemophilia A
| Turoctocog alfa | Advate® | |
|---|---|---|
| Mean (SD) | Mean (SD) | |
| F8-KO mice (280 IU/kg), N = 3–4 | ||
| t1/2 (h) | 7.2 (0.9) | 7.7 (1.4) |
| Clearance (mL/h/kg) | 11 (1) | 10 (2) |
| Mean residence time (h) | 10 (1.3) | 11 (2.1) |
| Haemophilia A dogs (100 IU/kg), N = 3 | ||
| t1/2 (h) | 8.9 (1.8) | 8.2 (0.2) |
| Clearance (mL/h/kg) | 3.6 (0.6) | 5.2 (0.7) |
| Mean residence time (h) | 12 (2) | 11 (0) |
| Humans (50 IU/kg), N = 20 | ||
| Incremental recovery (IU/mL)/(IU/kg) | 0.019 (0.002) | 0.019 (0.003) |
| AUC (h*IU/mL) | 12.97 (3.48) | 13.03 (4.25) |
| t1/2 (h) | 10.83 (4.95) | 11.19 (3.51) |
| Clearance (mL/h/kg) | 4.11 (1.06) | 4.17 (1.20) |
Values were based on FVIII:C measurements. Incremental recovery: FVIII activity 30 min after end of infusion relative to the administered dose. AUC, area under the plasma concentration curve; t1/2, terminal half-life.
Clinical safety and efficacy
The clinical safety and efficacy of turoctocog alfa was demonstrated in 213 previously treated patients with severe haemophilia A (FVIII activity ≤ 1%) and no history of inhibitors. None of these patients developed FVIII inhibitors, no safety concerns were identified and a prophylactic effect of turoctocog alfa was demonstrated 17,18. Overall, the median annualised bleeding rate was 3.0 bleeds per patient per year (estimated mean annualised bleeding rate 5.3 [95% CI: 3.9–7.3] bleeds per patient per year) in children and the median annualised bleeding rate was 3.7 bleeds per patient per year (estimated mean annualised bleeding rate 6.5 [95% CI: 5.3–8.0] bleeds per patient per year) in adults and adolescents. The majority of the bleeding episodes (95% in children and 89% in adults and adolescents) were controlled with 1–2 infusions of turoctocog alfa. Furthermore, the haemostatic responses during and after surgery were rated as excellent or good in all surgeries 43. These data confirm the clinical efficacy of turoctocog alfa.
Conclusions
Turoctocog alfa, a new rFVIII, has been carefully designed to achieve a safe, well-defined and homogeneous product and the molecular characterisation confirms the quality of the FVIII protein. The molecular properties of turoctocog alfa were supported by functional assays, which showed that turoctocog alfa is fully functional in haemophilia A plasma and blood. Furthermore, studies in F8-KO mice using bleeding and injury models of different severity all showed that turoctocog alfa was capable of normalising the bleeding phenotype. The data are in line with clinical results, confirming the haemostatic effect of turoctocog alfa and support the use of turoctocog alfa as FVIII replacement therapy in patients with haemophilia A.
Acknowledgments
This development programme was sponsored by Novo Nordisk A/S (Bagsvaerd, Denmark). Merete Pedersen, PhD, and Erik Andersen, MDSc, (Novo Nordisk A/S) provided editorial support during the manuscript preparation.
Author contributions
All authors gave input, reviewed and approved the manuscript.
Conflict of interest disclosure
All authors are employees of Novo Nordisk A/S.
References
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature. 1984;312:337–42. doi: 10.1038/312337a0. [DOI] [PubMed] [Google Scholar]
- Gitschier J, Wood WI, Goralka TM, Wion KL, Chen EY, Eaton DH, Vehar GA, Capon DJ, Lawn RM. Characterization of the human factor VIII gene. Nature. 1984;312:326–30. doi: 10.1038/312326a0. [DOI] [PubMed] [Google Scholar]
- Wood WI, Capon DJ, Simonsen CC, et al. Expression of active human factor VIII from recombinant DNA clones. Nature. 1984;312:330–7. doi: 10.1038/312330a0. [DOI] [PubMed] [Google Scholar]
- Toole JJ, Knopf JL, Wozney JM, Sultzman LA, Buecker JL, Pittman DD, Kaufman RJ, Brown E, Shoemaker C, Orr EC. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature. 1984;312:342–7. doi: 10.1038/312342a0. [DOI] [PubMed] [Google Scholar]
- Truett MA, Blacher R, Burke RL, Caput D, Chu C, Dina D, Hartog K, Kuo CH, Masiarz FR, Merryweather JP. Characterization of the polypeptide composition of human factor VIII: C and the nucleotide sequence and expression of the human kidney cDNA. DNA. 1985;4:333–49. doi: 10.1089/dna.1985.4.333. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ, Wasley LC, Dorner AJ. Synthesis, processing, and secretion of recombinant human factor VIII expressed in mammalian cells. J Biol Chem. 1988;263:6352–62. [PubMed] [Google Scholar]
- Burke RL, Pachl C, Quiroga M, Rosenberg S, Haigwood N, Nordfang O, Ezban M. The functional domains of coagulation factor VIII:C. J Biol Chem. 1986;261:12574–8. [PubMed] [Google Scholar]
- Toole JJ, Pittman DD, Orr EC, Murtha P, Wasley LC, Kaufman RJ. A large region (approximately equal to 95 kDa) of human factor VIII is dispensable for in vitro procoagulant activity. Proc Natl Acad Sci U S A. 1986;83:5939–42. doi: 10.1073/pnas.83.16.5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman DD, Alderman EM, Tomkinson KN, Wang JH, Giles AR, Kaufman RJ. Biochemical, immunological, and in vivo functional characterization of B-domain-deleted factor VIII. Blood. 1993;81:2925–35. [PubMed] [Google Scholar]
- Leyte A, Van Schijndel HB, Niehrs C, Huttner WB, Verbeet MP, Mertens K, van Mourik JA. Sulfation of Tyr1680 of human blood coagulation factor VIII is essential for the interaction of factor VIII with von Willebrand factor. J Biol Chem. 1991;266:740–6. [PubMed] [Google Scholar]
- Michnick DA, Pittman DD, Wise RJ, Kaufman RJ. Identification of individual tyrosine sulfation sites within factor VIII required for optimal activity and efficient thrombin cleavage. J Biol Chem. 1994;269:20095–102. [PubMed] [Google Scholar]
- Pipe SW. Functional roles of the factor VIII B domain. Haemophilia. 2009;15:1187–96. doi: 10.1111/j.1365-2516.2009.02026.x. [DOI] [PubMed] [Google Scholar]
- Thim L, Vandahl B, Karlsson J, et al. Purification and characterization of a new recombinant factor VIII (N8) Haemophilia. 2010;16:349–59. doi: 10.1111/j.1365-2516.2009.02135.x. [DOI] [PubMed] [Google Scholar]
- Stennicke HR, Kjalke M, Karpf DM, et al. A novel B-domain O-glycoPEGylated FVIII (N8-GP) demonstrates full efficacy and prolonged effect in hemophilic mice models. Blood. 2013;121:2108–16. doi: 10.1182/blood-2012-01-407494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen ML, Balling KW, Persson E, et al. Functional characteristics of N8, a new recombinant FVIII. Haemophilia. 2010;16:878–87. doi: 10.1111/j.1365-2516.2010.02333.x. [DOI] [PubMed] [Google Scholar]
- Kimchi-Sarfaty C, Schiller T, Hamasaki-Katagiri N, Khan MA, Yanover C, Sauna ZE. Building better drugs: developing and regulating engineered therapeutic proteins. Trends Pharmacol Sci. 2013;34:534–48. doi: 10.1016/j.tips.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Lentz SR, Misgav M, Ozelo M, et al. Results from a large multinational clinical trial (guardian™1) using prophylactic treatment with turoctocog alfa in adolescent and adult patients with severe haemophilia A: safety and efficacy. Haemophilia. 2013;19:691–7. doi: 10.1111/hae.12159. [DOI] [PubMed] [Google Scholar]
- Kulkarni R, Karim FA, Glamocanin S, et al. Results from a large multinational clinical trial (guardian™3) using prophylactic treatment with turoctocog alfa in paediatric patients with severe haemophilia A: safety, efficacy and pharmacokinetics. Haemophilia. 2013;19:698–705. doi: 10.1111/hae.12165. [DOI] [PubMed] [Google Scholar]
- Butler M, Meneses-Acosta A. Recent advances in technology supporting biopharmaceutical production from mammalian cells. Appl Microbiol Biotechnol. 2012;96:885–94. doi: 10.1007/s00253-012-4451-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg H, Almstedt A, Brandt J, Gray E, Holmquist L, Oswaldsson U, Sebring S, Mikaelsson M. Structural and functional characteristics of the B-domain-deleted recombinant factor VIII protein, r-VIII SQ. Thromb Haemost. 2001;85:93–100. [PubMed] [Google Scholar]
- Kjalke M, Heding A, Talbo G, Persson E, Thomsen J, Ezban M. Amino acid residues 721-729 are required for full factor VIII activity. Eur J Biochem. 1995;234:773–9. doi: 10.1111/j.1432-1033.1995.773_a.x. [DOI] [PubMed] [Google Scholar]
- Newell JL, Fay PJ. Acidic residues C-terminal to the A2 domain facilitate thrombin-catalyzed activation of factor VIII. Biochemistry. 2008;47:8786–95. doi: 10.1021/bi8007824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JJ, Bolt G, Kjærgard K, Kristensen C, Steenstrup TD. Generation of recombinant FVIII purification antibody for manufacturing of N8. J Thromb Haemost. 2013;7(Suppl 2):317–1168. [Google Scholar]
- Jankowski MA, Patel H, Rouse JC, Marzilli LA, Weston SB, Sharpe PJ. Defining ‘full-length’ recombinant factor VIII: a comparative structural analysis. Haemophilia. 2007;13:30–7. doi: 10.1111/j.1365-2516.2006.01388.x. [DOI] [PubMed] [Google Scholar]
- D’Amici GM, Timperio AM, Gevi F, Grazzini G, Zolla L. Recombinant clotting factor VIII concentrates: Heterogeneity and high-purity evaluation. Electrophoresis. 2010;31:2730–9. doi: 10.1002/elps.201000216. [DOI] [PubMed] [Google Scholar]
- Kristensen AK, Kjalke M, Klausen NK, Ezban M, Vad K. Structural comparison of a new recombinant factor VIII molecule, turoctocog alfa, and commercially available FVIII products. J Thromb Haemost. 2013;11(Suppl s2):1041–2. [Google Scholar]
- Shen BW, Spiegel PC, Chang CH, Huh JW, Lee JS, Kim J, Kim YH, Stoddard BL. The tertiary structure and domain organization of coagulation factor VIII. Blood. 2008;111:1240–7. doi: 10.1182/blood-2007-08-109918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo JC, Huang M, Roth DA, Furie BC, Furie B. Crystal structure of human factor VIII: implications for the formation of the factor IXa-factor VIIIa complex. Structure. 2008;16:597–606. doi: 10.1016/j.str.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Svensson LA, Thim L, Olsen OH, Nicolaisen EM. Evaluation of the metal binding sites in a recombinant coagulation factor VIII identifies two sites with unique metal binding properties. Biol Chem. 2013;394:761–5. doi: 10.1515/hsz-2012-0298. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Post-translational modifications required for coagulation factor secretion and function. Thromb Haemost. 1998;79:1068–79. [PubMed] [Google Scholar]
- Kannicht C, Ramstrom M, Kohla G, Tiemeyer M, Casademunt E, Walter O, Sandberg H. Characterisation of the post-translational modifications of a novel, human cell line-derived recombinant human factor VIII. Thromb Res. 2013;131:78–88. doi: 10.1016/j.thromres.2012.09.011. [DOI] [PubMed] [Google Scholar]
- Sandberg H, Almstedt A, Brandt J, et al. Structural and functional characterization of B-domain deleted recombinant factor VIII. Semin Hematol. 2001;38(2 Suppl 4):4–12. doi: 10.1016/s0037-1963(01)90103-9. [DOI] [PubMed] [Google Scholar]
- Macher BA, Galili U. The Galalpha1,3Galbeta1,4GlcNAc-R (alpha-Gal) epitope: a carbohydrate of unique evolution and clinical relevance. Biochim Biophys Acta. 2008;1780:75–88. doi: 10.1016/j.bbagen.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen PF, Bak S, Vandahl B. Characterization of tyrosine sulphation in rFVIII (turoctocog alfa) expressed in CHO and HEK-293 cells. Haemophilia. 2012;18:e397–8. doi: 10.1111/j.1365-2516.2012.02881.x. [DOI] [PubMed] [Google Scholar]
- Grancha S, Navajas R, Maranon C, Paradela A, Albar JP, Jorquera JI. Incomplete tyrosine 1680 sulphation in recombinant FVIII concentrates. Haemophilia. 2011;17:709–10. doi: 10.1111/j.1365-2516.2010.02454.x. [DOI] [PubMed] [Google Scholar]
- Elm T, Karpf DM, Ovlisen K, Pelzer H, Ezban M, Kjalke M, Tranholm M. Pharmacokinetics and pharmacodynamics of a new recombinant FVIII (N8) in haemophilia A mice. Haemophilia. 2012;18:139–45. doi: 10.1111/j.1365-2516.2011.02608.x. [DOI] [PubMed] [Google Scholar]
- Karpf DM, Kjalke M, Thim L, Agerso H, Merricks EP, Defriess N, Nichols TC, Ezban M. Pharmacokinetics and ex vivo whole blood clot formation of a new recombinant FVIII (N8) in haemophilia A dogs. Haemophilia. 2011;17:e963–8. doi: 10.1111/j.1365-2516.2011.02580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowitz U, Bjerre J, Brand B, Klamroth R, Misgav M, Morfini M, Santagostino E, Tiede A, Viuff D. Bioequivalence between two serum-free recombinant factor VIII preparations (N8 and ADVATE®)–an open-label, sequential dosing pharmacokinetic study in patients with severe haemophilia A. Haemophilia. 2011;17:854–9. doi: 10.1111/j.1365-2516.2011.02495.x. [DOI] [PubMed] [Google Scholar]
- Viuff D, Barrowcliffe T, Saugstrup T, Ezban M, Lillicrap D. International comparative field study of N8 evaluating factor VIII assay performance. Haemophilia. 2011;17:695–702. doi: 10.1111/j.1365-2516.2010.02481.x. [DOI] [PubMed] [Google Scholar]
- Hemker HC, Giesen P, Al Dieri R, Regnault V, de Smedt E, Wagenvoord R, Lecompte T, Béguin S. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb. 2003;33:4–15. doi: 10.1159/000071636. [DOI] [PubMed] [Google Scholar]
- Kjalke M, Ezban M, Monroe DM, Hoffman M, Roberts HR, Hedner U. High-dose factor VIIa increases initial thrombin generation and mediates faster platelet activation in thrombocytopenia-like conditions in a cell-based model system. Br J Haematol. 2001;114:114–20. doi: 10.1046/j.1365-2141.2001.02870.x. [DOI] [PubMed] [Google Scholar]
- Pastoft AE, Lykkesfeldt J, Ezban M, Tranholm M, Whinna HC, Lauritzen B. A sensitive venous bleeding model in haemophilia A mice: effects of two recombinant FVIII products (N8 and Advate®. Haemophilia. 2012;18:782–8. doi: 10.1111/j.1365-2516.2012.02780.x. [DOI] [PubMed] [Google Scholar]
- Santagostino E, Lentz SR, Misgav M, Brand B, Chowdary P, Savic A, Kilinc Y, Tuddenham EG, Lindblom A. Surgery with Turoctocog Alfa: Efficacy and Safety in Bleeding Prevention During Surgical Procedures-Results From the guardian™ Trials. ASH Annual Meeting Abstracts. 2012;120:2228. [Google Scholar]
- Karpf D, Kjalke M, Pelzer H, Thim L, Agerso H, Merricks E, Franck H, Raymer R, Nichols T, Ezban M. Pharmacokinetic (PK) and pharmacodynamic (PD) properties of a new recombinant FVIII (N8) in hemophilia a dogs. J Thromb Haemost. 2009;7(suppl 2):512. [Google Scholar]