

Abstract
RNA polymerase II transcribes genes encoding proteins and a large number of small stable RNAs. While pre-mRNA 3′-end formation requires a machinery ensuring tight coupling between cleavage and polyadenylation, small RNAs utilize polyadenylation-independent pathways. In yeast, specific factors required for snRNA and snoRNA 3′-end formation were characterized as components of the APT complex that is associated with the core complex of the cleavage/polyadenylation machinery (core-CPF). Other essential factors were identified as independent components: Nrd1p, Nab3p and Sen1p. Here we report that mutations in the conserved box D of snoRNAs and in the snoRNP-specific factor Nop1p interfere with transcription and 3′-end formation of box C/D snoRNAs. We demonstrate that Nop1p is associated with box C/D snoRNA genes and that it interacts with APT components. These data suggest a mechanism of quality control in which efficient transcription and 3′-end formation occur only when nascent snoRNAs are successfully assembled into functional particles.
Keywords: 3′-end formation, Nop1p, Saccharomyces cerevisiae, snoRNA, snoRNP assembly
Introduction
Genes transcribed by RNA polymerase II encode polyadenylated mRNAs as well as poly(A)− transcripts. Among the latter transcripts are the spliceosomal snRNAs and the nucleolar snoRNAs that guide 2′-0-ribose methylation (box C/D) and pseudouridylation (box H/ACA) of target RNAs (Kiss, 2002). The machinery for 3′-end formation acts quite differently, depending on the type of transcript. For pre-mRNAs, cleavage is coupled to polyadenylation. The poly(A) tail has several important functions: it stabilizes the 3′ end of the mRNA (Beelman and Parker, 1995), it facilitates transport of the mRNA to the cytoplasm (Huang and Carmichael, 1996) and it increases the efficiency of translation initiation (Tarun and Sachs, 1996). In the case of snRNAs and snoRNAs, cleavage must be uncoupled from polyadenylation (Fatica et al, 2000; Steinmetz et al, 2001; Morlando et al, 2002). Nonadenylated ends are required to produce entry sites for 3′–5′ trimming performed by the exosome (Allmang et al, 1999; Van Hoof et al, 2000) and to allow correct 3′-end maturation of the small RNAs. Even if such entry sites are often generated by the Rnt1p endonuclease (Chanfreau et al, 1998a, 1998b), in most of the cases processing starts from the 3′ end of the primary transcript.
The protein factors that catalyze yeast pre-mRNA 3′-end formation were initially defined by fractionation of whole-cell extracts (Chen and Moore, 1992): cleavage factors IA and IB (CFIA and CFIB), cleavage factor II (CFII), polyadenylation factor (PFI) and poly(A) polymerase (Pap1p) are sufficient for cleavage and polyadenylation in vitro (Zhao et al, 1999). In vivo, the CFII, PFI and Pap1p proteins are contained within a single complex, the cleavage and polyadenylation factor CPF (now referred to as core-CPF) (Ohnacker et al, 2000; Nedea et al, 2003). More recently, additional polypeptides were found associated with core-CPF (Dichtl et al, 2002; Gavin et al, 2002; He et al, 2003; Nedea et al, 2003). Of these, Pti1p, Glc7p, Ref2p, Swd2p, Ssu72p, Syc1p and the core-CPF component Pta1p were proposed to form the APT complex (associated with Pta1p; Nedea et al, 2003).
Polyadenylation-dependent and -independent 3′-end formation appears to require an overlapping but not identical set of trans-acting factors. Both processes are dependent on CFIA factors (Rna15p, Rna14p and Pcf11p) (Fatica et al, 2000; Morlando et al, 2002). In contrast, mutations in components of core-CPF (Yhh1p, Ysh1p, Pfs2p and Yth1p) did not interfere with correct 3′-end formation of snoRNAs and snRNAs in vivo (Morlando et al, 2002). However, several recent reports indicated that subunits of the APT complex (Ssu72p, Pti1p and Ref2p) are required for 3′-end formation on snoRNA coding genes (Dheur et al, 2003; Ganem et al, 2003; Nedea et al, 2003). In particular, Pti1p and Ref2p, which are required to prevent transcriptional read-through into downstream genes, were suggested to function in uncoupling cleavage and polyadenylation (Dheur et al, 2003). The APT complex may therefore mediate polyadenylation-independent 3′-end formation on specific subpopulations of pol II transcripts, which include snoRNAs and snRNAs.
In addition to this complex array of factors, other proteins are specifically required for 3′-end formation of snoRNAs: the RNA-binding proteins Nrd1 and Nab3 and the presumptive RNA helicase Sen1 (Steinmetz et al, 2001).
Box C/D snoRNAs are a very well characterized class of small noncoding RNAs with peculiar gene organization, biosynthesis and function (Kiss, 2002). Snu13p, Nop1p, Nop58p and Nop56p proteins have been described as integral components of mature snoRNPs (Lafontaine and Tollervey, 1999, 2000; Watkins et al, 2000); their association with the RNA is essential for snoRNA stability as well as for subcellular localization and function of the particle (Filipowicz and Pogacic, 2002). Nop1p carries out the site-specific methyltransferase reaction on 2′-0-ribose positions within target RNA (rRNAs and snRNAs) residues (Galardi et al, 2002; Omer et al, 2002). In vertebrates, it has been shown that the Snu13p homolog p15.5K is the primary binding protein, whose association is essential for the assembly of the remaining snoRNP proteins (Watkins et al, 2002): the conserved C and D box elements together with the internal stem (stemII), which forms a specific structural motif (K-turn), were shown to be required for such binding.
snoRNP assembly was also shown to be a prerequisite for the release of the intron-encoded U18 snoRNA from its host pre-mRNA, suggesting that formation of the snoRNP can occur very early. In this case, a specific interaction between Nop1p and the Rnt1p endonuclease, necessary for the release of U18 from the intron, was demonstrated (Giorgi et al, 2001).
In this study, we have asked whether the ability to assemble functional snoRNPs is crucial for the biosynthesis of independently transcribed snoRNA genes and found evidence that efficient and correct 3′-end formation of box C/D snoRNAs requires the Nop1 snoRNP factor.
Results
Mutations in the conserved box D affect snoRNA biosynthesis
Conserved sequence elements within box C/D snoRNAs are required for snoRNP assembly. To figure out whether these sequence elements are required for the early steps of snoRNA biosynthesis, that is, transcription and 3′-end formation, we produced a series of reporter constructs. The pG plasmids used here contained a tagged snoRNA coding region, followed by a poly(G) cassette and the 3′ downstream regions of either SNR13 (pG13) or SNR50 (pG50) snoRNA genes (Figure 1A). Both 3′ regions were shown to contain two closely spaced 3′-end formation elements: the first element (CS) resembles an Nrd1-binding site and is flanked by CT-rich elements responsive to the Sen1 protein (Rasmussen and Culbertson, 1998; Steinmetz et al, 2001; Morlando et al, 2002). The second element (pA) contains sequences characteristic of pre-mRNA polyadenylation sites. The bD derivatives (pG13bD and pG50bD) contained mutations in the conserved box D (CUGA → CUCC), which are known to prevent binding of snoRNP factors and which interfere with the assembly of a functional particle (Watkins et al, 2002). Therefore, such mutants were expected to produce highly unstable transcripts; in these cases, the poly(G) tract should protect the transcripts from rapid 3′–5′ exonucleolytic trimming (Decker and Parker, 1993) and, together with the cap structure at the 5′ end, should allow the accumulation/visualization of the otherwise unstable bD snoRNAs. Panels B and C of Figure 1 show that transcripts extending to the poly(G) cassette (G bands) and to the mature 3′ end (3′ bands) of the snoRNA can be visualized at steady-state conditions with wild-type constructs (lanes 1); this indicated that the G cassette did not affect the overall transcriptional and 3′-processing activities and provided an efficient barrier to exonucleolytic trimming. On the contrary, bD transcripts could not be detected either in the isogenic wild-type strain (lanes 2) or in the strain lacking the decapping enzyme (lanes 4). Similar results were obtained when the same constructs were transformed into strains rrp6Δ and rrp44-1, which contain mutations affecting the function of the 3′–5′ exosome (Mitchell et al, 1997; Allmang et al, 1999). The results indicate that while the wild-type constructs show increased accumulation of both the G and 3′ species (lanes 5, 7–9), the bD derivatives do not display any snoRNA accumulation (lanes 6, 10–12). The presence of *molecules in rrp6Δ lanes is diagnostic for the altered exonucleolytic activity; in fact, it was previously shown that in this strain box C/D snoRNAs accumulate as species with three-nucleotide extensions (Allmang et al, 1999). This is also confirmed by the control hybridizations to the endogenous snR13 RNA (lower panel). The mutant phenotype of the rrp6Δ and rrp44-1 strains was also tested by the accumulation of 7SRNA precursors (data not shown).
Figure 1.
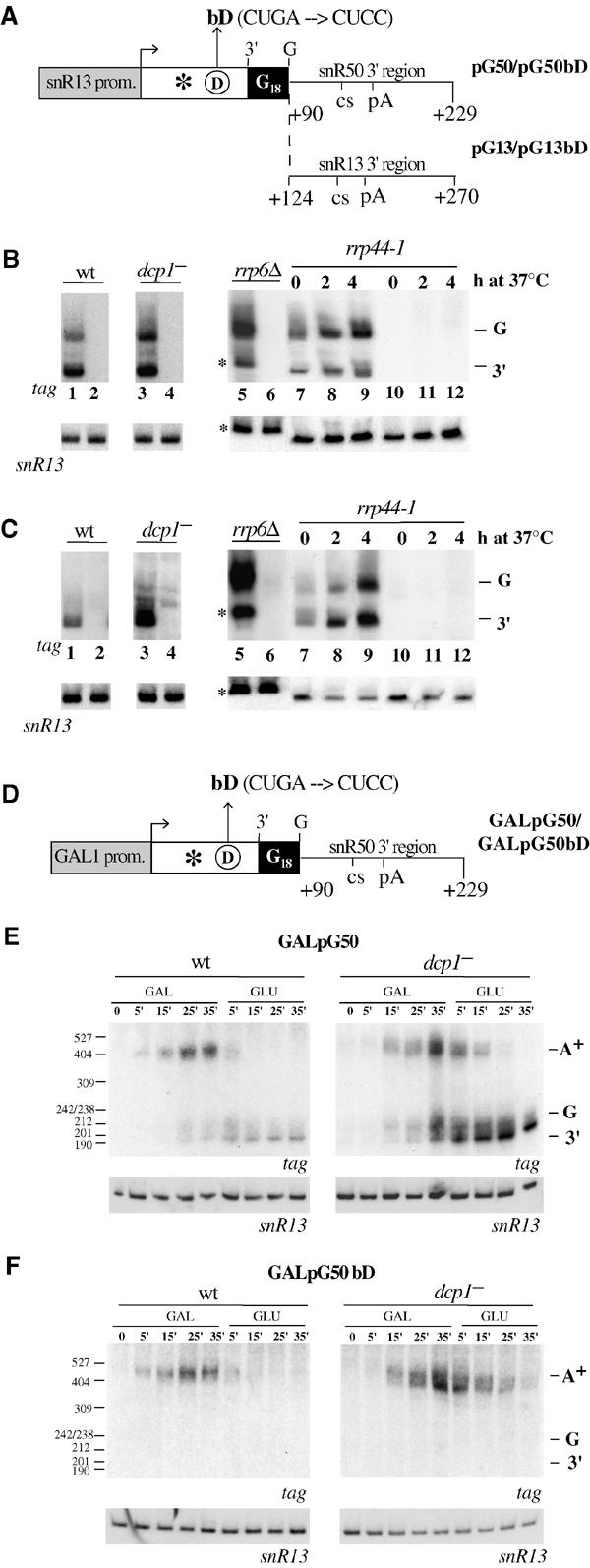
snoRNA biosynthesis is affected by mutations in the conserved box D. A decapping-deficient strain (YRP 1200, lanes dcp1−) and its isogenic wild-type strain (YRP 683, lanes wt) were transformed with the constructs schematically shown in (A) and (D). In parallel, the exo− strains (rrp6Δ and rrp44-1) were transformed with the constructs shown in (A). Promoter regions are represented by gray boxes, while snoRNA coding regions are represented by white boxes. The asterisk symbolizes the tag sequence (5′-CGGCATCCAGGCAGTCCGCA-3′) and the circled D indicates the conserved box D (the bD mutation is described above). The poly(G) cassette is represented in black. The numbers of the 3′ downstream regions indicate the distance from the transcriptional start site of the corresponding gene. ((B) (C) (E) and (F)) show Northern blots of RNA extracted from the indicated strains transformed with the following constructs: (B) pG50 (lanes 1, 3, 5, 7, 8 and 9) and pG50bD (lanes 2, 4, 6, 10, 11 and 12); (C) pG13 (lanes 1, 3, 5, 7, 8 and 9) and pG13bD (lanes 2, 4, 6, 10, 11 and 12). For the ts strain rrp44-1, cells were grown at 25°C (time 0) and then shifted to the nonpermissive temperature for 2 and 4 h. In the rrp6Δ strain, *molecules represent species with three-nucleotide extensions. In the transcriptional pulse-chase experiments of (E) and (F), cells were grown in 2% raffinose and 0.08 % glucose; then they were shifted to 2% galactose and, after 35 min, to 4% glucose. The plasmids and the strains utilized are indicated above the lanes. The times of galactose induction (GAL lanes) and glucose repression (GLU lanes) are also indicated. Polyadenylated species (A+), transcripts extending to the poly(G) cassette (G) and to the mature snoRNA 3′ end (3′) are indicated on the right side of each panel. On the left side of the gels, the size marker (pBR322 MspI-digested) is shown. In all experiments, control hybridizations were performed with the αsnR13 probe, which recognizes the untagged endogenous snR13 snoRNA (lower panels).
In order to test whether a strong promoter would result in detectable amounts of the very short-lived transcripts, we substituted the snoRNA promoter of constructs pG50 and pG50bD with the GAL1 promoter (constructs GalpG50 and GalpG50bD, panel D) and performed a transcriptional pulse-chase experiment. Also in this case, 3′-processed species (G and 3′) accumulated only in the strain containing the wild-type construct (panel E, lanes wt), while they were absent in the bD mutant (panel F, lanes wt). The same transcriptional pulse-chase experiment was performed in the decapping-deficient strain: even if a general increase in RNA stability was observed with the wild-type construct (panel E, lanes dcp1−), no 3′-processed species were detected with the bD construct (panel F, lanes dcp1−). The stabilization of nuclear transcripts (snoRNAs) in the decapping-deficient strain (dcp1−) is in agreement with recent findings reporting the presence of this protein also in the nucleus (L Milligan and D Tollervey, personal communication). The poly(A)+ species, visible as slower migrating bands (bands A+, panels E and F), originated from the utilization of the pA site and were produced only when transcription was driven by the GAL1 promoter. The reason for this observation is not clear at the moment; it would be very interesting to understand whether this was due to specific features of the GAL1 promoter that is normally tuned to direct efficient polyadenylation (see Discussion). Poly(A)+ snoRNAs were previously shown to be unable to form functional snoRNPs and to be very unstable (Fatica et al, 2000). This was confirmed here by their rapid disappearance after a few minutes shift to glucose. Taken together, the results of Figure 1 indicated that the absence of a functional box D interfered with early steps of snoRNA biosynthesis, which may include pol II transcription and/or 3′-end formation.
Run-on analysis on mutant transcriptional units
In order to verify the possible defects in transcription or 3′-end processing, transcriptional run-on (TRO) assays were performed on the YPG 1 strain. This strain, carrying a deletion of the endogenous SNR13 gene, was transformed with the plasmids shown in Figure 2A. snR13 wt contains the wild-type SNR13 gene, while the bD derivative (snr13 bD) carries the CUGA to CUCC mutation in box D. Both constructs have 146 nucleotides of 3′ downstream region. This region was previously shown to be sufficient for appropriate and efficient 3′-end formation (Morlando et al, 2002; Steinmetz and Brow, 2003). Figure 2A shows a representative autoradiography of a TRO assay and the histogram summarizes the densitometric values obtained from three independent experiments. In comparison with wild type, the bD mutant construct displayed a decrease in polymerase density in the snR13 coding region, together with an increased read-through into the downstream vector sequences (compare the hybridization signals of oligo #1 and #2).
Figure 2.
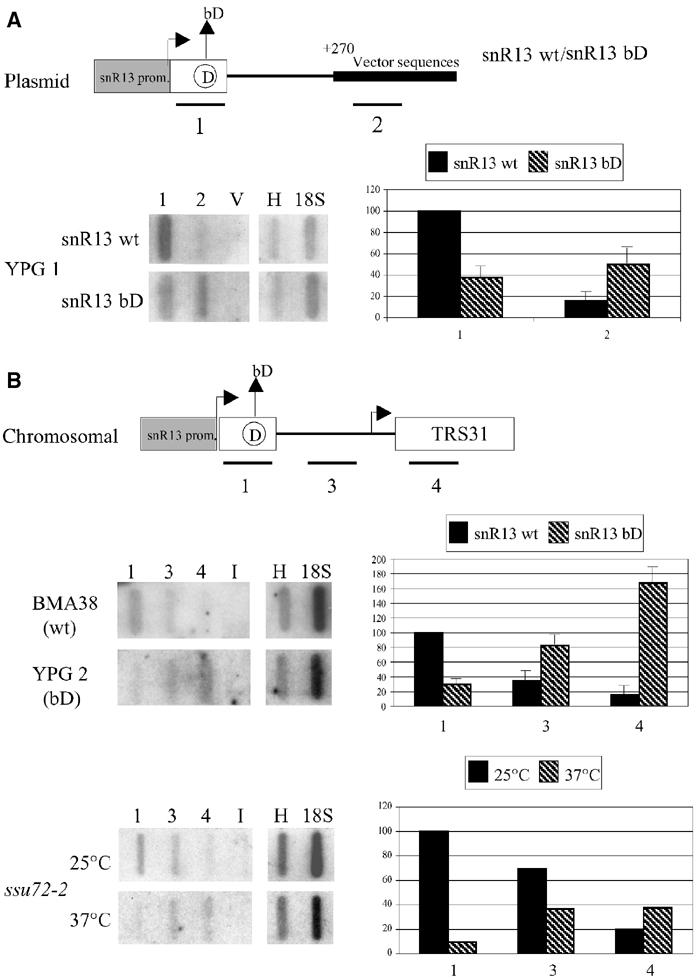
snoRNA transcription and 3′ processing/termination are impaired in box D mutants. The constructs utilized for the TRO assays are shown at the top of (A) and (B) and are represented as in Figure 1. The strains utilized are indicated on the left of the panels. Oligonucleotide probes (approx. 90 nucleotides) are shown below their locations. Representative slot blots of run-on analysis of wild-type and mutant strains are shown; slots V contain a nontranscribed region of the plasmid vector, while slots I contain an intergenic region from chromosome V (see Materials and methods). Slots H and 18S contain probes specific for histone H2A and 18S rRNA genes, respectively. The ssu72-2 strain was incubated at 37°C for 2 h prior to TRO analysis. Quantification of the transcripts was performed by phosphorimage analysis and the counts were normalized to the H and 18S probes. In (A), background (V) was subtracted from each value. Error bars represent s.d. (n=3).
To test whether this feature applied also to chromosomal sequences, we have raised a strain in which the endogenous copy of the SNR13 gene has been substituted by a copy containing the bD mutation (strain YPG 2). In this case, oligo #3 maps in the 3′ snR13 downstream region, while oligo #4 is complementary to the TRS31 gene (see schematic representation of the chromosomal locus in Figure 2B). In the wild-type strain, the signal corresponding to oligo #4 identified the basal level of transcription in the TRS31 coding region. Densitometric analysis on three independent run-on experiments showed that in the wild-type strain the hybridization signal decreases from oligo #1 to #4. This is in line with the notion that the SNR13 gene is transcribed at higher levels than TRS31. In the YPG 2 strain, in contrast, fewer polymerases were loaded on the snoRNA coding region, while the signal increased in the downstream regions. A control TRO assay was performed in an ssu72-2 strain grown at the permissive and nonpermissive temperatures. This mutation was previously shown to produce a read-through phenotype to genes located downstream of snoRNA coding genes (Ganem et al, 2003; Nedea et al, 2003). The lower part of panel B shows that read-through into the TRS31 region was observed at the nonpermissive temperature, with a hybridization pattern similar to that observed in the YPG 2 strain. These results indicated that the bD mutation resulted in two distinct deficiencies: (i) decreased transcription on the SNR13 gene and (ii) increased transcriptional read-through into the downstream gene.
The C/D box factor Nop1p interacts with APT components
Since the mutation in box D affects snoRNP assembly, we asked whether any of the snoRNP-specific factors are involved in the control of snoRNA transcription/3′-end formation. We tested by GST-pull-down experiments whether the snoRNP core proteins Snu13p and Nop1p interact with any of the proteins involved in snoRNA 3′-end formation. Figure 3 shows that no specific interaction was found between the GST–Snu13p fusion protein and any of the tested factors. In contrast, binding between GST–Nop1p and the Ref2p component of the APT complex was detected (Dheur et al, 2003; Nedea et al, 2003). Only very low levels were detected with the Ssu72p factor. No specific interaction was observed between GST–Nop1p and Swd2p, Ptip or CFIA components (Clp1p, Pcf11p, Rna14p and Rna15p).
Figure 3.
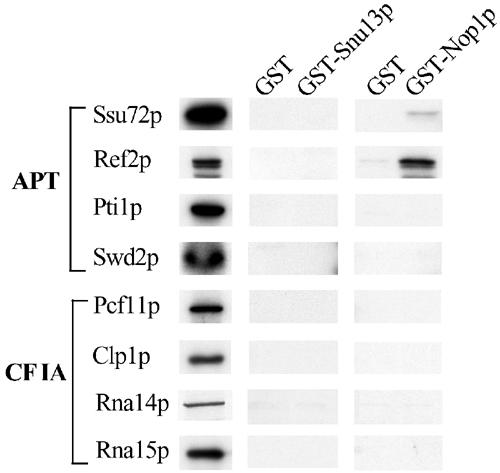
The snoRNP core protein Nop1p interacts with the APT complex. Recombinant GST-Snu13p or recombinant GST–Nop1p were bound to glutathione–sepharose beads and then incubated with in vitro-translated 35S-labelled proteins: Ssu72p, Ref2p, Pti1p and Swd2p belonging to the APT complex, and Pcf11p, Clp1, Rna14p and Rna15p, which are part of the CFIA complex (Gross and Moore, 2001). As control, GST alone was also utilized (lanes GST). After RNase treatment, the proteins bound to GST, GST–Snu13p or GST–Nop1p were eluted by boiling in loading buffer and separated on a 10% polyacrylamide–SDS gel in parallel with 25% of the input proteins (see Materials and methods). Radioactive signals were detected by autoradiography.
Nop1p is important for preventing read-through to snoRNA downstream genes
The Ref2p factor has been previously described to be involved in snoRNA biogenesis and 3′-end formation; ts derivatives thereof have a mutant phenotype characterized by the appearance of snoRNA read-through products into downstream genes (Dheur et al, 2003).
We investigated the possibility that Nop1p could be the trans-acting factor responsible for the mutant phenotype of bD derivatives and that it may be important for the control of 3′-end formation of snoRNAs. The production of snoRNA read-through products was tested in a strain, in which the endogenous copy of NOP1 was under the control of the GAL 1 promoter (GAL∷Nop1; Tollervey et al, 1991), before and after shift to glucose (Figure 4). Growth of this strain on glucose-containing medium efficiently represses NOP1 mRNA expression already after 4 h, as indicated in the Northern of panel A. The protein levels are strongly reduced at 8 h and are undetectable at 16 h (panel B). Nop1p depletion parallels the decrease of box C/D snoRNA accumulation, which appears very evident after 12 h of glucose repression, while it does not affect the levels of box H/ACA snoRNAs and of the U6 snRNA (panel C). By using primer extension analysis to identify read-through fusion transcripts, we found that RNA extracted from the GAL∷Nop1 strain grown in glucose (panel D: lanes 8, 12 and 16 h) gave read-through when compared with control RNA grown in galactose (lanes 0) at three box C/D snoRNA loci (SNR50, SNR71 and SNR13). The very low level of transcripts at 12 and 16 h after shift to glucose is due to the fact that cells almost stop duplicating and a general decrease of the major cellular biosyntheses, such as transcription, occurs. No read-through phenotype was observed with two control box H/ACA snoRNA loci (SNR5 and SNR189). Primer extension on the U6 snRNA, a pol III transcript, controlled the amount of RNA utilized in the reactions (panel U6). The read-through products obtained after depletion of Nop1p were the same as those obtained in ssu72-2 mutants (lanes ssu72-2) as previously reported (Ganem et al, 2003; Nedea et al, 2003). The lower levels of read-through products in the Nop1-depleted strain, with comparison to ssu72-2, might be due to: (i) lower stability, as expected for C/D box-containing transcripts in the absence of Nop1p; (ii) decreased transcription, as shown by the TRO analysis of Figure 2.
Figure 4.
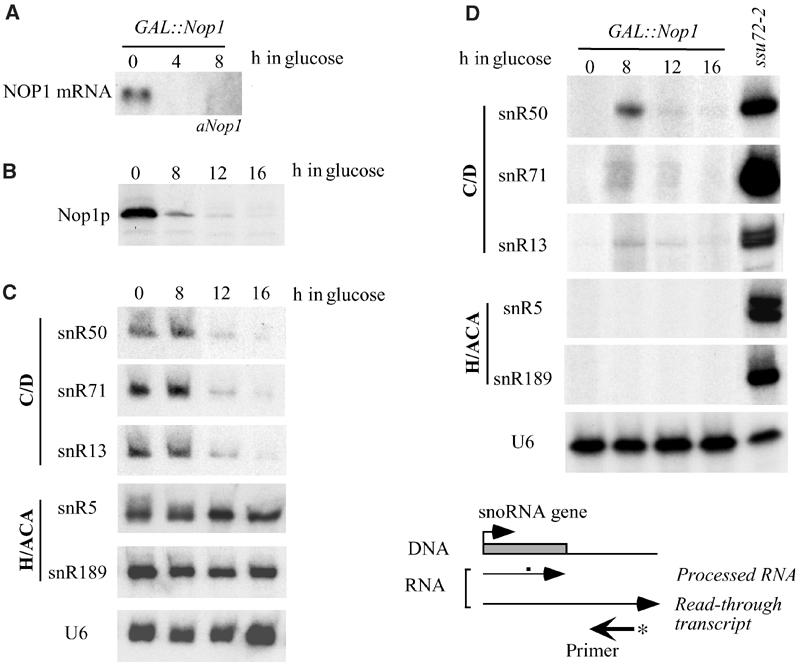
Analysis of read-through products in a Nop1p-depleted strain. Total RNA or proteins were extracted from 0.5 OD of D255 cells grown in galactose medium (lanes 0) and from cells shifted to glucose for the time indicated above each lane. (A) Northern-blot hybridization for the detection of NOP1 mRNA (GAL∷Nop1 lanes). (B) Western analysis of Nop1p depletion. (C) Northern analysis of snoRNAs (box C/D: snR50, snR71 and snR13; box H/ACA: snR5 and snR189) and of the control U6 snRNA. (D) Primer extension analysis was performed on total RNA extracted from the following strains: ssu72-2, grown at the restrictive temperature for 2 h (lanes ssu72-2), D255 (GAL∷Nop1 lanes), grown in galactose (lanes 0), and then shifted to glucose for the time indicated above each lane. The primers utilized (αsnR50, αsnR71, αTRS31, αsnR5 and αsnR189) are complementary to the downstream regions of SNR50, SNR71, SNR13, SNR5 and SNR189, respectively. A schematic representation of the position of the primer with respect to the snoRNA coding region is shown below. Primer extension with an oligo internal to the U6 snRNA coding region was performed as control for RNA recovery (lanes U6).
These data suggested the requirement of Nop1p for box C/D snoRNA 3′-end formation.
Nop1p associates with the site of snoRNA transcription
Even if several indirect evidences pointed to an early interaction of Nop1 with snoRNA precursors (Giorgi et al, 2001; Watkins et al, 2002), we tested by chromatin immunoprecipitation (ChIP) the possibility that Nop1p binds to transcriptionally active C/D snoRNA genes. A yeast strain containing a chromosomal TAP-tagged version of the NOP1 gene (strain YAF 1) was utilized to confirm the association of Nop1p with genes that encode box C/D snoRNAs. The level of crosslinking of Nop1p on SNR13 and EFB1, the host gene of the U18 snoRNA, was analyzed as described previously (Komarnitsky et al, 2000) (Figure 5A). As control, the SNR189 box H/ACA snoRNA gene was analyzed. Additional controls included a chromatin preparation of an isogenic strain lacking the TAP tag (panel BO). The box C/D snoRNA genes showed high efficiency of crosslinking: between seven-fold (snR13) and sixteen-fold (U18) higher than that obtained with the H/ACA gene (Figure 5B). These results suggested that Nop1p associated specifically with actively transcribed box C/D snoRNA coding genes.
Figure 5.
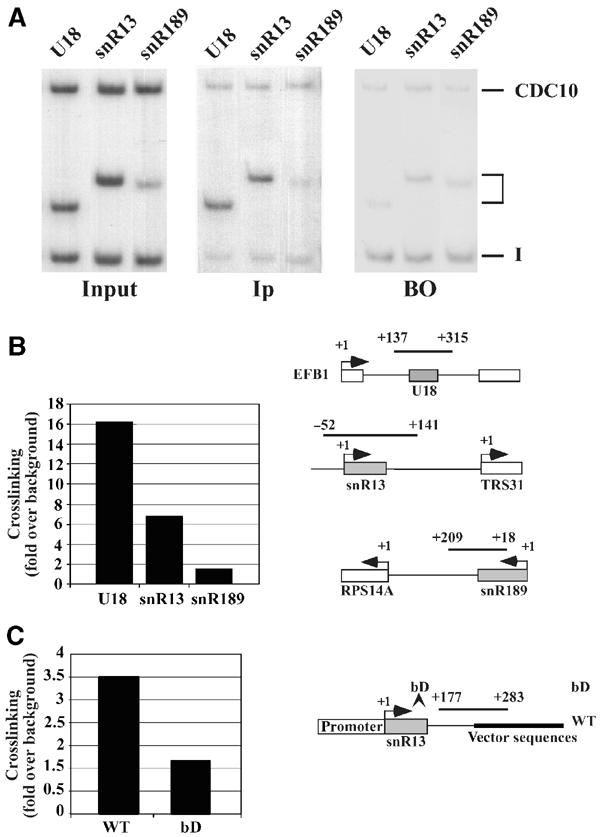
Nop1p is associated with the site of snoRNA transcription. (A) Crosslinked chromatin from a strain containing a TAP-tagged version of Nop1p (YAF 1) was PCR amplified before (panel Input) and after immunoprecipitation (panel Ip). The products were analyzed in parallel to an immunoprecipitation reaction performed with a chromatin preparation from an isogenic strain lacking the TAP tag (panel BO). The oligos utilized were specific for: SNR13, EFB1 and SNR189 genes (see schematic representation). A nontranscribed intergenic region of chromosome V (bands I) and a pol II-transcribed region localized in the CDC10 open reading frame (bands CDC10) were used as internal controls by co-amplification with each of the gene-specific primers. The middle bands (indicated by parentheses) identify the amplification products of the snoRNA coding regions specified above each lane. (B) Histogram displaying the degree of Nop1p crosslinking of the various genes, relative to the control intergenic region. (C) Histogram summarizing the degree of Ref2p crosslinking relative to the control region from the ampicillin gene of the wt and bD construct. For both histograms, normalization was performed by taking into account also the background level of panel BO. Each point is the average (s.d. <20%) of the signals of five PCR reactions, performed on five different preparations of immunoprecipitated DNA, normalized against the controls.
In order to analyze whether Ref2p recruitment is affected by Nop1p, a ChIP experiment was performed on a strain carrying a tagged version of Ref2p, transformed with plasmids containing either the wild-type SNR13 gene or the bD derivative. The Ref2p association on wt construct is two-fold higher than the bD derivative (Figure 5C), indicating that the assembly of functional snoRNP enhances Ref2p recruitment.
Discussion
It is meanwhile well accepted that transcription by RNA polymerase II and the subsequent processing of primary transcripts are intimately connected events (Bentley, 2002; Proudfoot et al, 2002). RNA polymerase II is thought to impact on pre-mRNA processing mainly via its C-terminal domain that may act as a platform to assemble processing components co-transcriptionally. Likewise, there is considerable evidence that pre-mRNA processing factors influence transcription in a reverse flow of information (Manley, 2002).
Here we add another interesting facet to the coupling of different steps in gene expression of RNA pol II transcription. We suggest that components that specifically assemble on stable transcripts to form functional snoRNPs profoundly influence both transcription and processing. We established a requirement for the box C/D-associated Nop1p protein in transcription and processing of snoRNAs. This requirement is based on the following results: (i) a functional box D sequence is required in cis for efficient transcription and 3′-end formation of box C/D snoRNAs; (ii) cellular depletion of the trans-acting factor Nop1p reproduces the defects of the mutation in cis and also results in deficient transcription and 3′-end formation of box C/D snoRNAs; (iii) GST-pull-down experiments showed that Nop1p physically interacts with specific components of the 3′-end formation machinery; and (iv) Nop1p associates with the sites of transcription box C/D snoRNA genes and enhances Ref2p recruitment.
Until now, only sequences downstream to the coding regions were shown to be required for box C/D snoRNA 3′-end formation. In all, 120 nucleotides of the SNR13 3′ downstream region are sufficient for correct and efficient 3′-end formation of the transcribed snoRNA (Fatica et al, 2000; Steinmetz et al, 2001; Morlando et al, 2002). This region contains sequence elements responsive to two well-characterized factors: Sen1p (Rasmussen and Culbertson, 1998) and Nrd1p (Steinmetz et al, 2001). Another element, the pA site, has several features of a cleavage/polyadenylation site; nevertheless, we have observed that it directs polyadenylation only when combined to a canonical mRNA promoter such as GAL1. This site has also a weak match to the Nrd1-binding consensus and it was indicated as also responsive to this factor (Steinmetz and Brow, 2003). Nrd1p was shown to physically interact with the C-terminal domain of pol II (CTD), with the RNA-binding protein Nab3p and with the CTD kinase Ctk1p (Conrad et al, 2000). Recent data have proven that the Nrd1 protein, together with Nab3p, Sen1p and the C-terminal domain of pol II, is an essential factor for poly(A)-independent 3′-end formation of snRNAs and snoRNAs (Steinmetz et al, 2001), suggesting the hypothesis of a specific mRNA factory being loaded on the nascent transcript. Besides these complexes, other general factors are required for correct 3′-end formation/termination of snoRNAs. They are brought to place by the cleavage–polyadenylation machinery. Among these factors are the Ssu72 protein, which was suggested to be required for efficient elongation and termination (Dichtl et al, 2002; Ganem et al, 2003; Steinmetz and Brow, 2003), and Pti1p and Ref2p, which were suggested to play a role in uncoupling cleavage from polyadenylation (Dheur et al, 2003). The cis- and trans-acting mutations in snoRNP components studied here summarize both the effect of ssu72-2 and ref2-2 (defective transcriptional elongation and transcriptional read-through), suggesting that Nop1p participates with these factors in common pathways.
A major open question that awaits clarification is how polyadenylation-dependent and -independent 3′-end formation occurs on different gene classes although a considerable overlap exists in the trans-acting factors that are involved in these processes. We can envision at least two different ways as to how this specificity is achieved: (i) the pol II-dependent cleavage and polyadenylation machinery is directed by different CPF-associated complexes specialized for the different classes of pol II genes and (ii) a single holo-CPF factor exerts specific functions on different classes of transcripts by recognition of distinct cis-acting signals. Several lines of evidence favor the first hypothesis. Distinct core-CPF and holo-CPF complexes were suggested to exist on the basis of TAP-purification procedures in which substoichiometric amounts of APT-specific factors were obtained when compared with the core-CPF components (Nedea et al, 2003). Therefore, it was proposed that only a subpopulation of core-CPF interacts with the APT proteins to form holo-CPF (Nedea et al, 2003). In addition, the APT components Pti1p and Ref2p, even if identified as new components associated with the core-CPF (Dichtl et al, 2002; Dheur et al, 2003; Nedea et al, 2003), were nonetheless shown to be required primarily for 3′ formation of snoRNAs and to be dispensable for pre-mRNA processing in vitro (Dheur et al, 2003). It was proposed that a factor (snCF) containing already characterized CPF subunits and at least Pti1p and Ref2p could specifically participate in snoRNA 3′-end formation in combination with CFIA (Dheur et al, 2003). Since they were described as the first CPF subunits with this specific feature, they were proposed to function in uncoupling cleavage from polyadenylation (Dheur et al, 2003). If this model is correct, how will the subpopulations of CPF complexes recognize the different target genes? For other snoRNA-specific factors, such as Sen1p and Nrd1p, specific sequence elements were found inside the snoRNA downstream regions (Rasmussen and Culbertson, 1998; Steinmetz et al, 2001; Morlando et al, 2002), thus explaining how these factors can be recruited on the correct transcriptional unit. The finding that Nop1p interacts with Ref2p and enhances its association with the C/D box snoRNA coding region led us to suggest a possible mechanism by which the recruitment of the 3′-end formation machinery occurs during transcription. Nop1p, after assembly on the nascent snoRNA, can interact with Ref2p and may recruit the holo-CPF complex; in turn, this would allow efficient 3′ processing and termination. Even if only a very weak physical interaction between Nop1p and Ssu72p was found, the similar phenotypes of Nop1p depletion and Ssu72p mutations suggest that Nop1p may have an unexpected role also in transcriptional elongation. Besides co-transcriptional recruitment, distinct 3′-end formation complexes may already be pre-assembled on RNA pol II at specific gene promoters to determine the mode of 3′-end formation on a specific transcription unit. This possibility is underscored by our observation that transcription of the SNR13 gene with a GAL1 promoter resulted in the accumulation of significant amounts of poly(A)+ RNAs (Figure 1).
Alternatively to the idea of distinct 3′-end formation complexes, the possibility remains that there is one multi-functional 3′-end formation complex that performs specific activities on distinct transcripts. In such a model, the pre-mRNA and the snoRNA primary transcripts would attract a specific set of RNA-binding proteins within the complex. This could be Yhh1p/Ydh1p/Yth1p, which interact with pre-mRNA poly(A) site sequences and which are involved in pre-mRNA processing but dispensable for snoRNA 3′-end formation (Barabino et al, 2000; Dichtl and Keller, 2001; Dichtl et al, 2002; Morlando et al, 2002; Kyburz et al, 2003). Ref2p and Pti1p, in contrast, may act to recognize snoRNA transcriptional units. Thus, specific RNA–protein and protein–protein interactions may produce polarity in the conformational assembly of the 3′-end formation complex on the substrate, which may interfere with the kind of reaction that takes place (i.e. polyadenylation yes or no). The Nop1p–Ref2p interaction, for example, may contribute to the ‘suppression' of polyadenylation at snoRNA cleavage sites and, or conversely other kinds of interaction may ‘stimulate' the occurrence of polyadenylation at poly(A) sites. Although it remains unclear as to how such effects could be brought about, they may include proteins that directly interact with poly(A) polymerase. Interestingly, FIR1 was found to interact in a two-hybrid screen with REF2 as bait (Russnak et al, 1996) and Fir1p itself interacts with poly(A) polymerase (del Olmo et al, 1997). Therefore, it will be interesting to test whether poly(A) polymerase may be subject to negative regulation at snoRNA cleavage sites. Clearly, additional work is required to elucidate the mechanisms of snoRNA 3′-end formation.
A prediction from our work on box C/D snoRNAs may be that factors, which specifically associate with box H/ACA snoRNAs, affect transcription and 3′-end formation in a way similar to Nop1p. Likewise, proteins associated with other pol II transcribed stable RNAs (e.g. snRNAs) may be required for the expression of distinct transcription units. Therefore, we would like to suggest that snoRNP proteins are required for a mechanism of quality control, in which the functional assembly of snoRNAs (and possibly other stable RNAs) is monitored during transcription. In this sense, it seems reasonable that stable RNAs will be efficiently transcribed and processed only when their binding partners are available and correctly assembled.
Materials and methods
Strains
Strains used in this study are listed in Table I. Standard techniques were used for growth and maintenance of Saccharomyces cerevisiae. Yeast strains were transformed with the appropriate plasmids as already described (Villa et al, 2000) and grown in the appropriate selective media. The induction experiments were performed as previously reported (Fatica et al, 2000) both for the wild-type and mutant strains. YPG 1 strain was obtained by disruption of the SNR13 gene with the Kanamycin gene by homologous recombination. The same technique was utilized to insert an SNR13 copy, carrying the mutations in the conserved box D, in the YPG1 strain; the resulting strain was called YPG 2.
Table 1.
List of strains
| Strain | Genotype | Source |
|---|---|---|
| YRP 683 | MATa, leu2-3, 112, lys2-201, his4-539, trp1-1, ura3-52 | Wendy et al (1997) |
| YRP 1200 | MATa, leu2-3, 112, lys2-201, his4-539, trp1-1, ura3-52, DCP1∷URA3 | Anderson and Parker (1998) |
| BMA 38 | MATa, his3 Δ200,leu2-3, 112, ura3-1,trp1Δ, ade2-1 | Baudin et al (1993) |
| YPG 1 | MATa, his3 Δ200,leu2-3, 112, ura3-1,trp1Δ, ade2-1, SNR13∷KAN | This study |
| YPG 2 | MATa, his3 Δ200,leu2-3, 112, ura3-1,trp1Δ, ade2-1, SNR13∷snR13bD∷URA | This study |
| W 303 | MATa, ura3-1, trp1-1, ade2-1, leu2-3,112, his3-11,15, can1-100 | Thomas and Rothstein (1989) |
| ssu72-2 | MATa, ura3-1, trp1-1, ade2-1, leu2-3,112, his3-11,15, ssu72-2 | Dichtl et al (2002) |
| D255 | MATa, ura3-52, leu2-3, 112, ade1-100, his4-519, URA3-pGAL10∷NOP1 | Tollervey et al (1991) |
| YAF 1 | MATα, trp1.Δ, his.Δ, ura3.52, lys2.801, ade2.101, URA3∷U24, NOP1∷TAP∷TRP1 | A Fatica (unpublished) |
| YCA 12 | MATa, ade2-1 his3-200 leu2-3, 112 trp1-1 ura3-1 can1-100 RRP6∷K1 TRP1 | Allmang et al (1999) |
| D348 | MATa, ura3-52, lys2, rrp44-1 | Bousquet-Antonelli et al (2000) |
Plasmid construction
pG50 and pG50bD constructs were derived from C/D50 (Morlando et al, 2002) by removing the GAL1 promoter by SacI–BamHI digestion and substituting it with a fragment obtained by amplification with oligos R13a1 and R13b1. The bD mutation and the 18 nt poly-G insertion were obtained by inverse PCR using the following oligos: 50bDa and 50bDb; 50pGa, 50pGb and 50pGbDb. Inverse PCR with the same oligonucleotides was also used to obtain GAL-pG50 and GAL-pG50bD constructs from plasmid C/D50. pG13 and pG13bD constructs were made by cloning PCR products (obtained with oligos R13a2 R13b2) into EcoRI–KpnI sites of pG50 and pG50bD plasmid. snR13wt and snR13bD constructs used in the run-on experiment were obtained by PCR amplification of the SNR13 gene (from position −355 to +270, with respect to the beginning of snR13 coding region) with oligonucleotides R13a1 and R13b2 and then cloned into the SacI/KpnI sites of p416 vector. The bD derivative was obtained by inverse PCR on snR13wt using R13bDa and R13bDb oligonucleotides.
Oligonucleotides
Sequences of the oligonucleotides used for the different cloning steps are (5′–3′) as follows:
 |
Oligos used for RNA analyses by Northern hybridization or primer extension are (5′–3′) as follows:
 |
RNA and protein analysis
Total RNA was extracted by the hot-phenol method (Schmitt et al, 1990). Northern blot and primer extension experiments were performed as previously described (Morlando et al, 2002). Total proteins from 0.5 OD cultures were resolved on a 10% SDS–PAGE gel and analyzed by Western analysis with anti-fibrillarin antibodies (R47) that specifically recognize the Nop1p protein (E Caffarelli, unpublished results).
Transcription run-on analysis
The single-stranded probes, 90-nucleotide long, possess 50 nucleotides of complementarity to the genomic region of interest and 40 nt of random sequence. Their positions from the 5′ end of the corresponding genes are +47/+99 (oligo 1), +346/+396 (oligo 2), +191/+241 (oligo 3), +20/+70 (oligo 4), +130/+180 (oligo H2A) and +480/+530 (oligo 18S). The coordinates of the intergenic region (oligo I) on chromosome V are 9777–9827.
The TRO procedure was carried out as described (Birse et al, 1998), with the only difference that 100 ml cultures at an OD600 of 0.15 were utilized. The TRO reaction was allowed to proceed for 5 min and then washed briefly with AE buffer (50 mM NaAc/10 mM EDTA, pH 5.5).
GST fusion proteins and in vitro interaction assays
The expression and purification of GST–Snu13p and GST–Nop1p as well as GST pull-down assays were carried out as previously described (Giorgi et al, 2001). In all, 31% of the total in vitro translation reaction was utilized for each interaction assay. The amount of input loaded as control corresponds to 1/4 of the 35S proteins tested in the pull-down experiment.
Chromatin immunoprecipitation
The YAF 1 strain was grown to an OD600 of 0.6–0.8 and processed essentially as previously described (Komarnitsky et al, 2000). The chromatin preparation was incubated overnight at 4°C with rabbit IgG-agarose beads (Sigma) prewashed in TE buffer. The immunoprecipitate was washed with 275–400 mM NaCl and the recovered chromatin and the input chromatin were de-crosslinked and amplified by PCR as already described (Komarnitsky et al, 2000). All the primers were designed as 24–27-mers with 50% GC content. Their location covers the following positions indicated with respect to the transcriptional initiation site: −52/+141 (SNR13), +137/+315 (U18/EF-1), +18/+209 (SNR189) and +177/+283 (wt and bD). [32P]dATP was added to the PCR reaction (1 μCi/12.5 μl). ChIP results were quantified as described by Nedea et al (2003): PCR signals were analyzed by PhosphorImager and ImageQuant software (Molecular Dynamics, Sunnyvale, CA) and normalized for amplification efficiency and subtraction of background. Each value in the histogram is the average of the signals of five PCR reactions performed on five different preparations of immunoprecipitated DNA.
Acknowledgments
We are grateful to Lionel Minvielle-Sebastia and David Tollervey for generously providing yeast strains, to Kate Abruzzi and Francesco Chiani for helpful suggestions on ChIP analysis and to Alessandro Fatica for discussion. We thank M Arceci and G Ricci for skilful technical help. This work was partially supported by grants from MURST (FIRB—p.n. RBNE015MPB and RBNE01KXC9, PRIN-Cofin and ‘Centro di Eccellenza BEMM').
References
- Allmang C, Kufel J, Chanfreau G, Mitchell P, Petfalski E, Tollervey D (1999) Functions of the exosome in rRNA, snoRNA and snRNA synthesis. EMBO J 19: 5399–5410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JSJ, Parker R (1998) The 3′ to 5′ degradation of yeast mRNAs is general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J 17: 1497–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabino SM, Ohnacker M, Keller W (2000) Distinct roles of two Yth1p domains in 3′-end cleavage and polyadenylation of yeast pre-mRNAs. EMBO J 19: 3778–3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C (1993) A simple and efficient method for direct gene depletion in Saccharomyces cerevisiae. Nucleic Acid Res 21: 3329–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beelman CA, Parker R (1995) Degradation of mRNA in eukaryotes. Cell 81: 179–183 [DOI] [PubMed] [Google Scholar]
- Bentley D (2002) The mRNA assembly line: transcription and processing machines in the same factory. Curr Opin Cell Biol 14: 336–342 [DOI] [PubMed] [Google Scholar]
- Birse CE, Minvielle-Sebastia L, Lee BA, Keller W, Proudfoot NJ (1998) Coupling termination of transcription to messenger RNA maturation in yeast. Science 280: 298–301 [DOI] [PubMed] [Google Scholar]
- Bousquet-Antonelli C, Presutti P, Tollervey D (2000) Identification of a regulated pathway for nuclear pre-mRNA turnover. Cell 102: 765–775 [DOI] [PubMed] [Google Scholar]
- Chanfreau G, Legrain P, Jacquier A (1998a) Yeast RNase III as a key processing enzyme in small nucleolar RNA metabolism. J Mol Biol 284: 975–988 [DOI] [PubMed] [Google Scholar]
- Chanfreau G, Rotondo G, Legrain P, Jacquier A (1998b) Processing of a dicistronic small nucleolar RNA precursor by the RNA endonuclease Rnt1. EMBO J 17: 3726–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Moore C (1992) Separation of factors required for cleavage and polyadenylation of yeast pre-mRNA. Mol Cell Biol 12: 3470–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad NK, Wilson SM, Steinmetz EJ, Patturajan M, Brow DA, Swanson MS, Corden JL (2000) A yeast heterogeneous nuclear ribonucleoprotein complex associated with RNA polymerase II. Genetics 154: 557–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker CJ, Parker R (1993) A turnover pathway for both stable and unstable mRNAs in yeast: evidence for a requirement for deadenylation. Genes Dev 7: 1632–1643 [DOI] [PubMed] [Google Scholar]
- del Olmo M, Mizrahi N, Gross S, Moore CL (1997) The Uba2 and Ufd1 proteins of Saccharomyces cerevisiae interact with poly(A) polymerase and affect the polyadenylation activity of cell extracts. Mol Gen Genet 255: 209–218 [DOI] [PubMed] [Google Scholar]
- Dheur S, Vo LTA, Voisinet-Hakil F, Minet M, Schmitter JM, Lacroute F, Wyers F, Minvielle-Sebastia L (2003) Pti1p and Ref2p found in association with the mRNA 3′ end formation complex direct snoRNA maturation. EMBO J 22: 2831–2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichtl B, Blank D, Ohnacker M, Friedlein A, Roeder D, Langen H, Keller W (2002) A role for SSU72 in balancing RNA polymerase II transcription elongation and termination. Mol Cell 10: 1139–1150 [DOI] [PubMed] [Google Scholar]
- Dichtl B, Keller W (2001) Recognition of polyadenylation sites in yeast pre-mRNAs by cleavage and polyadenylation factor. EMBO J 20: 3197–3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A, Morlando M, Bozzoni I (2000) Yeast snoRNA accumulation relies on a cleavage-dependent/polyadenylation-independent 3′ processing apparatus. EMBO J 13: 6218–6229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipowicz W, Pogacic V (2002) Biogenesis of small nucleolar ribonucleoproteins. Curr Opin Cell Biol 14: 319–327 [DOI] [PubMed] [Google Scholar]
- Galardi S, Fatica A, Bachi A, Scaloni A, Presutti C, Bozzoni I (2002) Purified box C/D snoRNPs are able to reproduce site-specific 2′-O-methylation of target RNA in vitro. Mol Cell Biol 19: 6663–6668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem C, Devaux F, Torchet C, Jacq C, Quevillon-Cheruel S, Labesse G, Facca C, Faye G (2003) Ssu72 is a phosphatase essential for transcription termination of snoRNAs and specific mRNAs in yeast. EMBO J 22: 1588–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415: 141–147 [DOI] [PubMed] [Google Scholar]
- Giorgi C, Fatica A, Nagel R, Bozzoni I (2001) Release of U18 snoRNA from its host intron requires interaction of Nop1p with the Rnt1p endonuclease. EMBO J 20: 6856–6865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross S, Moore C (2001) Five subunits are required for reconstitution of the cleavage and polyadenylation activities of Saccharomyces cerevisiae cleavage factor I. Proc Natl Acad Sci USA 98: 6080–6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Khan AU, Cheng H, Pappas DLJ, Hampsey M, Moore CL (2003) Functional interactions between the transcription and mRNA 3′ end processing machineries mediated by Ssu72 and Sub1. Genes Dev 17: 1030–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Carmichael GG (1996) Role of polyadenylation in nucleocytoplasmic transport of mRNA. Mol Cell Biol 16: 1534–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T (2002) Small nucleolar RNA: an abundant group of noncoding RNAs with diverse cellular functions. Cell 109: 145–148 [DOI] [PubMed] [Google Scholar]
- Komarnitsky P, Cho EJ, Buratowski S (2000) Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev 14: 2452–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyburz A, Sadowski M, Dichtl B, Keller W (2003) The role of the yeast cleavage and polyadenylation factor subunit Ydh1p/Cft2p in pre-mRNA 3′-end formation. Nucleic Acids Res 31: 3936–3945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontaine DLJ, Tollervey D (1999) Nop58p is a common component of the box C+D snoRNPs that is required for snoRNA stability. RNA 5: 455–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontaine DLJ, Tollervey D (2000) Synthesis and assembly of the box C+D small nucleolar RNPs. Mol Cell Biol 20: 2650–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley JL (2002) Nuclear coupling: RNA processing reaches back to transcription. Nat Struct Biol 9: 790–791 [DOI] [PubMed] [Google Scholar]
- Mitchell P, Petfalski E, Shevchenko A, Mann M, Tollervey D (1997) The exosome: a conserved eukaryotic RNA processing complex containing multiple 3′ → 5′ exoribonucleases. Cell 91: 457–466 [DOI] [PubMed] [Google Scholar]
- Morlando M, Greco P, Dichtl B, Fatica A, Keller W, Bozzoni I (2002) Functional analysis of yeast snoRNA and snRNA 3′-end formation mediated by uncoupling of cleavage and polyadenylation. Mol Cell Biol 22: 1379–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedea E, He X, Kim M, Pootoolal J, Zhong G, Canadien V, Hughes T, Buratowski S, Moore CL, Greenblatt J (2003) Organization and function of APT, a sub-complex of the yeast cleavage and polyadenylation factor involved in the formation of mRNA and snoRNA 3′ ends. J Biol Chem 278: 33000–33010 [DOI] [PubMed] [Google Scholar]
- Ohnacker M, Barabino SM, Preker PJ, Keller W (2000) The WD-repeat protein pfs2p bridges two essential factors within the yeast pre-mRNA 3′-end-processing complex. EMBO J 19: 37–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omer AD, Ziesche S, Ebhardt H, Dennis PP (2002) In vitro reconstitution and activity of a C/D box methylation guide ribonucleoprotein complex. Proc Natl Acad Sci USA 99: 5289–5294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot NJ, Furger A, Dye MJ (2002) Integrating mRNA processing with transcription. Cell 108: 501–512 [DOI] [PubMed] [Google Scholar]
- Rasmussen TP, Culbertson MR (1998) The putative nucleic acid helicase Sen1p is required for formation and stability of termini and for maximal rates of synthesis and levels of accumulation of small nucleolar RNAs in Saccharomyces cerevisiae. Mol Cell Biol 18: 6885–6896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russnak R, Pereira S, Platt T (1996) RNA binding analysis of yeast REF2 and its two-hybrid interaction with a new gene product, FIR1. Gene Expr 6: 241–258 [PMC free article] [PubMed] [Google Scholar]
- Schmitt ME, Brown TA, Trumpower BL (1990) A rapid simple method for preparation of RNA from Saccharomyces cerevisiae. Mol Cell Biol 20: 1311–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz EJ, Brow DA (2003) Ssu72 protein mediates both poly(A)-coupled and poly(A)-independent termination of RNA polymerase II transcription. Mol Cell Biol 23: 6339–6349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz EJ, Conrad NK, Brow DA, Corden JL (2001) RNA-binding protein Nrd1 directs poly(A)-independent 3′-end formation of RNA polymerase II transcripts. Nature 413: 327–331 [DOI] [PubMed] [Google Scholar]
- Tarun SZ Jr, Sachs AB (1996) Association of the yeast poly(A) tail binding protein with translation initiation factor eIF-4G. EMBO J 15: 7168–7177 [PMC free article] [PubMed] [Google Scholar]
- Thomas BJ, Rothstein R (1989) The genetic control of direct-repeat recombination in Saccharomyces: the effect of rad52 and rad1 on mitotic recombination at GAL10, a transcriptionally regulated gene. Genetics 123: 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollervey D, Lehtonen H, Carmo-Fonseca M, Hurt EC (1991) The small nucleolar RNP protein NOP1 (fibrillarin) is required for pre-rRNA processing in yeast. EMBO J 10: 573–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoof A, Lennertz P, Parker R (2000) Yeast exosome mutants accumulate 3′-extended polyadenylated forms of U4 small nuclear RNA and small nucleolar RNAs. Mol Cell Biol 20: 441–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa T, Ceradini F, Bozzoni I (2000) Identification of a novel element required for processing of intron-encoded box C/D small nucleolar RNAs in Saccharomyces cerevisiae. Mol Cell Biol 20: 1311–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins NJ, Dickmanns A, Luhrmann R (2002) Conserved stem II of the box C/D motif is essential for nucleolar localization and is required, along with the 15.5 K protein, for the hierarchical assembly of the box C/D snoRNP. Mol Cell Biol 22: 8342–8352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins NJ, Segault V, Charpentier B, Nottrott S, Fabrizio P, Bachi A, Wilm M, Rosbash M, Branlant C, Luhrmann R (2000) A common core RNP structure shared between the small nucleoar box C/D RNPs and the spliceosomal U4 snRNP. Cell 103: 457–466 [DOI] [PubMed] [Google Scholar]
- Wendy MO, Muhlrad D, Parker R (1997) Analysis of the yeast genome: of new non-coding and small ARF-containing RNAs. Nucleic Acid Res 25: 4619–4625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Hyman L, Moore C (1999) Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev 63: 405–445 [DOI] [PMC free article] [PubMed] [Google Scholar]