

Abstract
Genome-wide association studies (GWAS) have become a powerful tool in the identification of disease-associated variants. Unfortunately, many of these studies have found that the estimated variability in cardiovascular disease risk cannot be fully explained by traditional paradigms of genetic variation in protein coding genes. Moreover, traditional views do not sufficiently explain the well-known link between cardiovascular disease and environmental influence. We posit that epigenetics, defined as chromatin-based mechanisms important in the regulation of gene expression that do not involve changes in the DNA sequence per se, represents the missing link. The nuclear-based mechanisms that contribute to epigenetic gene regulation can be broadly separated into three unique but highly interrelated processes: DNA methylation and hydroxymethylation; histone density and post-translational modifications; and RNA-based mechanisms. Together they complement the cis/trans perspective on transcriptional control paradigms in blood vessels. Moreover, it provides a molecular basis for understanding how the environment impacts the genome to modify cardiovascular disease risk over the lifetime of a cell and its offspring. This review provides an introduction to epigenetic function and cardiovascular disease, with a focus on endothelial cell biology. Additionally, we highlight emerging concepts on epigenetic gene regulation that are highly relevant to atherosclerosis and coronary artery disease.
Keywords: ANRIL, cardiovascular disease, DNA methylation, epigenetics, endothelial cells, eNOS, histone posttranslational modifications and density, long noncoding RNA
Introduction
According to a report by the American Health Association, in 2009, 1 in 3 deaths in the United States of America (USA) was attributed to cardiovascular disease (CVD), which translates to 1 death every 40 seconds[1]. CVD as a whole is estimated to cost the USA $312.4 billion that year, an amount that exceeded costs of any other disease group. Moreover, this burden is expected to worsen due to an aging population and a shift in environmental risk factors in developing and third-world populations toward diets with higher sodium and fat content. Unique regions of the human vasculature affected include the coronary arteries, carotid arteries, and the peripheral arterial network[2]. In particular, coronary artery disease (CAD), which disrupts oxygen-rich blood flow to the heart, is the most common type of heart disease and is the most common cause of death in both men and women in the US. Based on 2009 USA statistics, CAD alone accounts for approximately 1 out of every 6 deaths, which projects to an estimated 386,000 deaths due to CAD [1].
CAD manifests from the progressive narrowing and either acute or chronic occlusion of blood vessels due to the gradual process of plaque formation, a process known as atherosclerosis[2]. As a result, stenosis of the vessel or thrombosis can occur and lead to angina pectoris and/or myocardial infarction (MI). Atherosclerosis is a complex inflammatory disease with well-defined genetic determinants as well as environmental risk factors supported by discordance between monozygotic twins, sexual dimorphism, parent-of-origin dependent clinical differences, and a relatively late age of onset of disease manifestation[3]. The environmental factors likely influence progressive stages of atherosclerotic plaque formation through their interaction with the endothelium, the innermost lining of the vasculature system, causing endothelial dysfunction. Endothelial cell (EC) “dysfunction” represents a net liability to the host through the disruption of its homeostatic functions including vasomotor tone, leukocyte trafficking, and hemostasis[4]. Typically, endothelial dysfunction and lesion initiation is thought to be triggered by risks factors such as elevated blood pressure, diabetes, dyslipidemia, and smoking. Although these risk factors affect the circulatory system in a systemic manner, the development of lesions displays a focal distribution[5]. This pattern is not arbitrary as numerous studies have identified a role for hemodynamics in promoting atherosclerosis, particularly in CAD patients[6]. Investigating both the genetic and environmental basis of CAD will allow us to design new and more effective therapeutic strategies.
Heritability vs. Environment – Implications for Future Generations
Our knowledge of CAD has evolved in parallel with advances in our understanding of atherosclerosis as a whole. Atherosclerotic disease of the carotid and peripheral vasculature system share common determinants. Traditional risk factors that increase the risk of CAD are well established and include hypertension, dyslipidemia, and diabetes. It is still unclear, however, the molecular mechanisms that promote CAD in individuals affected by these environmental factors. Surprisingly, these environmental changes can have transgenerational effects. Furthermore, numerous studies have revealed a genetic predisposition for CAD, in particular twin studies that suggest a heritable component for the disease[7]. However, studies have revealed that approximately 8–13% of the estimated CAD heritability is attributed to genetic variation alone[8]. We argue that the impact of gene-environment interactions have been underestimated and that the heritable component of CAD is not fully defined, but these challenges can potentially be resolved by applying the epigenetic theory of complex, non-Mendelian diseases.
The implementation of classical twin studies has proved to be a valuable tool in assessing the genetic contribution in many diseases, including CAD[7, 9]. The fundamental principle of these studies is that monozygotic twins (MZ) are nearly genetically identical, whereas dizygotic twins (DZ) only share approximately 50% of their genome. Therefore, if the concordance rate for a trait/disease is greater in MZ twins compared to DZ twins, a genetic component can be hypothesized for the disease phenotype. A 1994 study on 10,502 Swedish twin pairs revealed a genetic component in CAD as the mortality of a co-twin led to doubling of the relative hazard for death by CAD in MZ twins relative to DZ twins[7]. Similarly, a separate study on Danish twins also observed a strong genetic influence on mortality caused by CAD[10]. With respect to CAD, heritability of frailty (or liability to death) was found to be 0.55 and 0.53 for males and females, respectively. Although a genetic component is apparent, there was considerable discordance among MZ twins for death due to CAD. How can this discordance for CAD within MZ twin pairs be explained? Here we argue that gene-environment interactions are also important. Surprisingly, our understanding of the mechanistic links between environmental risk factors and CAD is poorly understood[9].
The application of genome-wide association studies (GWAS) has further expanded what we know about the role for genetics in the development of CAD. The ultimate purpose of these studies is to evaluate the association between genomic variation and phenotypic variation in a disease population, by identifying single-nucleotide polymorphisms (SNPs) or single-nucleotide variants (SNVs) that are correlated with the disease[11]. Although GWAS are powerful tools for exploring the genetic architecture of non-Mendelian diseases in an unbiased manner, they do have some limitations. They are dependent on the “common disease, common variant” hypothesis which postulates that common diseases are attributable to common risk variants found within at least 1–5% of the total population[11]. However, for most common human diseases or complex traits, susceptibility risk variants only account for a modest increase in risk (1–1.5 fold) and explain only a small proportion of the estimated variability. For example, Maremberg et al. demonstrated that 30–60% of inter-individual variability for CAD risk is attributable to genetic factors[7]. What accounts for this “missing heritability” in GWASs of complex diseases, such as CAD? We argue that epigenetic theory provides an alternative paradigm that accounts for, at least in part, the missing heritability of complex diseases. Interestingly, using a method designed to identify genomic susceptibility loci has instead identified epigenetic effectors as the major determinants of CAD liability. In 2007, multiple research groups identified a genomic susceptibility locus for CVD heritability within the intergenic non-coding region of chromosome 9p21 (Ch9p21)(reviewed in [12, 13]). This locus contains a long non-coding RNA (lncRNA), referred to as ANRIL (further discussed below), and is the most replicated marker of CAD & MI, independent from most traditional risk factors and its expression is correlated with atherosclerotic lesions[14]. The finding that a long noncoding RNA may be relevant to CAD pathogenesis was surprising.
The evolving study of transgenerational epigenetics addresses the capacity of an offspring’s epigenetic profile to be influenced by environmental factors from preceding generations (Figure 1)[15]. Epigenetics is defined as chromatin-based mechanisms important in the regulation of gene expression that do not involve changes in the DNA sequence per se [3]. Epigenetic processes are known to be triggered by changes in the environment, allowing cells to rapidly adapt to their dynamic surroundings. These modifications can persist in subsequent generations and contribute to disease risk in individuals[15]. Additionally, gene imprinting refers to chromatin-based mechanisms that regulate the monoallelic expression of either the paternal or maternal gene in parent-of-origin fashion. There is evidence that transgenerational diseases can disrupt genomic imprinting and subsequently enhance risk for disease in future generations. The Dutch Hunger Winter Famine of 1944–45 is a prominent example of the influence of transgenerational epigenetics and gene imprinting on disease risk inheritance[16]. In offspring of mothers who were malnourished during this period, whole blood extracts were found to display reduced DNA methylation at the maternal insulin-like growth factor 2 (IGF2) gene. Interestingly, the offspring of these affected individuals developed CAD earlier than healthy subjects[17]. This evidence suggests that environmental factors from previous generations can confer susceptibility for diseases in the present day, perhaps through epigenetic mechanisms that are passed on through meiosis.
Figure 1. Comparison of key facets of the genetic and epigenetic codes.

Within a multicellular organism all cells show an identical genetic code, the static A-T-C-G bases, which are conserved with cellular replication in mitosis and meiosis. This has been conserved throughout evolution from single cells to multicellular organisms. Therapy for defects in the static DNA code is complicated due to the complexity of gene therapy in multicellular organisms. The epigenetic code is semi-conserved in meiosis and mitosis and shows rapid divergence across species. Notably, it is different in all cells of multicellular organisms, and responsive to the environment, making it a candidate target for pharmacologic intervention.
In conclusion, these three perspectives on CAD – discordance of MZ twins for CAD, missing heritability in GWASs, and transgenerational effects of the environment – demonstrate the limitations of using traditional disease models to understand the risk factors for CAD.
Gene Expression in Vascular Endothelium – The influence of environment, specifically hemodynamics, on EC phenotype
Epigenetics is an alternative perspective that may enable us to fully understand the genetic and environmental basis of CAD. Our present environment is abundant in risk factors that influence vascular health and disease and can initiate CVD in general, and CAD specifically. Within the context of CAD, the endothelium is a key intermediary between environmental stimuli and the progression of atherosclerosis. Due to their location along the blood vessel wall, ECs are constantly exposed to circulating humoural factors, blood cellular constituents, other vessel wall types and the physical forces of the circulation[18]. Endothelial cells are able to sense and respond to these distinct stimuli rapidly and respond accordingly to maintain vascular homeostasis and hemostasis. A good example is the ability of EC to release vasoconstrictors and vasodilators that control local vasomotor tone[19]. The development of atherosclerosis typically begins with the onset of inflammation due to a range of risk factors, such as elevated levels of low-density lipoprotein (LDL) cholesterol, high glucose, or smoking[5]. Chronic exposure to these environmental factors can result in “endothelial dysfunction” which is primarily characterized by a reduction in endothelial nitric oxide synthase (eNOS, NOS3) expression (further discussed below) and the bioavailability of nitric oxide (NO). Given NO’s anti-inflammatory and anti-atherosclerotic properties, its reduction can exacerbate the inflammatory response which sets the stage for atherosclerosis[5]. Furthermore, endothelial dysfunction is associated with changes in other important genes which contribute to a pro-atherosclerotic phenotype. For example, there is a downregulation of several genes associated with “atheroprotection” including Kruppel-like factor 2 (KLF2), as well as the upregulation of genes that are pro-inflammatory such as endothelin-1[20, 21].
Although common risk factors for CAD, such as diabetes or smoking, affect the entire vasculature, there is compelling evidence in human and animal models that atherosclerotic lesions display a site-specific distribution[5]. This unexpected observation appears to be due to changes in the physical forces of the blood circulation, or “hemodynamics”. The endothelium is exposed to different forms of hemodynamic forces, but most attention has been on the influence of shear rate and shear stress on EC biology (Figure 2). The movement of blood can be modeled as a series of concentric layers called laminae. Shear rate (expressed as sec-1) is defined as the rate at which adjacent layers of fluid move with respect to each other[22]. On the other hand, shear stress (expressed as dynes/cm2) is the tangential frictional force per area generated at the surface of the endothelium by blood flow. With the assumption that blood is a Newtonian fluid, shear stress is dependent on the level of shear rate and viscosity of the fluid[21]. The relationship between hemodynamics and site propensity of atherosclerosis is centered on two unique patterns of arterial blood flow – atheroprotective and atheroprone flow – both of which have been modeled in vitro [20, 23]. Atheroprotective flow typically occurs in straight segments of the arterial network (e.g. descending thoracic aorta), where blood flow is laminar and associated with physiological levels of shear rate and shear stress. In contrast, atheroprone flow is characterized by turbulent flow that occurs at geometrical irregularities, such as bifurcations or curvatures (e.g. lesser curvature of aortic arch), resulting in flow reversal and low levels of shear rate/stress. Importantly, human coronary arteries are known to experience atheroprone flow[23]. These distinct types of flow have a significant impact on EC gene expression and phenotype, and most importantly, inflammation. In response to low shear stress and/or oscillatory flow, ECs exhibit a dysfunctional phenotype [20]. There is a lack of understanding in how ECs respond and adapt to changes in environmental conditions, in particular the changes in blood flow, and we believe that the remarkable plasticity of ECs can be attributed to epigenetic mechanisms.
Figure 2. Flow and endothelial phenotypes in the aortic arch.
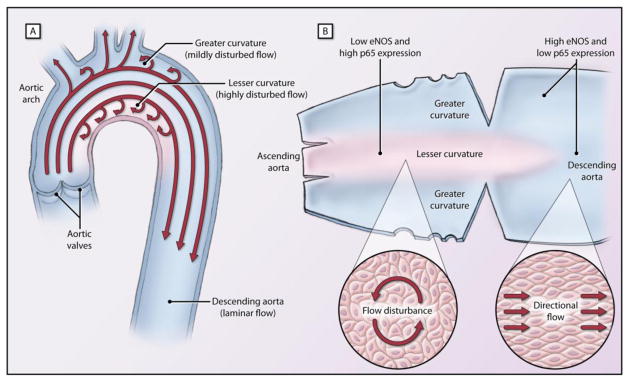
(A) Flow patterns of the aortic arch display both laminar flow in the greater curvature, and disturbed flow in the lesser curvature. (B) Dissection of the aortic arch reveals interesting phenotypic changes in endothelial cells (ECs). ECs of the greater curvature experiences laminar flow, exhibit eNOS expression and decreased p65 expression, an inflammatory mediator in ECs, and are less prone to atherosclerosis. These cells show a directional cell shape which correspond with directional flow. ECs of the lesser curvature of the aortic arch are prone to atherosclerosis, decreased eNOS expression and p65 enrichment. These ECs also experience disturbed flow and exhibit a disorganized pattern.
Epigenetics: Basic Mechanisms and the Endothelium
The haploid human genome is made up of approximately 3.3 billion DNA base pairs. Size variation reflects interindividual differences in copy number variation and sex-specific differences (i.e. x versus y chromosome length). Given the size of an individual base pair, the DNA strand from a single cell would span approximately two meters in length. The compaction of genomic DNA into the eukaryotic cell nucleus is a marvelous example of the packing of DNA into a DNA-protein complex referred to as chromatin. The human nucleosome, 146 bp of DNA wrapped around an octamer of core histone proteins, is composed of two molecules each of H2A, H2B, H3, and H4. Adjacent nucleosomes are linked by short stretches of linker DNA and the less conserved histone H1. The epigenetic theory constitutes the molecular effectors that regulate the structure and accessibility of chromatin without changing the underlying A-C-G-T genetic code per se. They constitute a highly integrated and evolutionarily conserved system for regulated gene expression (Figure 3) and will be individually elaborated in the following sections.
Figure 3. Epigenetics can be separated into three distinct but highly interrelated processes: DNA methylation; histone density and post-translational modifications; and RNA-based mechanisms.
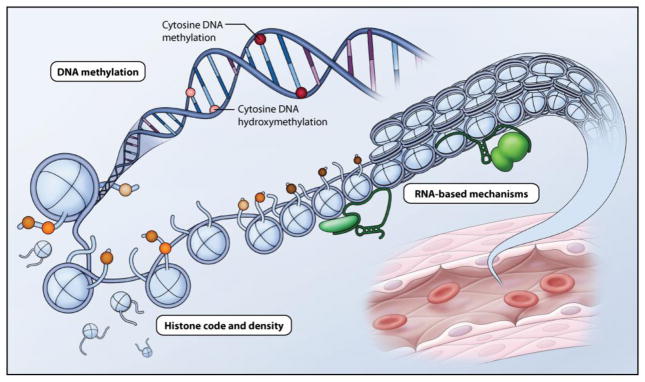
DNA methylation refers to the addition of a methyl group to the 5-position of cytosine and is a repressive mark which occurs symmetrically almost exclusively in the context of CpG dinucleotides in mammals. 5-hydroxymethylation also occurs at CpG dinucleotides and is thought to act as an intermediate for active demethylation. Histone density and post-translational modifications refers to the presence of histones and N-terminal histone tail post-translational modifications, respectively. These changes alter histone protein-DNA interactions and change chromatin structure to affect accessibility of transcription factors, modulating gene transcription. RNA-based mechanisms including long noncoding RNAs (lncRNAs) interact with chromatin and chromatin modifying complexes in cis or trans, to modulate gene expression.
DNA methylation/hydroxymethylation
DNA methylation refers to the covalent modification of the 5-position of cytosine to generate 5-methylcytosine (5mC), commonly referred to as the “fifth base of DNA”[24]. The understanding of the establishment and function of DNA methylation has increased since its initial discovery in mammals by Hotchkiss in 1948[25]. Early work found that 5-methylcytosine continues to base pair with guanine, is passed on semi-conservatively with DNA replication, and highly restricted to CpG dinucleotides in mammals. Although evolution of the mammalian genome has left it relatively CpG depleted due to spontaneous deamination of the methylated cytosine to thymine, the genome does contain regions that are not CpG depleted[26]. These are referred to as CpG islands. DNA methylation is traditionally viewed as a repressive mark associated with inhibition of transcriptional initiation when found adjacent to promoters. It is therefore important in silencing many developmental elements involved in genomic imprinting, X chromosome inactivation, mammalian embryogenesis and cellular differentiation. It also serves as a protective mechanism to silence repetitive DNA elements, which make up 25–40% of mammalian genomes, depending on how one defines “repetitive”. In general DNA methylation is a relatively stable covalent DNA modification[24].
In mammals, the DNA methyltransferase (DNMT) family of enzymes catalyze the addition of a methyl group from S-adenosyl methionine to the 5-position of cytosine. The ability of 5-methylcytosine to be propagated throughout mitotic divisions is due to the maintenance methyltransferase, DNMT1[27]. DNMT1 shows preferentially binding to hemimethylated DNA, typically at the DNA replication fork, and methylates the complementary cytosine. Notably, DNMT1 has been shown to be relatively inefficient at maintaining the methylation of CpG, especially in CpG dense regions. In order to establish new methylation profiles after replication, the other two family members, DNMT3A and DNMT3B, are recruited to de novo methylate CpGs. They are thought to exert their main function in establishing methylation patterns on unmethylated DNA during embryogenesis and early development[27]. Although a stable mark, removal of DNA methyl groups can occur, especially during development. Passive DNA demethylation can occur in primordial germ cells and pre-implantation embryos, where a reduction in DNMT activity may be observed[28]. An area of more active research is on the site-specific removal of cytosine methylation, termed active DNA demethylation. This has been noted when DNA methylation levels change quickly, sometimes within hours. These are DNA replication-independent cellular responses[29]. Active DNA demethylation has been proposed to occur through several pathways, but the most exciting is the recent discovery of the oxidative formation of 5-hydroxymethylcytosine, now known as the “sixth base of DNA”[30]. The ten-eleven translocation (TET) enzyme family (TET1, TET2 and TET3) act on 5-methylcytosine to form 5-hydroxymethylation with the subsequent oxidative products 5-formylcytosine or 5-carboxycytosine being produced[31]. The TET family may work in conjunction with the family of base excision repair glycosylases, to regenerate the initial cytosine residue[32]. The function of 5-hydroxymethylcytosine is currently under active study as it was initially thought to be an intermediate in demethylation. However, accumulation in various cell types and its specific recognition by protein complexes may indicate that it also serves a purpose in gene regulation[33].
How does DNA methylation affect gene expression? In general, it is a repressive transcriptional mark. DNA methylation participates in gene repression currently via three distinct mechanisms. First, the presence of the methyl group is thought to directly inhibit DNA binding of some transcription factors such as CTCF, c-Myc, and HIF-1α[24]. Although seemingly important, few transcription factors have been identified to be affected by DNA methylation therefore, this remains an area for future focus. Second, the methyl group can also influence other aspects of epigenetic pathways, namely histone modifications. Recent evidence suggests that DNA methylation inhibits the formation of the activating histone 3 lysine 4 (H3K4) di- and tri-methyl marks. Lastly, methyl binding proteins (MBP) can bind specifically to methylated CpGs. The recruitment of these proteins is directly mediated by the presence of DNA methylation. Although the mechanism of repression is not fully understood, the members of this family (MBD1, MBD2, MBD3, MeCP2 and Kaiso) are thought to bind repressor proteins and histone deacetylases which mediate inactive chromatin signatures[24, 34]. Methylation is a stable repressive mark and shows semi-conservation with DNA replication, important for disease heritability in both parent and offspring.
Histone post-translational modifications
The second layer of epigenetic gene regulation resides in the histone proteins around which the DNA double helix is wound. These evolutionarily conserved proteins are comprised of a globular domain and histone tails. In particular the amino termini histone tails, provide a robust platform for a myriad of post-translational modifications that can inform gene expression[35]. To date, more than one hundred distinct modification sites have been characterized [36] Examples of well studied histone post-translational modifications include lysine acetylation and lysine methylation, among others (Table 1)[35]. Many of these marks are mutually exclusive for any given histone amino acid residue. For example, histone 3 lysine 9 (H3K9) can either be acetylated or methylated, but not both.
Table 1.
Well studied epigenetic modifications and their effect on gene transcription.
| Mechanism | Transcriptional Effect |
|---|---|
| DNA modifications | |
| Methylation (CpG dinucleotides) | ↓ |
| Hydroxymethylation (CpG dinucleotides) | ? |
| Histone posttranslational modifications | |
| Histone H3 | |
| Acetylation (K9, K14) | ↑ |
| Methylation | |
| K4 | ↑ |
| K9 | ↓ |
| K27 | ↓ |
| K36 | ↑ |
| Phosphorylation (S10) | ↑/↓ |
| Histone H4 | |
| Acetylation (K5, K8, K12, K16) | ↑ |
| Methylation (K20) | ↓ |
| Long noncoding mRNA (lncRNA) mechanisms | |
| Repressive lncRNAs e.g. XIST, ANRIL, HOTAIR | ↓ |
| Activating lncRNAs e.g. HOTTIP | ↑ |
Abbreviations: K, lysine; S, serine
Histone post-translational modifications are postulated to regulate gene expression through two broad mechanisms. First, histone post-translational modifications can alter the physical structure of chromatin and its accessibility to DNA-binding proteins, such as transcriptional regulators[37]. For example, the higher-order compaction of chromatin is prevented by histone lysine acetylation, whereby acetyl groups neutralize the basic charge of lysine residues making chromatin more open and accessible to DNA-binding proteins[37]. More important than the direct physical effect on the structure of chromatin, perhaps, is the regulatory information contained within the specific combinations of histone marks. The “histone code hypothesis” provides the mechanistic concept that a given combination of modifications is “read” by a combination-specific protein or protein complex to effectuate a specific gene expression outcome[38]. Given the myriad of histone modifications, there are many permutations therefore deciphering the histone code remains a daunting, yet top priority for the epigenetics research community.
A series of histone code “readers”, “writers” and “erasers” are known[35]. Each modification is catalyzed by an increasingly well-characterized group of “writers” that can then be interpreted by a distinct group of “readers” [3, 35]. In mammalian cells, lysine acetylation is catalyzed by three families of histone acetyltransferases (HATs). Indeed, the demonstration that classical transcriptional coactivators possess intrinsic HAT activity helped to establish histone lysine acetylation as a permissive, epigenetic mark strongly correlated with transcriptional activation[35]. The removal of histone lysine acetylation is accomplished by four families of histone deacetylases (HDAC classes I-IV) or “erasers”. Experimentally, these families of HDACs are further categorized by their sensitivity to inhibition by the pharmacological agent, trichostatin A (TSA). Class I and II HDACs are TSA-sensitive, while class III and IV HDACs are TSA-insensitive. Importantly, HDAC inhibitors have found early clinical utility in the treatment of various cancers, neurodegenerative disease and inflammation [39]. Interestingly, HATs and HDACs demonstrate poor specificity for individual lysine residues and are recruited to target promoters in large, multiprotein complexes through a group of acetyl-lysine “readers” that contain a bromodomain[35].
Recent studies on histone lysine methylation underscore the tremendous complexity of epigenetic gene regulation. Unlike histone lysine acetylation, the downstream effects of lysine methylation on gene expression are residue-specific and context-dependent[3, 35, 37]. For example, histone H3 lysine 4 (H3K4) is strongly associated with transcriptional activation, while histone H3 lysine 9 (H3K9) is a classical stable repressive mark associated with heterochromatin formation and transcriptional silencing. Once again, these epigenetic modifications are controlled by distinct groups of “readers”, “writers” and “erasers”. Lysine methylation biology is even more finessed. An individual histone lysine residue can be mono-, di-, or trimethylated with profoundly distinct effects on mammalian gene expression. The functional relevance of these nuanced modifications is supported by their differential localization in the genome; for example, it has been suggested that H3K4 monomethylation preferentially localizes to enhancers and H3K4 trimethylation to active promoters.[3, 40].
In addition to considerable complexity of histone post-translational modifications, important roles for nucleosome density, ATP-dependent chromatin remodeling, and the regulated, replication-independent incorporation of histone variants in the control of mammalian gene expression have been noted[40, 41]. These additional layers of epigenetic control provide further complexity and sophisticating the highly responsive system superimposed on the static genetic code. Altogether, tremendous strides have been made in understanding the impact of specific histone post-translational and density modifications in the control of mammalian gene expression.
Epigenetic Model - eNOS
Given the role of EC dysfunction in the initiation, progression and clinical phenotype of atherosclerosis, investigating how gene expression is regulated in these cells and how these processes are perturbed in disease will provide newer insight into the pathogenesis of CAD. A prominent feature of endothelial dysfunction is reduced eNOS expression in the neointimal coverings of advanced atheromatous plaques (Figure 4)[5, 42–44]. There are numerous risk factors that contribute to EC dysfunction through changes in eNOS [5, 42–44]. Mechanistically, important transcriptional and post-transcriptional mechanisms for the control of eNOS mRNA levels have been identified[41, 46–48]. Although a critical biological process due to blood flow, hemodynamic forces at characteristic regions within the vasculature, such as arterial curvatures and bifurcations, also leads to differential expression of eNOS [5]. A decrease in eNOS activity and an increase in priming of the NF-κB signal transduction cascade exist in mouse aortas at regions predisposed to atherosclerosis [5, 43].
Figure 4. Large vessel occlusion is a major mechanism of coronary arterty disease.

Proper gene expression in endothelial cells in healthy vasculature maintains an antithrombotic and antiatherogenic cellular phenotype. Epigenetics may play a role, as chromatin configuration, mediated by environmental influences can alter expression of key genes in endothelial cells. An open chromatin configuration would allow transcription of atheroprotective genes, such as eNOS, in healthy endothelium. In contrast, a dysfunctional endothelium could be characterized by a closed chromatin configuration with repressive epigenetic profile.
After the cloning of the eNOS gene, the promoter was characterized and examined for mechanisms of endothelial cell-restricted expression [49–51]. The initial discovery of the role of epigenetics in eNOS gene expression came from the use of episomal eNOS promoter-reporter constructs. Interestingly, in vitro all cell types examined were able to express the construct, although the native eNOS gene was EC restricted[48]. This contrasted with stably integrated eNOS promoter-reporter transgenic mice that demonstrated a similar expression pattern to eNOS in humans[52]. Direct evidence of eNOS epigenetic regulation was observed in that the eNOS promoter showed DNA hypomethylation in ECs, but dense methylation in non-expressing cells, such as VSMC[48]. This DNA methylation was shown to be functionally relevant. This idea of endothelial-enriched expression due to differential DNA methylation was recently expanded to encompass several other important EC-enriched genes such as CD31, vWF and ICAM-2[53]. Further examination of the eNOS promoter revealed the presence of activating histone marks such as acetyl H3 and H4 and trimethylated H3K4, while non-EC lacked these marks[47]. The histone marks at the eNOS promoter can be regulated by the environment. Hypoxia, a classical cellular stress, causes a decrease in histone activating marks such as acetylation and H3K4 methylation under acute conditions, and additionally histone eviction. Under chronic hypoxia, the histone octamers returned to normoxic levels, but the basal activating marks were not present[41]. It is also worth highlighting that hypoxia upregulates the expression of an antisense eNOS gene, sONE (eNOS antisense, NOS3AS) that overlaps the 3′ end of the eNOS gene[54]. sONE has been shown to regulate eNOS levels at the post-transcriptional level[46, 54, 55]. Previous data have demonstrated that laminar flow induces eNOS expression through enhanced mRNA production, processing and stability[56]. Recent studies have also identified various epigenetic processes that are responsive to flow. These epigenetic change may play an active role in regulating eNOS expression. In response to laminar flow, there is increased acetylation of histones H3 and H4 at the promoter which is indicative of transcriptional activation[57]. Furthermore, HDACs can indirectly induce KLF2 and eNOS transcription in laminar shear stress[58]. Taken together, these data suggest that epigenetic mechanisms play a role in regulating eNOS expression in response to shear stress.
RNA-based mechanisms
The discovery of an extensive catalog of long RNA transcripts that do not code for proteins, termed long noncoding RNAs (lncRNAs), provides a new perspective on the importance of RNA in gene regulation. Long non-coding RNAs encompass the third layer of epigenetic regulation. Although the field is in its infancy and the nuclear function of lncRNAs is the least understood epigenetic regulator, the discovery of lncRNAs has made us appreciate the other aspects of RNA function, mainly RNA transcription and secondary structure. LncRNAs can mediate activation or repression of target genes through DNA methylation and histone posttranslational modifications, as described above[59]. LncRNAs are functionally distinct from small non-coding RNAs, namely microRNAs, which primarily mediate posttranscriptional repression in the cytoplasm. Interestingly, unlike protein-coding genes, lncRNAs lack strong conservation across species, suggesting that they may be under rapid evolutionary divergence and key for speciation[59].
One of the earliest characterized lncRNAs is X-inactive specific transcript (XIST), which is involved in female X-chromosome inactivation in mammals. This lncRNA works in conjunction with repressive histone marks and DNA methylation to silence one of the two X chromosomes in female somatic cells[60]. Initially, Xist and other long non-coding RNAs were thought to be rare examples of this functional group of RNAs. A global and unbiased approach using chromatin state mapping identified unique signatures of H3K4 trimethylation at promoters and H3K36 trimethylation, termed K4–K36 domains at actively transcribed genomic regions. This led to the identification of 1,600 human intergenic lncRNAs and has further expanded to more than 8,000 lncRNAs that may exist near or antisense to protein coding genes since 2007 [59, 61]. Most early work has focused on the functional identification of lncRNAs and their interaction with other epigenetic modifying machinery, but the effects of the genomic context has also become a prominent area of research[59, 62, 63].
Two excellent examples that encompass all the aspects of lncRNA function come from the HOX gene clusters. HOTAIR is expressed from the HOXC cluster on chromosome 12 in humans and acts in trans to regulate the expression of developmental genes in the HOXD locus on chromosome 2. It silences the HOXD cluster, in part, by recruiting polycomb repressive complex 2 (PRC2) which facilitates the addition of H3K27 trimethylation marks. In general, this is a labile repressive histone mark. The mechanism for how this targeting occurs remains unclear[62]. A second lncRNA found in the HOX gene cluster is HOTTIP, which is expressed at the distal tip of the HOXA locus and promotes transcription, in cis, of the adjacent HOXA genes. It activates these genes, in part, through its interaction with the histone methyltransferase MLL which mediates H3K4 trimethylation[63]. These two examples of lncRNAs provide good examples of lncRNAs with contrasting function, namely ones that act either in cis or trans to either activate or repress transcription, respectively. More complicated models exist, where the lncRNA overlaps or are present within introns of coding genes. Two notable examples exist in cardiovascular disease, namely, sONE and ANRIL, which will be discussed below. In general, the study of lncRNAs is just beginning. Their roles must be distinguished functionally from the biological effects of the adjacent protein coding genes.
Epigenetic Mediator - ANRIL
GWAS have identified lncRNAs that may be relevant to the genetic predisposition to CVD, especially ischemic stroke and CAD. This concept is important, as it was not previously appreciated that long non-coding RNAs were mechanistically important in CAD. An excellent example, and one we discuss in detail, is ANRIL. The lncRNA Antisense Non-coding RNA in the INK4 Locus (ANRIL, CDKN2B antisense RNA) is found within the INK/ARF gene cluster. It was first identified by a germ-line deletion in a melanoma-neural system tumor family in an unrelated discovery of the locus[64]. Only months afterward, the identification of several SNPs associated with a CAD risk were localized to a genomic region devoid of protein coding genes but in or near the ANRIL lncRNA[64–66]. One particular SNP (rs1333049) showed a modest effect size in heterozygotes (OR = 1.47 (95% CI 1.27–1.70) which was exaggerated in homozygotes (OR = 1.9 (95% CI 1.61–2.24)[64]. Two other SNPs in the locus were identified which exist in a cluster (rs10757274 and rs10757278). The former SNP is the strongest genetic predictor of early myocardial infarction and is not associated with established CAD risk factors such as lipoproteins or hypertension, making ANRIL a key candidate [66]. The latter SNP showed a modest effect size in myocardial infarction (OR = 1.26 (95% CI 1.16–1.36) in heterozygotes and is exaggerated in homozygotes (OR = 1.64 (95% CI 1.47–1.82). In CAD, rs10757278 shows a modest effect size (OR = 1.29 (95% CI 1.21–1.38), P=3.6×10−14)[65]. Moreover, this SNP was also identified in a GWAS studies for atherosclerotic diseases, a major component of CAD[14].
The ANRIL genomic locus spans approximately 42 kb on human chromosome 9p21. The molecular characterization of the encoded RNA has been complex due to the generation of multiple splice variants, both linear and circular[58]. Functional studies have found that ANRIL aids in cis recruitment of chromatin-modifying complexes, specifically, polycomb repressive complexes 1 and 2 (PRC1 and PRC2), to neighboring genes of the INK4b/ARF/INK4a locus. These protein-coding genes were known to serve as important regulators of cellular division and senescence[13]. ANRIL regulates these genes by recruiting PRC2 to add H3K27 trimethylation repressive methylation marks. H3K27 trimethylation is then recognized by PRC1 which establishes H2AK119 monoubiquitylation to silence transcription[67]. It is believed that ANRIL transcription, antisense to ARF/INK4b, and secondary RNA structure formation are able to interact and direct PRC1 and PRC2 to the adjacent INK4/ARF locus to mediate the establishment of these repressive marks[58]. The presence of these epigenetic marks is associated with chromatin compaction and transcriptional repression of these protein-coding genes[35, 67]. One model suggests that the presence of the disease-associated SNP allele changes the abundance or function of ANRIL splice variants, resulting in their reduced ability to regulate the INK4/ARF locus SNPs within the risk locus, or functionally important variability in linkage disequilibrium, may play a role in ANRIL function[58].
The clinical implications and the basic science discovery of lncRNAs is just emerging. ANRIL is a specific example of a lncRNA which was identified through unbiased GWAS studies and is associated with a non-Mendelian human disease and offers an exciting newer perspective on CAD pathophysiology. Moreover, many unbiased GWAS studies have identified a significant proportion of hits that fall into genomic regions with no protein coding genes, termed “gene deserts” [68]. This raises a key concept, that a gene can be implicated in the genetic cause of disease because it produces a functional RNA as an epigenetic modifier.
Future Directions
The use of epigenetic modifiers in the clinic has shown promise. DNMT inhibitors have been approved for myelodysplastic syndrome and acute myelogenous leukemia, while various HDAC inhibitors are being used for certain T-cell lymphomas[39]. Further use of epigenetic modifiers is bound to increase as our knowledge of epigenetics expands. Alternatively, epigenetic modifiers can also be used for prognostic purposes. For example, HOTAIR expression was found to be correlated with breast cancer metastasis and death, indicating that lncRNAs may serve a prognostic purpose and potentially a target in the future[59]. Importantly, there is work to suggest that HDAC inhibition may be of clinical use for CAD[39]. It would not be surprising if these findings may be further implicated in endothelial dysfunction and allow us to modulate important players in disease such as eNOS and ANRIL in diseased vasculature.
As noted by the famed physician William Osler in 1892, “Longevity is a vascular question, and has been well expressed in the axiom that ‘a man is only as old as his arteries.’”[69] As life expectancy increases, scientists must use new paradigms to help understand disease progression, especially in vascular diseases such as CAD. New therapies and approaches for CVD in general, and CAD specifically, are now urgently needed to help decrease the tremendous socioeconomic burden caused by this spectrum of diseases. Limitations, specifically the cis/trans paradigm, have arisen with classical models of transcription in vascular endothelial cells. It is well established that our environment has chronic effects on gene expression in endothelial cells. Epigenetic regulation in vascular endothelial cells has provided a molecular basis for understanding the environmental impacts on the genome and disease susceptibility[3, 70]. Most importantly, the dynamic nature of epigenetic modification in endothelial cells offers the possibility of therapeutic intervention for non-Mendelian diseases such as CAD.
Acknowledgments
Sources of Funding
PAM holds the Elisabeth Hofmann Chair in Translational Medicine at the University of Toronto and is supported by operating grants from CIHR MOP 79475 and NIH/HHLBI P01 HL076540-06A1. PJT and ANS gratefully acknowledge support from Queen Elizabeth II/Heart & Stroke Foundation of Ontario Scholarship in Science and Technology scholarship.
Abbreviations
- 5hmC
5-hydroxymethylcytosine
- 5mC
5-methylcytosine
- AID/APOBEC
activation-induced cytidine deaminase/apolipoprotein B mRNA-editing catalytic polypeptides
- ANRIL
antisense noncoding RNA in the INK4 locus
- BER
base excision repair
- CAD
coronary artery disease
- CVD
cardiovascular disease
- DNMT
DNA methyltransferase
- EC
endothelial cell
- eNOS
endothelial nitric oxide synthase
- GWAS
genome-wide association study
- GWAS
genome-wide association studies
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- HUVEC
human umbilical vien endothelial cells
- lncRNA
long noncoding RNA
- MI
myocardial infarction
- PRC1, 2
polycomb repressive complex 1, 2
- SNP
single-nucleotide polymorphism
- SNV
single-nucleotide variation
- TET
ten-eleven translocation
- TSA
trichostatin A
- VSMC
vascular smooth muscle cell
Footnotes
Broad area of epigenetics in cardiovascular disease with a focus on endothelial cells.
Disclosures
None.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Executive summary: heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013 Jan 1;127(1):143–52. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- 2.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. The New England journal of medicine. 2005 Apr 21;352(16):1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 3.Matouk CC, Turgeon PJ, Marsden PA. Epigenetics and stroke risk - beyond the static DNA code. Advances in Genomics and Genetics. 2012 Oct;2012(2):67–84. [Google Scholar]
- 4.Esper RJ, Nordaby RA, Vilarino JO, Paragano A, Cacharron JL, Machado RA. Endothelial dysfunction: a comprehensive appraisal. Cardiovascular diabetology. 2006;5:4. doi: 10.1186/1475-2840-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Won D, Zhu SN, Chen M, Teichert AM, Fish JE, Matouk CC, Bonert M, Ojha M, Marsden PA, Cybulsky MI. Relative reduction of endothelial nitric-oxide synthase expression and transcription in atherosclerosis-prone regions of the mouse aorta and in an in vitro model of disturbed flow. The American journal of pathology. 2007 Nov;171(5):1691–704. doi: 10.2353/ajpath.2007.060860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eshtehardi P, McDaniel MC, Suo J, Dhawan SS, Timmins LH, Binongo JN, Golub LJ, Corban MT, Finn AV, Oshinski JN, Quyyumi AA, Giddens DP, Samady H. Association of coronary wall shear stress with atherosclerotic plaque burden, composition, and distribution in patients with coronary artery disease. Journal of the American Heart Association. 2012 Aug;1(4):e002543. doi: 10.1161/JAHA.112.002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U. Genetic susceptibility to death from coronary heart disease in a study of twins. The New England journal of medicine. 1994 Apr 14;330(15):1041–6. doi: 10.1056/NEJM199404143301503. [DOI] [PubMed] [Google Scholar]
- 8.Peden JF, Farrall M. Thirty-five common variants for coronary artery disease: the fruits of much collaborative labour. Human molecular genetics. 2011 Oct 15;20(R2):R198–205. doi: 10.1093/hmg/ddr384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences of the United States of America. 2005 Jul 26;102(30):10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wienke A, Holm NV, Skytthe A, Yashin AI. The heritability of mortality due to heart diseases: a correlated frailty model applied to Danish twins. Twin research: the official journal of the International Society for Twin Studies. 2001 Aug;4(4):266–74. doi: 10.1375/1369052012399. [DOI] [PubMed] [Google Scholar]
- 11.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009 Oct 8;461(7265):747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holdt LM, Teupser D. Recent studies of the human chromosome 9p21 locus, which is associated with atherosclerosis in human populations. Arteriosclerosis, thrombosis, and vascular biology. 2012 Feb;32(2):196–206. doi: 10.1161/ATVBAHA.111.232678. [Research Support, Non-U.S. Gov’t Review] [DOI] [PubMed] [Google Scholar]
- 13.Congrains A, Kamide K, Ohishi M, Rakugi H. ANRIL: Molecular Mechanisms and Implications in Human Health. International journal of molecular sciences. 2013;14(1):1278–92. doi: 10.3390/ijms14011278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Mohlke KL, Ibrahim JG, Thomas NE, Sharpless NE. INK4/ARF transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis. PLoS One. 2009;4(4):e5027. doi: 10.1371/journal.pone.0005027. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whitelaw NC, Whitelaw E. Transgenerational epigenetic inheritance in health and disease. Current opinion in genetics & development. 2008 Jun;18(3):273–9. doi: 10.1016/j.gde.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 16.Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proceedings of the National Academy of Sciences of the United States of America. 2008 Nov 4;105(44):17046–9. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Painter RC, de Rooij SR, Bossuyt PM, Simmers TA, Osmond C, Barker DJ, Bleker OP, Roseboom TJ. Early onset of coronary artery disease after prenatal exposure to the Dutch famine. The American journal of clinical nutrition. 2006 Aug;84(2):322–7. doi: 10.1093/ajcn/84.1.322. quiz 466–7. [DOI] [PubMed] [Google Scholar]
- 18.Sumpio BE, Riley JT, Dardik A. Cells in focus: endothelial cell. The international journal of biochemistry & cell biology. 2002 Dec;34(12):1508–12. doi: 10.1016/s1357-2725(02)00075-4. [DOI] [PubMed] [Google Scholar]
- 19.Marsden PA, Brenner BM. Nitric oxide and endothelins: novel autocrine/paracrine regulators of the circulation. Seminars in nephrology. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. Review] 1991 Mar;11(2):169–85. [PubMed] [Google Scholar]
- 20.Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiological reviews. 2011 Jan;91(1):327–87. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. Journal of the American College of Cardiology. 2007 Jun 26;49(25):2379–93. doi: 10.1016/j.jacc.2007.02.059. [DOI] [PubMed] [Google Scholar]
- 22.Papaioannou TG, Stefanadis C. Vascular wall shear stress: basic principles and methods. Hellenic journal of cardiology: HJC = Hellenike kardiologike epitheorese. 2005 Jan-Feb;46(1):9–15. [PubMed] [Google Scholar]
- 23.Dai G, Kaazempur-Mofrad MR, Natarajan S, Zhang Y, Vaughn S, Blackman BR, Kamm RD, Garcia-Cardena G, Gimbrone MA., Jr Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proceedings of the National Academy of Sciences of the United States of America. 2004 Oct 12;101(41):14871–6. doi: 10.1073/pnas.0406073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J Cell Physiol. 2007 Nov;213(2):384–90. doi: 10.1002/jcp.21224. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review] [DOI] [PubMed] [Google Scholar]
- 25.Hotchkiss RD. The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J Biol Chem. 1948 Aug;175(1):315–32. [PubMed] [Google Scholar]
- 26.Scarano E, Iaccarino M, Grippo P, Parisi E. The heterogeneity of thymine methyl group origin in DNA pyrimidine isostichs of developing sea urchin embryos. Proceedings of the National Academy of Sciences of the United States of America. 1967 May;57(5):1394–400. doi: 10.1073/pnas.57.5.1394. [In Vitro] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010 Mar;11(3):204–20. doi: 10.1038/nrg2719. [Research Support, N.I.H., Extramural Review] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kagiwada S, Kurimoto K, Hirota T, Yamaji M, Saitou M. Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. Embo J. 2013 Feb 6;32(3):340–53. doi: 10.1038/emboj.2012.331. [Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oswald J, Engemann S, Lane N, Mayer W, Olek A, Fundele R, Dean W, Reik W, Walter J. Active demethylation of the paternal genome in the mouse zygote. Curr Biol. 2000 Apr 20;10(8):475–8. doi: 10.1016/s0960-9822(00)00448-6. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 30.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009 May 15;324(5929):930–5. doi: 10.1126/science.1170116. [Research Support, N.I.H., Extramural Research Support, N.I.H., Intramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011 Sep 2;333(6047):1300–3. doi: 10.1126/science.1210597. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011 Sep 16;146(6):866–72. doi: 10.1016/j.cell.2011.08.042. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yildirim O, Li R, Hung JH, Chen PB, Dong X, Ee LS, Weng Z, Rando OJ, Fazzio TG. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell. 2011 Dec 23;147(7):1498–510. doi: 10.1016/j.cell.2011.11.054. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeffery L, Nakielny S. Components of the DNA methylation system of chromatin control are RNA-binding proteins. J Biol Chem. 2004 Nov 19;279(47):49479–87. doi: 10.1074/jbc.M409070200. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 35.Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, Part I: Covalent histone modifications. Trends Mol Med. 2007 Sep;13(9):363–72. doi: 10.1016/j.molmed.2007.07.003. [Research Support, N.I.H., Extramural Review] [DOI] [PubMed] [Google Scholar]
- 36.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011 Sep 16;146(6):1016–28. doi: 10.1016/j.cell.2011.08.008. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kouzarides T. Chromatin modifications and their function. Cell. 2007 Feb 23;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [Review] [DOI] [PubMed] [Google Scholar]
- 38.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000 Jan 6;403(6765):41–5. doi: 10.1038/47412. [Research Support, U.S. Gov’t, P.H.S.] [DOI] [PubMed] [Google Scholar]
- 39.McKinsey TA. Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol Toxicol. 2012;52:303–19. doi: 10.1146/annurev-pharmtox-010611-134712. [Review] [DOI] [PubMed] [Google Scholar]
- 40.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007 Mar;39(3):311–8. doi: 10.1038/ng1966. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 41.Fish JE, Yan MS, Matouk CC, St Bernard R, Ho JJ, Gavryushova A, Srivastava D, Marsden PA. Hypoxic repression of endothelial nitric-oxide synthase transcription is coupled with eviction of promoter histones. J Biol Chem. 2010 Jan 8;285(2):810–26. doi: 10.1074/jbc.M109.067868. [Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilcox JN, Subramanian RR, Sundell CL, Tracey WR, Pollock JS, Harrison DG, Marsden PA. Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arterioscler Thromb Vasc Biol. 1997 Nov;17(11):2479–88. doi: 10.1161/01.atv.17.11.2479. [Comparative Study Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.] [DOI] [PubMed] [Google Scholar]
- 43.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proceedings of the National Academy of Sciences of the United States of America. 2000 Aug 1;97(16):9052–7. doi: 10.1073/pnas.97.16.9052. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stauss HM, Godecke A, Mrowka R, Schrader J, Persson PB. Enhanced blood pressure variability in eNOS knockout mice. Hypertension. 1999 Jun;33(6):1359–63. doi: 10.1161/01.hyp.33.6.1359. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 46.Fish JE, Matouk CC, Yeboah E, Bevan SC, Khan M, Patil K, Ohh M, Marsden PA. Hypoxia-inducible expression of a natural cis-antisense transcript inhibits endothelial nitric-oxide synthase. J Biol Chem. 2007 May 25;282(21):15652–66. doi: 10.1074/jbc.M608318200. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 47.Fish JE, Matouk CC, Rachlis A, Lin S, Tai SC, D’Abreo C, Marsden PA. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J Biol Chem. 2005 Jul 1;280(26):24824–38. doi: 10.1074/jbc.M502115200. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 48.Chan Y, Fish JE, D’Abreo C, Lin S, Robb GB, Teichert AM, Karantzoulis-Fegaras F, Keightley A, Steer BM, Marsden PA. The cell-specific expression of endothelial nitric-oxide synthase: a role for DNA methylation. J Biol Chem. 2004 Aug 13;279(33):35087–100. doi: 10.1074/jbc.M405063200. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 49.Marsden PA, Schappert KT, Chen HS, Flowers M, Sundell CL, Wilcox JN, Lamas S, Michel T. Molecular cloning and characterization of human endothelial nitric oxide synthase. FEBS Lett. 1992 Aug 3;307(3):287–93. doi: 10.1016/0014-5793(92)80697-f. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.] [DOI] [PubMed] [Google Scholar]
- 50.Marsden PA, Heng HH, Scherer SW, Stewart RJ, Hall AV, Shi XM, Tsui LC, Schappert KT. Structure and chromosomal localization of the human constitutive endothelial nitric oxide synthase gene. J Biol Chem. 1993 Aug 15;268(23):17478–88. [Research Support, Non-U.S. Gov’t] [PubMed] [Google Scholar]
- 51.Karantzoulis-Fegaras F, Antoniou H, Lai SL, Kulkarni G, D’Abreo C, Wong GK, Miller TL, Chan Y, Atkins J, Wang Y, Marsden PA. Characterization of the human endothelial nitric-oxide synthase promoter. J Biol Chem. 1999 Jan 29;274(5):3076–93. doi: 10.1074/jbc.274.5.3076. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 52.Teichert AM, Miller TL, Tai SC, Wang Y, Bei X, Robb GB, Phillips MJ, Marsden PA. In vivo expression profile of an endothelial nitric oxide synthase promoter-reporter transgene. Am J Physiol Heart Circ Physiol. 2000 Apr;278(4):H1352–61. doi: 10.1152/ajpheart.2000.278.4.H1352. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 53.Shirodkar AV, St Bernard R, Gavryushova A, Kop A, Knight BJ, Yan MS, Man HS, Sud M, Hebbel RP, Oettgen P, Aird WC, Marsden PA. A mechanistic role for DNA methylation in endothelial cell (EC)-enriched gene expression: relationship with DNA replication timing. Blood. 2013 Apr 25;121(17):3531–40. doi: 10.1182/blood-2013-01-479170. [Comparative Study Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robb GB, Carson AR, Tai SC, Fish JE, Singh S, Yamada T, Scherer SW, Nakabayashi K, Marsden PA. Post-transcriptional regulation of endothelial nitric-oxide synthase by an overlapping antisense mRNA transcript. J Biol Chem. 2004 Sep 3;279(36):37982–96. doi: 10.1074/jbc.M400271200. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 55.Ho JJ, Robb GB, Tai SC, Turgeon PJ, Mawji IA, Man HS, Marsden PA. Active stabilization of human endothelial nitric oxide synthase mRNA by hnRNP E1 protects against antisense RNA and microRNAs. Mol Cell Biol. 2013 May;33(10):2029–46. doi: 10.1128/MCB.01257-12. [Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weber M, Hagedorn CH, Harrison DG, Searles CD. Laminar shear stress and 3’ polyadenylation of eNOS mRNA. Circ Res. 2005 Jun 10;96(11):1161–8. doi: 10.1161/01.RES.0000170651.72198.fa. [Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, Non-P.H.S. Research Support, U.S. Gov’t, P.H.S.] [DOI] [PubMed] [Google Scholar]
- 57.Huddleson JP, Ahmad N, Srinivasan S, Lingrel JB. Induction of KLF2 by fluid shear stress requires a novel promoter element activated by a phosphatidylinositol 3-kinase-dependent chromatin-remodeling pathway. J Biol Chem. 2005 Jun 17;280(24):23371–9. doi: 10.1074/jbc.M413839200. [Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, P.H.S.] [DOI] [PubMed] [Google Scholar]
- 58.Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6(12):e1001233. doi: 10.1371/journal.pgen.1001233. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66. doi: 10.1146/annurev-biochem-051410-092902. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee JT, Bartolomei MS. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013 Mar 14;152(6):1308–23. doi: 10.1016/j.cell.2013.02.016. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review] [DOI] [PubMed] [Google Scholar]
- 61.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009 Mar 12;458(7235):223–7. doi: 10.1038/nature07672. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007 Jun 29;129(7):1311–23. doi: 10.1016/j.cell.2007.05.022. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, Wysocka J, Lei M, Dekker J, Helms JA, Chang HY. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011 Apr 7;472(7341):120–4. doi: 10.1038/nature09819. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pasmant E, Laurendeau I, Heron D, Vidaud M, Vidaud D, Bieche I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 2007 Apr 15;67(8):3963–9. doi: 10.1158/0008-5472.CAN-06-2004. [Research Support, Non-U.S. Gov’t] [DOI] [PubMed] [Google Scholar]
- 65.Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, Sigurdsson A, Baker A, Palsson A, Masson G, Gudbjartsson DF, Magnusson KP, Andersen K, Levey AI, Backman VM, Matthiasdottir S, Jonsdottir T, Palsson S, Einarsdottir H, Gunnarsdottir S, Gylfason A, Vaccarino V, Hooper WC, Reilly MP, Granger CB, Austin H, Rader DJ, Shah SH, Quyyumi AA, Gulcher JR, Thorgeirsson G, Thorsteinsdottir U, Kong A, Stefansson K. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007 Jun 8;316(5830):1491–3. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 66.McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, Boerwinkle E, Hobbs HH, Cohen JC. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007 Jun 8;316(5830):1488–91. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, Gil J, Walsh MJ, Zhou MM. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010 Jun 11;38(5):662–74. doi: 10.1016/j.molcel.2010.03.021. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences of the United States of America. 2009 Jun 9;106(23):9362–7. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Osler W. The principles and practice of medicine. New York: D. Appleton and Company; 1892. [Google Scholar]
- 70.Webster AL, Yan MS, Marsden PA. Epigenetics and cardiovascular disease. The Canadian journal of cardiology. 2013 Jan;29(1):46–57. doi: 10.1016/j.cjca.2012.10.023. [DOI] [PubMed] [Google Scholar]