

Abstract
Background
Outcomes of neonates with herpes simplex virus (HSV) encephalitis are worse after infection with HSV-2 when compared with HSV-1. The proteins herpes virus entry mediator (HVEM) and nectin-1 mediate HSV entry into susceptible cells. Prior studies have shown receptor-dependent differences in pathogenesis that depend on route of inoculation and host developmental age.
Methods
We investigated serotype-related differences in HSV disease and their relationship to entry receptor availability in a mouse model of encephalitis.
Results
Mortality was attenuated in seven-day-old wild-type (WT) mice inoculated with HSV-1(F) when compared with HSV-2(333). No serotype-specific differences were seen after inoculation of adult mice. HSV-1 pathogenesis was also attenuated relative to HSV-2 in newborn but not adult mice lacking HVEM or nectin-1. HSV-2 requires nectin-1 for encephalitis in adult but not newborn mice; in contrast, nectin-1 was important for HSV-1 pathogenesis in both age groups. Early viral replication was independent of age, viral serotype, or mouse genotype, suggesting host responses influence outcomes. In this regard, significantly greater amounts of inflammatory mediators were detected in brain homogenates from WT newborns 2 days after infection compared with adults and receptor-knockout newborns.
Conclusion
Dysregulation of inflammatory responses induced by infection may influence the severity of HSV encephalitis.
Introduction
Herpes simplex virus (HSV) is a common cause of infection, infecting more than 50% of US adults (1). The spectrum of disease from HSV is wide, ranging from asymptomatic infection to lethal dissemination and encephalitis (2). Newborns under one month of age are the human population at highest risk of severe HSV disease (3), and a variety of host and viral factors may contribute to this risk (4, 5). Multiple layers of immunity are involved in the host response to HSV infection, and differences in responses of newborns compared with older children and adults likely contribute to increased susceptibility (5). Additionally, host signals important in immunity are targeted by the virus for modulation (6), and it is not clear how HSV may manipulate these responses differently in the newborn. Finally, although the two serotypes of HSV are genetically closely related and share many clinical features (4), their ability to modulate host responses can differ substantially; for example, the virion host shutoff (vhs) function of HSV-2 is 40-fold more active than that of HSV-1 (7). Most of our understanding of the interaction between HSV and an infected host is based on studies using HSV-1, and there are few recent studies which directly compare host responses and pathogenesis of disease after HSV-1 infection relative to HSV-2 (8, 9). Importantly, clinical studies suggest that HSV-2 may confer worse neurologic outcomes than HSV-1 in human newborns with central nervous system (CNS) disease (10–12).
Mouse models of HSV infection recapitulate important aspects of human HSV disease and have contributed greatly to our understanding of HSV pathogenesis in both adults (13) and newborns (14, 15). The mouse entry receptors for HSV are orthologous to the human entry receptors (16), allowing mice mutated for specific HSV entry receptors to be studied as models for HSV entry requirements in disease. Such studies have identified a requirement for the HSV glycoprotein D entry receptor nectin-1 in the development of lethal encephalitis with HSV-2 in adult mice (17). However, in newborn mice nectin-1 was not required for lethal encephalitis due to HSV-2 (14).
We undertook the current experiments to test the hypothesis that viral serotype alters CNS pathogenesis in newborn mice, and to test the hypothesis that entry receptor requirements for HSV-1 would be similar to those of HSV-2. We identified differences between HSV-1 and HSV-2 in disease of newborn but not adult mice. Additionally, HSV-1 had a strong dependence on nectin-1 to mediate CNS disease in newborns, unlike prior observations with HSV-2. Significant differences in the production of several inflammatory mediators were measured in the brains of wild-type (WT) newborn mice when compared with either adult mice or single receptor-knockout mice, supporting the concept that a dysregulated inflammatory response contributes to HSV pathogenesis in the newborn CNS.
Results
Mortality in newborn mice is attenuated after IC inoculation with HSV-1 compared with HSV-2
Encephalitis from HSV leads to more severe clinical outcomes in newborn humans infected with HSV-2 as compared with HSV-1 (10–12). HSV-2 as a cause of encephalitis in older children and adult humans is much less common than HSV-1 (18–20), though neurologic outcomes among older survivors of HSV encephalitis are also often poor (19, 20). To compare the relative pathogenicity of HSV-1 and HSV-2 in the CNS of mice in different age groups, we inoculated newborn and adult mice IC with identical amounts of either HSV-1(F) or HSV-2(333) and followed for clinical disease and mortality. Seven-day-old WT mice had a significant delay in time to mortality, and lower overall mortality, after HSV-1 infection as compared with HSV-2 (Figure 1A). In contrast, IC infection with either HSV serotype led to comparable mortality in adult (10–12 week old) WT mice (Figure 1B), using a level of virus leading to about 50% mortality in the WT adult cohort.
Figure 1.

Survival of mice of different ages and genotypes infected with either HSV-1 (solid lines) or HSV-2 (dashed lines) by the intracranial route. A. Seven-day-old C57BL/6 were inoculated IC with 1000 PFU HSV-1(F) (n=24 from five separate litters) or HSV-2(333) (n=9 from three separate litters) and followed for signs indicating imminent mortality. Survival analysis by the Gehan-Breslow test indicated statistically significantly different time to mortality for the two viruses (p=0.003). Median survival times were 4 days after HSV-1 inoculation compared with 2 days for HSV-2. B. Adult C57BL/6 mice between 10 and 13 weeks old were inoculated IC with 10000 PFU of either HSV-1(F) (n=10) or HSV-2(333) (n=9). Time to mortality did not differ for the different viruses (p=0.65), with median survival times of 8.5 days for HSV-1 and >10 days for HSV-2. C, D. Same as A, B, except for HVEM KO mice inoculated with the same viruses. Significant differences were calculated for time to mortality in newborns (p<0.0001) but not adults (p=0.85). Median survival was 5 days for newborns infected with HSV-1 (n=19 from four separate litters), 3 days for newborns infected with HSV-2 (n=7 from two separate litters), 10 days for adults infected with HSV-1 (n=8), and 8.5 days for adults infected with HSV-2 (n=10). E, F. Same as A, B, except for nectin-1 KO mice inoculated with the same viruses (n=5–6 per group from two to four separate litters for newborns and 9–10 for adults). Significant differences were calculated for time to mortality in newborns (p=0.038) but not adults (p=0.34). Median survival was >10 days for newborns infected with HSV-1 (n=16), 8 days for newborns infected with HSV-2 (n=5), and >10 days for adults infected with either HSV-1 (n=10) or HSV-2 (n=9).
Previous experiments in adult mice inoculated IC with HSV-2 demonstrated a dependence on nectin-1 for development of disease (17), while our prior studies in newborns showed that nectin-1 was not required for HSV-2 to cause disease after IC inoculation (14). To evaluate whether there was a similar dependence on entry receptor expression for encephalitis after IC inoculation with HSV-1, we infected HVEM knockout (KO) and nectin-1 KO mice IC with identical amounts of either HSV-1 or HSV-2. Similar to results in WT mice, in both of the single entry receptor KO genotypes of newborn mice, there was longer time to death and less overall mortality with HSV-1 infection as compared with HSV-2 (Figures 1C, 1E). In contrast, we did not observe serotype-dependent differences in time to death or overall mortality in either genotype of single entry receptor KO adult mice (Figures 1D, 1F). As we previously observed for HSV-2, expression of nectin-1 was not required for HSV-1 to cause mortality in newborns. However, there was a strong dependence of both HSV-1 and HSV-2 on this receptor for newborn disease, as mortality of nectin-1 KO newborns was significantly delayed relative to WT newborns (Figures 1A, 1E; p=0.0005 for WT vs. nectin-1 KO after HSV-1 infection, and p=0.006 for WT vs. nectin-1 KO after HSV-2 infection). In adults, nectin-1 appeared to be required for disease from both serotypes, at least for the inoculum tested (Figure 1F). This is consistent with prior data from experiments using IC inoculation with HSV-2 in adult mice lacking nectin-1 (17).
Viral replication in brains of neonatal mice infected IC is similar for HSV-1 and HSV-2
Differences in mortality between groups of newborn mice inoculated IC with different serotypes of HSV could be explained by relative differences in the ability of the viruses to replicate in brain tissue. To assess HSV replication over time in brains of newborn mice, we inoculated separate groups of seven-day-old mice with either HSV-1(F) or HSV-2(333), euthanized them at different times after inoculation, and performed plaque assays on homogenates of brain tissue. A general trend of higher HSV-1 replication relative to HSV-2 was observed in all genotypes; however, we found no statistical differences in viral titer in WT or HVEM KO newborns when comparing replication of HSV-1 with HSV-2 over the first two days after inoculation (Figure 2A-B). In nectin-1 KO newborns, there was a statistical difference in titer on both days (p<0.05), with HSV-1 replication higher than HSV-2. Replication of HSV-1 and HSV-2 was also statistically similar when comparing HVEM KO and nectin-1 KO newborns with WT newborns the first two days after infection. In comparison, viral replication in brains of adult WT mice (inoculated with a higher amount of virus) led to generally lower mean titers relative to WT newborns, with statistically significant differences detected for HSV-1 on day 1 and HSV-2 on day 2.
Figure 2.
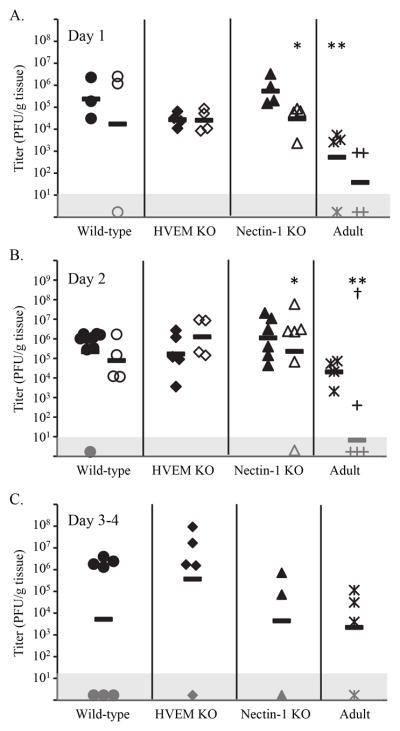
Viral titers in brain tissue at different times after IC inoculation with either HSV-1(F) (● wild-type newborns, ◆ HVEM KO newborns, ▲ nectin-1 KO newborns, * wild-type adults) or HSV-2(333) (○ wild-type newborns, ◇ HVEM KO newborns, △ nectin-1 KO newborns, + wild-type adults), in newborn mice of different genotypes or adult wild-type mice. Each symbol represents PFU per g tissue from an individual mouse, with horizontal bars indicating geometric means for each group. Symbols plotted in the gray box represent values below the detection limit of the assay. Virus was not detected in any samples from control mice inoculated in an identical manner with vehicle (PBS-GCS). Differences in titer between newborn samples for the same serotype of virus did not statistically differ based on genotype. A. Mice were sacrificed one day after inoculation (for each group of newborns, n=3–4 from a single litter). *p=0.04 vs. nectin-1 KO newborn infected with HSV-1; **p=0.05 vs. wild-type newborn infected with HSV-1. B. Mice were sacrificed two days after inoculation (for each group of newborns, n=4–9 from two separate litters). *p=0.03 vs. nectin-1 KO newborn infected with HSV-1; **p=0.002 vs. wild-type newborn infected with HSV-2, †p=0.004 vs. wild-type adult infected with HSV-1. C. Mice were sacrificed three to four days after inoculation (for each group of newborns, n=3–7 from 1–2 litters). No statistical differences were identified for replication within or between any groups.
We additionally measured viral titers from mice infected with HSV-1 which survived for three or four days following inoculation (Figure 2C). A similar analysis was unable to be conducted for HSV-2, due to mortality generally occurring in WT newborns prior to that time. As at earlier time points, there were no statistical differences in the titer of virus recovered from newborn brains for the different genotypes of mice. There was also no statistical difference in viral titer between WT newborn mice and WT adult mice.
Together, these data indicate that pathogenesis of HSV encephalitis in newborn mice does not correlate with the ability of the virus to replicate in the brain early after inoculation. Additionally, HSV-1 replication was evident in brains of nectin-1 KO newborns, despite the prior observation that limited mortality was observed with this virus in this genotype.
Production of inflammatory mediators in brains of IC inoculated mice varies with mouse age, genotype, and viral serotype
The lack of correlation between mortality and viral replication after IC inoculation of newborn mice suggested that differences in host responses in the brain might contribute to differences in pathogenesis. Prior in vitro studies reported that cells obtained from newborns generated diminished amounts of type I interferons (IFNs) in response to HSV when compared with cells from adults (21), and additional evidence in the literature suggests that the generation of type I IFNs is more effectively suppressed by HSV-2 than HSV-l strains (22, 23). We applied a multiplex ELISA assay to measure the production of different inflammatory mediators in brain tissue of infected mice two days after inoculation. We noted significantly higher levels of both IL-1β (Figure 3A) and IFN-γ (Figure 3B) in brain tissue from WT newborns infected with HSV-1 when compared with tissue from WT newborns infected with HSV-2. Serotype-related differences in these and other inflammatory mediators were not observed when comparing similarly inoculated receptor KO newborns or adult mice, likely attributable to the overall low levels detected in these samples.
Figure 3.

Chemokine levels in brain homogenates two days after IC inoculation with either HSV-1(F) (gray bars) or HSV-2(333) (black bars), in newborn mice of different genotypes or adult wild-type mice. Bars represent mean pg/g brain tissue from agroup of mice, with standard deviations indicated. *p<0.05 when compared with HSV-1 infection of WT newborns. †p<0.05 when compared with HSV-2 infection of WT newborns. Each group of newborn mice included 2–3 separate litters and 4–7 mice. A. IL-1β. B. IFN-γ . C. IL-6. D. KC/Gro.
We also noted significantly higher levels of certain cytokines and chemokines in brain tissue from WT newborn mice two days after inoculation when compared with adults or receptor KO newborns (Figure 3). In different comparisons, IFN-γ, IL-1β, IL-6, and KC/Gro were higher in brain tissue from WT newborns than other test groups two days after inoculation with HSV-1 or HSV-2. Similar perturbations in cytokine responses have been associated with CNS pathogenesis in STAT1-deficient mice at different times after infection (24), suggesting that in some cases improper regulation of the inflammatory response may contribute to disease in CNS infection with HSV.
Discussion
The major finding in this study is that there are age-dependent differences in the ability of HSV-1 and HSV-2 to cause mortality after neurologic infection of mice, with attenuation in mortality due to HSV-1 in newborn but not adult mice. Differences in pathogenesis of HSV-1 and HSV-2 in experimental models have been reported going back as far as the 1920’s (25), when Lipshutz noted less frequent keratitis and a lower incidence of progression to encephalitis after corneal infection of rabbits with HSV-2 compared with HSV-1. That other investigators of that era did not note such differences has been attributed to differences in the passage history of the different virus strains tested. In our study, viruses tested had limited passage in vitro (including our F strain, which was derived from the original isolate). In general, variable differences in mortality based on HSV serotype have been reported in different animal models of HSV infection (25). No studies have demonstrated statistically significant differences in mortality from CNS disease in human newborns based on HSV serotype, though such assessments are limited by patient numbers (12). However, as noted above, HSV serotype has been related to neurologic outcome in survivors of newborn HSV encephalitis (10–12).
Our observation of serotype-dependent differences in pathogenesis in newborn but not adult mice held for both wild-type and single receptor KO mice, with relatively attenuated mortality from HSV-1 as compared with HSV-2. Although we did not include double KO mice (lacking both HVEM and nectin-1) in the current study, all experimental models to date have shown complete resistance of double KO mice to HSV disease and replication (14, 17, 26, 27), suggesting that mice do not have alternative receptors capable of mediating HSV infection. In the ocular model, serotype-dependent differences in pathogenesis have been related to entry receptor expression, with HVEM important for efficient corneal replication and clinical disease after HSV-1 but not HSV-2 infection (9). In the current study, we have identified a greater dependence on nectin-1 for HSV-1 to mediate disease in the CNS of newborn mice. This increase in disease was independent of viral replication, which was similar for HSV-1 and HSV-2 in brains of newborn WT and HVEM KO mice, and slightly higher for HSV-1 in nectin-1 KO newborn mice (Figure 2). Together, the suggestion from these observations is that HVEM-expressing cells in the CNS of newborns support infection and replication equally or better for HSV-1 than for HSV-2, but serotype-related differences in the responses of these cells to infection may alter the outcome of encephalitis in the newborn brain. Studies are ongoing to identify the CNS cells which express HVEM, how they may change during CNS development, and the effects of HSV infection on these cells, which may provide mechanistic understanding of differences in newborn and adult pathogenesis.
As implied in the previous paragraph, our observation that there were few differences in viral replication in the CNS based on either viral serotype or entry receptor expression in the host, despite the observed differences in mortality, suggests that host responses contribute to mortality. Indeed, we found that HSV infection stimulated production of several inflammatory mediators in the CNS at varying amounts that depended on viral serotype, developmental age of the host, and expression of HSV entry receptors in the brain. We focused on the second day after inoculation, representing a time point prior to HSV-2-induced mortality but during symptomatic disease in newborns. In WT newborns, HSV-1 infection led to higher amounts of IL-1β and IFN-γ relative to HSV-2. In comparison, Zheng et al. inoculated adult mice with either HSV-1 or HSV-2 by the ocular or vaginal routes, and generally found higher replication of HSV-2 at both peripheral sites and in the nervous system (8). Higher HSV-2 replication was associated with generally increased levels of inflammatory chemokines and increased effector cell infiltration into the spinal cord. These results highlighted the divergent nature of the host response after infection with these related viruses, though it was noted that the passage number of the viruses studied was not controlled and may have influenced these observations (8).
In addition to serotype-related differences in production of inflammatory mediators, we also found that infection in WT newborns led to significantly higher levels of certain chemokines and cytokines in the CNS than in either the adult CNS or in single receptor knockout newborns. The increased susceptibility to HSV infection of newborn relative to adult mice has long been noted (28), and age-dependent differences in IFN response after HSV infection have been reported for nearly as long (25). Although it may be counterintuitive that higher levels of these inflammatory mediators were measured in WT newborn mice compared with adults, it is notable that STAT1 deficiency, which confers a defect in type I IFN signaling, has also been associated with dysregulated production of inflammatory mediators after CNS infection of mice with HSV, including significantly enhanced production of IL-6 and IL-1β (24). Our observations suggest further investigation of specific inflammatory pathways altered by HSV in the newborn CNS compared with adults are warranted.
Reasons for dampened inflammation in the single receptor KO newborns relative to WT newborns remain an area of investigation. HVEM mediates inflammatory signals in immune cells (29), and engagement of HVEM by HSV may affect early inflammatory signals in mucosal epithelia (30), generally resulting in suppression of inflammatory mediators. Whether HVEM expression in the CNS generally alters inflammation in this compartment has not been studied. Nectin-1 is a cell surface protein that interacts with other members of the nectin family to mediate structural interactions between cells, including neurons (31). Although several lines of evidence suggest that cell-to-cell spread of HSV relies on nectin-1 (32, 33), it is not clear that such a limitation exists in the newborn brain, especially given the equivalent replication we observed. There are also no reports of nectin-1 involvement in immune signaling.
Limitations in this study include the difficulty in extrapolating from animal models to human disease, the application of a non-physiologic route of inoculation, and the need to use different inocula in the different age groups. Importantly, mice infected with HSV demonstrate many phenotypic similarities to humans, including comparable skin and genital lesions, neuroinvasion, and establishment of viral latency. As in humans, peripheral inoculation of newborn mice with HSV leads to visceral dissemination and lethal encephalitis (14, 15). We used direct intracranial inoculation to avoid confounding our studies of CNS-related mortality and host response with the effects of visceral infection, which is observed after peripheral inoculation in newborn mice (14, 15). Dissemination in the newborn after IC inoculation is not likely to confound our observations, as plaque assays of visceral tissues from selected IC-inoculated animals failed to detect virus at any time after inoculation. Lastly, we chose inocula in the adult cohort expected to result in approximately 50% mortality in WT mice, resulting in about 10-fold more virus being used in adult mice than newborns. Although this limits to some extent our ability to directly compare replication and host response in the two age groups, the comparable levels of replication and dramatic differences in production of inflammatory mediators support the concept that there are important differences in the host control of CNS infection that relate to developmental age of the animal.
In summary, we have found age-dependent differences in pathogenesis of HSV encephalitis that are related to both viral serotype and entry receptor expression. We have also found dysregulated production of inflammatory mediators in the CNS of newborn mice, consistent with prior observations in immune-deficient adult mice (24). Although the extent to which these observations in mice translate to human disease is not known, an improved understanding of age-dependent differences in the host response to HSV in the CNS could ultimately lead to interventions that improve outcomes in this susceptible population.
Methods
Cells and viruses
Vero cells were cultured in Dulbecco's modification of Eagle's (DME) medium plus 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin, and were used for plaque assays and propagation of virus. Plaque titrations were performed by standard methods.
HSV-1 strain F was originally obtained from a facial lesion and isolated in Hep-2 cells (34). HSV-2 strain 333 was originally isolated from a genital lesion and underwent limited passage in human cells (35). Viruses were plaque-purified and passaged no more than three times in Vero cells.
Mouse HSV infection and postinfection procedures
Animal care and use in this study were in accordance with institutional and NIH guidelines, and all studies were approved by the Northwestern University Animal Care and Use Committee. The mouse strains used have been previously described (27), including C57BL/6 (WT), HVEM KO (Tnfrsf14−/−), and nectin-1 KO (Pvrl−/−) mice. Knockout strains were on the C57BL/6 genetic background. Mice were maintained in specific-pathogen-free conditions until transfer to a containment facility just prior to infection.
Breeding pairs were regularly monitored, with males separated from gravid females prior to delivery. Pups were inoculated at seven days of age, which from an immunologic perspective corresponds most closely to humans at birth (36). Virus was diluted in PBS containing 1% heat-inactivated calf serum and 0.1% glucose (PBS-GCS) to deliver a target intracranial (IC) inoculum of 1×103 PFU/pup. Infections of 10–12 week old (adult) mice were included for comparison with newborn infections, with target inocula of 1×104 PFU/mouse. To minimize numbers of animals used and because we had comparative data from prior studies using HSV-2 (14), more newborn mice per group were included for experiments using HSV-1 than HSV-2.
For IC inoculation of either adult or newborn mice, a positive displacement syringe with a 26-gauge needle and a needle guard was used to inoculate 5 3L total volume into one hemisphere of the brain in the approximate region of the hippocampus, equidistant between the bregma and lambdoid sutures, and lateral to the sagittal suture through the parietal bone. Experiments also included control mice injected IC in an identical manner using the same volume PBS-GCS.
Infected mice were monitored daily for signs of neurologic disease, including lethargy, motionlessness, and hunched posture. Mice displaying severe signs of illness were immediately euthanized; other groups were euthanized at indicated time points for measurements of viral spread. Brains were harvested from infected and control mice at indicated times after infection. Tissues were weighed, homogenized in DMEM with 5% inactivated calf serum and 1% ciprofloxacin, and sonicated. Tissue homogenates were stored at −70°C until analysis.
Multiplex enzyme-linked immunosorbent assay (ELISA)
Tissue levels of selected cytokines and chemokines were measured by multiplex ELISA according to the manufacturer's instructions (V-Plex MSD Multi-spot system, Meso Scale Discovery, Rockville, MD). Samples were loaded in 96-well plates pre-coated with target analyte antibodies for two hours at room temperature. Plates were washed, detection antibodies containing electrochemiluminescent tags added, plates were again washed, and a voltage applied to the plates resulted in emission of light by the captured labels. The intensity of light emitted allowed a quantitative measure of analyte concentrations in the samples using a standard curve. Initial assays of a subset of samples measured IL-6, KC/Gro, IL-1β, IFN-γ, IL-10, IL-12, and TNF-α, with only IL-6, KC/Gro, IL-1β, and IFN-γ measured in subsequent assays. Levels of IL-10, IL-12, and TNF-α were below the detection limit of the assay in all of the initial samples tested.
Statistical Tests
Kaplan-Meier survival analysis was performed using the Gehan-Breslow test. Log-transformed values for viral infectious units in brain tissue were compared using the unpaired Student's t test. Mean values for chemokine and cytokine levels in tissue homogenates were also compared by unpaired Student's t test.
Acknowledgments
The authors wish to thank Nanette Susmarski for providing cell culture expertise and assistance in viral plaque assays. We also appreciate helpful comments and guidance from Patricia Spear, Richard Longnecker, Gregory Smith, Anne Rowley, and members of the Longnecker and Smith Labs.
Statement of financial support: This work was supported by National Institutes of Health (USA) grant K08AI089942 to WJM.
Footnotes
Potential/perceived conflicts of interest: None
References
- 1.Bradley H, Markowitz LE, Gibson T, McQuillan GM. Seroprevalence of herpes simplex virus types 1 and 2--United States, 1999–2010. J Infect Dis. 2014;209:325–333. doi: 10.1093/infdis/jit458. [DOI] [PubMed] [Google Scholar]
- 2.Roizman B, Knipe DM, Whitley RJ. Herpes Simplex Viruses. In: Knipe DM, Howley PM, editors. Fields' Virology. Lippincott Williams & Wilkins; Philadelphia, PA: 2007. pp. 2502–2576. [Google Scholar]
- 3.Kimberlin DW. Neonatal herpes simplex infection. Clin Microbiol Rev. 2004;17:1–13. doi: 10.1128/CMR.17.1.1-13.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schiffer JT, Corey L. Herpes Simplex Virus. In: Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases. Churchill Livingstone; Philadelphia, Pennsylvania: 2009. pp. 1943–1962. [Google Scholar]
- 5.Gantt S, Muller WJ. The immunologic basis for severe neonatal herpes disease and potential strategies for therapeutic intervention. Clin Dev Immunol. 2013;2013:369172. doi: 10.1155/2013/369172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melchjorsen J, Matikainen S, Paludan SR. Activation and evasion of innate antiviral immunity by herpes simplex virus. Viruses. 2009;1:737–759. doi: 10.3390/v1030737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Everly DN, Jr, Read GS. Mutational analysis of the virion host shutoff gene (UL41) of herpes simplex virus (HSV): characterization of HSV type 1 (HSV-1)/HSV-2 chimeras. J Virol. 1997;71:7157–7166. doi: 10.1128/jvi.71.10.7157-7166.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng M, Conrady CD, Ward JM, Bryant-Hudson KM, Carr DJ. Comparison of the host immune response to herpes simplex virus 1 (HSV-1) and HSV-2 at two different mucosal sites. J Virol. 2012;86:7454–7458. doi: 10.1128/JVI.00702-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karaba AH, Kopp SJ, Longnecker R. Herpesvirus entry mediator is a serotype specific determinant of pathogenesis in ocular herpes. Proc Natl Acad Sci U S A. 2012;109:20649–20654. doi: 10.1073/pnas.1216967109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corey L, Whitley RJ, Stone EF, Mohan K. Difference between herpes simplex virus type 1 and type 2 neonatal encephalitis in neurological outcome. Lancet. 1988;1:1–4. doi: 10.1016/s0140-6736(88)90997-x. [DOI] [PubMed] [Google Scholar]
- 11.Whitley R, Arvin A, Prober C, et al. Predictors of morbidity and mortality in neonates with herpes simplex virus infections. The National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. N Engl J Med. 1991;324:450–454. doi: 10.1056/NEJM199102143240704. [DOI] [PubMed] [Google Scholar]
- 12.Kimberlin DW, Lin CY, Jacobs RF, et al. Natural history of neonatal herpes simplex virus infections in the acyclovir era. Pediatrics. 2001;108:223–229. doi: 10.1542/peds.108.2.223. [DOI] [PubMed] [Google Scholar]
- 13.Mester JC, Rouse BT. The mouse model and understanding immunity to herpes simplex virus. Rev Infect Dis. 1991;13 (Suppl 11):S935–945. doi: 10.1093/clind/13.supplement_11.s935. [DOI] [PubMed] [Google Scholar]
- 14.Kopp SJ, Karaba AH, Cohen LK, Banisadr G, Miller RJ, Muller WJ. Pathogenesis of neonatal herpes simplex 2 disease in a mouse model is dependent on entry receptor expression and route of inoculation. J Virol. 2013;87:474–481. doi: 10.1128/JVI.01849-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kern ER, Overall JC, Jr, Glasgow LA. Herpesvirus hominis infection in newborn mice. I. An experimental model and therapy with iododeoxyuridine. J Infect Dis. 1973;128:290– 299. doi: 10.1093/infdis/128.3.290. [DOI] [PubMed] [Google Scholar]
- 16.Spear PG, Eisenberg RJ, Cohen GH. Three classes of cell surface receptors for alphaherpesvirus entry. Virology. 2000;275:1–8. doi: 10.1006/viro.2000.0529. [DOI] [PubMed] [Google Scholar]
- 17.Kopp SJ, Banisadr G, Glajch K, et al. Infection of neurons and encephalitis after intracranial inoculation of herpes simplex virus requires the entry receptor nectin-1. Proc Natl Acad Sci U S A. 2009;106:17916–17920. doi: 10.1073/pnas.0908892106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nahmias AJ, Whitley RJ, Visintine AN, Takei Y, Alford CA., Jr Herpes simplex virus encephalitis: laboratory evaluations and their diagnostic significance. J Infect Dis. 1982;145:829–836. doi: 10.1093/infdis/145.6.829. [DOI] [PubMed] [Google Scholar]
- 19.Whitley RJ. Herpes simplex encephalitis: adolescents and adults. Antiviral Res. 2006;71:141–148. doi: 10.1016/j.antiviral.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 20.Mateen FJ, Miller SA, Aksamit AJ., Jr Herpes simplex virus 2 encephalitis in adults. Mayo Clin Proc. 2014;89:274–275. doi: 10.1016/j.mayocp.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 21.Sullender WM, Miller JL, Yasukawa LL, et al. Humoral and cell-mediated immunity in neonates with herpes simplex virus infection. J Infect Dis. 1987;155:28–37. doi: 10.1093/infdis/155.1.28. [DOI] [PubMed] [Google Scholar]
- 22.Murphy JA, Duerst RJ, Smith TJ, Morrison LA. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J Virol. 2003;77:9337–9345. doi: 10.1128/JVI.77.17.9337-9345.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J Exp Med. 1999;189:663–672. doi: 10.1084/jem.189.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pasieka TJ, Cilloniz C, Carter VS, Rosato P, Katze MG, Leib DA. Functional genomics reveals an essential and specific role for Stat1 in protection of the central nervous system following herpes simplex virus corneal infection. J Virol. 2011;85:12972–12981. doi: 10.1128/JVI.06032-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nahmias AJ, Dowdle WR. Antigenic and biologic differences in herpesvirus hominis. Prog Med Virol. 1968;10:110–159. [PubMed] [Google Scholar]
- 26.Karaba AH, Kopp SJ, Longnecker R. Herpesvirus entry mediator and nectin-1 mediate herpes simplex virus 1 infection of the murine cornea. J Virol. 2011;85:10041–10047. doi: 10.1128/JVI.05445-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor JM, Lin E, Susmarski N, et al. Alternative entry receptors for herpes simplex virus and their roles in disease. Cell Host Microbe. 2007;2:19–28. doi: 10.1016/j.chom.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson RT. The Pathogenesis of Herpes Virus Encephalitis. Ii. A Cellular Basis for the Development of Resistance with Age. J Exp Med. 1964;120:359–374. doi: 10.1084/jem.120.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sedy JR, Spear PG, Ware CF. Cross-regulation between herpesviruses and the TNF superfamily members. Nat Rev Immunol. 2008;8:861–873. doi: 10.1038/nri2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoon M, Kopp SJ, Taylor JM, Storti CS, Spear PG, Muller WJ. Functional Interaction between Herpes Simplex Virus Type 2 gD and HVEM Transiently Dampens Local Chemokine Production after Murine Mucosal Infection. PLoS One. 2011;6:e16122. doi: 10.1371/journal.pone.0016122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takai Y, Ikeda W, Ogita H, Rikitake Y. The immunoglobulin-like cell adhesion molecule nectin and its associated protein afadin. Annu Rev Cell Dev Biol. 2008;24:309–342. doi: 10.1146/annurev.cellbio.24.110707.175339. [DOI] [PubMed] [Google Scholar]
- 32.Even DL, Henley AM, Geraghty RJ. The requirements for herpes simplex virus type 1 cell-cell spread via nectin-1 parallel those for virus entry. Virus Res. 2006;119:195–207. doi: 10.1016/j.virusres.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 33.Sakisaka T, Taniguchi T, Nakanishi H, et al. Requirement of interaction of nectin-1alpha/HveC with afadin for efficient cell-cell spread of herpes simplex virus type 1. J Virol. 2001;75:4734–4743. doi: 10.1128/JVI.75.10.4734-4743.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ejercito PM, Kieff ED, Roizman B. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J Gen Virol. 1968;2:357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 35.Westmoreland D, Rapp F. Host range temperature-sensitive mutants of herpes simplex virus type 2. J Virol. 1976;18:92–102. doi: 10.1128/jvi.18.1.92-102.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nat Rev Immunol. 2004;4:553–564. doi: 10.1038/nri1394. [DOI] [PubMed] [Google Scholar]