

Abstract
Background
Loss of function COQ2 mutations results in primary CoQ10 deficiency. Recently, recessive mutations of the COQ2 gene have been identified in two unrelated Japanese families with multiple system atrophy (MSA). It has also been proposed that specific heterozygous variants in the COQ2 gene may confer susceptibility to sporadic MSA. To assess the frequency of COQ2 variants in patients with MSA, we sequenced the entire coding region and investigated all exonic copy number variants of the COQ2 gene in 97 pathologically-confirmed and 58 clinically-diagnosed MSA patients from the United States.
Results
We did not find any homozygous or compound heterozygous pathogenic COQ2 mutations including deletion or multiplication within our series of MSA patients. In two patients, we identified two heterozygous COQ2 variants (p.S54W and c.403 + 10G > T) of unknown significance, which were not observed in 360 control subjects. We also identified one heterozygous carrier of a known loss of function p.S146N substitution in a severe MSA-C pathologically-confirmed patient.
Conclusions
The COQ2 p.S146N substitution has been previously reported as a pathogenic mutation in primary CoQ10 deficiency (including infantile multisystem disorder) in a recessive manner. This variant is the third primary CoQ10 deficiency mutation observed in an MSA case (p.R387X and p.R197H). Therefore it is possible that in the heterozygous state it may increase susceptibility to MSA. Further studies, including reassessing family history in patients of primary CoQ10 deficiency for the possible occurrence of MSA, are now warranted to resolve the role of COQ2 variation in MSA.
Electronic supplementary material
The online version of this article (doi:10.1186/1750-1326-9-44) contains supplementary material, which is available to authorized users.
Keywords: COQ2, Multiple system atrophy, Genetics, CoQ10 deficiency
Background
Multiple system atrophy (MSA) is a rare sporadic neurodegenerative disorder, with an estimated prevalence ranging from approximately 2–5 cases per 100,000 people [1]. This devastating and rapidly progressive disease is characterized clinically by combinations of poor levodopa responsive parkinsonism, autonomic failure, cerebellar ataxia, and pyramidal symptoms [1, 2]. Widespread presence of glial cytoplasmic inclusions is the neuropathologic hallmark of MSA [3]. To date, there are no effective therapies for MSA and our understanding of the disease etiology remains limited.
Recently, a homozygous mutation p.M128V-V393A/p.M128V-V393A (referred to by Tsuji and colleagues as p.M78V-V343A/p.M78V-V343A) and compound heterozygous mutations p.R387X/p.V393A (referred to by Tsuji and colleagues p.R337X/p.V343A) in the coenzyme Q2 4-hydroxybenzoate polyprenyltransferase gene (COQ2; OMIM 609825) were identified in affected members of two unrelated Japanese families with MSA [4]. This is the first report of recessive COQ2 pathogenic mutations in adults, however recessive COQ2 mutations are known to cause primary coenzyme Q10 (CoQ10) deficiency-1 (COQ10D1; OMIM 607426) which includes phenotype of infantile multisystem disorder or nephrotic syndrome [5–12]; COQ2 is involved in the synthesis of CoQ10. Interestingly, it was also reported that specific heterozygous variation (p.V393A) in the COQ2 gene may affect susceptibility to MSA [4]. Given these findings, we screened the Mayo Clinic Florida series of 155 MSA patients to assess the frequency of COQ2 variants in disease. Given the proposed loss-of-function mechanism we also investigated exon dosage as a possible disease-related phenomenon.
Results
Result of sanger sequencing and copy number assay
In this study, our sequence analysis of COQ2 in 155 MSA patients identified eleven variants including one intronic, six synonymous, three non-synonymous and one nonsense variants (Table 1), however exon dosage assays did not detect any deletion/multiplication. Four variants are common, p.R22X, p.L66V, p.D298D and p.S330S, and present in the Exome Variant Server (EVS) and 1000 Genomes; there was no significant minor allele frequency (MAF) difference with our patients, EVS data, or 1000 Genomes data (Table 1; Additional file 1). We found one homozygous and five heterozygous carriers of p.R22X in 155 MSA patients, but there was no significant difference between patients and control subjects or patients and EVS data or 1000 Genomes data (Table 1; Additional file 1). This variant is incorporated into one of the longer COQ2 transcripts (Figure 1); given the frequency of this variant and the presence of homozygotes both in our study and the EVS, it is unlikely this variant is of clinical relevance in primary CoQ10 deficiency or MSA. Indeed, Schottlaender and Houlden recently reported that p.R22X was more common in controls than cases [13].
Table 1.
Variants of COQ2 observed in this study
| Exon | Exon 1 | Exon 1 | Exon 1 | Exon 1 | Intron 2 | Exon 2 | Exon 2 | Exon 5 | Exon 5 | Exon 6 | Exon 7 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs number | rs183012002 | rs112033303 | rs376396608 | rs6818847 | rs121918233 | rs199581249 | rs6535454 | rs1129617 | rs141431344 | |||||||
| Genotypea | c.30G > A | c.64A > T | c.161C > G | c.196G > T | c.403 + 10G > T | c.426A > G | c.437G > A | c.801G > A | c.894 T > C | c.990C > T | c.1107C > T | |||||
| Amino acid changeb | p.R10R | p.R22X | p.S54W | p.L66V | Exon1 + 10 | p.P142P | p.S146N | p.A267A | p.D298D | p.S330S | p.Y369Y | |||||
| Cases | Cases | Cont. | Cases | Cont. | Cases | Cont. | Cases | Cont. | Cases | Cases | Cont. | Cases | Cases | Cases | Cases | |
| Major allele | 154 | 149 | 339 | 154 | 360 | 87 | 175 | 154 | 360 | 154 | 154 | 360 | 154 | 85 | 89 | 154 |
| Heterozygous allele | 1 | 5 | 20 | 1 | 0 | 54 | 146 | 1 | 0 | 1 | 1 | 0 | 1 | 64 | 59 | 1 |
| Minor allele | 0 | 1 | 1 | 0 | 0 | 14 | 39 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 7 | 0 |
| Total | 155 | 155 | 360 | 155 | 360 | 155 | 360 | 155 | 360 | 155 | 155 | 360 | 155 | 155 | 155 | 155 |
| MAF (%) | 0.3 | 2.3 | 3.1 | 0.3 | 0 | 26.5 | 31.1 | 0.3 | 0 | 0.3 | 0.3 | 0 | 0.3 | 24.5 | 23.5 | 0.3 |
| OR (95% CI) | NA | 0.65 (0.0.21, 1.71) | NA | 0.74 (0.50, 1.10) | NA | NA | NA | NA | NA | NA | NA | |||||
| P-value | NA | 0.51 | 0.30 | 0.12 | 0.30 | NA | 0.30 | NA | NA | NA | NA | |||||
| PolyPhen-2 | NA | NA | Benign | Benign | NA | NA | Probably damaging | NA | NA | NA | NA | |||||
| SIFT | Tolerated | NA | Tolerated | Tolerated | NA | Tolerated | Damaging | Tolerated | Tolerated | Tolerated | Tolerated | |||||
acDNA reference (NM_015697.7) from NCBI gene was used to annotate the position of genomic DNA. bAccession number NP_056512.5 from NCBI protein were used to annotate the position of amino acid. P-values result from Fisher’s exact test comparing the frequency of carriers of the minor allele between MSA patients and controls. Estimation of an odds ratio (and 95% confidence interval) is not possible when the rare allele is not observed in one of the comparison groups, and therefore NA is given for these quantities for three of the variants for which the rare allele was observed in MSA patients but not in controls. After applying a Bonferroni adjustment for multiple testing, p-values ≤0.01 (5 tests) were considered as statistically significant. Abbreviation: Cont. Control subjects, MAF minor allele frequency, NA not available, OR odds ratio, CI confidence interval.
Figure 1.
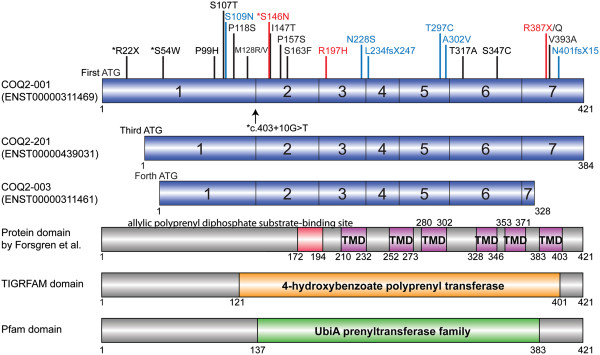
COQ2 variants and transcripts. The upper three panels in blue represents the protein coding transcripts of the COQ2 gene and the exons are numbered (transcripts which cause nonsense mediated decay or transcripts which produce no protein are not shown). Transcripts and protein domains are referred to Ensemble (http://useast.ensembl.org/index.html) and 1000 Genomes (http://www.1000genomes.org/home). The lower three panels represent the protein domains and motifs by Forsgren and colleagues [14], TIGRFAMs database (http://www.tigr.org/TIGRFAMs) [15] and Pfam database (http://pfam.xfam.org/) [16]. There were no transcripts which start from second ATG. Red box indicates the allylic polyprenyl diphosphate substrate-binding site. Purple Boxes indicate the six transmembrane domains (TMD) predicted. The orange box and green box show the areas of 4-hydroxybenzoate polyprenyl transferase (Accession: TIGR01474) and UbiA prenyltransferase family (Accession: PF01040) respectively. Potential risk variants and p.R22X shown in black were identified in multiple system atrophy (MSA) [4, 13, 17]. p.R22X only exists on the longest isoform, COQ2-001, and does not associated with MSA. †ENST00000439031 was missing in the latest Ensembl release 77 - October 2014. *Asterisks indicate variants found in our MSA series. Arrow shows an intronic variant, c403 + 10G > T (Exon1 + 10). Mutations in blue were identified in primary coenzyme Q10 (CoQ10) deficiency [5–12]. Mutations in red (p.S146N, p.R197H and p.R387X) were identified both in MSA and primary CoQ10 deficiency [4, 6, 8, 13].
Rare heterozygous variants: p.R10R, p.S54W, c.403 + 10G > T (Exon1 + 10), p.P142P, p.S146N, p.A267A and p.Y369Y were observed in one patient each but p.S54W, c.403 + 10G > T (Exon1 + 10) and p.S146N were not found in 360 control subjects. Sequencing for p.R10R, p.P142P, p.A267A, p.D298D, p.S330S and p.Y369Y was not assessed in controls because they are synonymous variants. In total, we identified three COQ2 variants with unknown significance; p.S54W, c.403 + 10G > T (Exon1 + 10) and p.S146N. The COQ2 p.S146N [6–8] has been shown to affect COQ2 function [7] and was not identified in 500 control chromosomes in a previous report [6].
Statistical analysis for nine COQ2 mutations found in CoQ10 deficiency-1
Primary CoQ10 deficiency-1 is extremely rare; only 11 patients and nine COQ2 mutations have been identified so far (Table 2) [5–12]. We investigated the publicly available data (EVS) and these nine variants (p.S109N, p.S146N, p.R197H, p.N228S, p.L234fsX247, p.T297C, p.A302V, p.R387X and p.N401fsX15) were not found in more than 4000 European American subjects. If we perform a meta-analysis of the results from our study, the other case–control studies [4, 13, 17, 18] and EVS data, pathologically-confirmed MSA is associated with COQ2 mutations previously found in primary CoQ10 deficiency-1 (2/361 vs 0/6356, p value =0.0029, Table 3); clinical MSA did not associate with those nine mutations. (1/1907 vs 0/6356, p value =0.23, Table 3).
Table 2.
COQ2 mutations previously found in primary CoQ10 deficiency-1
| # | Amino acid change | Age at onset | Age at death | Clinical phenotype | Reference |
|---|---|---|---|---|---|
| 1 | p.S109N/p.S109N | 3 week | 5 month | Seizures, hypertrophic cardiomegaly, nephrotic syndrome | [5] |
| 2 | p.S146N/p.S146N | 5 day | 6 month | Nephrotic syndrome, seizures, encephalopathy, hypotonia, respiratory failure | [6] |
| 3 | p.S146N/p.S146N | Not-available | Not-available | Not available | [7] |
| 4 | p.S146N/p.R387X | birth | 2 month | Respiratory failure, lactic acidosis, hypertrophic cardiomyopathy, necrotizing enterocolitis, encepharopathy | [8] |
| 5 | p.R197H/p.N228S | 18 month | alive at 29 month | Nephrotic syndrome, generalized edema | [6] |
| 6 | p.N228S/p.L234fsX247 | 24 month | Not-available | Nephrotic syndrome, acute renal failure | [9] |
| 7 | p.T297C/p.T297C (proband) | 12 month | alive at 33 month | Nephrotic syndrome, hypotonia, optic atrophy, psychomotor regression, seizures, hemiplegia | [10] |
| 8 | p.T297C/p.T297C (sister) | 9 month | Not-available | Nephrotic syndrome | [10] |
| 9 | p.A302V/p.A302V (twin, male) | birth | 6 month | Respiratory failure, generalized edema, seizures, hypotonia, dystonic-hyper kinetic movement, lactic acidosis, retinopathy | [11] |
| 10 | p.A302V/p.A302V (twin, female) | birth | 5 month | Respiratory failure, generalized edema, seizures, hypotonia, dystonic-hyper kinetic movement, lactic acidosis, intracranial hemorrhage | [11] |
| 11 | p.N401fsX15/p.N401fsX15 | birth | 12 day | Neurologic distress, liver failure, nephrotic syndrome, pancytopenia, diabetes, cytolysis, seizures, lactic acidosis | [12] |
Table 3.
Frequency of nine COQ2 mutations found in CoQ10 deficiency-1
| Study | Country | Mutation | Frequency | P value vs. controls | |||||
|---|---|---|---|---|---|---|---|---|---|
| Clinical MSA N (%) | Pathologically-confirmed MSA N (%) | Total MSA N (%) | Control N (%) | Clinical MSA | Pathologically-confirmed MSA | Total MSA | |||
| This study | USA | p.S146N | 0/58 (0) | 1/97 (1.0) | 1/155 (0.6) | NA | NA | NA | NA |
| Tsuji et al. [4] | 2mixed | p.R387X | 1/762 (0.1) | 0/2 (0.0) | 1/764 (0.1) | 0/1129 (0) | 0.40 | NA | 0.40 |
| Jeon et al. [17] | Korea | None | 0/299 (0) | NA | 0/299 (0) | 0/365 (0) | 1.00 | NA | 1.00 |
| Sharma et al. [18] | Europe | None | 0/788 (0) | NA | 0/788 (0) | 0/600 (0) | 1.00 | NA | 1.00 |
| Schottlaender et al. [13] | Europe | p.R197H | NA | 1/300 (0.3) | 1/300 (0) | 0/262 (0) | NA | 1.00 | 1.00 |
| 1EVS | USA | None | NA | NA | NA | 0/4000 (0) | NA | NA | NA |
| Combined | Combined | p.S146N, p.R197H or p.R387X | 1/1907 (0.05) | 2/361* (0.5) | 3/2268 (0.1) | 0/6356 (0) | 0.23 | 0.0029 | 0.019 |
Total nine COQ2 mutations previously found in primary CoQ10 deficiency-1, p.S109N, p.S146N, p.R197H, p.N228S, p.L234fsX247, p.T297C, p.A302V, p.R387X and p.N401fsX15, were not found in more than 4000 subjects of EVS. 1Data of European American population from Exome Variant Server. 2Mixed indicates Japan, Europe and North America. Abbreviation: EVS, Exome Variant Server, MSA, multiple system atrophy, NA, not available. *Thirty eight out of 97 our patients were included in the study of Schottlaender et al. P-values result from Fisher’s exact test. Because only two pathologically-confirmed MSA patients were included in the study by Tsuji et al., we did not make a comparison of pathologically confirmed MSA patients vs. controls for that study.
In silico analysis
For the c.403 + 10G > T (Exon1 + 10) variant, in silico analysis, Human Splicing Finder predicted this variant to change exon 1 splicing site including 8 intronic nucleotides between exon 1 and 2. We performed RNA/cDNA study to validate whether the heterozygous c.403 + 10G > T (Exon1 + 10) caused a splicing effect in the border between exon 1 and 2.
For the COQ2 protein domain, TIGRFAMS domain showed 4-hydroxybenzoate polyprenyl transferase (Accession: TIGR01474) and Pfam database showed UbiA prenyltransferase family (Accession: PF01040) respectively (Figure 1). Main roles of those domains are biosynthesis of cofactors, prosthetic groups, and carriers. We did not find any specific relationship between COQ2 variants and protein domains (Figure 1), although there is a tendency that the most of variants found in CoQ10 deficiency-1 exist in those of domains found in TIGRFAM and Pfam.
RNA/cDNA sequencing
RNA/cDNA sequencing showed normal sequence between exon 1 and 2 (after p.R22X) from the cerebellum tissue with homozygous or heterozygous p.R22X (data not shown), suggesting the isoform including first ATG (ENST00000311469) does not play major role in MSA patients (Figure 1). Mitsui and Tsuji reported that the majority of the transcripts in human tissues start near the third or fourth ATG codon [19] and p.R22X lies upstream of the third ATG codon (Figure 1). Analysis of the transcript generation for the carrier of the c.403 + 10G > T (Exon1 + 10) variants did not show any alternate band length and sequencing of the amplified cDNA product did not show any alternate nucleotide inclusion.
Clinical and pathological presentation of a patient with heterozygous COQ2 p.S146N variant
This patient was a Caucasian male who had been clinically diagnosed with MSA-C (cerebellar variant of MSA) during his life. There is no family history of any similar disorder or other neurological disorders. His initial symptoms were staggering gait and frequent falls at the age of 54 years. A brain MRI showed marked atrophy of the pontine and cerebellar vermis at the age of 55 years. He had deterioration of his coordination, with not only worsening of his gait but also of his limbs and speech difficulties. Autonomic dysfunction included difficulty emptying the bladder, necessitating self-catheterization. Other autonomic dysfunction was otherwise suggested by several syncopal episodes. He was wheelchair bound at the age of 60 years. Muscle strength was normal. Deep tendon reflex were abnormally brisk at all sites and bilateral Babinski sign was positive. Severe ataxia was seen on finger to nose to finger test and heel-knee-shin testing. His treatment was limited to speech and physical therapy. He died of septicemia at the age of 68 years.He had an autopsy examination (Figure 2). The fixed tissue of the case weighted 680 grams and the calculated brain weight was 1360 grams. The infratentorial tissues were remarkable for severe atrophy of the pontine base and the inferior olive (Figure 2A). Horizontal sections of the midbrain, pons and medullar were remarkable for extremely marked atrophy. The substantia nigra had loss of pigmentation. The cerebrum peduncle was severely atrophic (Figure 2B, arrowheads). Microscopic analysis showed the neocortex was unremarkable in most areas. The pontine base and the inferior olive had severe neuronal loss and gliosis. In addition there were GCIs (Figure 2F & G). The cerebellum had severe diffuse loss of Purkinje cells with many axonal torpedoes. The internal granular cell layer had diffuse depopulation. The deep cerebellar white matter was markedly attenuated and gliotic with GCIs (Figure 2H). The pathological findings were of MSA (striatonigral and olivopontocerebellar degeneration), though the degree of the α-synuclein pathology was more severe than for typical MSA cases.
Figure 2.
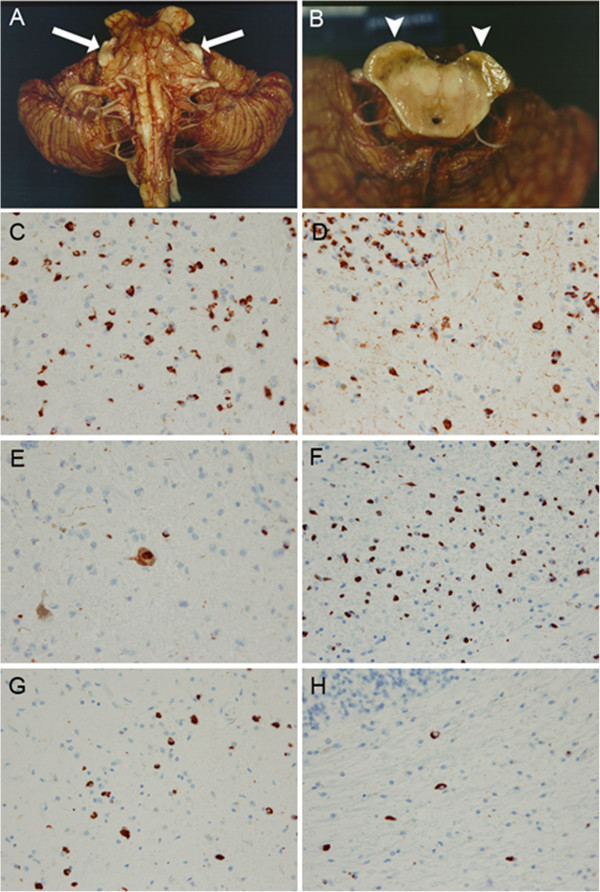
Macroscopic features and α-synuclein pathology of a patient with heterozygous COQ2 p.S146N variant (A-H). The infratentorial tissues were remarkable for severe atrophy of the pontine base and the inferior olive (A). The trigeminal nerve was unusually prominent due to the extreme pontine atrophy (arrows). Horizontal sections of the midbrain were remarkable for extremely marked atrophy (B). The substantia nigra had loss of pigmentation. The cerebrum peduncle was severely atrophic (B, arrowheads). Immunohistochemical staining for α-synuclein (LB509, mouse monoclonal; 1:100) in the globuls pallidus (C), putamen (D), substantia nigra (E), pontine nucleus (F), inferior olive (G) and the cerebellar white matter (H) (magnification × 400).
Discussion
Our study examined the relevance of COQ2 variation (and subsequent CoQ10 deficiency) in susceptibility to MSA. The absence of pathogenic recessive COQ2 mutations or copy number variants within our series of clinical and pathologically-confirmed MSA patients means further screening of case–control series and functional studies are likely required to confirm the role of this candidate gene in MSA. We identified two heterozygous COQ2 variant, p.S54W and c.403 + 10G > T, of unknown significance, although cDNA study for c.403 + 10G > T did not show any alternate splicing. Recently, three independent case–control studies also reported an absence of recessive COQ2 mutations in MSA supporting the rarity of this as a potential cause of disease [13, 17, 18]. In addition, Jeon and colleagues noted the Asian-specific genetic risk factor p.V393A was observed at the same frequency in cases and controls (~2.6%). These reports question the role of COQ2 variants in risk of MSA but do not rule out the potential for rare variants in disease.
Interestingly, we found one heterozygous COQ2 p.S146N variant. Clinical and pathological presentation of our Caucasian patient harboring the heterozygous p.S146N variant was MSA-C with severe pathological changes. This mutation has been previously reported as a pathogenic homozygous mutation in infantile multisystem disorder (a major phenotype of primary CoQ10 deficiency-1) [6, 7] or as part of a compound heterozygous state [8] (Table 2). The pathogenicity of this mutation is supported by the high conservation at this amino acid position, by the prediction as probably damaging by PolyPhen-2 and SIFT, and by the finding of abnormal mitochondria proliferation and low CoQ10 levels in the skeletal muscle of carriers [6].
The study of Bujan and colleagues demonstrated that a patient carrying the homozygous p.S146N mutation showed significant CoQ10 deficiency in fibroblasts and presented deficient CoQ10 biosynthesis [7]. Collectively, these reports suggest that the p.S146N substitution functionally impairs CoQ10 activity and therefore it is possible that in the heterozygous state p.S146N might increase susceptibility to MSA. Our patient with heterozygous p.S146N was clinically diagnosed as MSA-C. Fitting with this, it was reported that patients with COQ2 mutations have increased frequency of MSA-C compared to MSA-P (the parkinsonian variant of MSA) and the cerebellum is more vulnerable to compromised COQ2 function than other regions of the central nervous system [4]. A previous study has also revealed that the cerebellum in both rats and humans contains the lowest concentration of CoQ10 in the brain [20]. Recessive COQ2 mutations are known to cause primary CoQ10 deficiency-1, including infantile multisystem disorder, it remains unresolved why the same gene would cause MSA in an adult [5–12]. One possible explanation would be that the decrease in COQ2 activity associated with the recessive mutations in patients with MSA is milder than that observed in patients with primary CoQ10 deficiency-1 [4].
This report is the third time a mutation known to cause primary CoQ10 deficiency-1 has been observed in an MSA case. Recently Tsuji and colleagues found R387X (referred to R337X by Tsuji et al.) in one of the original multiple families (Family 12) and Schottlaender and Houlden (MSA Brain Bank Collaboration) reported one heterozygous p.R197H carrier in 300 pathologically-confirmed MSA [13]. COQ2 mutations found in CoQ10 deficiency-1 were associated with MSA (3/2268 vs 0/6356, p value =0.019, Table 3), especially pathologically-diagnosed MSA (p value =0.0029). Although primary CoQ10 deficiency-1 due to COQ2 mutations is rare, it may be worth reassessing family history in these patients for the possible increased occurrence of MSA.
Previously, several genetic risk factors for MSA have been reported. For instance, a common variant in the α-synuclein gene (SNCA rs11931074) was associated with MSA in an initial study (Odds ratio [OR] = 6.2, 95% confidence interval [CI]: 3.4 – 11.2, p value under recessive model =5.5 × 10−12) [21] and this result was subsequently replicated in an independent set of pathologically-confirmed MSA cases (OR: 4.7, 95% CI: 1.0 – 21.7, p value =0.06) [22]. Another common variant, the microtubule-associated protein tau (MAPT) H1 haplotype (rs1052553) has also been associated with risk of MSA (OR: 1.9, 95% CI: 1.1 – 3.2, p =0.016) [23]. On the other hand, rare variants (MAF < 1%) have not been reported to be associated with MSA, probably due to the low disease frequency of MSA. Copy number loss of “src homology 2 domain containing-transforming protein 2 gene” was reported as a risk of MSA [24] but a replication study from another institute failed to show the association [25]. Further studies of rare variants and copy number variation are warranted to elucidate further genetic risks for MSA.
Conclusions
MSA due to recessive COQ2 mutations (including exon dosage) was not observed in our study. We did detect the presence of a single heterozygous pathogenic COQ2 variant, which causes infantile multisystem disorder (primary CoQ10 deficiency-1) in a homozygous condition. Given the lack of therapeutic options in MSA, more work is warranted to resolve the role, if any, of COQ2 variants in disease. Primary CoQ10 deficiencies, with early treatment, respond well to CoQ10 supplementation [26]. Whether this is a viable option for treatment or prevention of MSA, or within in a small subset of individuals carrying COQ2 mutations, remains to be determined. Further studies on the etiology of MSA will lead to more rational and improved drug discovery.
Methods
Subjects
A total of 97 pathologically-confirmed MSA patients, 58 clinically-diagnosed MSA patients, and 360 control subjects from the United States were included in this study (Table 4). Thirty eight pathologically-confirmed MSA cases were included in a previous study as part of the MSA brain collaboration study [13]. The diagnosis of MSA was made using current consensus criteria [2]. In MSA patients, mean age was 61.6 ± 9.1 years, and 93 patients (60%) were male. Mean age at evaluation in controls was 70.0 ± 13.4 years, and 142 control subjects (39.4%) were male. All control individuals were free of personal or familial history suggestive of parkinsonism, cerebellar ataxia or autonomic failure. All individuals were unrelated within and between sample groups. The Mayo Clinic Institutional Review Board approved the study, and all living subjects provided informed consent. Informed consent for pathologically-confirmed cases was obtained from the next-of-kin.
Table 4.
The demographic, pathologic and clinical details of the MSA patients
| Mayo clinic MSA series | |||
|---|---|---|---|
| Pathologically-confirmed MSA | Clinically- diagnosed MSA | Total | |
| Total number | 97 | 58 | 155 |
| Male/Female | 59/38 | 34/24 | 93/62 |
| Positive family history of MSA | 0 | 2 | 2 |
| Positive family history of PD | 1 | 10 | 11 |
| Mean age at onset, SD | 59.1 ± 8.7 (39–78) | 64.7 ± 8.8 (48–83) | 61.6 ± 9.1 (39–83) |
| Mean age at death, SD | 67.5, 8.5 (47–86) | NA | NA |
| MSA-P/MSA-C/MSA-PC/Unclassified* | 36/13/47/1 | 49/9/0/0 | 85/22/47/1 |
| Ethnicity (Race), % | |||
| White (Caucasian) | 93 | 47 | 140 |
| African-American | 1 | 4 | 5 |
| Asian | 2 | 0 | 2 |
| Hispanic | 1 | 0 | 1 |
| More than One Race/NA | 0 | 7 | 7 |
Abbreviation: MSA-P parkinsonian variant of multiple system atrophy, MSA-C cerebellar variant of multiple system atrophy, MSA-PC mixed type of multiple system atrophy with parkinsonism and cerebellar ataxia, NA not available, PD Parkinson’s disease, SD standard deviation. *One MSA patient was unclassified.
Genetic analysis
For direct sequence analysis, each exon was amplified by polymerase chain reaction (PCR) using published primers for COQ2 [4]. DNA was extracted from frozen brain or whole blood samples using standard protocols. Electropherograms were analyzed with SeqScape v2.5 using 3730 DNA Analyzer (ABI, Applied Biosystems, Foster City, CA, USA). Specific genetic variants observed (p.R22X, p.S54W, p.L66V, Exon1 + 10 and p.S146N) on exon 1 and 2 were screened through a series of US Caucasian healthy control subjects (n = 360) but synonymous (silent) variants were not screened. Of note, COQ2 gene has multiple start codons and a number of alternative isoforms are expressed. Herein, we have used the consensus RefSeq accession NM_015697.7 to number all variants within the COQ2 gene and protein; however this does not necessarily indicate this long isoform is the one related to the MSA or primary CoQ10 deficiency phenotype.
Exonic deletion or multiplication of COQ2 gene was assessed in all patients with MSA. To determine exon dosage genomic copy number probes were used for each individual exon (n = 7; Applied Biosystems) and details are available on request. TaqMan Copy Number Reference Assay RNase P (human) was used as an endogenous control (ABI). Quantitative PCR was carried out using TaqMan expression chemistry protocol as previously described [27]. All assays were performed in triplicate on the ABI 7900HT Fast Real-Time PCR System and analyzed with ABI SDS 2.2.2 Software (ABI).
RNA/cDNA sequencing
RNA was extracted from brain tissue (cerebellum) using standard protocol with TRIzol reagent (Life Technologies, Foster City, CA, USA). For carriers of specific variation, p.R22X and c.403 + 10G > T (Exon1 + 10), cDNA was made by using High-Capacity cDNA Reverse Transcription Kit (ABI) and sequencing was performed. Primers for cDNA sequencing for the border between COQ2 exon 1 and 2 are 5'-CACGGTGGTGACTTGCAG-3' (Forward primer sequence) and 5'-TGCTCCACGCATCAGAATAG-3' (Reverse primer sequence).
In-silico analyses
For prediction of the functional consequences of variants on COQ2 protein sequence, we used software programs available on the internet, namely PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) [28] and SIFT (http://sift.jcvi.org/) [29].
The results were shown in Table 1. Splice site prediction of intronic variants identified in COQ2 were calculated by using the online bioinformatics tools, Human Splicing Finder (version 2.4.1) [30]. See the URL. (http://www.umd.be/HSF/HSF.html).
The protein domain search tools, TIGRFAMs database (http://www.tigr.org/TIGRFAMs) [15] and Pfam database (http://pfam.xfam.org/) [16] were used to elucidate the association between variants and protein domains (Figure 1). We used an online software of DOG (Domain Graph, version 2.0.1; http://dog.biocuckoo.org/) to prepare publication-quality figures of protein domain structures [31].
Statistical analysis
For each COQ2 mutation, comparisons of the frequency of carriers of the minor allele between MSA patients and (1) controls from our study, (2) EVS data, and (3) 1000 Genomes data were made using Fisher’s exact test. We utilized a Bonferroni correction to adjust for multiple testing separately for these three sets of comparisons, after which p-values ≤0.01 (MSA patients vs. controls, 5 tests), ≤0.0071 (MSA patients vs. EVS data, 7 tests), and ≤0.0071 (MSA patients vs. 1000 Genomes data, 7 tests) were considered as statistically significant. Note that only COQ2 variants for which information was obtained for both MSA patients and the given comparison group of interest were utilized when making comparisons. Additionally, when combining data from our study and previously published or publicly available data, we compared the frequency of carriers of any COQ2 mutation found in primary CoQ10 deficiency-1 (p.S109N, p.S146N, p.R197H, p.N228S, p.L234fsX247, p.T297C, p.A302V p.R387X, and p.N401fsX15; Table 2) [5–12] between MSA patient and control groups using Fisher’s exact test (Table 3). All statistical analysis was performed using R Statistical Software (version 2.14.0; R Foundation for Statistical Computing, Vienna, Austria).
Electronic supplementary material
Additional file 1: For variants of COQ2 observed in this study, comparisons of MSA patients with controls, with EVS data, and with 1000 Genomes data. (DOCX 21 KB)
Acknowledgements
We would like to thank all those who have contributed to our research, particularly the patients and families who donated Brain tissue and DNA samples for this work. This work was supported by the National Institutes of Health [grant numbers R01 NS078086 (OAR), Morris K. Udall Parkinson's Disease Research Center of Excellence P50 NS072187 (OAR, RR, RU, ZKW and DWD), Mayo Clinic Center for Regenerative Medicine (ZKW), The Michael J. Fox Foundation for Parkinson's Research (ZKW), the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch (ZKW and SF).
Abbreviations
- COQ2
Coenzyme Q2 4-hydroxybenzoate polyprenyltransferase
- CoQ10
Coenzyme Q10
- EVS
Exome Variant Server
- MAF
Minor allele frequency
- MSA
Multiple system atrophy
- PD
Parkinson’s disease.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KO carried out the molecular genetic studies and drafted the manuscript. MGH, SR, AIS-O, CL, RLW, OLB, XW and YA carried out the molecular genetic studies and data analysis. MGH performed all statistical analysis. SF, NG-R, RU, RR, WPC, ZKW and DWD made substantial contributions to acquisition of patient material and data. OAR conceived of the study, obtained study funding, participated in its design and coordination and drafted the manuscript. All authors participated in the interpretation of results, read and approved the final manuscript.
Contributor Information
Kotaro Ogaki, Email: ogaki.kotaro@mayo.edu.
Shinsuke Fujioka, Email: shinsuke@cis.fukuoka-u.ac.jp.
Michael G Heckman, Email: heckman.michael@mayo.edu.
Sruti Rayaprolu, Email: sruti.rayaprolu@ufl.edu.
Alexandra I Soto-Ortolaza, Email: ortolaza.alexandra@mayo.edu.
Catherine Labbé, Email: labbe.catherine@mayo.edu.
Ronald L Walton, Email: walton.ronald@mayo.edu.
Oswaldo Lorenzo-Betancor, Email: lorenzobetancor.oswaldo@mayo.edu.
Xue Wang, Email: wang.xue@mayo.edu.
Yan Asmann, Email: asmann.yan@mayo.edu.
Rosa Rademakers, Email: rademakers.rosa@mayo.edu.
Neill Graff-Radford, Email: graffradford.neill@mayo.edu.
Ryan Uitti, Email: uitti@mayo.edu.
William P Cheshire, Email: cheshire@mayo.edu.
Zbigniew K Wszolek, Email: wszolek.zbigniew@mayo.edu.
Dennis W Dickson, Email: dickson.dennis@mayo.edu.
Owen A Ross, Email: ross.owen@mayo.edu.
References
- 1.Stefanova N, Bucke P, Duerr S, Wenning GK. Multiple system atrophy: an update. Lancet Neurol. 2009;8:1172–1178. doi: 10.1016/S1474-4422(09)70288-1. [DOI] [PubMed] [Google Scholar]
- 2.Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, Wood NW, Colosimo C, Durr A, Fowler CJ, Kaufmann H, Klockgether T, Lees A, Poewe W, Quinn N, Revesz T, Robertson D, Sandroni P, Seppi K, Vidailhet M. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–676. doi: 10.1212/01.wnl.0000324625.00404.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome) J Neurol Sci. 1989;94:79–100. doi: 10.1016/0022-510X(89)90219-0. [DOI] [PubMed] [Google Scholar]
- 4.The Multiple-System Atrophy Research Collaboration Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med. 2013;369:233–244. doi: 10.1056/NEJMoa1212115. [DOI] [PubMed] [Google Scholar]
- 5.Scalais E, Chafai R, Van Coster R, Bindl L, Nuttin C, Panagiotaraki C, Seneca S, Lissens W, Ribes A, Geers C, Smet J, De Meirleir L. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2) Eur J Paediatr Neurol. 2013;17:625–630. doi: 10.1016/j.ejpn.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, Caridi G, Piemonte F, Montini G, Ghiggeri GM, Murer L, Barisoni L, Pastore A, Muda AO, Valente ML, Bertini E, Emma F. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol. 2007;18:2773–2780. doi: 10.1681/ASN.2006080833. [DOI] [PubMed] [Google Scholar]
- 7.Bujan N, Arias A, Montero R, Garcia-Villoria J, Lissens W, Seneca S, Espinos C, Navas P, De Meirleir L, Artuch R, Briones P, Ribes A. Characterization of CoQ biosynthesis in fibroblasts of patients with primary and secondary CoQ deficiency. J Inherit Metab Dis. 2014;37:53–62. doi: 10.1007/s10545-013-9620-4. [DOI] [PubMed] [Google Scholar]
- 8.Dinwiddie DL, Smith LD, Miller NA, Atherton AM, Farrow EG, Strenk ME, Soden SE, Saunders CJ, Kingsmore SF. Diagnosis of mitochondrial disorders by concomitant next-generation sequencing of the exome and mitochondrial genome. Genomics. 2013;102:148–156. doi: 10.1016/j.ygeno.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCarthy HJ, Bierzynska A, Wherlock M, Ognjanovic M, Kerecuk L, Hegde S, Feather S, Gilbert RD, Krischock L, Jones C, Sinha MD, Webb NJ, Christian M, Williams MM, Marks S, Koziell A, Welsh GI, Saleem MA, RADAR the UK SRNS Study Group Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2013;8:637–648. doi: 10.2215/CJN.07200712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quinzii C, Naini A, Salviati L, Trevisson E, Navas P, Dimauro S, Hirano M. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet. 2006;78:345–349. doi: 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jakobs BS, van den Heuvel LP, Smeets RJ, de Vries MC, Hien S, Schaible T, Smeitink JA, Wevers RA, Wortmann SB, Rodenburg RJ. A novel mutation in COQ2 leading to fatal infantile multisystem disease. J Neurol Sci. 2013;326:24–28. doi: 10.1016/j.jns.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 12.Mollet J, Giurgea I, Schlemmer D, Dallner G, Chretien D, Delahodde A, Bacq D, de Lonlay P, Munnich A, Rotig A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest. 2007;117:765–772. doi: 10.1172/JCI29089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schottlaender LV, Houlden H. Mutant COQ2 in multiple-system atrophy. N Engl J Med. 2014;371:81. doi: 10.1056/NEJMc1311763. [DOI] [PubMed] [Google Scholar]
- 14.Forsgren M, Attersand A, Lake S, Grunler J, Swiezewska E, Dallner G, Climent I. Isolation and functional expression of human COQ2, a gene encoding a polyprenyl transferase involved in the synthesis of CoQ. Biochem J. 2004;382:519–526. doi: 10.1042/BJ20040261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haft DH, Selengut JD, White O. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003;31:371–373. doi: 10.1093/nar/gkg128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeon BS, Farrer MJ, Bortnick SF. Mutant COQ2 in multiple-system atrophy. N Engl J Med. 2014;371:80. doi: 10.1056/NEJMc1311763. [DOI] [PubMed] [Google Scholar]
- 18.Sharma M, Wenning G, Kruger R. Mutant COQ2 in multiple-system atrophy. N Engl J Med. 2014;371:80–81. doi: 10.1056/NEJMc1311763. [DOI] [PubMed] [Google Scholar]
- 19.Mitsui J, Tsuji S. Mutant COQ2 in multiple-system atrophy. N Engl J Med. 2014;371:82–83. doi: 10.1056/NEJMc1311763. [DOI] [PubMed] [Google Scholar]
- 20.Naini A, Lewis VJ, Hirano M, DiMauro S. Primary coenzyme Q10 deficiency and the brain. Biofactors. 2003;18:145–152. doi: 10.1002/biof.5520180217. [DOI] [PubMed] [Google Scholar]
- 21.Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D, Melchers A, Paudel R, Gibbs JR, Simon-Sanchez J, Paisan-Ruiz C, Bras J, Ding J, Chen H, Traynor BJ, Arepalli S, Zonozi RR, Revesz T, Holton J, Wood N, Lees A, Oertel W, Wüllner U, Goldwurm S, Pellecchia MT, Illig T, Riess O, Fernandez HH, Rodriguez RL, Okun MS, Poewe W, Wenning GK, Hardy JA, Singleton AB, Del Sorbo F, Schneider S, Bhatia KP, Gasser T. SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol. 2009;65:610–614. doi: 10.1002/ana.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ross OA, Vilarino-Guell C, Wszolek ZK, Farrer MJ, Dickson DW. Reply to: SNCA variants are associated with increased risk of multiple system atrophy. Ann Neurol. 2010;67:414–415. doi: 10.1002/ana.21786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vilarino-Guell C, Soto-Ortolaza AI, Rajput A, Mash DC, Papapetropoulos S, Pahwa R, Lyons KE, Uitti RJ, Wszolek ZK, Dickson DW, Farrer MJ, Ross OA. MAPT H1 haplotype is a risk factor for essential tremor and multiple system atrophy. Neurology. 2011;76:670–672. doi: 10.1212/WNL.0b013e31820c30c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sasaki H, Emi M, Iijima H, Ito N, Sato H, Yabe I, Kato T, Utsumi J, Matsubara K. Copy number loss of (src homology 2 domain containing)-transforming protein 2 (SHC2) gene: discordant loss in monozygotic twins and frequent loss in patients with multiple system atrophy. Mol Brain. 2011;4:24. doi: 10.1186/1756-6606-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferguson MC, Garland EM, Hedges L, Womack-Nunley B, Hamid R, Phillips JA, Shibao CA, Raj SR, Biaggioni I, Robertson D. SHC2 gene copy number in multiple system atrophy (MSA) Clin Auton Res. 2014;24:25–30. doi: 10.1007/s10286-013-0216-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopez LC, Quinzii CM, Area E, Naini A, Rahman S, Schuelke M, Salviati L, Dimauro S, Hirano M. Treatment of CoQ(10) deficient fibroblasts with ubiquinone, CoQ analogs, and vitamin C: time- and compound-dependent effects. PLoS One. 2010;5:e11897. doi: 10.1371/journal.pone.0011897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajput A, Vilarino-Guell C, Rajput ML, Ross OA, Soto-Ortolaza AI, Lincoln SJ, Cobb SA, Heckman MG, Farrer MJ. Alpha-synuclein polymorphisms are associated with Parkinson's disease in a Saskatchewan population. Mov Disord. 2009;24:2411–2414. doi: 10.1002/mds.22795. [DOI] [PubMed] [Google Scholar]
- 28.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. DOG 1.0: illustrator of protein domain structures. Cell Res. 2009;19:271–273. doi: 10.1038/cr.2009.6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: For variants of COQ2 observed in this study, comparisons of MSA patients with controls, with EVS data, and with 1000 Genomes data. (DOCX 21 KB)