

Abstract
Multiple sclerosis (MS) is an inflammatory, demyelinating disease of the central nervous system (CNS) of presumed autoimmune origin. Of all the genetic factors linked with MS, MHC class-II molecules have the strongest association. Generation of HLA class-II transgenic mice has helped to elucidate the role of HLA class-II genes in chronic inflammatory and demyelinating diseases. We have shown that the human HLA-DRB1*0301 gene predisposes to proteolipid protein (PLP)-induced EAE, whereas HLA-DQβ1*0601 (DQ6) was resistant. We also showed that the DQ6 molecule protects from EAE in DRB1*0301.DQ6 double transgenic mice by producing anti-inflammatory interferon gamma (IFNγ). HLA-DQβ1*0302 (DQ8) transgenic mice were also resistant to PLP91-110-induced EAE, but production of pro-inflammatory IL-17 exacerbated disease in DRB1*0301.DQ8 mice. To further confirm the role of IFNγ in protection, we generated DRB1*0301.DQ8 mice lacking IFNγ (DRB1*0301.DQ8.IFNγ−/−). Immunization with PLP91-110 peptide caused atypical EAE in DRB1*0301.DQ8.IFNγ−/− mice characterized by ataxia, spasticity and dystonia, hallmarks of brain-specific disease. Severe brain specific inflammation and demyelination in DRB1*0301.DQ8.IFNγ−/− mice with minimal spinal cord pathology further confirmed brain-specific pathology. Atypical EAE in DRB1*0301.DQ8.IFNγ−/− mice was associated with increased encephalitogenicity of CD4 T cells and their ability to produce higher levels of IL-17 and GM-CSF compared to DRB1*0301.DQ8 mice. Further, areas with demyelination showed increased presence of CD68+ inflammatory cells, suggesting an important role for monocytes/microglia in causing brain pathology. Thus, our study supports a protective role for IFNγ in the demyelination of brain through down regulation of IL-17/GM-CSF and induction of neuro-protective factors in the brain by monocytes/microglial cells.
Keywords: Autoimmunity, experimental autoimmune encephalomyelitis, multiple sclerosis, HLA class II transgenic mice, cytokines, IFNγ, IL-17, microglia, macrophages, neuroimmunology, inflammation, demyelination, axon
Introduction
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the CNS of presumed autoimmune origin (1). Genetic predisposition to MS is associated with certain MHC class II genes, while environmental factors also contribute to disease (1-6). High polymorphism, linkage disequilibrium, and heterogeneity of the human population made it difficult in the past to decipher the exact role of the human HLA class II genes HLA-DQ and HLA-DR in disease pathogenesis. The generation of HLA class II transgenic mice has helped resolve some of these questions. Studies done in HLA class II transgenic mice indicate that transgenic mice expressing human HLA-DR2, DRB1*0301 and DR4 molecules are susceptible to CNS antigen-induced experimental autoimmune encephalomyelitis (EAE), an experimental model used to study MS (7, 8). Utilizing these HLA transgenic mice, we showed that HLA-DRB1*0301 transgenic mice were susceptible to PLP (91-110)-induced EAE (9), whereas DQ6 (DQB1*0601) and DQ8 (DQB1*0302) transgenic mice were resistant. Surprisingly, DQ6/DRB1*0301 double transgenic mice were resistant (10) whereas DQ8/DRB1*0301 mice showed higher disease incidence and severity than DRB1*0301 mice (11).
CNS antigen-specific CD4+ T cells secreting pro-inflammatory cytokines are responsible for disease onset as well as the chronic phase. Elegant studies in murine/rodent EAE have documented that encephalitogenic T cells are CD4+, T helper (Th1)-type cells secreting TNF-α/β and IFNγ (12-14). However, recent studies have indicated that another T-cell phenotype, Th17 secreting IL-17, IL-17F, IL-21, IL-22 and IL-23, plays an important role in the immunopathogenesis of EAE (15). Thus, the current hypothesis of EAE indicates that Th1 and/or Th17 cytokines play important roles in the immunopathogenesis of EAE. However, the exact role of IFNγ and IL-17 in disease pathogenesis is poorly understood because IFNγ is pathogenic in some models and protective in others. Further, the Th1/Th17 ratio might influence the CNS pathology because a high Th17-to-Th1 ratio leads to a severe brain pathology, whereas a high Th1-to-Th17 ratio leads to a severe spinal cord pathology (16, 17).
Our previous data indicate that MHC class II molecules modulate immune responses through activation of specific cytokine profiles (10, 11, 18). The protective effect of DQ6 in DRB1*0301.DQ6 mice was mediated by IFNγ (10), whereas the disease-exacerbating effect of DQ8 molecule was mediated by IL-17 (11). Thus, our studies indicated a pathogenic role for IL-17 and a protective role for IFNγ in the pathogenesis of EAE in certain HLA class-II transgenic mice. To further decipher the role of IFNγ in inflammation and demyelination, we generated HLA-DRB1*0301.DQ8 transgenic mice lacking IFNγ (DRB1*0301.DQ8.IFN-γ−/−). We observed that DRB1*0301.DQ8.IFN-γ−/− mice developed atypical EAE characterized by ataxia, abnormal gait, and dystonia without hind limb paralysis. Mice with EAE showed brain-specific pathology with minimal or no spinal cord pathology. Brain pathology was characterized by severe demyelination in the cerebellum, brain stem and subcortex/striatum region. Demyelinating regions showed relative preservation of axon and neurons with extensive presence of CD68+ cells suggesting an important pathogenic role for monocytes/microglial cells in demyelination. Thus, our study indicates that IFNγ plays a protective role in HLA-DRB1*0301.DQ8.IFNγ−/− HLA class- II transgenic mice.
Material and Methods
Transgenic (Tg) mice
The HLA- DQ8/DRB1*0301 double transgenic (Tg) mice [DQ8 (DQA1*0103, DQB1*0302).-DRB1*0301 (DRB1*0301] were produced as previously described (11, 19). Briefly, HLA class II transgenes were introduced into (B6 × SWR)F1 fertilized eggs. Positive offspring were backcrossed to B10.M mice for several generations. HLA class Tg mice were mated with MHC class II deficient mice (AE−/−) on B6 background (7) and intercrossed to generate the HLA-DRB1*0301.AE−/− and HLA-DQ8.AE −/− Tg lines. HLA-DRB1*0301.AE−/− and HLA-DQ8.AE −/− Tg mice were separately mated with IFNγ-deficient mice on B6 background (Jackson Laboratory) to generate HLA-DRB1*0301.AE−/−.IFNγ−/−, and HLA-DQ8.AE−/−.IFNγ−/− respectively. DRB1*0301.DQ8.AE−/−.IFNγ−/− Tg mice were produced by intercrossing HLA-DRB1*0301.AE−/−.IFNγ−/− with HLA-DQ8.AE−/−.IFNγ−/− mice. Majority of HLA class-II Tg mice have mixed B6/B10 background. All mice were bred and maintained in the pathogen-free Immunogenetics Mouse Colony of Mayo Clinic according to National Institutes of Health and institutional guidelines. All experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Mayo Clinic, Rochester.
Flow cytometry
Expression of HLA-DR and HLA-DQ molecules on peripheral blood leukocytes (PBLs), lymph node cells (LNCs), and splenocytes were analyzed by flow cytometry using monoclonal antibodies (mAbs) L227 and IVD12 specific for HLA-DR (20) and HLA-DQ (21), respectively. Surface expression of CD4 (GK1.5), CD8 (53.6.72), B cells (RA3-6B2), DCs (HL3), monocytes/macrophages (M1/70), NK cells (PK136), and neutrophils (7/4) were analyzed using fluorescent conjugated mAb from BD Biosciences (San Jose, USA). Intracellular levels of IL-6, IL-17, TNFα, GM-CSF and IFNγ were analyzed using directly conjugated antibodies form BD Biosciences (San Jose, USA).
Peptide
The twenty-amino acid-long synthetic peptide PLP91-110 (YTTGAVRQIFGDYKTTICGK) was synthesized at the peptide core facility of Mayo Clinic, Rochester, MN.
Disease induction and scoring
For disease induction, 12- to 16-week-old transgenic mice were immunized subcutaneously in both flanks with 100 μg of PLP91-110 emulsified in CFA containing Mycobacterium tuberculosis H37Ra (400 μg/mice) (11). Pertussis toxin (Sigma Chemicals, St. Louis, MO, USA; 100ng) was injected i.v. at day 0 and 2, post immunization. Mice of both genders were used. Since IFNγ- sufficient mice develop classical EAE characterized by ascending paralysis, these mice were scored for disease severity using standard EAE scores: 0, normal; 1, loss of tail tone; 2, hind-limb weakness; 3, hind-limb paralysis; 4, hind-limb paralysis and forelimb paralysis or weakness; 5, morbidity/death. Atypical EAE was graded as described previously (22) with slight modification. Mild head tilting/ataxia =1, severe ataxia =2, severe ataxia and/or gait problem =3, spasticity and/or dystonia =4. As the classical and atypical EAE were scored on different scales, mice were weighed mice as a common and quantitative measure of the disease in both groups.
Immunization and T-cell proliferation assay
Splenocytes and lymph nodes were collected from immunized mice and challenged with antigen in vitro (9). The results are presented as stimulation indices (CPM of test sample/CPM of the control). For in vitro inhibition experiments, mAbs specific for CD4 (GK1.5), CD8 (TIB 105), HLA-DQ (IVD12), and HLA-DR (L227) were added to LNCs challenged in vitro with human PLP91-110 (20 μg/ml). All of the neutralizing antibodies were generated in-house using the Mayo Monoclonal Hybridoma core facility.
In vitro antigen presentation assay
To study the antigen-presentation function, CD4+ T cells, C11b+ monocytes/macrophages, CD19+ B cells, and CD11c+ DCs were isolated from splenocytes and draining lymph nodes of PLP91-110 immunized HLA-DRB1*0301.DQ8 or DRB1*0301.DQ8.IFNγ−/− Tg mice by magnetic sorting with a cell-specific positive isolation kit according to manufacturer's protocol (Miltenyi Biotec). CD4+ T cells were plated at 1 × 105 cells/well in presence or absence of 20μg/ml of PLP91-110. Magnetically sorted C11b+ monocytes/macrophages, CD19+ B cells, and CD11c+ DCs from DRB1*0301.DQ8 or DRB1*0301.DQ8.IFNγ−/− Tg mice were irradiated and added at 0.2 × 105 cells/well to CD4 T cells cultures in 96-well plates. Two sets of experiments were run in parallel, with one set used for T-cell proliferation measurement and the other to collect supernatant for cytokine analysis.
Cytokine production
Draining LNs were collected 10 days post immunization and stimulated with PLP91-110 peptide as mentioned before in the T-cell proliferation section. Supernatants were collected from the culture 48 hrs after peptide stimulation. The concentration of cytokines was measured using the mouse cytokine 23-plex protein bead array system as per the manufacturer's instructions and analyzed with Bio-Plex manager 2.0 software (Bio-Plex; Bio-Rad Laboratories Ic., Hercules, CA). Some cytokines were measured by sandwich ELISA using pairs of relevant anti-cytokine monoclonal antibodies according to manufacturer's protocol (BD Biosciences, San Jose, CA).
Real time PCR
Expression of various cytokines, chemokines and chemokine receptors (supplemental table 1), were analyzed by Real-time PCR using commercial primer pairs (Realtimeprimrs.com, Elkins Park, PA). RNA was extracted from cells using RNAeasy columns (Qiagen) and cDNA was prepared using RNase H-reverse transcriptase (Invitrogen). cDNA was analyzed by real-time quantitative PCR in triplicates by using SYBR® GreenER™ qPCR reagent system (Invitrogen). The expression level of each gene was quantified using the threshold cycle (Ct) method normalized for the house keeping genes β-actin, GADPH and HPRT (11).
Pathology
Mice were perfused via intra-cardiac puncture with 50 ml of Trump's fixative (4% paraformaldehyde + 0.5% glutaraldehyde). The spinal cords and brains were removed and post-fixed for 24-48 hours in Trump's fixative in preparation for morphologic analysis. All grading was performed without knowledge of the experimental group.
Spinal cords were cut into 1 mm coronal blocks and every third block post fixed in osmium and embedded in glycol methacrylate. Two-micron sections were stained with a modified erichrome/cresyl violet stain. Morphological analysis was performed on 12 to 15 sections per spinal cord. Briefly, each quadrant from every coronal section of each spinal cord was graded for the presence or absence of inflammation and demyelination. The score was expressed as the percentage of pathologic abnormality in the spinal cord quadrants examined. A maximum score of 100 indicated a particular pathologic abnormality in each quadrant of each spinal cord section. Brain pathology was assessed following perfusion. Two coronal cuts in the intact brain (one section through the optic chiasm and a second section through the infundibulum) resulted in three paraffin-embedded blocks. This allowed analysis of the cortex, corpus callosum, hippocampus, brainstem, striatum, and cerebellum. The resulting slides were stained with hematoxylin and eosin. Each area of brain was graded on a 4-point scale: 0 = no pathology; 1 = no tissue destruction but minimal inflammation; 2 = early tissue destruction, demyelination and moderate inflammation; 3 = moderate tissue destruction (neuronal loss, demyelination, parenchymal damage, cell death, neurophagia, neuronal vacuolation); 4 = necrosis (complete loss of all tissue elements with associated cellular debris). Meningeal inflammation was graded as follows: 0 = no inflammation; 1 = one cell layer of inflammation; 2 = two cell layers of inflammation; 3 = three cell layers of inflammation; 4 = four or more cell layers of inflammation. The area with maximum tissue damage was used to assess each brain region.
Immunofluorescence staining
Brain and spinal cord tissues from animals were collected in optimal cutting temperature (OCT) compound (Sakura Finetek Tissue Tek, The Netherland) and immediately frozen at −80°C freezer. Ten μm sections were cut using cryostat, trasnfered to positively charged slides, fixed in cold acetone and stained with either fluorochrome-conjugated antibodies or primary antibody and followed by corresponding secondary antibody as per standard techniques. Stained sections were mounted using VECTASHIELD® HardSet Mounting Medium with DAPI (Vector Lab, , Burlingame, CA) and were analyzed using an Olympus Provis AX70 microscope (Leeds Precision Instruments, Inc., Minneapolis, MN, USA) fitted with a DP70 digital camera.
Statistical analysis
The statistical significance of the differences in functional and histological scores between groups was assessed by a one-way analysis of variance on ranks (Kruskall–Wallis test) when comparing more than two groups and by the Mann–Whitney rank-sum test when comparing only two groups. The differences in proliferation or in cytokine levels between groups was assessed by a one-way analysis of variance with multiple comparisons of the means when more than two groups were analyzed or by Student's t-test when only two groups were analyzed and their data were normally distributed.
Results
All Tg lines developed normally and showed no gross phenotypic abnormalities. Both HLA-DR and DQ were expressed on 35-50% of cell population in PBLs and splenocytes. No endogenous class-II expression was observed (data not shown). Thus both HLA-DR and HLA–DQ molecules were expressed at similar levels in DRB1*0301.DQ8 and DRB1*0301.DQ8.IFNγ −/− Tg mice.
Effect of IFNγ on development of EAE in HLA-DRB1*0301.DQ8 transgenic mice
To investigate the effect of IFNγ on disease susceptibility, EAE was induced in IFNγ-sufficient and -deficient strains using PLP91-110 as an antigen. The susceptibility and clinical features of IFNγ-sufficient and -deficient strains to PLP91-110-induced EAE are presented in Table I. Administration of PLP91-110 to DRB1*0301.DQ8 Tg mice led to chronic progressive disease in 96% (24/25) of Tg mice. The disease was characterized by weight loss as well as ascending paralysis (limp tail followed by hind-limb weakness followed by complete hind-limb paralysis). Interestingly, DRB1*0301.DQ8.IFNγ −/− Tg mice developed atypical EAE with disease incidence of 92% (23/25) characterized by ataxia, gait problems, shivering and tremors (dystonia), spasticity, moderate hind-limb weakness, and weight loss, features suggestive of brain-specific pathology. These mice also maintained their tail tonicity during the course of the disease. The ascending paralysis observed in IFNγ-sufficient DRB1*0301.DQ8 Tg mice has been associated with both brain- and spinal cord-specific pathology. The disease onset (10.3±1.5 Vs 13±1.5, p=ns) and incidence (96% vs. 92%, p=ns) was similar between IFNγ-sufficient and - deficient DRB1*0301.DQ8 Tg mice. Both DRB1*0301.DQ8.IFNγ −/− with atypical EAE and DRB1*0301.DQ8 with classical EAE showed similar weight loss (data not shown). DRB1*0301.DQ8 Tg mice showed progressive weight loss, and there was a direct correlation between the clinical disease score and weight loss. Although DRB1*0301.DQ8.IFNg−/− mice also showed progressive weight loss that correlated with disease score, animals started to recover clinically and gained weight around the fourth week (data not shown). The major difference between EAE observed in IFNγ-sufficient and -deficient mice was the course of EAE. IFNγ-sufficient DRB1*0301.DQ8 mice developed chronic progressive EAE characterized by ascending paralysis and never recovered from their symptoms. These mice were sacrificed after 3 to 4 weeks due to their severe paralysis and significant weight loss. In contrast, IFNγ-deficient DRB1*0301.DQ8.IFNγ−/− mice develop a resolving disease with disease onset at day 13 (±1.5), disease peak at week 3 or 4 and resolution by week 7 or 8. We followed these mice for 90 days post-immunization, and they remained in remission. We used DRB1*0301.IFNγ−/− DQ8.IFNγ−/− and AEº.IFNγ−/− (lacking mouse class II and IFNγ) as controls. In contrast to atypical EAE in DRB1*0301.DQ8.IFNγ−/− Tg mice, DRB1*0301.IFNγ −/− single Tg mice develop both classical as well as atypical EAE (Table 1). IFNγ sufficient DRB1*0301 transgenic mice develop classical EAE characterized by ascending paralysis and weight loss, however it was less severe compared to DRB1*0301.DQ8 double Tg mice (data not shown). Majority of the DRB1*0301 transgenic mice remain paralytic till end of the study but did not become quadriplegic as observed in DR3.DQ8 transgenic mice. No disease was observed in DQ8.IFNγ−/− or AEo.IFNγ−/− mice (Table 1). Thus our data indicates that disease-susceptible human HLA class II transgene DRB1*0301 is necessary for the development of disease whereas absence of IFNγ in DRB1*0301.DQ8 double Tg mice leads to atypical EAE disease characterized by head tilting/ataxia, gait, dystonia, and spasticity.
Table 1.
PLP91-110 induced EAE in IFNγ sufficient and deficient HLA Tg micea
| Mouse strain | Disease incidencea (%) | Mean onset of disease±SD (Days) | Classical EAE with paralysis | Atypical EAE | Limb weakness | ataxia | Gait problem | spasticity | Dystonia |
|---|---|---|---|---|---|---|---|---|---|
| DR3.DQ8.IFNg-/-AEo | 23/25 (100%) | 13±1.5 | 0/25 | 23/25 | 10/25 | 20/25 | 18/25 | 15/25 | 14/25 |
| IFNg-/-AEo | 0/10 (0%) | - | - | - | - | - | - | - | - |
| DR3.DQ8.AEo | 24/25 (96%) | 10.+1.5 | 24/25 | 0/25 | 24/25 | 0/25 | 0/25 | 0/25 | 0/25 |
| DR3.IFNg-/-AEo | 9/10(90%) | 16±1.5 | 4/10 | 5/10 | 6/10 | 5/10 | 4/10 | 4/10 | 4/10 |
| DR3.AEo | 8/10(80%) | 13±1.0 | 8/10 | 0/10 | 8/10 | 0/10 | 0/10 | 0/10 | 0/10 |
| DQ8.IFNg-/-AEo | 0/10 (0%) | - | - | - | - | - | - | - | - |
| DQ8.AEo | 0/10 (0%) | - | - | - | - | - | - | - | - |
| AEo | 0/10 (0%) | - | - | - | - | - | - | - | - |
IFNγ sufficient and deficient HLA Tg mice were immunized with 100μg of PLP91-110 and scored daily for disease (as stated in material and methods), and were examined over time for clinical signs of tail atony, limb weakness, limb paralysis, ataxia, dystonia, gate problem, and spasticity Results are representative of three separate experiments.
Absence of IFNγ in DRB1*0301.DQ8 double Tg mice leads to inflammation and demyelination restricted to the brain
The presence of atypical EAE in the absence of IFNγ in DRB1*0301.DQ8 transgenic lines (DRB1*0301.DQ8.IFNγ −/−) suggested a brain-centric pathology (23, 24) compared to classical EAE observed in IFNγ-sufficient mice, which is characterized by both brain- and spinal cord- specific pathology (11). To confirm this, we analyzed the brain and spinal cord tissue of mice with EAE from all single and double transgenic mice sufficient or deficient for IFNγ. Pathological analysis of CNS tissue showed that, whereas IFNγ sufficient DRB1*0301 single Tg as well as DRB1*0301.DQ8 double Tg mice developed both spinal cord and brain pathology, IFNγ-deficient DRB1*0301.DQ8 mice showed only brain-specific pathology. Although both IFNγ-sufficient and -deficient strains with EAE showed brain pathology, the pathology was more severe in DRB1*0301.DQ8.IFNγ −/− Tg mice compared to DRB1*0301.DQ8 Tg mice and was characterized by a higher degree of inflammation, and demyelination especially in the cerebellum, brain stem, striatum, and septal nuclei regions (Fig 1A-1P). IFNγ-deficient DRB1*0301.DQ8 Tg mice with disease showed extensive inflammation and demyelination in the septal nuclei area of forebrain regions (Fig 1E), whereas no pathology in this region was observed in IFNγ-sufficient DRB1*0301, and DRB1*0301.DQ8 mice with EAE (Fig 1A). Similarly, severe inflammation and demyelination was observed in the striatum region of IFNγ-deficient DRB1*0301.DQ8 Tg animals with atypical disease (Fig 1F) compared to IFNγ-sufficient DRB1*0301.DQ8 Tg animals with classical EAE (Fig 1B). Although DRB1*0301.DQ8 mice with EAE showed inflammation and demyelination in cerebellum (Fig 1C), the extent of inflammation and demyelination in the cerebellum of DRB1*0301.DQ8.IFNγ −/− Tg mice (Fig 1E) was more extensive. IFNγ deficient DRB1*0301.DQ8 animals with disease also showed a higher degree of inflammation in the brain stem region (Fig 1D) compared to the mild inflammation in IFNγ-sufficient DRB1*0301.DQ8 animals with disease (Fig 1H). Brain sections from DRB1*0301.DQ8.IFNγ−/− showed more parenchymal white matter loss compared to DRB1*0301.DQ8 mice. HLA-DRB1*0301.IFNγ−/− single Tg mice also showed increased brain pathology (Fig. 1I-1L) compared to IFNγ sufficient HLA-DRB1*0301 Tg mice (Fig. 1M-1P). However, the extent of pathology in IFNγ deficient HLA-DRB1*0301 single Tg mice was less severe compared to DRB1*0301.DQ8.IFNγ −/− double Tg mice indicating importance of DQ8 molecule in the brain pathology. Interestingly, despite severe brain pathology, IFNγ-deficient DRB1*0301.DQ8 mice with EAE showed a very mild or no pathology in the spinal cord. As expected, DRB1*0301.DQ8 mice with EAE showed significant inflammation and demyelination in the spinal cord compared with very mild pathology in DRB1*0301.DQ8.IFNγ −/− mice (Fig 2A). Quantitative analysis of spinal cord tissues showed that, on average, 38±10% of the spinal cord quadrants from DRB1*0301.DQ8 showed inflammation compared to only 2±3% in DRB1*0301.DQ8.IFNγ−/− mice (p<0.01,Fig. 2B). Similarly, IFNγ-sufficient DRB1*0301.DQ8 showed demyelination in 37.8±6% of spinal cord quadrants, compared with only 2±3% of the quadrants from IFNγ-deficient Tg mice (p<0.001, Fig. 2B). Both IFNγ sufficient and deficient DRB1*0301 single Tg mice showed similar level of spinal cord inflammation (26.8±7.2% vs 20.8±7.2%, p=ns) and demyelination (20.3±5.4% vs 15.7±6.6%, p=ns) (Fig 2B). Thus, the absence of IFNγ leads to brain-restricted inflammation and demyelination in DRB1*0301.DQ8.IFNγ−/− whereas both brain and spinal cord pathology was observed in DRB1*0301IFNγ−/− Tg mice.
Figure 1. Absence of IFNγ leads to severe inflammation and demyelination restricted to brain tissue.

Representative photomicrograph of inflammatory and demyelinating lesions in the brain of DRB1*0301.DQ8 Tg mice (A-D), DRB1*0301.DQ8.IFNγ−/− Tg mice (E-H), DRB1*0301 Tg mice (I-L), and DRB1*0301.IFNγ−/− Tg mice (M-P) with EAE. A) The photographs (4X) of brain sections show severe inflammation and demyelination in IFNγ- deficient DRB1*0301.DQ8.IFNγ−/− Tg mice compared with IFNγ-sufficient DRB1*0301.DQ8 Tg mice. DRB1*0301.IFNγ−/− Tg mice also showed severe brain pathology compared to DRB1*0301 Tg mice, however most severe brain pathology was observed in DRB1*0301.DQ8.IFNγ−/− Tg mice. LV denotes left ventricle whereas CTX denotes cortex region of the forebrain. These figures represent one of three experiments performed at different time points. EAE was induced in different strains and monitored for disease as described in methods. The animals were sacrificed at day 25 post-immunization for CNS histopathology. *p<0.05, **p<0.005 as compared to DRB1*0301.DQ8 Tg mice. The image was taken (4X magnification) using an Olympus Provis AX70 microscope (Leeds Precision Instruments, Inc., Minneapolis, MN, USA) fitted with a DP70 digital camera.
Figure. 2. IFNγ-deficient DRB1*0301.DQ8 show milder spinal cord pathology than IFNγ-sufficient DRB1*0301.DQ8 Tg mice.

A) Representative photomicrograph of inflammatory and demyelinating lesions in the spinal cord (10X and 40X) of DRB1*0301.DQ8, DRB1*0301.DQ8.IFNγ−/−, DRB1*0301, and DRB1*0301.DQ8.IFNγ−/− with EAE. DRB1*0301.DQ8.IFNγ−/− Tg mice with disease showed mild inflammation and demyelination in spinal cord in contrast to severe inflammation and demyelination observed in IFNγ-sufficient DRB1*0301.DQ8 Tg mice. However, the spinal cord pathology was similar between DRB1*0301 and DRB1*0301.IFNγ−/− mice with EAE. B) Quantitative analysis of spinal cord pathology also showed that IFNγ-deficient mice (n=10) with disease had a lower pathology score (percent of spinal cord quadrants showing inflammation and demyelination for each mouse (mean ± SD)) than DRB1*0301.DQ8 Tg mice (n=10). * p<0.01 compared to inflammation and demyelination in DRB1*0301.DQ8 Tg mice with disease. These figures represent one of three experiments performed at different time points. These figures represent one of three experiments performed at different time points. EAE was induced in different strains and monitored for disease as described in methods. The animals were sacrificed at day 25 post-immunization for CNS histopathology. The image was taken (10X and 40X magnification) using an Olympus Provis AX70 microscope (Leeds Precision Instruments, Inc., Minneapolis, MN, USA) fitted with a DP70 digital camera.
DRB1*0301.DQ8.IFNγ−/− mice with EAE show severe demyelination but relative preservation of axons
To confirm demyelination of axons, we performed Luxol Fast Blue (LFB) staining which stains lipoproteins in myelin and gives them a blue appearance under the microscope. LFB staining, of paraffin-embedded brain tissues showed severe loss of LFB staining indicating myelin loss in DRB1*0301.DQ8.IFNγ−/− compared to DRB1*0301.DQ8 Tg mice. Severe inflammation and demyelination was also observed in the brain stem of DRB1*0301.DQ8.IFNγ−/− mice with EAE compared to only mild inflammation in DRB1*0301.DQ8 mice with EAE (Fig 3A and B). As shown in Fig 3C, whereas DRB1*0301.DQ8 mice with EAE had perivascular inflammation and some demyelination in the cerebellum, DRB1*0301.DQ8.IFNγ−/− mice had myelin loss in significantly larger portions of the cerebellum (Fig 3D) and brain stem (Fig 3B). All the myelin was destroyed within some lobules of the cerebellum (Fig 3D). In addition to the cerebellum and brain stem, loss of myelin was also observed in the striatum, fornix, and sub-cerebrum (data not shown).
Figure 3. DRB1*0301.DQ8.IFNg−/− mice with EAE show severe demyelination but relative axonal preservation in the brain tissue.
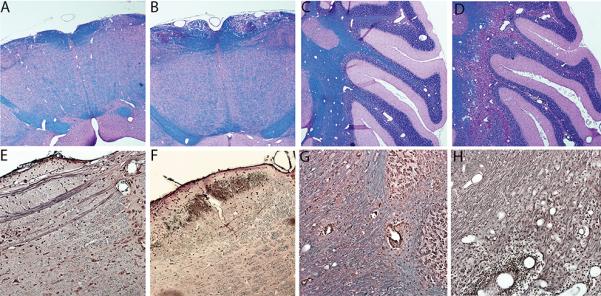
Representative photomicrograph showing demyelination (LFB stain) (A-D) and loss of axons (Bielchowsky silver impregnation) (E-H) in the brain of DRB1*0301.DQ8 and DRB1*0301.DQ8.IFNγ−/− Tg mice with EAE. IFNγ-deficient DRB1*0301.DQ8.IFNγ−/− with disease showed more demyelination (loss of blue color) in brain stem (B) and cerebellum (D) than IFNγ-sufficient DRB1*0301.DQ8 Tg mice (A and C). Despite severe demyelination, DRB1*0301.DQ8.IFNγ−/− Tg mice with disease showed axonal loss in the brain stem (F) but relative axonal preservation in the cerebellum (H) except in the areas with extensive inflammation (→). No axonal loss was observed in DRB1*0301.DQ8 Tg mice with EAE (E and G). EAE was induced in different strains and monitored for disease as described in Methods. Mice were sacrificed at day 25 post-immunization for CNS histopathology. The image was taken (10X magnification) using an Olympus Provis AX70 microscope (Leeds Precision Instruments, Inc., Minneapolis, MN, USA) fitted with a DP70 digital camera.
We also performed Bielschowsky (Biel) staining to investigate if demyelination was associated with axon loss in DRB1*0301.DQ8.IFNγ−/− mice. DRB1*0301.DQ8 mice with EAE showed preserved axon tracts in the brain stem (Fig 3E) and cerebellum in demyelinated areas (Fig 3G). IFNγ-deficient DRB1*0301.DQ8 animals with severe demyelination showed some axonal loss in the brain stem (Fig 3F), whereas axons in the cerebellum were preserved except in areas with extensive inflammation. Thus the data indicate that absence of IFNγ leads to severe brain pathology characterized by inflammation and demyelination but relatively preserved axons.
CD4+ T cells from IFNγ-deficient mice proliferate strongly and have a distinct cytokine and chemokine profile
Antigen-specific CD4+ T cells of both Th1 and Th17 subtype have been shown to be responsible for initiation of disease in the EAE model. Therefore, we analyzed T-cell proliferative responses and levels of different inflammatory cytokines in an attempt to identify the mechanism for this unique form of EAE in IFN-γ-deficient mice. Although the numbers of total cells were similar between the two groups, we observed increased PLP91-110 peptide-specific CD4 T-cell proliferation in IFNγ-deficient DRB1*0301.DQ8 mice (Fig 4A). HLA-DRB1*0301.IFNγ−/− single Tg mice also showed increased T cell proliferation compared to IFNγ sufficient HLA-DRB1*0301 Tg mice (supplemental Fig. 1A). However, among all the groups, the strongest T cell proliferative response was observed in DRB1*0301.DQ8.IFNγ −/− double Tg mice (supplemental Fig. 1A). We have previously shown that DRB1*0301.DQ8 mice with EAE produce both Th1 as well as Th17 cytokines; however IL-17 is the major cytokine as neutralization of IL-17 abrogated disease in DRB1*0301.DQ8 Tg mice (11). Cytokine analysis showed that both IFNγ-deficient DRB1*0301.DQ8.IFNγ−/− and IFNγ-sufficient DRB1*0301.DQ8 Tg mice produced Th17 cytokines such as IL-17 (Fig 4B), GMCSF (Fig 4B), IL-6, and IL-12 (data not shown). However, levels of these pro-inflammatory cytokines were significantly higher in DRB1*0301.DQ8.IFNγ−/− compared to DRB1*0301.DQ8 mice (Fig 4B and D), indicating that the absence of IFNγ caused increased levels of these pro-inflammatory cytokines. HLA-DRB1*0301.IFNγ−/− single Tg mice also showed higher levels of IL17 and GMCSF compared to IFNγ sufficient HLA-DRB1*0301 Tg mice (supplemental Fig. 1B and 1D). However, the levels of both IL17 and GMCSF were highest in DRB1*0301.DQ8.IFNγ −/− double Tg mice (supplemental Fig. 1B and 1D). To confirm that CD4+ T cells are the main source of IL-17 and IFNγ, we analyzed intracellular levels of these cytokines. Splenocytes from PLP91-110- immunized mice were stimulated in vitro in the presence or absence of the same antigen. After three days, cells were stimulated with PMA and ionomycin for 5 hours and followed by addition of Golgistop after 1 hour. Intracellular levels of IL-17 and IFNγ on CD4+ gated T cells were analyzed by flow cytometry. As shown in Fig 4E, CD4+ T cells from DRB1*0301.DQ8.IFNγ−/− mice produced higher levels of IL-17 (4.8±1.0 vs. 2.2±0.7, p<0.05) compared to DRB1*0301.DQ8 mice, but DRB1*0301.DQ8 mice produced significantly higher levels of IFNγ (6.0±1.6 vs. 1.0±0.1, p>0.01).
Figure. 4. Increased proliferation and production of pro-inflammatory mediators in antigen-specific CD4 T cells from IFNγ-deficient mice compared to IFNγ-sufficient mice.

A) IFNγ-deficient DRB1*0301.DQ8.IFNγ−/− showed stronger T-cell proliferation than IFNγ-sufficient DRB1*0301.DQ8 Tg mice. For measurement of antigen specific T cell recall response, splenocytes were isolated from PLP91-110 immunized DRB1*0301.DQ8 and DRB1*0301.DQ8.IFNγ−/− Tg mice peptide and then cultured with or without (control) PLP91-110 peptide for 48 h. The proliferative response was assessed by pulsing the cultures with [3H] thymidine for the last 16 h. The data are presented as the mean CPM ± SD and are the average of three independent experiments. DRB1*0301.DQ8.IFNγ−/− with EAE showed higher levels of IL-17 (B) and GM-CSF (D) than IFNγ-sufficient DRB1*0301.DQ8 Tg mice. As expected, only IFNγ-sufficient mice produce IFNγ (C), whereas no IFNγ was observed in DRB1*0301.DQ8.IFNγ−/− Tg mice. E) Flow-cytometry analysis for intra-cellular cytokines confirmed that CD4+ T cells from IFNγ-deficient mice produce higher levels of IL-17 than IFNγ-sufficient mice. At the same time, DRB1*0301.DQ8 mice produce higher levels of IFNγ. The accompanying bar diagram shows the average frequency of IL-17+ or IFNγ+ cells in DRB1*0301.DQ8 or DRB1*0301.DQ8.IFNγ−/− Tg mice (n=3). F) CD4+ T cells from DRB1*0301.DQ8.IFNγ−/− Tg mice showed higher protein levels of IL-1β, IL-3, IL-5, IL-6, IL-13, IL-17, G-CSF, GM-CSF, KC and TNF-α relative to CD4+ T cells from DRB1*0301.DQ8 mice, whereas CD4+ T cells from DRB1*0301.DQ8 Tg mice with classical EAE produced higher levels of IL-4, IL-12p40, IFNγ and MCP-1. Cytokines level in culture supernatant were determined using mouse cytokine 23-plex protein bead array system as described in methods. G) CD4+ T cells from DRB1*0301.DQ8.IFNγ−/− Tg mice showed higher expression levels of CCR4, CCR6, CXCR2 and CXCR4 relative to CD4+ T cells from DRB1*0301.DQ8 mice. Whereas CD4+ T cells from IFNγ-sufficient mice with classical EAE showed increase in levels of CCR5 and CXCR3. Cytokines and chemokine receptor expression were analyzed in RNA from CD4+ T cells isolated from IFNγ-deficient or sufficient mice using Real-time PCR, and quantified using the threshold cycle (Ct) method normalized for the house keeping genes β-actin, GADPH and HPRT.
To further characterize the CD4+ T cell specific cytokines and chemokines responsible for the atypical EAE and brain specific pathology, we isolated splenic-CD4+ T cells from IFNγ-deficient mice with atypical-EAE and IFNγ-sufficient mice with classical EAE. CD4+ T cells were stimulated in in-vitro cultures with PLP91-110 and levels of various cytokines, chemokines, chemokine receptors, and transcription factors were analyzed at protein level (Bio-plex) or mRNA level (Real-time RT-PCR). CD4+ T cells from DRB1*0301.DQ8.IFNγ−/− Tg mice with atypical EAE had higher protein levels of IL-1β, IL-3, IL-5, IL-6, IL-13, IL-17, G-CSF, GM-CSF, KC, and TNF-α (Fig. 4F), whereas CD4+ T cells from DRB1*0301.DQ8 Tg mice with classical EAE produced higher levels of IL-4, IL-12p40, IFNγ, and MCP-1 (Fig. 4F). Analysis of mRNA showed that CD4+ T cells from IFNγ-deficient mice showed higher levels of the above cytokines as well as an increase in IL-11, IL-12p40, IL-22, CCR4, CCR6, CXCR2, and CXCR4, whereas CD4+ T cells from IFNγ-sufficient mice with classical EAE showed an increase in IL-15, CCR5, and CXCR3 beside the inflammatory mediators observed at protein level (Fig. 4G and supplemental table 1). Thus, these data indicate that the severe brain specific disease in DRB1*0301.DQ8.IFNγ−/− mice is due to the increased ability of antigen-specific CD4+ T cells to proliferate as well as its ability to produce pro-inflammatory cytokines and express specific chemokine receptors.
Increased encephalitogenic capacity of IFNγ-deficient mice is due to CD4+ T cells, not APCs
CD4+ T cells from DRB1*0301.DQ8.IFNγ−/− mice with EAE showed increased proliferation of and higher Th17 cytokines, which can be due either to increased antigen presentation of IFNγ-deficient mice or increased proliferative and IL-17/GM-CSF-producing capacity of CD4+ T cells. To identify the cell population responsible for the increased encephalitogenic CD4+ T cells, we isolated CD4+ T cells, CD11b+ monocytes, CD19+B cells and CD11c+ DCs from IFNγ-sufficient or -deficient DRB1*0301.DQ8 Tg mice and performed an in vitro antigen-presentation assay in a crisscross manner. As shown in Fig 5A, when CD4+ T cells from DRB1*0301.DQ8 or DRB1*0301.DQ8.IFNγ−/− Tg mice with EAE were cultured with CD11c+ as APCs from either of these two strains, CD4+ T cells from IFNγ- deficient DRB1*0301.DQ8 mice (DRB1*0301.DQ8.IFNγ−/−) showed higher proliferation in response to recall antigens compared to those from IFNγ-sufficient mice. As monocytes/macrophages have been shown to play an important role in EAE pathogenesis, we isolated CD11b+ monocytes from IFNγ-sufficient or IFNγ-deficient mice and performed antigen presentation. As shown in Fig 5B, culturing of CD11b+ monocytes with CD4+ T cells from either IFNγ-sufficient or IFNγ-deficient mice induced similar T cell proliferation. We did not observe any difference in CD4+ T-cell proliferation when B cells from IFNγ-sufficient or IFNγ-deficient mice were used as APCs, (data not shown). Analysis of cytokines showed that CD4+ T cells from DRB1*0301.DQ8.IFNγ−/− mice produced higher levels of IL-17 and GM-CSF when cultured with CD11c+ DCs from either strains compared to CD4+ T cells from DRB1*0301.DQ8 mice (Fig 5C and D). Similar difference in the cytokine levels of IL-17 and GM-CSF between the two groups were observed, when CD11b+ monocytes or CD19+ B cells were used as antigen presenting cells (data not shown). Thus our antigen-presentation studies indicate that increased proliferative and IL-17/GM-CSF- producing capacity in IFNγ-deficient mice were due to the proliferating ability of CD4+ T cells rather than the increased antigen-presenting function of APCs such as B cells, CD11b+ monocytes, or CD11c+ DCs. To further confirm that atypical EAE and brain specific pathology in IFNγ-deficient mice is due to inability of immune cells to produce IFNγ, we performed bone marrow chimera experiments. IFNγ-sufficient DRB1*0301.DQ8 mice and IFNγ-deficient DRB1*0301.DQ8 mice were lethally irradiated (600 rads 4 hrs. apart) and reconstituted with BM cells from IFNγ-deficient or- sufficient mice respectively. Six week post adoptive transfer, reconstitution was confirmed and EAE was induced in mice using standard protocol. IFNγ-sufficient mice reconstituted with BM from IFNγ-deficient mice showed both classical and atypical EAE (Table II) with both severe brain and spinal cord pathology (data not shown). DRB1*0301.DQ8.IFNγ−/− Tg mice reconstituted with BM from IFNγ-sufficient mice develop classical EAE (Table II) with severe spinal cord pathology and mild brain pathology (data not shown). These data demonstrate that absence of IFNγ leads to atypical EAE and IFNγ induced pathways protect brain from severe pathology, whereas presence of IFNγ leads to the development of classical EAE with severe spinal cord pathology.
Figure. 5. Increased encephalitogenic capacity of IFNγ-deficient mice is due to CD4+ T cells and not APCs.

A) CD4+ T cells from DRB1*0301.DQ8.IFNγ−/− Tg mice showed significantly more proliferation than CD4+ T cells from DRB1*0301.DQ8 Tg mice. CD11c+ APCs (A) or CD11b+ APCs (B) from either IFNγ-sufficient or - deficient mice showed similar capacity to induce proliferation of CD4+ T cells. (C) CD4+ T cells from DRB1*0301.DQ8.IFNγ−/− Tg mice produced higher levels of IL-17 whereas CD11c+ APCs from both strains induce similar levels of IL-17 from CD4 T cells. (D) Similarly CD4+ T cells from DRB1*0301.DQ8 IFNγ−/− Tg mice produced higher levels of GM-CSF compared to DRB1*0301.DQ8 Tg mice, whereas there was no difference in ability of CD11c+ APCS from either IFNγ-sufficient or -deficient mice to induce GM-CSF. CD4+ T cells and CD11c+ DCs were isolated from PLP91-110 immunized DRB1*0301.DQ8 or DRB1*0301.DQ8.IFNγ−/− Tg mice at day 20 post-immunization and cultured in different combinations as depicted in the figures. The proliferative response was assessed by pulsing the cultures with [3H]thymidine for the last 16 h, whereas cytokine levels were measured using commercially available ELISA. The data are from three independent experiments combined.
Table II.
Incidence of atypical and classical EAE in BM chimera experimenta
| Host strain | Donor strain | Disease incidence (%) | Mean onset of disease ±SD (Days) | Classical EAE with paralysis | Atypical EAE | Classical and atypical EAE |
|---|---|---|---|---|---|---|
| DR3.DQ8.AEo | DR3.DQ8.IFNg-/-AEo | 8/10(80%) | 14±1.8 | 3/10 | 8/10 | 3/10 |
| DR3.DQ8.IFNg-/-AEo | DR3.DQ8.AEo | 9/10 (90%) | 11.±1.0 | 9/10 | 0/10 | 0/10 |
IFNγ sufficient and deficient DR3.DQ8 Tg mice were lethally irradiated (600 rads 4 hrs apart) and were reconstituted with BM from strain indicated in the table. Six weeks post-reconstitution, mice were immunized with 100μg of PLP91-110, and examined over time for clinical signs of classical or atypical EAE as stated in material and methods.
Increased CD11b+ monocytes and neutrophil in CNS of IFNγ-deficient mice compared with IFNγ-sufficient mice
CD4+ Th17 cells producing IL-17 attract polymorphic nuclear cells (PMNLs) such as brain neutrophils, leading to increased CNS pathology (25). Therefore, we determined the cellular profile of immune cells in both the spleen and central nervous systems in order to identify the cell population responsible for the brain- restricted pathology in DRB1*0301.DQ8.IFNγ−/− mice. Both IFNγ-sufficient and -deficient mice with EAE had similar numbers of cells in the spleen (Fig 6A); however, DRB1*0301.DQ8.IFNγ−/− mice had more brain-infiltrating leukocytes (BILs) than IFNγ-sufficient DRB1*0301.DQ8 mice (Fig 6B). Of the immune cells, we specifically observed increased frequency of CD4+ T cells, CD11b+ cells, and 7/4+ neutrophils in IFNγ-deficient mice compared with IFNγ-sufficient mice (Fig. 6C). DRB1*0301.DQ8.IFNγ−/− mice had fewer B220+ B cells and CD8+ T cells than DRB1*0301.DQ8 mice with EAE (Fig 6C). Spinal cord infiltrating cells (SCILs) from DR3.DQ8 mice with classical EAE had a cellular profile similar to BILs from the same strain except an increase in number of CD11b+ cells. The major difference between two groups was reduced number of B cells and CD8 T cells in IFNγ-deficient mice with atypical EAE. We were not able to get enough cells from DRB1*0301.DQ8.IFNγ−/− mice with atypical EAE to perform cellular profile analysis in SCILs. Cytokine analysis of BILs showed DRB1*0301.DQ8.IFNγ−/− mice with EAE have higher levels of IL-17 (Fig 6D) and GM-CSF (Fig 7E) than DRB1*0301.DQ8 mice with EAE (6D). We also performed intra-cellular analysis of BILs to confirm that CD4+ T cells are the main source of IL-17 and GM-CSF. BILs from PLP91-110- immunized mice were stimulated with PMA and ionomycin for five hours and followed by addition of Golgistop after 1 hour. Intracellular levels of IL-17, GM-CSF, IL-6, and TNF-α on CD4+ gated T cells were analyzed by flow cytometry. As shown in Fig 6F, BILs-CD4+ T cells from DRB1*0301.DQ8.IFNγ−/− mice produced higher levels of IL-17 (18±4.0 vs. 3.0±1.0, p<0.05), GM-CSF (20±3.6 vs. 3.5±1.5, p<0.05), and IL17+GM-CSF+ double positive cells (11±2.6 vs. 0.8±1.0, p<0.01) compared to IFNγ-sufficient mice. CD4+ T cells from BILs of IFNγ-deficient mice also showed higher levels of IL-6 and TNF-α (data not shown). However, spinal cord infiltrating cells from IFNγ-sufficient DRB1*0301.DQ8 mice produced higher levels of IFNγ (5.0±1.2 vs. 1.0±0.4, p>0.05) and IL-17 (6.0±1.6 vs. 1.5±0.6, p>0.05) compared to IFNγ-deficient mice (data not shown). Thus, the above data indicate that the severe brain specific disease in DRB1*0301.DQ8.IFNγ−/− mice is due to the increased ability of antigen-specific CD4+ T cells to proliferate as well as their ability to produce higher levels of both IL-17, and GM-CSF.
Figure. 6. Increased CD11b+ monocytes and neutrophil in CNS of IFNγ-deficient mice compared to IFNγ-sufficient mice.

(A) While number of splenocytes were similar between DRB1*0301.DQ8 or DRB1*0301.DQ8.IFNγ−/− Tg mice with EAE, higher number of brain-infiltrating leukocytes (BILs) were observed in DRB1*0301.DQ8.IFNγ−/− Tg mice with atypical EAE than in DRB1*0301.DQ8 mice with EAE (B). (C) Cellular subset analysis of BILs and SCILs (spinal cord infiltrating leukocytes) showed that IFNγ-deficient mice with EAE had a distinct cellular profile in brain with dominance of CD11b+ monocytes and A7/4+ neutrophils. Since, we could not get enough cells from spinal cord of IFNγ-deficient mice with atypical EAE; SCILs data only from DR3DQ8 is shown. DRB1*0301.DQ8.IFNγ−/− Tg mice with EAE produced higher levels of IL-17 (C) and GM-CSF (D) than DRB1*0301.DQ8 with EAE. (F) BILs from IFNγ deficient animals with disease had higher frequency of IL17+, GM-CSF+ and IL17+GM-CSF+ CD4+ T cells compared to IFNγ sufficient mice with EAE. G) Brain tissue from DRB1*0301.DQ8.IFNγ−/− Tg mice showed higher expression levels of MIP-1α, CCL7, CCL8, CCL11, CCL17 and CCL19 relative to brain tissue from DRB1*0301.DQ8 mice. Whereas spinal cord tissue from IFNγ-sufficient DRB1*0301.DQ8 mice with classical EAE showed increase in levels of CCL5, CXCL9, CXCL10 and CXCL12. EAE was induced in HLA-Tg mice and BILs /SCILs were isolated at day 25 post-immunization using percol gradient. Cellular subsets were analyzed using flow cytometry using specific antibodies. For cytokine analysis at protein levels, BILs were stimulated with or without PLP91-110 for 48 h in vitro culture and levels of IL-17 and GM-CSF were determined as described. Intracellular levels of IL-17 and GM-CSF were analyzed by flow-cytometry using cytokine specific antibodies as described in methods. Cytokines and chemokine receptor expression were analyzed using Real-time PCR in total RNA isolated from brain and spinal cord tissues of IFNγ-deficient or sufficient mice, and quantified using the threshold cycle (Ct) method normalized for the house keeping genes β-actin, GADPH and HPRT. The data represent three separate experiments done in triplicate.
Figure. 7. Demyelinating regions in IFNγ-deficient mice showed increased presence of CD68+ monocytes/microglia cells.
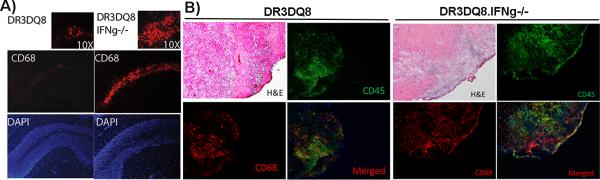
A) Representative photomicrograph showing CD68+ cells within the demyelinating region of the brain. DRB1*0301.DQ8.IFNγ−/− Tg mice with severe demyelination in the cerebellum region showed presence of CD68+ monocyte/microglial cells along the whole lobule of cerebellum. Whereas, DRB1*0301.DQ8 mice showed only perivascular presence of CD68+ cells. B) CD45 and CD68 dual staining showing that DRB1*0301.DQ8.IFNγ−/− Tg mice with severe demyelination had both CD45+CD68+ infiltrating monocytes and CD45-CD68+ microglia, whereas majority of CD68+ cells in the DRB1*0301.DQ8 mice with classical EAE were CD45+. DRB1*0301.DQ8.IFNγ−/− Tg mice also had larger area with inflammation and demyelination compared to DRB1*0301.DQ8 mice with severe demyelination. EAE was induced in HLA Tg mice, and at day 25 post-immunization mice were sacrificed. Brain tissue was isolated, snap frozen in liquid nitrogen and embedded in Tissue-Tek® OCT compound. 10 μm thick section were stained for CD68 as mentioned in material and methods. The stained sections were analyzed by Olympus Provis AX70 microscope (Leeds Precision Instruments, Inc., Minneapolis, MN, USA) fitted with a DP70 digital camera at 10X or 40X magnification.
Presence of specific chemokines in brain vs. spinal cord determined regional localization of inflammation and demyelination
To decipher the mechanism responsible for localization of brain specific pathology characterized with atypical EAE in IFNγ-deficient mice vs. classical EAE with spinal cord pathology in IFNγ-sufficient mice, we performed mRNA expression analysis of various chemokine ligands and cytokines in brain and spinal cord tissue. As shown in Fig 6G, higher levels of MIP-1α, CCL7, CCL8, CCL17, and CCL19 chemokines were observed in brain tissue of IFNγ-deficient mice compared to the brain tissue of IFNγ-sufficient mice. Whereas spinal cord tissue from DRB1*0301.DQ8 mice with classical EAE showed a relative increase in the expression levels of CCL5, CXCL9, CXCL10, and CXCL12 compared to IFNγ-deficient mice (Fig 6H). Levels of CXCL12 were similar between two groups within the brain tissue. We also analyzed BILs and SCILS from both groups and observed a relative increase in mRNA levels of IL-6, IL-11, IL-17, IL-22, IL-23, and TNF-α in BILs from IFNγ-deficient mice compared to IFNγ-sufficient mice, whereas the latter had increased levels of IL-1β, IL-15, and IFNγ (supplemental table 1). SCILs from IFNγ-sufficient mice showed higher transcript levels of IL-1β, IL-10, IL-12p35, IL-17 and IFNγ (supplemental table 1). These data demonstrate that the presence of specific cytokines and chemokines within the brain or spinal cord might determine the differential disease in IFNγ-sufficient vs deficient mice.
Immunohistochemistry of brain tissue confirms presence of monocytes/microglia in the CNS of IFNγ-deficient mice with EAE
To further characterize the cells present in demyelinating areas in the brain, frozen sections from both IFNγ-sufficient and -deficient mice with EAE were stained with antibodies specific for CD4, CD8, CD68 (monocyte/microglia) and A7/4 (neutrophil). CD4+ and CD8+ T cells were present around the perivascular inflammatory region and meningeal lining in both strains (data not shown). However, demyelinating regions in the cerebellum were specifically populated by CD68+ cells, suggesting an important role for monocytes/microglia in inducing demyelination (Fig 7A). We observed the presence of neutrophils along the borders of demyelinated regions; however, they were significantly less than CD68+ cells. To further analyze whether CD68+ cells are peripheral monocytes or brain resident glial cells, we stained with brain tissue with CD68 and CD45. While IFNγ KO mice with atypical EAE had large area with inflammation and demyelination, IFNγ sufficient mice had small areas with pathology (Fig 7B). Within the brain tissue with inflammation and demyelination, both CD45+CD68+ and CD45-CD68+ cell population were present indicating importance of both monocytes as well as microglia in the pathology (Fig 7B). In the brain of DR3DQ8 mice with classical EAE, majority of CD68+ cells were positive for CD45 indicating a role for only monocyte population. Thus, our immunohistochemistry data indicate that CD68+ monocytes/microglia are the major cells in the demyelinating lesions of IFNγ-deficient DRB1*0301.DQ8 mice.
Discussion
We have previously shown that IFNγ plays a protective role in PLP91-110-induced EAE in disease-susceptible HLA-DRB1*0301 mice (10). In this study, we have extended the findings by generating IFNγ-deficient HLA-DRB1*0301.DQ8 (HLA-DRB1*0301.DQ8.IFNγ−/−) Tg mice and showed that the absence of IFNγ leads to atypical EAE characterized by ataxia and gait abnormalities. HLA-DRB1*0301.DQ8.IFNγ−/− mice with EAE showed inflammation and demyelination restricted to brain, compared with the brain and spinal cord pathology observed in IFNγ-sufficient DRB1*0301.DQ8 Tg mice. The inflammation and demyelination in the brain was also more severe and widespread in HLA-DRB1*0301.DQ8.IFNγ−/− than in HLA-DRB1*0301.DQ8 Tg mice. The demyelination in our model correlated with relative preservation of axons. Antigen-presentation assays revealed that severe pathology in HLA-DRB1*0301.DQ8.IFNγ−/− Tg mice was due to increased encephalitogenicity of IFNγ-deficient antigen- specific CD4+ T cells. These CD4+ T cells caused an increase in inflammation and demyelination through production of pro-inflammatory cytokines IL-17, GM-CSF, IL-6, and TNF-α, which helped in the recruitment of inflammatory cells into CNS and/or the activation of resident microglia. The role of IFNγ in inflammatory and demyelinating diseases is not well understood and is hypothesized to have both a pathogenic as well as protective role. Our study indicates that i) IFNγ is not required for development of disease; ii) its absence leads to development of atypical EAE characterized by ataxia, gait problem, dystonia, and weight loss; iii) IFNγ-deficient mice with EAE, develop brain-specific inflammation and demyelination; and iv) CD68+ monocytes and microglia plan an important role in inducing the brain pathology.
Previous studies have shown that absence of either the IFNγ or IL-12 signaling pathway leads to more severe neurological disease (26, 27). Ferber et al (26) and Krakowski and Owens (27) used IFNγ- deficient mice with B10.PL (H-2u) and BALB/c (H-2d) backgrounds to show that the absence of IFNγ leads to the development of severe classical EAE. In contrast to these studies, we observed development of atypical EAE characterized by ataxia, circling (ataxia-rotation), gait problem, and dystonia in our IFNγ-deficient double Tg strains. Abromson-Leeman et al (22) reported the development of similar ataxia-rotation, atypical disease in BALB/c IFNγ KO animals on immunization with MBP Exon 2 peptide. Wensky et al. (24) also reported atypical EAE in MBP-specific, TCR transgenic mice lacking IFNγ. However, in both studies, atypical disease was attributed to extensive inflammation in the lateral medullary regions of brain. None of these studies mentioned or showed demyelination. Our study is the first to show that atypical EAE in IFNγ-deficient DRB1*0301.DQ8 mice correlates with demyelination in cerebellum, brain stem, as well as striatum and septal nuclei region of the forebrain. The maximal demyelination was observed in the cerebellum region as LFB staining revealed the destruction of approximately half of the myelin sheath within some lobules of the cerebellum. Myelin within the simple lobule and inferior semilunar lobule region of cerebellum showed complete loss in IFNγ-deficient mice compared to mild perivascular inflammation in IFNγ-sufficient mice. Despite the severe demyelination observed in IFNγ- deficient DRB1*0301.DQ8 Tg mice, the majority of the axons were preserved, and only the area with extensive inflammation showed axonal degeneration. Thus, our study confirms a strong neuro-protective role of IFNγ, whereas its absence leads to severe inflammation and demyelination in the unique parts of the brain. IFNγ deficient DRB1*0301 single Tg mice developed both atypical EAE as well as classical EAE and showed higher degree of inflammation and demyelination compared to IFNγ sufficient DRB1*0301 single Tg mice. However, the pathology was more severe in IFNγ deficient DRB1*0301.DQ8 double Tg mice compared to DRB1*0301.IFNγ−/− single Tg mice. This together with failure of the HLA-DQ8 Tg mice to develop EAE either in IFNγ sufficient or deficient mice indicate that whereas HLA-DR3 is required for susceptibility of disease, the presence of DQ8 molecule worsen the disease. We have previously shown that HLA-DQ8 restricted Th17 cells exacerbate EAE in the HLA-DRB1*0301.DQ8 Tg mice (11). As IFNγ is known to regulate Th17 response (28), absence of IFNγ might cause an increase in the DQ8 restricted Th17 cell response. The increased expansion of DQ8 restricted Th17 subset together with expansion of DR3-restricted encephalitogenic CD4 T cells, could lead to increased trafficking of inflammatory cells to CNS as well as production of pro-inflammatory cytokines and chemokines resulting in a severe brain pathology in DRB1*0301.DQ8.IFNγ−/− double Tg mice. Thus in the presence of disease susceptible HLA-class II molecule (e.g. HLA-DRB1*0301), other class II molecule (e.g. HLA-DQ8 in the present study) can influence the outcome of the disease through epistatic interaction in trans.
DRB1*0301.DQ8.IFNγ−/− with EAE had more brain-infiltrating cells (BILs) than IFNγ- sufficient DRB1*0301.DQ8 mice. Flow-cytometry analysis of BILs showed increased frequency of monocytes and neutrophils in IFNγ-deficient mice compared to IFNγ-sufficient DRB1*0301.DQ8 Tg mice. Increased levels of IL-17 and GM-CSF in BILs of DRB1*0301.DQ8.IFNγ−/− mice suggest an important role for these cytokines in recruiting monocytes and neutrophils to the brain. Recent studies have shown that high IL-17-to-IFNγ ratio leads to brain-specific pathology, whereas high IFNγ-to-IL-17 ratio leads to spinal cord pathology (17, 23). The increased brain-specific pathology in DRB1*0301.DQ8.IFNγ−/− mice was accompanied with the increased levels of IL-17, thus supporting a major role for IFNγ in localizing pathology in brain vs. spinal cord Previous studies have shown that neutrophil recruitment to inflammatory sites is a major function of the IL-17 cytokine (29). However, we observed an increased presence of CD68+ inflammatory monocytes in demyelinating areas, whereas neutrophils were present only around perivascular areas, which suggests that CD68+ monocytes/microglia might be the major pathogenic cells in our demyelinating model. Our CD45 and CD68 dual staining of the brain tissues indicated that both infiltrating monocytes and brain resident microglia play a role in inducing severe pathology in IFNγ deficient mice with atypical EAE. Both infiltrating monocytes and microglia have been shown to be involved in the CNS demyelination with monocytes derived inflammatory macrophages causing the destruction of myelin whereas microglia derived macrophages are responsible for clearing the debris (30). Presence of inflammatory monocytes in demyelinating region, is also in agreement with an earlier study from Lafaille's group (24), who showed that adoptive transfer of MBP specific TCR Tg T cells lacking IFNγ into RAG-deficient mice leads to development of atypical EAE characterized with increased presence of monocytes in brain. However, because they did not analyze levels of IL-17 and GM-CSF in their system, we cannot compare requirement of IL-17 and GM-CSF between these two models.
IFNγ also regulates T-cell response, specifically IL-17 levels, in the peripheral nervous system (28). Indeed, we observed higher T-cell proliferation and increased levels of IL-17 in vitro in response to PLP91-110 in IFNγ-deficient DRB1*0301.DQ8 Tg mice compared to IFNγ sufficient DRB1*0301.DQ8 Tg mice. The increased proliferation and IL-17 levels observed in IFNγ-deficient DRB1*0301.DQ8 mice were due to the ability of CD4+ T cells to proliferate more vigorously as antigen presentation by APCs from both IFNγ-sufficient and -deficient mice were similar. PLP91-110- specific CD4+ T cells from IFNγ-deficient mice also produced higher levels of GM-CSF. IFNγ can regulate peripheral immune response by keeping the antigen-specific CD4+ T-cell response in check through activation-induced death of T cells (31). Previous studies have shown that IFNγ loss results in the expansion of auto-reactive CD4 T cells and development of severe EAE (32). The increased IL-17 observed in the absence of IFNγ supports previous reports showing that IFNγ down-regulates the number and function of Th17-specific CD4+ T cells (28). Thus, the absence of IFNγ leads to increase in the frequency of encephalitogenic Th17 cells producing IL-17 and GM-CSF, which results in increased recruitment of inflammatory cells to the brain. We observed a high number of CD4+ T cells producing both IL-17 and GM-CSF (IL17+GM-CSF+ CD4+ T cells) in the brain infiltrate from IFNγ-deficient mice. Recently, it has been suggested that CD4+ T cells producing both IL17 and GM-CSF are more encephalitogenic than single IL17+ or GM-CSF+ cells (33). GM-CSF is required for induction of IL-6 and IL-23, which plays an important role in the generation and maintenance of encephalitogenic Th17 cells. We also observed higher levels of IL-3 and IL-12 expression in brain tissue as well as in BILs. Transgenic expression of CNS specific IL-3 (34) or IL-12 (35) has been shown to cause ataxia like symptoms with cerebellum specific pathology in animals. Thus in the absence of IFNγ-induced protective factors, increased expression of GM-CSF, IL-3, IL-6, and IL-12 play an important role in activation of the inflammatory cascade in the brain of IFNγ-deficient mice, which leads to severe demyelination. Additionally, we also observed higher of levels of IL-5 and IL-13 in CD4+ T cells from IFNγ-deficient mice, indicating their importance in disease pathogenesis. Both of these cytokines have been shown to have pro-inflammatory characteristics and can cause CNS specific inflammatory disease (24, 36).
Our data further demonstrates that presence of specific chemokines in the brain vs spinal cord determines the regional localization of pathology. We observed that brain tissue from IFNγ-deficient mice with atypical EAE had higher levels of chemokines such as CCL7, CCL8, CCL19, and CCL21, whereas spinal cord from IFNγ sufficient mice with classical EAE showed higher levels of CCL5, CXCL9, and CXCL10. CCL7 and CCL8 are known to bind CCR2 and attract inflammatory cells such as monocytes and/or polymorphic nuclear cells (PMNLs) (37). CCL19 and CCL21 attract memory CD4+ T cells especially Th17 cells through CCR7 receptors (38). CD4+ T cells and BILs from IFNγ-deficient mice had higher expression levels of CCR2, CCR4 and CCR7. In contrast, CD4+ T cells and SCILs from IFNγ-sufficient mice showed higher levels of CXCR3 and CCR5. IFNγ is known to induce CXCR3 (39) whereas IL-12/IFNγ induce CCR5 (40). Both these receptors are considered to be specific for Th1 cells. CCL5 attracts immune cells to inflammatory region by binding to CCR5, whereas CXCL9 and CXCL10 do so by binding to CXCR3 receptors. Thus, distinct chemokines/chemokine receptor combinations direct regional pathology in EAE. Once recruited to their specific tissue, cytokines and other chemical mediators such as reactive oxygen species (ROS) and nitric oxide (NO) determine the final outcome through modulation of the inflammatory cascade, leading to demyelination and tissue injury.
In normal circumstances, IFNγ interacts with brain resident cells to induce neurotrophins (brain- protective factors) such as glial cell line-derived neurotrophic factor (GDNF), insulin-like growth factor-1 (IGF-1), nerve growth factors (NGF), brain derived neurotropic factors (BDNF), and neurotrophic cytokines- such as ciliary neurotrophic factors (CNTF) and leukemia inhibitory factor (LTF). These factors protect the myelin sheaths and axons from inflammation-induced injuries (41-43). Previously, in a spinal cord-injury model, IFNγ treatment has been shown to enhance myelination through induction of neurotrophins such as glial cell line-derived neurotrophic factor (GDNF) or insulin-like growth factor-1 (IGF-1) (41). However, in the absence of IFNγ, IL-17-producing autoreactive CD4+ T cells expand and recruit monocytes, neutrophils, and other inflammatory cells from the periphery, which leads to increased brain-infiltrating inflammatory cells. These cells secrete inflammatory mediators such as ROS and NO, which, in the absence of brain-specific protective factors, attack and destroy the myelin coating on axons. This results in severe demyelination and the onset of neurological deficits.
In summary, we describe an animal model with severe inflammation and demyelination in DRB1*0301.DQ8.IFNγ KO mice. Our data suggest a protective role for IFNγ in peripheral immune cells as well as in the brain. In the periphery, IFNγ might regulate expansion of encephalitogenic IL-17/GM-CSF-producing CD4+ T cells, whereas in the CNS, IFNγ might induce glial cells to produce neurotrophin and neurotrophic cytokines that protect the myelin sheath and axons. Thus, our studies indicate an important protective role of IFNγ in brain inflammation and demyelination.
Supplementary Material
Acknowledgments
We thank Julie Hanson and her staff for breeding and maintaining the various HLA class II transgenic mice used for this study. We also thank Laurie Zoecklein, and Mabel Pierce for technical assistance and Lea Dacy for proof-reading and editing the manuscript.
The work in our lab was supported by National Institutes of Health grants NS52173.
References
- 1.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 2.Dyment DA, Herrera BM, Cader MZ, Willer CJ, Lincoln MR, Sadovnick AD, Risch N, Ebers GC. Complex interactions among MHC haplotypes in multiple sclerosis: susceptibility and resistance. Hum Mol Genet. 2005;14:2019–2026. doi: 10.1093/hmg/ddi206. [DOI] [PubMed] [Google Scholar]
- 3.Dyment DA, Sadovnick AD, Ebers GC. Genetics of multiple sclerosis. Hum Mol Genet. 1997;6:1693–1698. doi: 10.1093/hmg/6.10.1693. [DOI] [PubMed] [Google Scholar]
- 4.McDonald WI. Multiple sclerosis: epidemiology and HLA associations. Ann N Y Acad Sci. 1984;436:109–117. doi: 10.1111/j.1749-6632.1984.tb14781.x. [DOI] [PubMed] [Google Scholar]
- 5.Oksenberg JR, Begovich AB, Erlich HA, Steinman L. Genetic factors in multiple sclerosis. Jama. 1993;270:2362–2369. [PubMed] [Google Scholar]
- 6.Weinshenker BG, Santrach P, Bissonet AS, McDonnell SK, Schaid D, Moore SB, Rodriguez M. Major histocompatibility complex class II alleles and the course and outcome of MS: a population-based study. Neurology. 1998;51:742–747. doi: 10.1212/wnl.51.3.742. [DOI] [PubMed] [Google Scholar]
- 7.Madsen LS, Andersson EC, Jansson L, krogsgaard M, Andersen CB, Engberg J, Strominger JL, Svejgaard A, Hjorth JP, Holmdahl R, Wucherpfennig KW, Fugger L. A humanized model for multiple sclerosis using HLA-DR2 and a human T-cell receptor. Nat Genet. 1999;23:343–347. doi: 10.1038/15525. [DOI] [PubMed] [Google Scholar]
- 8.Mangalam AK, Rajagopalan G, Taneja V, David CS. HLA class II transgenic mice mimic human inflammatory diseases. Advances in immunology. 2008;97:65–147. doi: 10.1016/S0065-2776(08)00002-3. [DOI] [PubMed] [Google Scholar]
- 9.Mangalam AK, Khare M, Krco C, Rodriguez M, David C. Identification of T cell epitopes on human proteolipid protein and induction of experimental autoimmune encephalomyelitis in HLA class II-transgenic mice. Eur J Immunol. 2004;34:280–290. doi: 10.1002/eji.200324597. [DOI] [PubMed] [Google Scholar]
- 10.Mangalam A, Luckey D, Basal E, Behrens M, Rodriguez M, David C. HLA-DQ6 (DQB1*0601)-restricted T cells protect against experimental autoimmune encephalomyelitis in HLA-DR3.DQ6 double-transgenic mice by generating anti-inflammatory IFN-gamma. J Immunol. 2008;180:7747–7756. doi: 10.4049/jimmunol.180.11.7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mangalam A, Luckey D, Basal E, Jackson M, Smart M, Rodriguez M, David C. HLA-DQ8 (DQB1*0302)-restricted Th17 cells exacerbate experimental autoimmune encephalomyelitis in HLA-DR3-transgenic mice. J Immunol. 2009;182:5131–5139. doi: 10.4049/jimmunol.0803918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ando DG, Clayton J, Kono D, Urban JL, Sercarz EE. Encephalitogenic T cells in the B10.PL model of experimental allergic encephalomyelitis (EAE) are of the Th-1 lymphokine subtype. Cell Immunol. 1989;124:132–143. doi: 10.1016/0008-8749(89)90117-2. [DOI] [PubMed] [Google Scholar]
- 13.Kuchroo VK, Anderson AC, Waldner H, Munder M, Bettelli E, Nicholson LB. T cell response in experimental autoimmune encephalomyelitis (EAE): role of self and cross-reactive antigens in shaping, tuning, and regulating the autopathogenic T cell repertoire. Annu Rev Immunol. 2002;20:101–123. doi: 10.1146/annurev.immunol.20.081701.141316. [DOI] [PubMed] [Google Scholar]
- 14.Zamvil SS, Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol. 1990;8:579–621. doi: 10.1146/annurev.iy.08.040190.003051. [DOI] [PubMed] [Google Scholar]
- 15.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annual review of immunology. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 16.Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev. 2012;248:205–215. doi: 10.1111/j.1600-065X.2012.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008;14:337–342. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mangalam AK, Taneja V, David CS. HLA class II molecules influence susceptibility versus protection in inflammatory diseases by determining the cytokine profile. Journal of immunology. 2013;190:513–518. doi: 10.4049/jimmunol.1201891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Das P, Drescher KM, Geluk A, Bradley DS, Rodriguez M, David CS. Complementation between specific HLA-DR and HLA-DQ genes in transgenic mice determines susceptibility to experimental autoimmune encephalomyelitis. Hum Immunol. 2000;61:279–289. doi: 10.1016/s0198-8859(99)00135-4. [DOI] [PubMed] [Google Scholar]
- 20.Lampson LA, Levy R. Two populations of Ia-like molecules on a human B cell line. Journal of immunology. 1980;125:293–299. [PubMed] [Google Scholar]
- 21.Bradley DS, Nabozny GH, Cheng S, Zhou P, Griffiths MM, Luthra HS, David CS. HLA-DQB1 polymorphism determines incidence, onset, and severity of collagen-induced arthritis in transgenic mice. Implications in human rheumatoid arthritis. J Clin Invest. 1997;100:2227–2234. doi: 10.1172/JCI119760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abromson-Leeman S, Bronson R, Luo Y, Berman M, Leeman R, Leeman J, Dorf M. T-cell properties determine disease site, clinical presentation, and cellular pathology of experimental autoimmune encephalomyelitis. Am J Pathol. 2004;165:1519–1533. doi: 10.1016/S0002-9440(10)63410-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lees JR, Golumbek PT, Sim J, Dorsey D, Russell JH. Regional CNS responses to IFN-gamma determine lesion localization patterns during EAE pathogenesis. The Journal of experimental medicine. 2008;205:2633–2642. doi: 10.1084/jem.20080155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wensky AK, Furtado GC, Marcondes MC, Chen S, Manfra D, Lira SA, Zagzag D, Lafaille JJ. IFN-gamma determines distinct clinical outcomes in autoimmune encephalomyelitis. Journal of immunology. 2005;174:1416–1423. doi: 10.4049/jimmunol.174.3.1416. [DOI] [PubMed] [Google Scholar]
- 25.Carlson T, Kroenke M, Rao P, Lane TE, Segal B. The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J Exp Med. 2008;205:811–823. doi: 10.1084/jem.20072404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 27.Krakowski M, Owens T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol. 1996;26:1641–1646. doi: 10.1002/eji.1830260735. [DOI] [PubMed] [Google Scholar]
- 28.Berghmans N, Nuyts A, Uyttenhove C, Van Snick J, Opdenakker G, Heremans H. Interferon-gamma orchestrates the number and function of Th17 cells in experimental autoimmune encephalomyelitis. J Interferon Cytokine Res. 2011;31:575–587. doi: 10.1089/jir.2010.0137. [DOI] [PubMed] [Google Scholar]
- 29.Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC, Kidd G, Zorlu MM, Sun N, Hu W, Liu L, Lee JC, Taylor SE, Uehlein L, Dixon D, Gu J, Floruta CM, Zhu M, Charo IF, Weiner HL, Ransohoff RM. Differential roles of microglia and monocytes in the inflamed central nervous system. The Journal of experimental medicine. 2014 doi: 10.1084/jem.20132477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Refaeli Y, Van Parijs L, Alexander SI, Abbas AK. Interferon gamma is required for activation-induced death of T lymphocytes. J Exp Med. 2002;196:999–1005. doi: 10.1084/jem.20020666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sabatino JJ, Jr., Shires J, Altman JD, Ford ML, Evavold BD. Loss of IFN-gamma enables the expansion of autoreactive CD4+ T cells to induce experimental autoimmune encephalomyelitis by a nonencephalitogenic myelin variant antigen. Journal of immunology. 2008;180:4451–4457. doi: 10.4049/jimmunol.180.7.4451. [DOI] [PubMed] [Google Scholar]
- 33.El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugita Y, Zhao B, Shankar P, Dunbar CE, Doren S, Young HA, Schwartz JP. CNS interleukin-3 (IL-3) expression and neurological syndrome in antisense-IL-3 transgenic mice. J Neuropathol Exp Neurol. 1999;58:480–488. doi: 10.1097/00005072-199905000-00007. [DOI] [PubMed] [Google Scholar]
- 35.Krauthausen M, Ellis SL, Zimmermann J, Sarris M, Wakefield D, Heneka MT, Campbell IL, Muller M. Opposing roles for CXCR3 signaling in central nervous system versus ocular inflammation mediated by the astrocyte-targeted production of IL-12. Am J Pathol. 2011;179:2346–2359. doi: 10.1016/j.ajpath.2011.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sinha S, Kaler LJ, Proctor TM, Teuscher C, Vandenbark AA, Offner H. IL-13-mediated gender difference in susceptibility to autoimmune encephalomyelitis. Journal of immunology. 2008;180:2679–2685. doi: 10.4049/jimmunol.180.4.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:10480–10485. doi: 10.1073/pnas.1201836109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuwabara T, Ishikawa F, Yasuda T, Aritomi K, Nakano H, Tanaka Y, Okada Y, Lipp M, Kakiuchi T. CCR7 ligands are required for development of experimental autoimmune encephalomyelitis through generating IL-23-dependent Th17 cells. Journal of immunology. 2009;183:2513–2521. doi: 10.4049/jimmunol.0800729. [DOI] [PubMed] [Google Scholar]
- 39.Nakajima C, Mukai T, Yamaguchi N, Morimoto Y, Park WR, Iwasaki M, Gao P, Ono S, Fujiwara H, Hamaoka T. Induction of the chemokine receptor CXCR3 on TCR-stimulated T cells: dependence on the release from persistent TCR-triggering and requirement for IFN-gamma stimulation. European journal of immunology. 2002;32:1792–1801. doi: 10.1002/1521-4141(200206)32:6<1792::AID-IMMU1792>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 40.Iwasaki M, Mukai T, Gao P, Park WR, Nakajima C, Tomura M, Fujiwara H, Hamaoka T. A critical role for IL-12 in CCR5 induction on T cell receptor-triggered mouse CD4(+) and CD8(+) T cells. European journal of immunology. 2001;31:2411–2420. doi: 10.1002/1521-4141(200108)31:8<2411::aid-immu2411>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 41.Fujiyoshi T, Kubo T, Chan CC, Koda M, Okawa A, Takahashi K, Yamazaki M. Interferon-gamma decreases chondroitin sulfate proteoglycan expression and enhances hindlimb function after spinal cord injury in mice. J Neurotrauma. 2010;27:2283–2294. doi: 10.1089/neu.2009.1144. [DOI] [PubMed] [Google Scholar]
- 42.Linker R, Gold R, Luhder F. Function of neurotrophic factors beyond the nervous system: inflammation and autoimmune demyelination. Crit Rev Immunol. 2009;29:43–68. doi: 10.1615/critrevimmunol.v29.i1.20. [DOI] [PubMed] [Google Scholar]
- 43.Lu Z, Hu X, Zhu C, Wang D, Zheng X, Liu Q. Overexpression of CNTF in Mesenchymal Stem Cells reduces demyelination and induces clinical recovery in experimental autoimmune encephalomyelitis mice. Journal of neuroimmunology. 2009;206:58–69. doi: 10.1016/j.jneuroim.2008.10.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.