

Abstract
A novel Brønsted acid/Lewis acid dual catalyst system has been developed to promote an efficient C–C bond formation between a range of oxocarbenium precursors derived from chromene acetals and ethyl diazoacetate. The reaction proceeds under mild conditions and is tolerant of common functionalized 2H-chromene and isochromene acetals. In addition, an asymmetric variant of diazoacetate addition towards 2H-chromene acetal is described. Continued investigations include the further optimization of asymmetric induction towards the formation of diazo ester substituted 2H-chromene.
Keywords: Brønsted Acid, Lewis acid, Diazo compounds, Oxocarbenium, Enantioselectivity
Introduction
In situ generated, highly reactive oxocarbenium ions are useful intermediates for the synthesis of complex ether-containing compounds.1 Oxocarbenium chemistry offers numerous possibilities in the synthesis of natural products and pharmaceuticals.2 In particular, resonance-stabilized chromene and isochromane derivatives have been accessed by trapping of oxocarbenium precursors.3 Jacobsen and co-workers designed an elegant chiral anion binding approach for the thiourea catalyzed asymmetric addition of enoxysilanes to chloroisochromans.4 Schaus reported the reaction between boronates and 2H-chromene acetals to form 2-vinyl- and 2-aryl-2H-chromenes in high yield and enantioselectivity (Scheme 1).5 Rueping reported a synergistic catalytic system for the first asymmetric addition of aldehydes to in-situ generated prochiral oxocarbenium ions employing an enamine catalyst (Scheme 1).6 Furthermore, metal-mediated nucleophilic addition to oxocarbenium has also been reported. The recent work of Doyle illustrates a Ni(0) promoted C–C bond formation between 2H-chromene acetals and boronic acid in a racemic fashion, which tolerates a wide variety of boronic acids.7 Watson developed an enantioselective Cu(I)-catalyzed addition of terminal alkynes to isochroman acetal.8
Scheme 1.

2H-chromene acetal addition reaction.
BINOL-phosphoric acid has emerged as a powerful chiral Brønsted acid catalyst in many C–C bond-forming reactions. Akiyama9 and Terada10 pioneered the studied of chiral BINOL-derived phosphoric acids in Mannich-type reactions and aza-Friedel–Crafts alkylation. The development of novel organic transformations promoted by BINOL phosphoric acid and its derivatives has been continuously studied in recent years.11 List has utilized BINOL phosphate for a variety of asymmetric reactions.12 Boronate was utilized as a nucleophile by Antilla for asymmetric addition using BINOL-phosphoric acid as the catalyst.13 Several novel organic transformations were developed by using BINOL-phosphoric acid under various reaction condition.14 α-Unsubstituted diazocarbonyl compounds, which are generally considered as a carbene precursors,15 can be coupled with various electrophiles as nucleophiles,16 such as aldehydes17 and imines18 with the retention of diazo moiety under Brønsted acid catalyzed reaction condition. However, to the best of our knowledge, the use of nucleophilic diazoacetates additions to oxocarbeniums has not been reported.
Herein, we report the development of a new dual catalyst system using both BINOL-phosphoric acid and Lewis acid,19 which enables the reaction of diazoacetate with benzocyclic acetals. This methodology provides access to a class of C2-substituted chromenes, which are present in many bioactive natural products.20 The optimized reaction conditions are mild and can tolerate a variety of functional groups to generate the products in excellent yields. The diazoacetate addition can be rendered asymmetric utilizing chiral BINOL-phosphoric acid and decent enantioselectivity was achieved together with good yield of diazo ester substituted 2H-chromene.
Results and Discussion
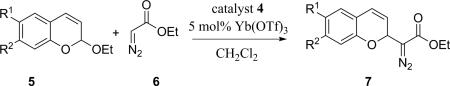
We initiated our study by investigating the addition of ethyl diazoacetate 6 to 2-ethoxy-2H-chromene 5a (Table 1). The diazoacetate addition product 7a can act as the synthetic intermediates for the synthesis of bioactive compounds, such as deoxyerythrostominone21 and (S,R,R,R)-nebivolol.22 The control reaction showed that the addition reaction could not occur without both Brønsted acid and Lewis acid (Table 1, entry 1). The use of previously successful tartaric acid in a racemic form, in combination of catalytic amount of Yb(OTf)3, provided the desired addition product 7 in modest yields (Table 1, entry 2). Amino-acid such as L-proline 2, which is a powerful catalyst in organocatalysis, only resulted in low reactivity (Table 1, entry 3). Thiourea catalyst 3 was more successful at promoting the diazo addition reaction affording the product in 66% yield (Table 1, entry 4). Alternatively, BINOL (1,1’-bi-2-naphthol) derived phosphoric acid (BINOL-phosphoric acid 4), together with Yb(OTf)3, provided the highest yield in the addition reaction (Table 1, entry 5) and was selected as the catalyst for further investigation. The addition reaction did not proceed in the absence of Brønsted acid, which indicates the crucial role of the BINOL-phosphoric acid in this reaction (Table 1, entry 6). Furthermore, the utilization of BINOL-phosphoric acid 4 without Yb(OTf)3 Lewis acid gave decent yield of the desired diazo chromene product (Table 1, entry 7). This observation suggested that the utilization of Yb(OTf)3 enhanced the reaction rate, which results high yield of the desired product. However, significant decrease in yield was observed when the reaction was run at 0 °C (Table 1, entry 8). As a result, the rest of this study was conducted under room temperature. It is concluded that the addition reaction was promoted by Brønsted acid and the reaction rate was further enhanced by the Lewis acid. n-Butyl and isopropyl derived acetals 5b and 5c were synthesized as alternative oxocarbenium precursors. However, only lower yields were observed under the standard reaction conditions of these bulkier acetals (Table 1, entries 9 and 10). Solvent effects were also investigated under the optimal reaction conditions with BINOL-phosphoric acid 4 and 2-ethoxy-2H-chromene acetal 5a. A solvent screen indicated that CH2Cl2 was the best solvent among other common organic solvents (Table 1, entries 4 and 11-14). Chloroform and toluene gave comparable yields (Table 1, entries 11 and 12), and THF and methanol gave lower yields, presumably due to unfavorable interactions with the catalysts or inhibition of oxocarbenium formation (Table 1, entries 13-14).
Table 1.
Reaction optimizations[a].

| |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Lewis acid | R | Solvent | Yield[b] |
| 1 | - | - | Et (5a) | CH2Cl2 | 0% |
| 2 | 1 | Yb(OTf)3 | Et (5a) | CH2Cl2 | 40% |
| 3 | 2 | Yb(OTf)3 | Et (5a) | CH2Cl2 | 11% |
| 4 | 3 | Yb(OTf)3 | Et (5a) | CH2Cl2 | 66% |
| 5 | 4 | Yb(OTf)3 | Et (5a) | CH2Cl2 | 82% |
| 6 | - | Yb(OTf)3 | Et (5a) | CH2Cl2 | <5% |
| 7 | 4 | - | Et (5a) | CH2Cl2 | 58% |
| 8[c] | 4 | Yb(OTf)3 | Et (5a) | CH2Cl2 | 42% |
| 9 | 4 | Yb(OTf)3 | nBu (5b) | CH2Cl2 | 74% |
| 10 | 4 | Yb(OTf)3 | iPr (5c) | CH2Cl2 | 63% |
| 11 | 4 | Yb(OTf)3 | Et (5a) | CHCl3 | 78% |
| 12 | 4 | Yb(OTf)3 | Et (5a) | PhCH3 | 80% |
| 13 | 4 | Yb(OTf)3 | Et (5a) | THF | 54% |
| 14 | 4 | Yb(OTf)3 | Et (5a) | CH3OH | 12% |
Reaction conditions: 0.5 mmol of acetal 5, 0.6 mmol of ethyl diazoacetate 6, 20 mol-% Brønsted acid catalyst, 0.2 M in the solvent for 12 h at room temperature.
Isolated yield.
at 0 °C
Encouraged by these preliminary results, we further extended this methodology to a variety of substituted 2H-chromene acetals. The optimized reaction proved to be general for a range of 2H-chromene acetals (Table 2). This reaction proceeded smoothly with chromene acetal 5a, furnishing diazoacetate substituted 2H-chromene 7a in good yield (Table 2, entry 1). Methyl substituted chromene acetal 5b provided the desired product in good yield (Table 2, entry 2). Reaction with electron-rich chromene acetal 5c preceded smoothly within 1 h, despites the fact that 7-methoxy substituted chromene acetal 5c tends to decompose in the presence of strong Brønsted acid over time (Table 2, entry 3). 6,7-dioxo-2H-chromene acetal 5d also underwent this reaction well with slightly compromised yield (Table 2, entry 4). Halogenated chromene acetals 5e and 5f were transformed to the desired diazo ester substituted chromenes in good yields (Table 2, entries 5 and 6). Electron-deficient chromene acetal 5g underwent the reaction in the same fashion, providing desired product 7g in good yield at extended reaction times under room temperature (Table 2, entry 7).
Table 2.
2H-chromene acetal evaluation[a].














Reaction conditions: 0.5 mmol of 5, 20 mol-% catalyst 4, 0.2 M in the solvent for 12 h at room temperature.
1 h.
24 h.
Isolated yield.
We next tested whether the reaction could be extended to the diazo addition of isochromene acetal 8. Under the optimal reaction condition for diazo oxocarbenium addition, the reaction with an isochromene acetal proceeded in a promising 76% yield (Figure 1). This result provides a preliminary indication of the utility of the catalytic methodology and the possibility to expand this strategy to other cyclic oxocarbenium ions.
Figure 1.

Nucleophilic addition to isochromene acetal 8.
At last, we sought to develop an asymmetric variant of the diazo addition reaction using a chiral Brønsted acid (Table 3). Chiral acid catalyst, such as (+)-tartaric acid or L-proline failed to afford any enantioselectivity. We initiated our investigation by evaluating the reaction of ethyl diazoacetate 6 with 2H-chromene acetal 5a in the presence of chiral BINOL-phosphoric acid catalysts. Unsubstituted BINOL-phosphoric acid afforded the product in very low er (Table 3, entry 1). 3,3’-Dibromo-substituted BINOL-phosphoric acid 10b afforded the product in good yield and modest enantioselectivity (Table 3, entry 2). 3,3’-Aryl-substituted catalysts 10c-f afforded similar or lower selectivities (Table 3, entries 3-6) Bulky catalysts with silyl and anthracenyl substitutions at C3 position were also evaluated but gave only marginal improvement in selectivity (Table 3, entries 8-9). Finally, the commercially available (S)-TRIP catalyst 10i afforded the desired product in the highest er (81:19, Table 3, entry 9) Further solvent screens indicated that toluene gave the highest selectivity (Table 3, entries 10-13).
Table 3.
The stereoselective nucleophilic addition reaction of 6 using chiral phosphoric acid.[a]

| ||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Yield[b] | e.r.[c] |
| 1 | 10a | PhCH3 | 81% | 52:48 |
| 2 | 10b | PhCH3 | 75% | 65:35 |
| 3 | 10c | PhCH3 | 71% | 55:45 |
| 4 | 10d | PhCH3 | 76% | 56:44 |
| 5 | 10e | PhCH3 | 72% | 56:44 |
| 6 | 10f | PhCH3 | 87% | 65:35 |
| 7 | 10g | PhCH3 | 66% | 55:45 |
| 8 | 10h | PhCH3 | 76% | 68:32 |
| 9 | 10i | PhCH3 | 82% | 81:19 |
| 10 | 10i | PhCF3 | 82% | 78:22 |
| 11 | 10i | CH2Cl2 | 83% | 63:37 |
| 12 | 10i | Et2O | 54% | 65:35 |
| 13 | 10i | THF | 55% | 75:25 |
Reaction conditions: 0.5 mmol of 5a, 20 mol-% chiral catalyst, 0.2 M in the solvent for 12 h at room temperature.
Isolated yield.
Determined by HPLC.
Conclusions
In summary, we have developed the first asymmetric addition of diazo acetate to prochiral oxocarbenium ions yielding valuable chiral 2H-chromene scaffolds. A catalytic system consisting of an achiral Lewis acid and a Brønsted acidic phosphoric acid catalyst was the key to affording the products in good yields. Asymmetric versions of this reaction are available by using commercially available chiral phosphoric acids. Further studies to expand the scope and utility of the reaction are ongoing.
Experimental Section
General Information
All 1H NMR, and 13C NMR spectra were recorded using Varian Unity Plus 400 or 500 MHz spectrometer at ambient temperature in CDCl3. Chemical shifts are reported in parts per million as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), coupling constant, and integration. Infrared spectra were recorded on a Nicolet Nexus 670 FT-IR ESP spectrophotometer. Optical rotations were recorded on an AUTOPOL III digital polarimeter at 589 nm, and were reported as [α]D (concentration in grams/100 mL solvent). Chiral HPLC analysis was performed using an Agilent 1100 series HPLC System with a diode array detector. Chiral columns include Chiralcel®OD (Chiral Technologies Inc., 25cm×4.6mm I.D.), Chiralpak®AD-H (Chiral Technologies Inc., 25cm × 4.6 mm I.D.) and (R,R)-Whelk-O (Regis®Technologies Inc., 25cm × 4.6 mm I.D.). Analytical thin layer chromatography was performed using EMD 0.25 mm silica gel 60-F plates. Flash column chromatography was performed on Sorbent Technologies 60 Å silica gel.
Procedure for the dual acid-catalyzed addition of ethyl diazoacetate 6 to 2-ethoxy-2H- chromene 5a
To a flame-dried 5 mL round-bottom flask equipped with stir bar and rubber septum under Ar was added Yb(OTf)3 (15.5 mg, 0.025 mmol, 5 mol%), BINOL-phosphoric acid (34.8 mg, 0.1 mmol, 20 mol%), ethyl diazoacetate 6 (78.4 mg, 0.6 mmol, 1.2 equiv. with 20% of CH2Cl2) and CH2Cl2 (2.5 mL). Then the chromene acetal 5a (88.0 mg, 0.5 mmol, 1.0 equiv.) was added and the reaction was stirred vigorously for 12 h at room temperature. The reaction was purified without work-up by flash chromatography over silica gel column (2% EtOAc in hexanes) to afford the diazo addition product 7a as a yellow oil (97 mg, 0.42 mmol, 83% yield). The product 7a can be stored at −20 °C in the dark for up to two months without decomposition of the structure.
Supplementary Material
Acknowledgements
Preliminary experiments were performed by Y.L. at Boston University. Completion of the work was accomplished at University of Science and Technology Beijing, China. S.E.S. and Y.L. gratefully acknowledge the NIH for support (NIGMS R01 GM078240). Y.L., Y.Q., H.Y.G. and Q.Q.M thank Beijing Natural Science Foundation (Grant No. 2144052) and the China Postdoctoral Science Foundation (2014T70036) for financial support. We also thank Dr. P. N. Moquist (International Knowledge Editing) for helpful discussions and English editing.
Footnotes
Supporting Information (see footnote on the first page of this article): General experimental remarks, experimental details and spectra of the products.
References
- 1.Braun M, Kotter W. Angew. Chem. Int. Ed. 2004;43:514–517. doi: 10.1002/anie.200352128. [DOI] [PubMed] [Google Scholar]
- 2.Han X, Peh GR, Floreancig PE. Eur. J. Org. Chem. 2013;7:1193–1208. [Google Scholar]
- 3.Wu YC, Li HJ, Liu L, Demoulin N, Liu Z, Wang D, Chen YJ. Adv. Synth. Catal. 2011;353:907–912. [Google Scholar]
- 4.a Reisman SE, Doyle AG, Jacobsen EN. J. Am. Chem. Soc. 2008;130:7198–7199. doi: 10.1021/ja801514m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Burns NZ, Witten MR, Jacobsen EN. J. Am. Chem. Soc. 2011;133:14578–14581. doi: 10.1021/ja206997e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kotke M, Schreiner PR. Tetrahedron. 2006;62:434–439. [Google Scholar]; d Kotke M, Schreiner PR. Synthesis. 2007;5:779–790. [Google Scholar]
- 5.Moquist PN, Kodama T, Schaus SE. Angew. Chem. Int. Ed. 2010;49:7096–7100. doi: 10.1002/anie.201003469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rueping M, Volla CMR, Atodiresei I. Org. Lett. 2012;14:4642–4645. doi: 10.1021/ol302084q. [DOI] [PubMed] [Google Scholar]
- 7.a Graham TJA, Doyle AG. Org. Lett. 2012;14:1616–1619. doi: 10.1021/ol300364s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sylvester KT, Wu K, Doyle AG. J. Am. Chem. Soc. 2012;134:16967–16970. doi: 10.1021/ja3079362. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Graham TJA, Shields JD, Doyle AG. Chem. Sci. 2011;2:980–984. [Google Scholar]
- 8.Maity P, Srinivas HD, Watson MP. J. Am. Chem. Soc. 2011;133:17142–17145. doi: 10.1021/ja207585p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akiyama T, Itoh J, Yokota K, Fuchibe K. Angew. Chem. Int. Ed. 2004;43:1566–1568. doi: 10.1002/anie.200353240. [DOI] [PubMed] [Google Scholar]
- 10.a Uraguchi D, Terada M. J. Am. Chem. Soc. 2004;126:5356–5357. doi: 10.1021/ja0491533. [DOI] [PubMed] [Google Scholar]; b Uraguchi D, Sorimachi K, Terada M. J. Am. Chem. Soc. 2004;126:11804–11805. doi: 10.1021/ja046185h. [DOI] [PubMed] [Google Scholar]
- 11.Zamfir A, Schenker S, Freunda M, Tsogoeva SB. Org. Biomol. Chem. 2010;8:5262–5276. doi: 10.1039/c0ob00209g. [DOI] [PubMed] [Google Scholar]
- 12.a Coric I, Kim JH, Vlaar T, Patil M, Thiel W, List B. Angew. Chem. Int. Ed. 2013;52:3490–3493. doi: 10.1002/anie.201209983. [DOI] [PubMed] [Google Scholar]; b Vellalath S, Coric I, List B. Angew. Chem. Int. Ed. 2010;49:9749–9752. doi: 10.1002/anie.201005347. [DOI] [PubMed] [Google Scholar]
- 13.a Jain P, Antilla JC. J. Am. Chem. Soc. 2010;132:11884–11886. doi: 10.1021/ja104956s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jain P, Wang H, Houk KN, Antilla JC. Angew. Chem. Int. Ed. 2012;51:1391–1394. doi: 10.1002/anie.201107407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a Rono LJ, Yayla HG, Wang DY, Armstrong MF, Knowles RR. J. Am. Chem. Soc. 2013;135:17735–17738. doi: 10.1021/ja4100595. [DOI] [PubMed] [Google Scholar]; b Wang ZB, Law KW, Sun JW. Org. Lett. 2013;15:5964–5966. doi: 10.1021/ol402797v. [DOI] [PubMed] [Google Scholar]; c Yamanaka M, Hoshino M, Katoh T, Mori K, Akiyama T. Eur. J. Org. Chem. 2012;24:4508–4514. [Google Scholar]
- 15.a Ye T, McKervey MA. Chem. Rev. 1994;94:1091–1160. [Google Scholar]; b Li H, Zhang Y, Wang JB. Synthesis. 2013;45:3090–3098. [Google Scholar]
- 16.Facklam T, Hoffmann K-L, Regitz M. Chem. Ber. 1987;120:1397–1402. [Google Scholar]
- 17.a Trost BM, Malhotra S, Koschker P, Ellerbrock P. J. Am. Chem. Soc. 2012;134:2075–2084. doi: 10.1021/ja206995s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Trost BM, Malhotra S, Fried BA. J. Am. Chem. Soc. 2009;131:1674–1675. doi: 10.1021/ja809181m. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Trost BM, Malhotra S, Ellerbrock P. Org. Lett. 2013;15:440–443. doi: 10.1021/ol303011f. [DOI] [PubMed] [Google Scholar]; d Krishna PR, Prapurna YL, Alivelu M. Eur. J. Org. Chem. 2011;26:5089–5095. [Google Scholar]; e Yao WG, Wang JB. Org. Lett. 2003;5:1527–1530. doi: 10.1021/ol0343257. [DOI] [PubMed] [Google Scholar]
- 18.a Hashimoto T, Kimura H, Nakatsu H, Maruoka K. J. Org. Chem. 2011;76:6030–6037. doi: 10.1021/jo2005999. [DOI] [PubMed] [Google Scholar]; b Uraguchi D, Sorimachi K, Terada M. J. Am. Chem. Soc. 2005;127:9360–9361. doi: 10.1021/ja051922a. [DOI] [PubMed] [Google Scholar]
- 19.a Yang T, Ferrali A, Sladojevich F, Campbell L, Dixon DJ. J. Am. Chem. Soc. 2009;131:9140–9141. doi: 10.1021/ja9004859. [DOI] [PubMed] [Google Scholar]; b Terada M. Synthesis. 2010;12:1929–1982. [Google Scholar]; c Akiyama T. Chem. Rev. 2007;107:5744–5758. doi: 10.1021/cr068374j. [DOI] [PubMed] [Google Scholar]
- 20.a Harrity JPA, La DS, Cefalo DR, Visser MS, Hoveyda AH. J. Am. Chem. Soc. 1998;120:2343–2351. [Google Scholar]; b Wipf P, Weiner WS. J. Org. Chem. 1999;64:5321–5324. doi: 10.1021/jo990352s. [DOI] [PubMed] [Google Scholar]
- 21.Kittakoopa P, Punya J, Kongsaeree P, Lertwerawat Y, Jintasirikul A, Tanticharoen M, Thebtaranonth Y. Phytochemistry. 1999;52:453–457. [Google Scholar]
- 22.a Carreño MC, Hernández-Torres G, Urbano A, Colobert F. Eur. J. Org. Chem. 2008:2035–2038. [Google Scholar]; b Chandrasekhar S, Reddy M. Venkat. Tetrahedron. 2000;56:6339–6344. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.