

Abstract
Mechanisms of neuroendocrine tumor (NET) proliferation are poorly understood and therapies that effectively control NET progression and metastatic disease are limited. We found amplification of a putative oncogene, RABL6A, in primary human pancreatic NETs(PNETs) that correlated with high level RABL6A protein expression. Consistent with those results, stable silencing of RABL6A in cultured BON-1 PNET cells revealed that it is essential for their proliferation and survival. Cells lacking RABL6A predominantly arrested in G1 phase with a moderate mitotic block. Pathway analysis of microarray data suggested activation of the p53 and retinoblastoma (Rb1) tumor suppressor pathways in the arrested cells. Loss of p53 had no effect on the RABL6A knockdown phenotype, indicating RABL6A functions independent of p53 in this setting. By comparison, Rb1 inactivation partially restored G1 to S phase progression in RABL6A knockdown cells although it was insufficient to override the mitotic arrest and cell death caused by RABL6A loss. Thus, RABL6A promotes G1 progression in PNET cells by inactivating Rb1, an established suppressor of PNET proliferation and development. This work identifies RABL6A as a novel negative regulator of Rb1 that is essential for PNET proliferation and survival. We suggest RABL6A is a new potential biomarker and target for anticancer therapy in PNET patients.
Keywords: pancreatic neuroendocrine tumor, RABL6A, ARF, Retinoblastoma protein (Rb1), p53
Introduction
Neuroendocrine tumors (NETs) are rare malignancies that have risen five-fold in incidence over the last 30 years and area significant clinical challenge(1). They arise in many organs and are a heterogenous group of slowly growing neoplasms with diverse morphologies and biological manifestations (2, 3). NETs are classified as “functional” if they produce hormones/peptides that elicit systemic effects while “non-functional” tumors have symptoms related only to tumor malignancy. The disease is typically associated with non-specific symptoms and often eludes diagnosis for many years, consequently 50% of patients have liver metastases at diagnosis (1, 3). Sadly, there are few effective treatment options for these patients since NETs are resistant to standard anticancer therapies. Most treatments seek to limit metastatic tumor progression with somatostatin analogs, surgical debulking, hepatic embolization, or peptide radioreceptor therapy but cure is unusual and recurrent disease usually develops(4-8). Biomarker identification and an improved understanding of key pathways underlying the disease are needed to improve NET diagnosis and treatment.
Recent work has begun to expand our limited molecular understanding of pancreatic NET (PNET) pathogenesis. For instance, exome sequencing of sporadic PNETs revealed common mutations in MEN1, DAXX and ATRX genes, as well as genes in the mammalian target of rapamycin (mTOR) pathway (9). Excitingly, inhibitors of mTOR signaling, such as everolimus, were recently approved for treating PNETs and are showing significant promise clinically(10-12). Other major pathways implicated in PNET development involve the tumor suppressor genes INK4a/ARF, RB1 and TP53. The INK4a/ARF locus encodes two unrelated tumor suppressors, p16INK4a (Inhibitor of Cdk4/6) and ARF (Alternative Reading Frame), via overlapping reading frames in shared exons(13, 14). Whereas p16INK4a enforces retinoblastoma (Rb1) anti-proliferative activity in G1 phase, ARF promotes p53-mediated cell cycle arrest and apoptosis (15). ARF can also act through p53-independent mechanisms (16) and was found to inhibit PNET angiogenesis and progression in mice lacking functional p53(17). Notably, primary PNETs from patients contain wild-type INK4a/ARF, TP53 and RB1 genes (9), yet their function is compromised in a high percentage of tumors due to gene amplification of negative regulators (for p53 and Rb1) (18, 19) or promoter silencing (for p16INK4a and ARF) (9, 20, 21).
RABL6A is a novel RAB-like GTPase of unknown function that we previously discovered binds ARF (22). The RABL6 gene (also called Parf [Partner of ARF], RBEL1 or c9orf86) encodes four isoforms (A-D), all of which retain GTPase activity(23, 24) while only the largest form, RABL6A, contains the ARF binding domain(22). Recent work suggests RABL6A binds to p53, as well as its antagonist, Mdm2, and through those interactions RABL6A impairs p53 stability and transactivation activity (25). Such data imply an oncogenic role for RABL6A. In fact, RABL6A is overexpressed in the majority of primary breast tumors (23, 26) and pancreatic ductal adenocarcinomas (27), and its expression is associated with worse survival in those patients(26, 27). We and others have shown that silencing RABL6A in cultured tumor cells reduces their survival, proliferation and malignant growth in nude mice(24, 26, 27). In contrast to its tumor promoting activities in transformed cells, analyses of RABL6A in non-transformed fibroblasts showed it can prevent centrosome amplification and chromosomal instability (28). Together, the cumulative data suggest RABL6A may be an important regulator of chromosomal stability in normal cells whose elevated expression in neoplastic cells has tumor-promoting consequences including increased tumor cell survival and proliferation.
RABL6A protein is most highly expressed in the pancreas compared to other tissues(27). Here, we investigated the biological significance and mechanisms of action of RABL6A in PNET biology. We found that RABL6A is amplified in the majority of patient PNETs and is essential for PNET cell survival and cell cycle progression. Its ability to promote G1 progression is independent of ARF and p53 but requires Rb1 inactivation. These findings demonstrate the importance of RABL6A and the Rb1 pathway in regulating PNET proliferation, and identify RABL6A as a novel oncogenic inhibitor of Rb1.
Materials and Methods
Cell culture
Human BON-1 PNET cells (kind gift to Dr. Sue O'Dorisio from Dr. Courtney Townsend, who originally established the line and authenticated their origin) (29) were maintained in Dulbecco's modified Eagle's medium (DMEM) / F12 containing 10% fetal bovine serum (FBS), 4 mM glutamine, and 100 μg/mL penicillin / streptomycin. Human Qgp-1 PNET cells were purchased from the Japanese Collection of Research Bioresources (JCRB0183, National Institute of Biomedical Innovation, Japan) and maintained in RPMI 1640 medium containing 10% FBS, 4 mM glutamine, and 100 μg/mL penicillin / streptomycin. Human embryonic kidney (HEK) 293T cells were grown in DMEM with the same supplements.
RNA interference, virus production and infection
Human RABL6A and p53 shRNA constructs in the pLKO.1 lentiviral vector (Open Biosystems, Huntsville, AL) have been described(27, 30). Similarly, pLKO-based shRNA constructs targeting human Rb1 and p27 (Open Biosystems) were used. The HPV-16 E7 LXSN vector and constitutively producing PA317 packaging line, including virus collection and transduction, have also been described (31, 32). Stable BON-1 cells expressing E7 were selected with 0.75 mg/mL neomycin for 2 weeks followed by maintenance in 0.375 mg/mL neomycin. Lentiviruses encoding human p53 and RABL6A shRNAs were produced in 293T cells, and BON-1 or Qgp-1 cells infected exactly as described(27). Cells were harvested 3, 6 or 8 days after infection for assays depending on the study. For dual knockdown of p53 and RABL6A, cells were sequentially infected with p53 shRNA viruses followed by RABL6A shRNA viruses. For long-term culture, cells infected with RABL6A shRNA viruses were selected with 1 μg/mL puromycin for 2 days and maintained in 0.5 μg/mL puromycin.
Analyses of RABL6A status in human PNETs
Fresh frozen and formalin-fixed, paraffin embedded (FFPE) PNET specimens used for quantitative PCR (qPCR) and immunohistochemical (IHC) analyses of RABL6A copy number and protein expression, respectively, were obtained from patients with progressive disease who provided informed consent to participate in the Iowa Neuroendocrine Tumor Registry (approved by the University of Iowa Institutional Review Board). For qPCR, genomic DNA was isolated using the Puregene Blood Kit (Qiagen) from samples stored in RNA Later (Qiagen, Valencia, CA). PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems by LIFE Technologies, Carlsbad, CA) with data collected in triplicates from three independent experiments on CFX Connect™ Real time System thermal cycler (Bio-Rad, Hercules, CA). Human RABL6A values were normalized to β-globin values, which we established to be diploid in these tumors by other cytogenetic methods. Relative gene copy number was calculated using a comparative Ct method (33), and RABL6A levels in tumors compared to levels observed in matched normal tissue from the same patient. Primers included: RABL6A forward: 5′- ACAAGAGTAAGTGTGGTGGGTG -3′; RABL6A reverse: 5′- GAACTCTTCGGGGAGACCC-3′; β-globin forward: 5′- ACCATGGTGCATCTGACTCC -3′; β-globin reverse: 5′ CTGTCTCCACATGCCCAGT -3′.
Methods for IHC staining of RABL6A, including use of an IgG isotype control, have been described (27). IHC was performed on a human pancreatic cancer tissue array containing normal pancreas, 2 non-functional PNETs, 1 insulinoma and 2 solid-pseudopapillary specimens (US Biomax, Rockville, MD; catalog T142), as well as 5 FFPE PNETs from the University of Iowa NET Registry.
Cell proliferation and survival assays
Cell number and viability following RABL6A knockdown were measured by Trypan blue staining (1:1 v/v) and counting using a hemocytometer at 3 to 8 days after infection with RABL6A shRNA viruses. For growth curves, cells were passed, counted and re-plated at identical numbers every 3 to 4 days for up to 4 weeks. DNA content was measured by flow cytometry and analyzed using MOD Fit LT software (Verity Software House, Topsham, ME) (27). To measure DNA synthesis, infected cells were plated onto coverslips and the next day incubated for 5 hours with 10 μM bromodeoxyuridine (BrdU). Cells were stained for BrdU incorporation and with 4′,6-diamidino-2-phenylindole (DAPI) to detect nuclei(34). Fluorescence images were captured by confocal microscopy (Zeiss LSM 710, Germany) and quantified (100 or more cells per sample) from 3 or more separate experiments.
Expression Microarray and Data Analysis
Total RNA was isolated using RNeasy (Qiagen) from 3 separately generated sets of BON-1 cells expressing RABL6A shRNAs or control vector. RNA preparation for hybridization to Illumina-Human H12 v4 BeadChips (BD-103-0204) was performed at the University of Iowa DNA Facility (see Supplementary methods for more detail). Beadchips were scanned with the Illumina iScan System and data collected using the GenomeStudio software v2011.1.
Illumina array data were exported from GenomeStudio to the Partek GS software (Partek Incorporated v6.6, St. Louis, MO) and the Partek “gene expression workflow” was employed to detect differentially expressed genes using an ANOVA test. ANOVA factors included status as vector control (CON), KD1 or KD2. Gene expression differences between each individual knockdown experiment and the control as well as the two knockdowns together versus control were determined using a false discovery rate (FDR) of 0.05 and a minimum fold change of -2 or +2. A list of differentially expressed genes between the RABL6A knockdowns and vector was created, imported into Ingenuity Pathway Analysis (IPA) software tool (Ingenuity® Systems, Redwood City, CA) and subjected to an IPA Core Analysis. The “Disease and Functions” component of the core analysis assessed enrichment for genes in cell cycle regulation and checkpoint signaling. Significant enrichment was defined as reaching a p value < 0.01 after a Benjamini-Hochberg multiple testing correction. The “Upstream Analysis” component of the core analysis assessed for enrichment of genes involved in p53 and Rb1 pathways.
Western Analyses
Frozen cells were lysed directly in SDS-PAGE loading buffer and identical cell equivalents were electrophoresed through polyacrylamide gels, followed by western blotting and protein detection with enhanced chemiluminescence (ECL, Amersham, Buckinghamshire, UK), as described (35). Antibodies used included those against RABL6A (rabbit polyclonal at 1.5 μg/mL and mouse monoclonal at 1:100 dilution) (22, 27), total Rb1 including its differently phosphorylated forms (BD Pharmingen, 554136, 1:100), p21 (BD Pharmingen, 554228, 1:100), cyclin D1 (Santa Cruz, sc-753, 1:200), p53 (Santa Cruz, sc-126 [DO-1], 1:200), p27 (Abcam, 54563, 1 μg/mL), cleaved caspase-3 (Cell Signaling, D175, 1:1,000), PUMA (Cell Signaling, 4976, 1:1,000) and GAPDH (Abcam, Ab8245, 1:20,000).
Results
RABL6A is amplified in primary PNETs
Earlier studies revealed that RABL6A mRNA and protein are more highly expressed in normal pancreas than other tissues(22, 27), implying an important role in pancreas physiology and pathologies. We performed immunohistochemical staining of RABL6A on a commercial pancreatic tissue microarray (TMA) to assess RABL6A protein expression within the normal human pancreas and pancreatic tumors. The monoclonal antibody used specifically recognizes the human RABL6A isoform(27). Moderate levels of RABL6A were seen in epithelial and acinar cells in the normal pancreas whereas robust expression was found in the islets of Langerhans (Fig. 1A). The TMA contained three islet-derived pancreatic neuroendocrine tumors (PNETs), including two non-functional tumors and one insulin-secreting insulinoma. RABL6A expression was high in the non-functional tumors and moderate in the insulinoma (Fig. 1A). By comparison, RABL6A was undetectable in two pancreatic solid-pseudopapillary tumors, uncommon cystic neoplasms of low malignant potential associated with long patient survival(36).
Figure 1. High expression of RABL6A in human PNETs.
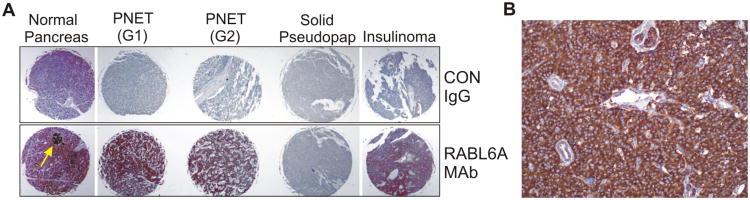
A, immunohistochemical staining of a pancreatic TMA with RABL6A monoclonal antibody (MAb) or IgG control in the indicated specimens. Yellow arrow, islet with high RABL6A levels in the normal pancreas. Two solid-pseudopapillary (pseudopap) tumors stained negatively for RABL6A, one is shown. G, tumor grade. B, representative immunohistochemical stain of RABL6A in a primary PNET specimen obtained at the University of Iowa Neuroendocrine Tumor Clinic. No staining was detected in negative controls in which buffer was substituted for the primary antibody.
PNETs are rare cancers that are poorly represented on TMAs, therefore we studied additional primary PNET specimens that were collected at the Iowa Neuroendocrine Tumor Clinic (37). Moderate to high RABL6A protein expression was again observed by immuno-staining in all (n=5) formalin-fixed paraffin embedded primary PNETs examined (Fig. 1B). In addition, qPCR analyses of RABL6A gene status in fresh frozen PNETs (11 primary and matched metastatic specimens, as well as control normal tissue for each patient) revealed RABL6A amplification in the majority of primary PNETs (6 of 11), which was sustained in the paired metastatic lesions (Table 1 and supplementary Fig. S1).
Table 1. RABL6A gene status measured by qPCR in human PNETs relative to matched normal tissue.
| Primary | Metastases* | |
|---|---|---|
| Loss | 1 of 11 (9%) | 1 of 10 (10%) |
| Gain | 6 of 11 (55%) | 5 of 10 (50%) |
Paired metastases for each primary PNET
RABL6A is required for PNET cell proliferation
To investigate the biological significance of RABL6A in PNET cell proliferation and survival, endogenous RABL6A was silenced in BON-1 PNET cells using RNA interference. Two different short hairpin RNAs (shRNAs) named KD1 and KD2 were used to target RABL6A (Fig. 2A), as described previously (27). KD1 targets all four RABL6 isoforms while KD2 selectively silences RABL6A, the most abundant isoform expressed in cells (27). Compared to an empty vector control virus, lentiviruses expressing the shRNAs effectively down-regulated RABL6A protein expression and caused a significant reduction in cell number by 3 days post-infection (Fig. 2B). Flow cytometric analyses showed a pronounced G1 phase arrest and moderate G2/M arrest in RABL6A depleted cells (Fig. 2C). Remarkably, the anti-proliferative effect of RABL6A knockdown was long-term and sustained over a 3-week time period, as long as shRNA expression was preserved by antibiotic selection (Fig. 2D). There was no evidence of senescence in the RABL6A depleted cells, as indicated by an absence of senescence-associated β-galactosidase expression and lack of morphological changes (flattening and enlargement) normally adopted by senescent cells. Together, these data establish an essential role for RABL6A in PNET cell proliferation, particularly in G1 phase progression.
Figure 2. RABL6A is required for PNET cell proliferation.

A, Schematic of RABL6 isoforms and RABL6A mRNA regions targeted by KD1 and KD2 shRNAs. B, western blot showing effective RABL6A knockdown in BON-1 cells expressing KD1 and KD2 shRNAs compared to vector control (CON). Graph shows reduced cell numbers following RABL6A knockdown relative to CON cells (*, p<0.05). C, flow cytometric analyses of DNA content show RABL6A depletion causes cell cycle arrest predominantly in G1 phase along with a moderate G2/M arrest. D, infected BON-1 cells were kept under antibiotic selection to maintain shRNA expression and cells counted at the indicated days. A representative growth curve from more than three independent experiments shows RABL6A loss causes sustained inhibition of proliferation.
RNA microarray analyses were performed in RABL6A knockdown and control cells to identify genes and signaling pathways affected by RABL6A loss. Approximately 750 genes were differentially regulated (>2-fold change, p<0.05 with FDR) by RABL6A knockdown (Supplementary Table S1). Ingenuity Pathway Analysis (IPA), which identifies functional relationships, mechanisms and pathways of predicted relevance from gene expression data, revealed that RABL6A depletion significantly altered the expression of genes involved in cell cycle regulation and checkpoint signaling (Fig. 3A). This included genes classified into the broad categories of cell cycle, DNA replication / recombination / repair, cellular growth and proliferation, and cell death and survival, among others. Within those categories, genes that participate either directly or indirectly in activated p53 and/or Rb1 tumor suppressor pathways were prominently affected (p53 p value overlap = 8.91×10-20, Rb1 p value overlap = 4.90×10-16) (Fig. 3B and supplementary Fig. S2). Few transcriptional targets of p53 were actually identified in the analysis. However, in many cases, regulators within the implicated pathways affect or are affected by both p53 and Rb1, suggesting that activation of one or both of those tumor suppressors may mediate the cell cycle arrest caused by RABL6A loss.
Figure 3. RABL6A depletion in BON-1 PNET cells significantly alters cell cycle regulatory and checkpoint pathway genes.

A, Ingenuity Pathway Analysis (IPA) of microarray data shows a statistically significant effect of RABL6A knockdown on gene expression within the indicated pathways. B, heat map shows the effect of RABL6A knockdown (from three independent BON-1 cell infections designated A-C) on genes that function in p53 signaling, Rb1 signaling or both pathways. Genes were categorized according to IPA software and reflect those with two-fold or greater changes in expression (p<0.05) in RABL6A knockdown cells. Red, relatively increased expression; blue, relatively decreased expression.
RABL6A promotes G1 progression by inactivating Rb1
A recent study reported that RABL6A inhibits p53 expression and transcriptional activity(25), which would be consistent with our microarray data suggesting p53 pathway activation in RABL6A depleted cells. As shown in Fig. 4A, RABL6A knockdown led to increased accumulation of the p21 protein, a p53 transcriptional target and cell cycle inhibitor that blocks cyclin-dependent kinase (Cdk)-mediated phosphorylation of Rb1 (38). KD1 cells, which display the greatest down-regulation of RABL6A, also showed a modest increase in p53 levels (Fig. 4A). These results, when considered with the microarray pathway analysis, supported the possibility that RABL6A loss might activate p53. On the other hand, depletion of RABL6A in KD2 cells had no effect on p53 levels and examination of individual gene expression from the microarray revealed that well-established transcriptional targets of p53 (including CDKN1A (p21), MDM2, PCNA, DDB2, GADD45α, TRIM22, FAS, BAX, BIRC5 (survivin) and PUMA, among others) were not differentially expressed at the mRNA level after RABL6A loss in KD1 and KD2 cells. In agreement, qRT-PCR for p21 showed it is not transcriptionally regulated in RABL6A knockdown cells (Supplementary Fig. S3).
Figure 4. Anti-proliferative effects of RABL6A loss in PNET cells are independent of p53.

The indicated PNET cells (BON-1 or Qgp-1) were infected with control (CON) or RABL6A shRNA (KD1 and KD2) viruses and analyzed either 4 days (A-C) or 8 days (D and E) later. A, western blots of RABL6A, p53, p21 and GAPDH expression in BON-1 infected cells. B, BON-1 cells were sequentially infected with empty vector (EV) or p53 shRNA (shp53) viruses followed by control (CON) or RABL6A shRNA (KD1 and KD2) viruses, and expression of RABL6A, p53, p21, cyclin D1 and GAPDH assessed by western blotting. The relative cell number of each population is indicated below each lane. C, representative histograms of DNA content in each population show p53 silencing has no effect on the cell cycle arrest phenotype of RABL6A knockdown cells. The percent of cells in each cell cycle phase is denoted. D, Western blots of the indicated proteins in BON-1 and Qgp-1 cells with sustained RABL6A knockdown. Relative cell number of each population is denoted. E, Quantification of percent cell death, as measured by trypan blue staining, in BON-1 and Qgp-1 cells with sustained RABL6A knockdown. Error bars are the deviation from the mean for data from three separate experiments (*, p<0.05).
To directly test if p53 mediates the anti-proliferative effects of RABL6A loss, BON-1 cells were sequentially infected with p53 shRNA (shp53) or empty vector (EV) viruses followed by RABL6A shRNA viruses. Cells analyzed 3 days after RABL6A depletion showed efficient silencing of both p53 and RABL6A (Fig. 4B). As anticipated, RABL6A silencing in EV control cells caused p21 upregulation, modest p53 elevation in KD1 cells, and reduced cell numbers (Fig. 4B, lanes 1-3). These changes coincided with G1 arrest (Fig. 4C) and accumulation of cyclin D1 (Fig. 4B, lanes 1-3), a G1 phase regulator whose elevated levels in non-proliferating cells is a useful marker of G1 arrest. Notably, p53 silencing failed to reverse the RABL6A knockdown phenotype. Loss of RABL6A in shp53 cells still caused p21 upregulation and cyclin D1 accumulation (Fig. 4B, lanes 4-6), as well as G1 arrest (Fig. 4C).
Similar studies of Qgp-1 cells, a p53-null PNET-derived cell line, likewise showed that p53 is dispensable for p21 upregulation and growth inhibition in RABL6A depleted cells (Fig. 4D). Interestingly, the sustained loss of RABL6A over an 8-day period caused significant cell death in both BON-1 and Qgp-1 cells (Fig. 4E), which was associated with elevated expression of PUMA and cleaved caspase-3 (Fig. 4D). Thus, p53 does not control the cell cycle arrest, cell death or upregulation of growth inhibitory (p21) and pro-apoptotic (PUMA, cleaved caspase-3) factors that is triggered by RABL6A silencing, establishing that RABL6A function is independent of p53 in PNET cells. Moreover, both BON-1 and Qgp-1 cells lack the p53 activator, ARF (Fig. 4D), revealing that the growth promoting effects of RABL6A in PNET cells are also ARF-independent.
The microarray data also implicated Rb1 activation in mediating the RABL6A knockdown arrest phenotype. Rb1 is a potent inhibitor of G1 progression whose hyper-phosphorylation by Cdks abolishes its growth suppressive activity(38, 39). Western analysis of Rb1 protein expression and phosphorylation showed it is hyper-phosphorylated in proliferating BON-1 CON cells, as expected, while RABL6A down-regulation caused a striking loss in expression (Fig. 5A). The remaining Rb1 protein expressed in RABL6A depleted cells was predominantly the hypo-phosphorylated, growth inhibitory form. Identical results were seen in Qgp-1 cells (not shown). Two approaches were used to ablate Rb1 function and test if its hypo-phosphorylation helped mediate the G1 arrest caused by RABL6A knockdown. First, we generated stable BON-1 cells expressing the E7 viral oncoprotein, which inhibits Rb1 expression and activity through multiple mechanisms (40). Compared to LXSN vector control cells, E7 cells expressed less phosphorylated Rb1 and cyclin D1 did not accumulate following RABL6A knockdown (Fig. 5B), predicting a reduced G1 phase arrest. Indeed, E7 promoted increased DNA synthesis (Fig. 5C) and greater S phase entry (Fig. 5D) in RABL6A depleted cells. Importantly, silencing of Rb1 using three different shRNAs (supplementary Fig. S4) yielded similar results. Rb1 downregulation partially rescued the RABL6A knockdown phenotype, increasing both cell number and S phase entry in RABL6A depleted cells (Figs. 5E and 5F). These results demonstrate that RABL6A normally promotes G1-S phase progression in PNET cells through a mechanism at least partially dependent upon Rb1 inactivation.
Figure 5. G1 arrest induced by RABL6A depletion is partially Rb1-dependent.

A, western blots of RABL6A, total and phosphorylated Rb1 (bracket indicates hyper-phosphorylated forms, bar indicates hypo-phosphorylated form), cyclin D1 (marker of G1 accumulation) and GAPDH (loading control) in parental BON-1 cells 3 days after infection with vector control (CON) or RABL6A shRNA (KD1 and KD2) viruses. B-D, stable BON-1 lines expressing LXSN vector or HPV E7 were infected with CON, KD1 or KD2 viruses and assayed three days later. B, RABL6A, Rb1, cyclin D1 and GAPDH were measured by western blotting. C, percent BrdU-positive cells in each population was quantified, showing increased DNA synthesis in BON-1 RABL6A knockdown cells expressing E7 relative to LXSN controls (*, p<0.05). D, representative histograms of DNA content in each BON-1 cell population show E7 expression increases S phase entry in RABL6A depleted cells (arrows), partially rescuing the G1 arrest caused by loss of RABL6A. E, relative cell numbers, normalized to the number of CON cells, following RABL6A knockdown in BON-1 cells infected with empty vector (EV) or Rb1 shRNAs (*, p=0.00014; **, p=0.0004). F, fold increase in S phase in RABL6A knockdown cells following Rb1 knockdown (+), as normalized to EV control (-) cells (*, p=0.0049; **, p=0.0013). Rb1 knockdown results shown in E and F were compiled from data using 2 or 3 different shRNAs, and error bars in panels C, E and F represent the deviation from the mean for data obtained in 3 independent experiments.
We tested if RABL6A overexpression would influence PNET proliferation and Rb1 status (supplementary Fig. S5). Elevated expression of RABL6A in BON-1 cells had a minimal, albeit reproducible and statistically significant effect on proliferation, increasing cell number 1.1-fold relative to vector control (p=0.0032). PNET tumors and derived cells express significant levels of RABL6A protein; therefore the modest effect of its overexpression on proliferation suggests that RABL6A proliferative activity is already nearly maximal in these cells. Interestingly, increased RABL6A expression had no impact on p53 levels but it did cause a marked rise in total Rb1 expression and phosphorylation that coincided with decreased p21 (Fig. S5A). The changes in p21 and Rb1 protein levels, which did not correlate with altered transcription (Fig. S5B), directly mirror the opposing effects of RABL6A knockdown on those proteins (i.e., p21 upregulation and decreased Rb1 phosphorylation and expression), Together, these results are consistent with our findings that RABL6A promotes PNET proliferation, at least in part, via Rb1 regulation.
RABL6A is required for PNET cell mitosis and survival
To determine if Rb1 inactivation could rescue the sustained growth inhibition of PNETs cells caused by RABL6A loss (see Fig. 2D), we examined cell number, percent of cell death, and mitotic progression of RABL6A knockdown in stable BON-1 E7 expressing cells at early and later time points. Analyses were performed at either day 3 (as in Figs. 2-5, except Figs. 4D and 4E) or day 6 following transduction with RABL6A shRNAs. At day 3, E7 expression transiently prevented the decrease in cell number caused by RABL6A downregulation in KD2 but not KD1 cells (Fig. 6A, left). This difference could be attributed to more efficient silencing of RABL6A by the KD1 shRNA or that KD2 selectively silences RABL6A while KD1 targets all RABL6 isoforms(27). Regardless, E7 was insufficient to promote continuous cell cycling in the face of RABL6A loss, as indicated by the equivalent reduction in cell number for both KD1 and KD2 knockdown lines at day 6 (Fig. 6A, right). E7 expression was also unable to prevent the marked increase in cell death of RABL6A depleted cells at day 6 (Fig. 6B). Interestingly, E7 did increase the percentage of RABL6A knockdown cells in G2/M phase at day 6 compared to day 3 (Fig. 6C). The mitotic arrest of RABL6A depleted cells is consistent with the significant down-regulation of established mitotic regulators, including BUB1, BUB1B, CENPF, MAD2L1, AURKB, CCNB1, PLK1 and NEK2, among other genes (Fig. 3B and Table S1), and with previous work showing RABL6A is required for mitotic progression in pancreatic ductal adenocarcinoma cells (27). The data here show E7-mediated Rb1 inactivation facilitates G1-S progression but does not enable mitotic progression of cells lacking RABL6A. It can therefore be concluded that sustained RABL6A loss causes increased mitotic arrest and death that are independent of Rb1.
Figure 6. Sustained RABL6A depletion causes increased cell death and mitotic arrest that is Rb1-independent.

Stable BON-1 lines expressing LXSN vector (open bars) or HPV E7 (black bars) were infected with CON, KD1 or KD2 viruses and assayed at either day 3 or day 6 post-infection. Error bars represent the mean for data from three or more independent experiments. A, cell numbers in each population were quantified and normalized to results for cells infected with shRNA control vector (dashed line). E7 expression reversed the decrease in cell number caused by RABL6A loss in KD2 cells at day 3 (p<0.001) but not day 6. B, the percent of cell death in each population was measured by trypan blue staining and counting. *, p<0.025 for KD1 and KD2 cells relative to CON cells expressing LXSN or E7. C, the percent of cells in G2/M phase for each population was determined by flow cytometry. E7 expression promoted G2/M accumulation of KD1 and KD2 cells at day 6 compared to day 3 (p<0.001).
Discussion
There is currently a lack of reliable biomarkers for the diagnosis of NETs. Our data show that RABL6A is amplified in primary and metastatic PNETs and highly expressed at the protein level in those tumors, suggesting RABL6A may be a useful diagnostic biomarker for PNETs. RABL6A is a new, largely unstudied protein so relatively little is currently known about its function and significance in cancer. Early work suggested its mRNA is increased in approximately 20 to 70% of tumors for certain malignancies including breast, uterine, colon, stomach and ovarian cancers(23). More recently, we and others showed that RABL6A protein is overexpressed in the majority of breast and pancreatic adenocarcinomas and this is associated with poor patient survival (26, 27). Thus, RABL6A status in PNETs could have both diagnostic and prognostic significance. Correlative analyses of patient clinicopathologic data and RABL6A status in a significantly expanded number of PNETs will address this important possibility.
The potential value of RABL6A as a PNET biomarker is heightened by its essential role in PNET cell proliferation and survival. We showed that RABL6A loss causes a robust cell cycle arrest, predominantly in G1 phase but also in G2/M, that precedes apoptotic cell death. Microarray analyses revealed prominent effects of RABL6A depletion on cell cycle regulatory and checkpoint pathways, which suggested a possible role for pathways involving p53 and/or Rb1 in mediating the arrest phenotype. We initially suspected p53 may play a role since p21 protein was upregulated in RABL6A knockdown cells and recent work suggested RABL6A can inhibit p53 (25). Specifically, RABL6A was reported to interact with Mdm2 and p53, promote p53 ubiquitination and impair p53 transcriptional activity; however, the importance of p53 to RABL6A biological activity was never examined(25). We directly assessed the role of p53 in the arrest caused by RABL6A knockdown and found that RABL6A controls p21 levels and proliferation independent of p53. In agreement with that finding, we have tested but found no evidence for a physical interaction between endogenous RABL6A and p53.
By comparison, our work uncovers a novel mechanistic link between RABL6A and the Rb1 tumor suppressor. We found that RABL6A is required for Rb1 hyperphosphorylation and inactivation, an event that is essential for G1 phase progression and cell proliferation. Normally, the sequential phosphorylation of Rb1 during G1 phase by cyclin D-dependent kinases, Cdk4 and/or Cdk6, and cyclin E/Cdk2 causes release of E2F transcription factors from Rb complexes and activation of genes required for S phase (39). Conversely, increased expression of Cdk inhibitors, including those in the Cip/Kip family (e.g., p21 and p27) and the INK4 family (e.g., p16INK4a), potently blocks Cdk-mediated Rb1 phosphorylation and enforces a G1 phase arrest(41). Our data showed that in the absence of RABL6A, Rb1 levels decrease and the hypo-phosphorylated form becomes predominant, which actively induces G1 arrest. In keeping with Rb1 contributing to the arrest, the microarray results show that 23 E2F1-regulated genes are down-regulated in RABL6A knockdown cells (see Table S1). The mechanisms by which RABL6A controls Rb1 levels are currently unknown although it is clear that the minimal decrease in Rb1 mRNA caused by RABL6A knockdown (Fig. S4) cannot account for the significant decrease in its protein levels. The fact that RABL6A overexpression enhances Rb1 protein levels without affecting its mRNA expression (Fig. S5) further supports the likelihood that post-transcriptional mechanisms are involved.
The effect of RABL6A knockdown and overexpression on Rb1 phosphorylation is likely due to altered expression of p21 and/or p27. Both Cdk inhibitors are upregulated in RABL6A knockdown cells while p21 is down-regulated in RABL6A over-expressing cells (supplementary Figs. S5 and S6). Since p21 mRNA expression is not affected by changes in RABL6A expression, it is probable that p21 and p27 levels are controlled post-transcriptionally. In RABL6A depleted cells, CKS1B mRNA is significantly reduced (3.3 fold, p=0.0000019), which may lead to reduced ubiquitination and proteasomal degradation of p21 and p27 by the SCFSkp2 (Skp1-Cul1-FBox protein-Skp2) ubiquitin ligase since CKS1B is required for its activity(42-44). Either p21 or p27 could promote the Rb1-mediated G1 arrest in RABL6A depleted cells although the dramatic rise in p27 expression and fact that p27-p18INK4c null mice develop islet cell hyperplasia(45) predict a more significant role for p27 in RABL6A signaling and PNET proliferation. We examined the effects of p27 knockdown on RABL6A function and found that p27 loss promotes S phase entry in cells arrested by RABL6A depletion (Fig. S6), although similar to Rb1 inactivation, the rescue was not sufficient to enable sustained proliferation. Those findings indicate p27 contributes to the G1 arrest caused by RABL6A loss. Studies of PNET cells lacking p21 or p27 versus both factors together will establish their relative importance to RABL6A regulation of Rb1 and G1-S progression. Importantly, the PNET cells used in this study lack INK4a/ARF, consequently neither p16INK4a nor ARF contribute to RABL6A function in this context.
Several lines of evidence suggest Rb1 inactivation is a key mechanism underlying PNET cell proliferation. Early studies showed that RIP-Tag2 mice, which lack both Rb1 and p53 function due to SV40 T antigen expression in pancreatic beta cells, develop insulinomas (46). The importance of Rb1 loss for tumor development in that system has been supported by studies showing frequent alteration of Rb1 regulators in PNETs. For instance, Cdk4 or Cdk6 genes are amplified in roughly 20% of PNETs and Cdk4 protein expression is elevated in nearly 60% of the tumors (19). Other work has shown silencing of the INK4a/ARF locus in up to 50% of gastroenteropancreatic NETs (20, 21). Interestingly, mice engineered to express a mutant form of Cdk4 (R24C allele), which cannot be inhibited by p16INK4a, develop PNETs spontaneously (47, 48). Our findings identify a new negative regulator of Rb1 signaling, RABL6A, that is amplified in primary PNETs and required for PNET cell proliferation. These data, in addition to evidence that RABL6A overexpression in other tumors is associated with poor patient survival (26, 27), strongly suggest RABL6A is a novel oncoprotein.
Since Rb1 remains wild-type in patient PNETs, pharmacological re-activation of Rb1 suppressive activity in tumors with elevated RABL6A and/or Cdk4/6 could be therapeutically beneficial. In that regard, treatment with the selective Cdk4/6 inhibitor, PD 0332991, effectively abolished the in vitro and in vivo growth of PNET cells that over-express Cdk4(19). Combination therapies that include PD 0332991, which is currently in clinical trials for other cancers(49, 50), will likely hold the greatest potential in the clinic. Indeed, PD 0332991 acts synergistically with the mTOR inhibitor, rapamycin, to provoke G1 arrest in cultured BON-1 cells (19). This is relevant because everolimus, another mTOR inhibitor, is an important component of current PNET therapy(10-12). Intriguingly, our microarray data suggest RABL6A normally promotes Akt activation(Table S1 and Fig. S2) and preliminary molecular evidence reveals RABL6A is required for Akt-mTOR signaling (unpublished data, J. Hagen and D.E. Quelle). We speculate that RABL6A may be a master regulator of PNET proliferation that functions through multiple mechanisms that includes Rb1 inactivation as well as Akt-mTOR activation and possibly other cancer pathways implicated in our microarray data. That would concur with our finding that RABL6A is not only required for G1-S phase progression but also mitosis. When considered with the frequent amplification and elevated expression of RABL6A in PNETs, this work suggests that RABL6A could be an important new target for anticancer therapy in PNET patients.
Supplementary Material
Acknowledgments
We thank Dr. M. Sue O'Dorisio for providing BON-1 cells and advice, and Dr. Steve Berberich for p53 shRNA constructs. We are grateful for assistance from Donghong Wang, various core facilities within the University of Iowa College of Medicine and Holden Comprehensive Cancer Center (Flow Cytometry, Pathology, Central Microscopy and DNA Core), the Shivanand R. Patil Cytogenetics and Molecular Laboratory, and Holden Comprehensive Cancer Center. We also thank the Iowa Neuroendocrine Tumor Registry and Neuroendocrine Tumor Fund.
Grant Support: This work was supported by NIH grants R01-CA090367 [D.E.Q.], R21-CA127031 [D.E.Q.] and 5T32-CA148062 [S.K.S.] as well as the Holden Comprehensive Cancer Center [D.E.Q., B.W.D.].
Footnotes
Conflicts of interest: None
References
- 1.Vinik AI, Woltering EA, Warner RRP, Caplin M, O'Dorisio TM, Wiseman GA, et al. NANETS Consensus Guidelines for the Diagnosis of Neuroendocrine Tumor. Pancreas. 2010;39:713–34. doi: 10.1097/MPA.0b013e3181ebaffd. [DOI] [PubMed] [Google Scholar]
- 2.Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, et al. One Hundred Years After “Carcinoid”: Epidemiology of and Prognostic Factors for Neuroendocrine Tumors in 35,825 Cases in the United States. J Clin Oncol. 2008;26:3063–72. doi: 10.1200/JCO.2007.15.4377. [DOI] [PubMed] [Google Scholar]
- 3.Frilling A, Sotiropoulos GC, Li J, Kornasiewicz O, Plöckinger U. Multimodal management of neuroendocrine liver metastases. HPB. 2010;12:361–79. doi: 10.1111/j.1477-2574.2010.00175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ehehalt F, Saeger HD, Schmidt CM, Grützmann R. Neuroendocrine Tumors of the Pancreas. The Oncologist. 2009;14:456–67. doi: 10.1634/theoncologist.2008-0259. [DOI] [PubMed] [Google Scholar]
- 5.Bilimoria KY, Tomlinson JS, Merkow RP, Stewart AK, Ko CY, Talamonti MS, et al. Clinicopathologic features and treatment trends of pancreatic neuroendocrine tumors: Analysis of 9,821 patients. Journal of Gastrointestinal Surgery. 2007;11:1460–7. doi: 10.1007/s11605-007-0263-3. [DOI] [PubMed] [Google Scholar]
- 6.Falconi M, Bartsch DK, Eriksson B, Kloeppel G, Lopes JM, O'Connor JM, et al. ENETS Consensus Guidelines for the Management of Patients with Digestive Neuroendocrine Neoplasms of the Digestive System: Well-Differentiated Pancreatic Non-Functioning Tumors. Neuroendocrinology. 2012;95:120–34. doi: 10.1159/000335587. [DOI] [PubMed] [Google Scholar]
- 7.Sherman SK, Howe JR. Translational Research in Endocrine Surgery. Surg Oncol Clin N Am. 2013;22:857–+. doi: 10.1016/j.soc.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Carbonero R, Capdevila J, Crespo-Herrero G, Diaz-Perez JA, Martinez del Prado MP, Alonso Orduna V, et al. Incidence, patterns of care and prognostic factors for outcome of gastroenteropancreatic neuroendocrine tumors (GEP-NETs): results from the National Cancer Registry of Spain (RGETNE) Ann Oncol. 2010;21:1794–803. doi: 10.1093/annonc/mdq022. [DOI] [PubMed] [Google Scholar]
- 9.Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR Pathway Genes Are Frequently Altered in Pancreatic Neuroendocrine Tumors. Science. 2011;331:1199–203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kulke MH, Anthony LB, Bushnell DL, de Herder WW, Goldsmith SJ, Klimstra DS, et al. NANETS Treatment Guidelines: Well-Differentiated Neuroendocrine Tumors of the Stomach and Pancreas. Pancreas. 2010;39:735–52. doi: 10.1097/MPA.0b013e3181ebb168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raut CP, Kulke MH. Targeted Therapy in Advanced Well-Differentiated Neuroendocrine Tumors. The Oncologist. 2011;16:286–95. doi: 10.1634/theoncologist.2010-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, et al. Everolimus for Advanced Pancreatic Neuroendocrine Tumors. N Engl J Med. 2011;364:514–23. doi: 10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 14.Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 15.Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Current Opinion in Genetics & Development. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 16.Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–73. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- 17.Ulanet DB, Hanahan D. Loss of p19Arf Facilitates the Angiogenic Switch and Tumor Initiation in a Multi-Stage Cancer Model via p53-Dependent and Independent Mechanisms. PLoS ONE. 2010;5:e12454. doi: 10.1371/journal.pone.0012454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu W, Feng Z, Modica I, Klimstra DS, Song L, Allen PJ, et al. Gene Amplifications in Well-Differentiated Pancreatic Neuroendocrine Tumors Inactivate the p53 Pathway. Genes & Cancer. 2010;1:360–8. doi: 10.1177/1947601910371979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang LH, Contractor T, Clausen R, Klimstra DS, Du YCN, Allen PJ, et al. Attenuation of the Retinoblastoma Pathway in Pancreatic Neuroendocrine Tumors Due to Increased Cdk4/Cdk6. Clin Cancer Res. 2012;18:4612–20. doi: 10.1158/1078-0432.CCR-11-3264. [DOI] [PubMed] [Google Scholar]
- 20.Liu L, Broaddus RR, Yao JC, Xie S, White JA, Wu TT, et al. Epigenetic alterations in neuroendocrine tumors: methylation of RAS-association domain family 1, isoform A and p16 genes are associated with metastasis. Mod Pathol. 2005;18:1632–40. doi: 10.1038/modpathol.3800490. [DOI] [PubMed] [Google Scholar]
- 21.Simon B, Lubomierski N. Implication of the INK4a/ARF Locus in Gastroenteropancreatic Neuroendocrine Tumorigenesis. Ann N Y Acad Sci. 2004;1014:284–99. doi: 10.1196/annals.1294.033. [DOI] [PubMed] [Google Scholar]
- 22.Tompkins V, Hagen J, Zediak VP, Quelle DE. Identification of novel ARF binding proteins by two-hybrid screening. Cell Cycle. 2006;5:641–6. [PubMed] [Google Scholar]
- 23.Montalbano J, Jin WX, Sheikh MS, Huang Y. RBEL1 is a novel gene that encodes a nucleocytoplasmic ras superfamily GTP-binding protein and is overexpressed in breast cancer. J Biol Chem. 2007;282:37640–9. doi: 10.1074/jbc.M704760200. [DOI] [PubMed] [Google Scholar]
- 24.Montalbano J, Lui K, Sheikh MS, Huang Y. Identification and Characterization of RBEL1 Subfamily of GTPases in the Ras Superfamily Involved in Cell Growth Regulation. J Biol Chem. 2009;284:18129–42. doi: 10.1074/jbc.M109.009597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lui K, An J, Montalbano J, Shi J, Corcoran C, He Q, et al. Negative regulation of p53 by Ras superfamily protein RBEL1A. Journal of Cell Science. 2013;126:2436–45. doi: 10.1242/jcs.118117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li YY, Fu S, Wang XP, Wang HY, Zeng MS, Shao JY. Down-Regulation of C9orf86 in Human Breast Cancer Cells Inhibits Cell Proliferation, Invasion and Tumor Growth and Correlates with Survival of Breast Cancer Patients. PLoS ONE. 2013;8:e71764. doi: 10.1371/journal.pone.0071764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muniz VP, Askeland RW, Zhang X, Reed SM, Tompkins VS, Hagen J, et al. RABL6A Promotes Oxaliplatin Resistance in Tumor Cells and Is a New Marker of Survival for Resected Pancreatic Ductal Adenocarcinoma Patients. Genes & Cancer. 2013;4:273–84. doi: 10.1177/1947601913501074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X, Hagen J, Muniz VP, Smith T, Coombs GS, Eischen CM, et al. RABL6A, a Novel RAB-Like Protein, Controls Centrosome Amplification and Chromosome Instability in Primary Fibroblasts. PLoS ONE. 2013;8:e80228. doi: 10.1371/journal.pone.0080228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Townsend CM, Ishizuka J, Thompson JC. Studies of Growth Regulation in a Neuroendocrine Cell Line. Acta Oncol. 1993;32:125–30. doi: 10.3109/02841869309083900. [DOI] [PubMed] [Google Scholar]
- 30.Kelley KD, Miller KR, Todd A, Kelley AR, Tuttle R, Berberich SJ. YPEL3, a p53-Regulated Gene that Induces Cellular Senescence. Cancer Res. 2010;70:3566–75. doi: 10.1158/0008-5472.CAN-09-3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halbert CL, Demers GW, Galloway DA. The E6-gene and E7-gene of human papillomavirus type-6 have weak immortalizing activity in human epithelial-cells. Journal of Virology. 1992;66:2125–34. doi: 10.1128/jvi.66.4.2125-2134.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Darbro BW, Lee KM, Nguyen NK, Domann FE, Klingelhutz AJ. Methylation of the p16(INK4a) promoter region in telomerase immortalized human keratinocytes co-culturedwith feeder cells. Oncogene. 2006;25:7421–33. doi: 10.1038/sj.onc.1209729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research. 2001;29 doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korgaonkar C, Zhao L, Modestou M, Quelle DE. ARF function does not require p53 stabilization or Mdm2 relocalization. Mol Cell Biol. 2002;22:196–206. doi: 10.1128/MCB.22.1.196-206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.di Tommaso A, Hagen J, Tompkins V, Muniz V, Dudakovic A, Kitzis A, et al. Residues in the alternative reading frame tumor suppressor that influence its stability and p53-independent activities. Experimental Cell Research. 2009;315:1326–35. doi: 10.1016/j.yexcr.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klimstra DS, Wenig BM, Heffess CS. Solid-pseudopapillary tumor of the pancreas: A typically cystic carcinoma of low malignant potential. Seminars in Diagnostic Pathology. 2000;17:66–80. [PubMed] [Google Scholar]
- 37.Dahdaleh FS, Calva-Cerqueira D, Carr JC, Liao J, Mezhir JJ, O'Dorisio TM, et al. Comparison of Clinicopathologic Factors in 122 Patients with Resected Pancreatic and Ileal Neuroendocrine Tumors from a Single Institution. Ann Surg Oncol. 2012;19:966–72. doi: 10.1245/s10434-011-1997-4. [DOI] [PubMed] [Google Scholar]
- 38.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 39.Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes & Development. 2000;14:2393–409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- 40.Klingelhutz AJ, Roman A. Cellular transformation by human papillomaviruses: Lessons learned by comparing high- and low-risk viruses. Virology. 2012;424:77–98. doi: 10.1016/j.virol.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G(1)-phase progression. Genes & Development. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 42.Cardozo T, Pagano M. The SCF ubiquitin ligase: Insights into a molecular machine. Nature Reviews Molecular Cell Biology. 2004;5:739–51. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- 43.Spruck C, Strohmaier H, Watson M, Smith AP, Ryan A, Krek TW, et al. A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol Cell. 2001;7:639–50. doi: 10.1016/s1097-2765(01)00210-6. [DOI] [PubMed] [Google Scholar]
- 44.Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M, et al. The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat Cell Biol. 2001;3:321–4. doi: 10.1038/35060126. [DOI] [PubMed] [Google Scholar]
- 45.Franklin DS, Godfrey VI, O'Brien DA, Deng CX, Xiong Y. Functional collaboration between different cyclin-dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Mol Cell Biol. 2000;20:6147–58. doi: 10.1128/mcb.20.16.6147-6158.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hanahan D. Heritable formation of pancreatic beta-cell tumors in transgenic mice expressing recombinant insulin simian virus-40 oncogenes. Nature. 1985;315:115–22. doi: 10.1038/315115a0. [DOI] [PubMed] [Google Scholar]
- 47.Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- 48.Sotillo R, Dubus P, Martin J, de la Cueva E, Ortega S, Malumbres M, et al. Wide spectrum of tumors in knock-in mice carrying a Cdk4 protein insensitive to INK4 inhibitors. Embo Journal. 2001;20:6637–47. doi: 10.1093/emboj/20.23.6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–66. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 50.Flaherty KT, LoRusso PM, DeMichele A, Abramson VG, Courtney R, Randolph SS, et al. Phase I, Dose-Escalation Trial of the Oral Cyclin-Dependent Kinase 4/6 Inhibitor PD 0332991, Administered Using a 21-Day Schedule in Patients with Advanced Cancer. Clin Cancer Res. 2012;18:568–76. doi: 10.1158/1078-0432.CCR-11-0509. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.