

Abstract
Malignant Rhabdoid Tumors (MRTs), a pediatric cancer that most frequently appears in the kidney and brain, generally lack SNF5 (SMARCB1/INI1), a subunit of the SWI/SNF chromatin-remodeling complex. Recent studies have established that multiple SWI/SNF complexes exist due to the presence or absence of different complex members. Therefore, the effect of SNF5 loss upon SWI/SNF complex formation was investigated in human MRT cells. MRT cells and primary human tumors exhibited reduced levels of many complex proteins. Furthermore, re-expression of SNF5 increased SWI/SNF complex protein levels without concomitant increases in mRNA. Proteomic analysis, using mass spectrometry, of MRT cells before and after SNF5 re-expression indicated the recruitment of different components into the complex along with the expulsion of others. IP-Western blotting confirmed these results and demonstrated similar changes in other MRT cell lines. Finally, reduced expression of SNF5 in normal human fibroblasts led to altered levels of these same complex members. These data establish that SNF5 loss during MRT development alters the repertoire of available SWI/SNF complexes, generally disrupting those associated with cellular differentiation. These findings support a model where SNF5 inactivation blocks the conversion of growth promoting SWI/SNF complexes to differentiation inducing ones. Therefore, restoration of these complexes in tumors cells provides an attractive approach for the treatment of malignant rhabdoid tumors.
Implications
SNF5 loss dramatically alters SWI/SNF complex composition and prevents formation of complexes required for cellular differentiation.
Keywords: Malignant Rhabdoid Tumor (MRT), SNF5/INI1, PBRM1/BAF180, SWI/SNF Complex
Introduction
Malignant Rhabdoid Tumors (MRT) are a highly aggressive pediatric cancer commonly found in the kidneys (RTK) and brain (AT/RT). These tumors have a median onset of 11 months and 80–90% of children die from the disease within a year of diagnosis (1). MRTs are characterized by the loss of SNF5/BAF47/INI1/SMARCB1, a core member of the SWI/SNF complex, hereafter referred to as SNF5 (2). Interestingly, SNF5 loss does not affect genetic stability in MRT, but causes epigenetic instability (3). Thus, this consistent and specific molecular change, SNF5 inactivation, provides a unique model for understanding the role of epigenetics in human tumor development. Numerous studies have shown that SNF5 loss affects multiple signaling pathways (4). However, little else is known regarding the etiology of this cancer.
The SWI/SNF complex has multiple variants and is classified by their mutually exclusive ATPase subunits, BRM and BRG1. BRG1 containing SWI/SNF complexes can be subdivided into two mutually exclusive groups- BAF180/PBRM1 and BAF250A/ARID1A containing complexes. While SNF5 appears in all the SWI/SNF complexes, little is known about the effects of SNF5 loss upon SWI/SNF activity. One previous study by Doan et al. showed that the SWI/SNF complex appeared intact in MRT cell lines despite the loss of SNF5 (5). They also demonstrated that BRG1-dependent genes did not show altered expression in MRT cell lines (5). However, they did not address the stoichiometry of the complex components in MRT cell lines or the interaction of the complex with chromatin. Other studies have indicated that loss of a SWI/SNF complex member could affect the stability of other complex members in mammalian cells and in flies (6–12). Furthermore, a growing number of studies have identified multiple SWI/SNF complexes defined by the presence or absence of different components (4, 13–15). Importantly, these different complexes can promote either growth or differentiation depending upon their composition (4, 13–15).
In order to address whether SNF5 loss affects SWI/SNF complex composition during MRT development, we examined the effects of SNF5 expression in MRT cell lines. Our studies demonstrate that re-expression of SNF5 in MRT cell lines altered total protein levels of many components while knock down of SNF5 in normal human fibroblasts showed the opposite effect without altering mRNA expression. After SNF5 re-expression, many components were recruited into SWI/SNF complexes that are generally associated with induction of differentiation. Therefore, SNF5 loss may promote MRT development by preventing a switch from SWI/SNF complexes that promote proliferation to those associated with differentiation. These studies emphasize the need to resolve the scope and composition of SWI/SNF complexes among different tissues and may account for the low incidence of these deadly childhood cancers.
Materials and Methods
Cell lines
Our laboratory previously described the A204.1, D98OR, and G401.6 cell lines (16–18). The MCF-7, A673, Jurkat, DAOY and RD cell lines were obtained from the American Type Tissue Collection (ATCC). The TTC642 and TTC549 cell lines were kindly provided from Dr. Timothy Triche, Children’s Hospital of Los Angeles. Dr. Peter Houghton, Nationwide Childrens Hospital, kindly provided the BT-12 AT/RT cell line. 293FT cells were kindly provided by Dr. Inder Verma, Salk Institute. All cell lines were cultured in RPMI 1640 plus 10% fetal bovine serum (Gibco, Grand Island, NY or Benchmark FBS, Gemini Biosciences, Sacramento, CA) and were used within 30 passages of their initial arrival to minimize chances of cross-contamination.
Adenovirus infection
The Ad/pAdEasyGFPINI-SV+ adenoviral vectors expressing hSNF5 and coexpressing green fluorescent protein (GFP; designated Ad-hSNF5) and the Ad/pAdEasyGFP expressing GFP (designated Ad-GFP) were previously published (19). To generate adenovirus expressing HA-tagged SNF5, we used the pAdEasy system (20). We first subcloned the insert from the pMDK225 lentivirus (21) expressing SNF5 with a c-terminal triple-HA tag into the pShuttle vector. The UNC Vector Core generated Ad-SNF5-HA following the pAdEasy system protocol. The SNF5-HA was sequenced to verify the wild-type gene and the triple HA tag at the c-terminus. To achieve infection of over 90% of cells, we infected at a multiplicity of infection (MOI) of 20 for the A204.1 cell line, 10 for the G401 cell lines, 10 for the TTC549 cell line, and 200 for the TTC642 cell line. Infection of cell lines were carried out as previously described (22).
Protein extracts and Western blotting
Isolation of nuclear protein and Western blotting was carried out as described previously (16, 23). Western blot analyses for protein expression were carried out using the antibodies listed in Supplementary Table 1. Densitometry was carried out using the BioRad Imagelab 4.1 software.
RNA extraction and quantitative real-time reverse transcription–PCR analysis
RNA was extracted using the RNeasy mini kit (Qiagen), and 1 μg was used for cDNA synthesis primed with random primers (Invitrogen). cDNA was analyzed using TaqMan (Applied Biosystems) quantitative real-time reverse transcription–PCR (qPCR) analysis, with β-ACTIN as the reference gene in each reaction. Reactions were performed on an ABI 7900 HT sequence detection system (Applied Biosystems), and relative quantification was determined using the 2−ΔΔCt method (Chai et al., 2005a). The TaqMan gene expression assay primer/probe sets used in these studies are listed in Supplementary Table 2.
Immunoprecipitation
Cells were infected with adenovirus expressing either HA tagged SNF5 (Ad-SNF5-HA) or empty vector (Ad-CMV). Proteins were isolated from the infected cells using IP buffer (50mM Tris, 400 mM NaCl, 2mM EDTA, 10% Glycerol, 1% NP-40, 0.5% sodium deoxycholate, 0.1 mM PMSF, 4 mM sodium fluoride, 40 nM sodium orthovanadate, 1x complete Mini protease inhibitor cocktail (Roche Diagnostics)). The isolated protein was quantified and adjusted to 1mg/ml. One mg of protein from each sample was incubated with BRG-1 (A303-877A; Bethyl), or normal rabbit IgG (sc-2027; Santa Cruz Biotechnology) rotating overnight at 4°C with 30 μl of a 50% slurry of protein A/G Sepharose beads. The beads were then washed 3 times with IP wash buffer (1x PBS, 10% glycerol, 1% Triton) and then suspended in 1x Nupage LDS loading buffer supplemented with 125mM DTT and boiled for 5 minutes. The supernatants were then run on 4–12% Bis-Tris polyacrylamide gel, transferred to PVDF membranes and probed with anti-SNF5, anti-BRG1, anti-BAF155, anti-BAF180, anti-BAF200, anti-BAF250A, anti-BAF170, anti-BAF60A, or anti-BAF57 as described above (Supplementary Table 1).
Mass spectrometry
The determination and quantification of SWI/SNF complex components by mass spectrometry was carried out on immunoprecipitated samples prepared as in the immunoprecipitation protocol above following our previously published protocols (24). The resulting mass spectra were searched against the SwissProt sequence database (downloaded 07/23/13) using MaxQuant (Version 1.4.1.2) (25) with a static carbamidomethyl modification on cysteines, variable oxidation of methionine, variable acetylation of N-termini, and a fully-tryptic digestion. MaxQuant’s LFQ algorithm was used for label-free quantification. Experiments with and without doxycycline treatment were each performed in biological duplicate, with each biological sample analyzed in technical duplicate. Technical replicates’ LFQ intensities were averaged. Ratios for each batch of biological duplicates were determined by the LFQ intensity of the doxycycline treated sample divided by the LFQ intensity of the untreated control sample.
Lentiviral procedures and small hairpin RNA
Lentivirus was generated using 293FT cells following the protocol of Kafri and colleagues (Xu et al., 2001). Either pLKO.1, a nontargeting small hairpin RNA (shRNA) control vector (SHC002; Sigma), or SNF5 shRNA lentiviral transduction particles (TRCN 39585 and 39587) were cotransfected with the packing construct ΔNRF [from Dr. Tal Kafri, University of North Carolina; (26)] and the VSV-G envelope expression plasmid (pMDK64; from Dr. Matthias Kaeser, Salk Institute) into 293FT cells using calcium phosphate transfection. pLKO.1 is a negative control containing an inert sequence that does not target any human or mouse gene but will activate the RNAi pathway. For infection, cells were incubated with lentiviral particles and polybrene and then selected with puromycin.
Generation of inducible SNF5 cell lines
Flag tagged SNF5 was amplified from an existing plasmid, pcDNA3-fSNF5, by PCR (T7 promoter F primer, hSNF5 ORF R primer – TTA CCA GGC CGG CGT GTT) (16). The resulting PCR product was TOPO-cloned into a Gateway vector using the pCR®8/GW/TOPO® TA cloning kit (45-0642, Invitrogen). The fSNF5 was transferred into the pINDUCER20 vector that provided a c-terminal HA tag using Gateway® LR Clonase ™ II Plus Enzyme Mix (12538-120, Invitrogen). The identity of the fSNF5HA insert was confirmed by DNA sequencing.
β-galactosidase staining
To identify β-galactosidase positive cells, we used the Senescence β-Galactosidase Staining kit (Cell Signaling #9860). Cells were prepared and stained following the kit protocol as previously described (9).
Inhibition of protein degradation
To inactivate the proteasome, cells were treated with 10μM MG132 (1748, Boston Biochem) or DMSO vehicle control for 6 hours. Cells were harvested and prepared lysates were analyzed by immunoblotting, as described above.
Results
SWI/SNF Complex Components Are Post-Transcriptionally Regulated in MRT Cell Lines
We first examined whether component protein levels differed between SNF5-deficient MRT and AT/RT cell lines and SNF5-positive non-MRT cell lines including pediatric (A673 Ewing sarcoma, DAOY medulloblastoma, and RD rhabdomyosarcoma) and adult (D98OR, a HeLa cell derivative, Jurkat acute T cell lymphoma and MCF-7 breast cancer) tumor cell lines. As expected, we found a clear loss of SNF5 protein in the four MRT cell lines and one AT/RT cell line in comparison to non-MRT cancers (Figure 1A) (23). We also found no expression of BRM/SMARCA2 protein or mRNA, consistent with previous reports (Figure 1A & B) (27, 28) while levels of the other ATPase, BRG1/SMARCA4, appeared similar among all cell lines. Surprisingly, the other component proteins showed generally lower levels in MRTs compared to other pediatric and adult tumor cell lines ranging from nearly complete absence (ARID1A, SMARCC2 and SMARCD2) to moderate reduction (ARID2) (Figure 1A).
Figure 1. Decreased SWI/SNF complex protein but not mRNA in MRT cell lines.

(A) Total cellular proteins (30 μg) isolated from MRT and non-MRT cell lines were separated on a 4% to 12% SDS-polyacrylamide gel, transferred to PVDF membranes and probed with antiserum against SWI/SNF complex members. We used Ku80 as the loading control. (B) RNA was extracted from the indicated cell lines. The mRNA levels were measured for each gene by qPCR and normalized to D98OR β-actin expression. Blue- MRT cell lines; Red- non-MRT cell lines. Columns – mean of four independent experiments; Error bars – standard error.
We next asked whether the reduced protein levels reflected changes in mRNA expression. Thus, we assessed component mRNA levels using quantitative PCR (qPCR) normalized to the HeLa-derived D98OR cell line, a cell line often used for the purification of SWI/SNF complex members (29, 30). Consistent with the western blot data, SNF5 mRNA expression was undetectable in MRT cell lines, with the exception of TTC642, a cell line that contains a nonsense mutation in the SNF5 gene, (23). In contrast, other than SMARCA2/BRM, we could not observe consistent changes in the mRNAs of other complex components in MRT cell lines that could account for their lower protein levels (Figure 1B)
To extend these findings to primary MRTs, we examined protein expression in nuclear extracts from a series of primary tumors derived from MRTs as well as other prototypical childhood cancers, including Wilm’s tumors (WT), and rhabdomyosarcomas (RMS) (23). Due to limited amount of starting material, we could only assess a subset of SWI/SNF components. Protein levels in these nuclear extracts were normalized to histone H3 protein levels and aggregated by MRT vs. non-MRT tumor type. Loss of SNF5 protein again was apparent in the MRTs compared to the other tumor types (Figure 2A). The average ratio of BAF180 and BAF57 to H3 in MRTs was significantly lower than in non-MRT samples, while the BRG1 levels remained similar (Figure 2B). Thus, the primary tumor data for MRTs recapitulate the findings derived from cell lines.
Figure 2. Decreased SWI/SNF complex proteins in primary tumor samples.

(A) Nuclear protein (3 μg) isolated from primary tumors were separated on a 4 to 12% Bis-Tris polyacrylamide gel, transferred to PVDF membranes and probed with anti-BRG1, anti-BAF57, anti-SNF5, anti-BAF180, or anti-H3. WT = Wilms’ tumor; RMS = Rhabdomyosarcoma; MRT = Malignant Rhabdoid Tumor. (B) Densitometry was carried out using Bio-Rad Imagelab 4.1. All values expressed as a ratio to H3. The values were then aggregated by tumor types and averaged. Columns, mean of ratio of each tumor type; Error bars – standard error *, P < 0.05 relative to MRT; #, P > 0.05 relative to MRT using Student’s T-test.
hSNF5 Re-expression in MRT Cell Lines Increases SWI/SNF Complex Component Levels
We next examined the effects of SNF5 re-expression on SWI/SNF complex members in MRT cell lines. To carry out these experiments, we utilized adenovirus to re-express SNF5 in 3 representative MRT cell lines. We focused on the MRT cell lines, G401, TTC549 and TTC642 and omitted the soft tissue derived A204 to maintain our focus on renal-derived MRTs. We examined SWI/SNF complex component protein expression by Western blotting and mRNA levels by qPCR before and after 24 hours after infection with adenovirus. As shown in Figure 3A, we observed a consistent increase in the protein level BAF180 as a result of SNF5 re-expression in all MRT cell lines. In contrast, BRG1 and BAF200 protein levels did not change significantly after SNF5 re-expression. Other complex members showed differential changes with BAF155 increasing in the G401 and TTC549 cell lines, BAF250A showing an increase in the TTC549 cell line and BAF60A and BAF57 showing increases in the TTC642 cell line. These findings appear consistent with the observations in primary tumors (Figure 2A) in which SNF5 negative MRTs demonstrated decreased BAF180 and BAF57 levels but not BRG1 in comparison to WT and RMS. Similar to the results in Figure 1A&B, changes in protein levels did not result from altered mRNA expression (Figure 3B), consistent with post-transcriptional regulation.
Figure 3. hSNF5 Re-expression in MRT Cell Lines Increases SWI/SNF Complex Component Levels.

(A) Cells were harvested at time 0 and 24 hours after infection with Ad-CMV, Ad-GFP or Ad-SNF5-GFP. Total cellular proteins (30 μg) were separated on a 4% to 12% Bis-Tris polyacrylamide gel, transferred to PVDF membranes and probed with indicated antibody. Ku served as the loading control. UN, uninfected control. (B) Cells were harvested at time 0 and 24 hours after infection with Ad-CMV, Ad-GFP or Ad-SNF5-GFP and RNA extracted from the indicated cell lines. The mRNA levels were measured for each gene by qPCR and normalized to β-ACTIN expression at time 0. Because of the undetectable levels of SNF5 in the G401 and TTC549 cell lines, a log scale was used to graph SNF5 expression. Blue – G401; Red – TTC549; Green – TTC642; Columns, mean of four independent experiments; Error bars- standard error.
hSNF5 Expression Increases the Complexity of SWI/SNF Complexes
Our data thus far demonstrated that global SWI/SNF complex component levels increase following SNF5 over-expression. Next, we determined if this global change is mirrored within the SWI/SNF complex. To carry out these experiments, we developed a tet-inducible SNF5 vector (pIND20-fSNF5-HA) using the pINDUCER system (31) to develop MRT cell lines with inducible SNF5 expression (Figure 4). As shown in Figure 4, the tet-inducible cell lines validated the results from the adenovirus infections studies in the previous section (Figure 3A).
Figure 4. Characterization of SNF5-inducible MRT cell lines.
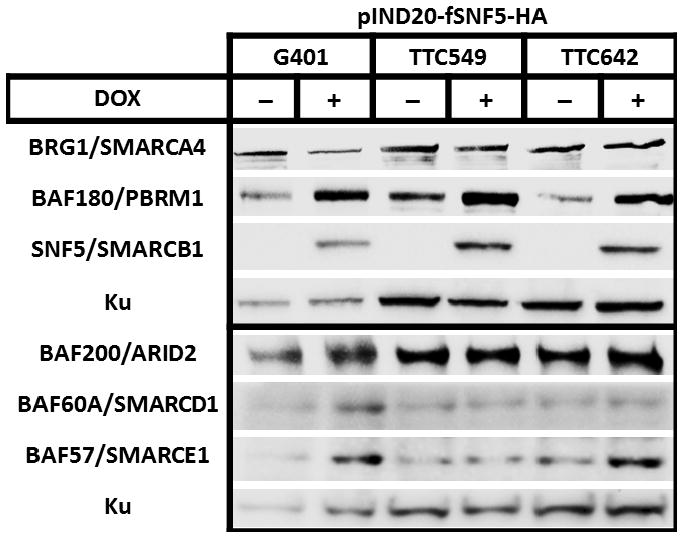
Cells were induced with doxycycline (DOX) at Time 0 and samples were harvested at 24 hours after induction. Total cellular proteins (30 μg) were separated on a 4% to 12% Bis-Tris polyacrylamide gel, transferred to PVDF membranes and probed with indicated antibody. KU served as the loading control. UN, untreated control
To determine the composition of the SWI/SNF complex in the presence and absence of SNF5, we initially carried out label-free quantitative mass spectrometry on the TTC642 cell line. We isolated SWI/SNF complexes from this cell line +/− SNF5 from whole cell lysates by immunoprecipitation with an antibody against BRG1. Because the MRT cell lines used in this study do not express the mutually exclusive BRM ATPase (Figure 1A), functional complexes must possess the BRG1 ATPase. As shown in Figure 5A, re-expression of SNF5 resulted in significant changes in the components associated with BRG1.
Figure 5. SWI/SNF complex composition changes after SNF5 re-expression.

(A) The relative abundance of SWI/SNF complex components in the absence (untreated) and presence (Dox) of SNF5 in the TTC642 cell line was determined by label-free quantitative mass spectrometry. The identity of BCL7B could not unambiguously be determined versus BCL7C due to shared peptides. (B) The indicated cell lines (A–C) were induced with doxycycline, and the samples were immunoprecipitated with either rabbit IgG or anti-BRG1. IP samples were then separated on a 4% to 12% Bis-Tris polyacrylamide gel, transferred to PVDF membranes and probed with the indicated antibodies.
In order to confirm the semi-quantitative mass spectrometry results, we assessed individual component protein levels by Western blotting after immunoprecipitation with our BRG1 antibody. Because BRG1/SMARCA4 levels did not change or are decreased after SNF5 re-expression (Figures 3 and 4), we used the ratio of complex subunits to BRG1 to measure changes in component levels within the complex. Whole cell protein extracts from MRT cell lines, G401, TTC549 and TTC642 cells, either untreated or induced with doxycycline for 24 hours, were harvested, immunoprecipitated with α-IgG or α-BRG1 antibodies and Western blotted to determine the protein levels of BRG1 and other complex members.
While the overall levels of most component proteins increased, BRG1 levels either remained the same or decreased after SNF5 re-expression (compare Input lanes in each cell line in Figure 5B). Furthermore, without the confounding issue of viral infection, we found increased expression of BAF57 and BAF170 in all cell lines. As shown in Figure 5B, the ratio of most component proteins to BRG1 increased in all MRT cell lines after SNF5 re-expression. Surprisingly, we could not detect several components, such as BAF250A/ARID1A and BAF170/SMARCC2, in the complex in the absence of SNF5. Other components showed a differential pattern among the cell lines. For example, the complexes in the TTC549 and TTC642 cell lines contain BAF200/ARID2 after SNF5 re-expression while it is absent in complexes from the G401 cell line. Based on these data, the increased component protein levels observed in MRT cell lines after SNF5 re-expression coincides with increased association with SWI/SNF complex members.
Inhibition of SNF5 Expression in Normal Human Fibroblasts Causes Reduced Complex Component Expression
While our studies indicated that restoration of SNF5 in MRT cell lines increased expression of SWI/SNF complex components, they did not address whether SNF5 loss in normal cells would lead to decreased protein levels. To address this issue, we infected normal human fibroblasts (NHF-1) with 2 different shRNAs against SNF5 and assessed component mRNA and protein levels. Because the cell of origin for MRTs remains unknown, we used NHFs as a well-characterized normal human cell line model. As shown in Figure 6A, we achieved an approximately 80% decrease in SNF5 expression 48 hours after lentiviral infection with 2 different shRNAs. The SNF5 reduction corresponded to decreases in all SWI/SNF complex components with the exception of BRG1, ARID200 and BAF155. The non-targeting shRNA control, pLKO.1, did not show any effect with the exception of a moderate decrease in ARID1A levels. Thus, the decreases in component protein levels after SNF5 loss correlated well the opposite changes observed in the MRT cell lines after SNF5 reexpression. However, similar to our results with the MRT cell lines, the change in component protein levels did not mirror its mRNA expression (Supplementary Figure 1). However, using β-galactosidase staining, we observed replicative senescence only in the cells with SNF5 knockdown (Supplementary Figure 2).
Figure 6. Reduction of SWI/SNF complex protein levels after inhibition of SNF5 in normal human fibroblasts and the effects of MG132.

(A) SNF5 knockdown cells were generated by infecting normal human fibroblasts (NHF-1) with lentivirus expressing 2 different shRNAs against SNF5 (TRCN 39585 and TRCN 39587) or a non-targeting shRNA control (pLKO.1) and immediate selection with puromycin. After 72 hrs. on selective medium, total cellular protein (30 μg), was separated on a 4% to 12% Bis-Tris polyacrylamide gel, transferred to PVDF membranes and probed with the indicated antibodies. Ku served as the loading control. (B) NHF-1 hTERT cells were infected with indicated lentivirus and placed in selective media for 3 days. Six hours before harvesting for protein isolation, half of the samples were treated with the proteasome inhibitor MG132. After protein isolation, levels for the indicated proteins were detected by SDS-PAGE and Western blotting. c-FOS served as a positive control for proteasome inhibition while Ran, unaffected by MG132 treatment, served as a negative control. Ku served as the loading control.
Proteasome inhibitors can increase protein levels of SWI/SNF complex components
The levels of many cellular proteins are regulated through ubiquitination and subsequent degradation via the proteasome machinery (32). To assess whether a posttranscriptional mechanism might contribute to changes in component levels in the MRT cell lines, we examined the effects of MG132, a potent proteasome inhibitor, on protein expression in the TTC642, G401 and TTC549 cell lines (33). We treated the cell lines with DMSO or 10uM MG132. After 6 hours, we assessed representative SWI/SNF complex members as well as c-FOS (positive control) and RAN (negative control/loading control) protein levels by Western blotting. As shown in Figure 7, MG132 treatment increased protein levels of many components as well as or better than SNF5 reexpression, suggesting post-transcriptional regulation.
Figure 7. Proteasome inhibition causes increase protein levels for a subset of SWI/SNF complex components in MRT cell lines.

The TTC642 pINDUCER20-fSNF5-HA, G401 pINDUCER20-fSNF5-HA and TTC549 pINDUCER20-fSNF5-HA cell lines, were treated with DMSO, 1μg/ml doxycycline or 10uM MG132 for 6 hrs. After protein isolation, levels for the indicated proteins were detected by SDS-PAGE and Western blotting as previously described (Chai et al., 2005a). c-FOS served as a positive control for proteasome inhibition while Ran, unaffected by MG132 treatment, served as a negative control. Ku served as the loading control.
We then asked whether MG132 treatment would restore expression of SWI/SNF components after SNF5 knockdown in NHF-1 hTERT cells. The pLKO.1 non-targeting lentivirus served as a negative control. We used the hTERT expressing cells because they abrogate the problem of the limited lifespan of normal human fibroblasts in culture. In general, we saw good agreement between the effects of SNF5 KD on NHF-1 and NHF-1 hTERT (Figure 6). However, some differences appeared including the levels of ARID2, BAF155 BAF60A. We again saw differential effects on protein stability after MG132 treatment with some components increasing (BAF180, BAF60B & BAF57) while BAF60A demonstrated decreased expression (Figure 6B). BRG1 levels were unaffected, consistent with the previous results, while BAF250A was not detected. These results implicate a proteasome-dependent mechanism in lowering some component protein levels after SNF5 knockdown.
Discussion
Previous reports have shown the effects of loss of individual SWI/SNF complex members upon the organization and stability of the remaining complex components. For example, studies by Chen and Archer posited that BAF155 serves as a scaffolding element required for stability of other SWI/SNF complex including BRG1 (34) and BAF57 (6). Another report demonstrated that ARID2/BAF200 regulated BAF180 expression at the level of mRNA expression (35). Other reports have shown increased expression of BRM upon loss of BRG1 expression (36–38). Recent evidence has also shown decreased BAF57 and SNF5 levels in BAF155 deficient cells lines (9). Only a few studies have addressed the effects on complex formation in the absence of different complex members (5). Doan et al,. previously showed assembly of the complex in the absence of SNF5 with no effect on expression of BRG1-dependent genes (5). Our data demonstrate increases in SWI/SNF complex members binding to the complex after SNF5 ectopic expression, implying that SNF5 plays a role in the stability of the complex members but not the corresponding mRNA. This role does not contradict prior research but further expands the functions of SNF5 within the SWI/SNF complex.
We have previously shown that SNF5 re-expression in MRT cell lines causes increased p21WAF1/CIP1 expression associated with preferential recruitment of SNF5 to the p21 TSS (39). This information, in the context of our data, suggests that the constant presence of BRG1 containing complexes lacking SNF5 along the p21 promoter maintains transcription but cannot remodel nucleosomes at the TSS. This obstacle could prevent the elongation of RNA polymerase II resulting in promoter pausing and minimal p21 expression (40, 41). Similar effects have been observed for the regulation of developmental genes in Drosophila (12, 42). SNF5 re-expression could stabilize the SWI/SNF complex resulting in targeting to TSSs and increased gene transcription. Alternatively, our results could support a model where SNF5-deficient complexes are already recruited to TSSs and maintain a basal level of gene expression. However, gene expression levels remain low due to the short half-life of most complex members in the absence of SNF5 or the type of SWI/SNF complex present at the promoter. SNF5 expression would stabilize the complex or recruit a different type of SWI/SNF complex causing increased gene expression.
Our finding that a subset of SWI/SNF complex components appears degraded in a proteasome-dependent manner seems consistent with previous reports indicating their instability in the absence of other complex members (6, 34). However, SNF5 loss correlates with degradation of multiple complex members, in contrast to the limited numbers affected by loss of other complex components (6, 34, 43). The identity of the proteasome pathway responsible for the degradation of complex components and whether it acts directly or indirectly remains unknown. The observation that BAF60A levels decrease after MG132 treatment in the NHFs supports an indirect mechanism (Figure 6B). However, our observations appear consistent with previous results that cells maintain tight control over the protein levels of SWI/SNF complex members (36, 44, 45).
The mechanisms by which SNF5 loss initiates MRT development remain unresolved. Recent reports have identified at least 9 different forms of the SWI/SNF complex, based upon protein composition, that promote diverse biological functions including growth and differentiation (4, 15). Our current study implicates changes in SWI/SNF complex composition after SNF5 inactivation as a mechanism for MRT development. This makes an attractive model because it accounts for several hallmarks of this cancer. Presumably, the transition from a growth promoter configuration of the SWI/SNF complex to a differentiating inducing one occurs within a narrow window of development. Therefore, SNF5 loss would only exert an effect if it happened within this time frame. This strict requirement for timing could account for the relative paucity of these tumors. Second, if MRTs arise from retention of growth promoting complexes, affecting gene expression, one would expect little genomic instability in these tumors. In agreement with this notion, a recent report from Lee et al. demonstrates a lack of significant changes in MRTs (46). The dramatic effects on ARID1A protein expression in the presence or absence of SNF5 compared to changes in BAF180 suggest that PBAF complexes remain more stable during MRT development than BAF complexes. Finally, while the loss of SNF5 from a stem-like population may prevent differentiation, KD on SNF5 in differentiated cells may result in SWI/SNF complexes with growth inhibitory properties. This would account for the paradoxical result that SNF5 reexpression in MRT cell lines and SNF5 KD in primary NHFs both cause growth arrest (16, 47, 48).
The changes in gene expression observed after SNF5 re-expression in MRT cell lines or its inactivation in normal cells may arise from differential binding of the SWI/SNF complexes present under each condition (49, 50). Therefore, future ChIP-seq experiments should identify additional unique and mutual binding sites for SWI/SNF complex members with and without SNF5. These data, in conjunction with gene expression analyses, will allow for further insights into the role of SWI/SNF complex activities after SNF5 inactivation in MRT development. It will also be important to investigate the activities of SWI/SNF complexes that do form in the absence of SNF5. These complexes may account for the observation of Roberts and colleagues that lymphoma development after SNF5 inactivation requires BRG1 expression (38).
The existing protocols for treatment of MRT include tumor resection, followed by adjuvant chemotherapy and/or radiation (2). These current protocols suffer from several inadequacies, including the difficulty of resection due to the tumor size and the contraindication of radiation in young patients (2). Understanding the molecular mechanisms behind MRT development and growth will allow for improved patient treatment and survival rate. For example, the posttranscriptional regulation of SWI/SNF complex components suggests that they can be potential targets for therapeutic intervention. Further screening of inhibitors of the 20S proteasome could prove fruitful in finding a drug candidate to stabilize the complex in the absence of SNF5. Additionally, identifying the pathways regulated by the SWI/SNF complexes present in the absence of SNF5 should reveal additional therapeutic targets. Furthermore, our results, showing that BAF180 and BAF250 stability inextricably depends upon SNF5’s presence, suggests a potential mechanistic link between SNF5 loss in MRT and BAF180 and BAF250A losses in renal cell carcinoma and ovarian carcinomas, respectively. Future studies will further our understanding of chromatin remodeling functions and provide knowledge for translation into the clinic for improving the outcomes of patients with MRTs and other cancers with SWI/SNF complex mutations.
Supplementary Material
Acknowledgments
We thank the members of the Weissman lab for their help, advice, and support, Drs. Ramon Parsons, Dale Ramsden, Jiandie Lin, and Zhuoxian Meng for antibodies, Drs. Dennis Simpson, William Kaufmann, Peter Houghton, Inder Verma and Tim Triche for cell lines and Dr. Guang Hu, NIEHS, for the kind gift of the pINDUCER20 vector. We also thank the UNC Vector Core for generation of the adenovirus expressing SNF5-HA. The studies received financial support, in part, from Lineberger Comprehensive Cancer Center development funds, the Sidney Kimmel Foundation for Cancer Research (Scholar Award) (M.B.M.) and the National Institutes of Health 1-DP2-OD007149-01 (M.B.M.), R01CA91048 (B.E.W.) and T32ES007126 (D.W.).
Footnotes
Conflict of Interest
The authors declare that they have no conflicts of interest relating to these studies.
References
- 1.Roberts CW, Orkin SH. The SWI/SNF complex--chromatin and cancer. Nat Rev Cancer. 2004;4:133–42. doi: 10.1038/nrc1273. [DOI] [PubMed] [Google Scholar]
- 2.Biswas A, Goyal S, Puri T, Das P, Sarkar C, Julka PK, et al. Atypical teratoid rhabdoid tumor of the brain: case series and review of literature. Child’s nervous system: ChNS: official journal of the International Society for Pediatric Neurosurgery. 2009;25:1495–500. doi: 10.1007/s00381-009-0903-x. [DOI] [PubMed] [Google Scholar]
- 3.McKenna ES, Sansam CG, Cho Y-J, Greulich H, Evans\ JA, Thom CS, et al. Loss of Epigenetic Tumor Suppressor SNF5 Leads to Cancer without Genomic Instability. Molecular and Cellular Biology. 2008;28:6223–33. doi: 10.1128/MCB.00658-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei D, Weissman BE. eLS. Vol. 2014 John Wiley & Sons Ltd; Chichester: 2014. Genetics and Genomics of Malignant Rhabdoid Tumors. [Google Scholar]
- 5.Doan DN, Veal TM, Yan Z, Wang W, Jones SN, Imbalzano AN. Loss of the INI1 tumor suppressor does not impair the expression of multiple BRG1-dependent genes or the assembly of SWI/SNF enzymes. Oncogene. 2004;23:3462–73. doi: 10.1038/sj.onc.1207472. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Archer TK. Regulating SWI/SNF subunit levels via protein-protein interactions and proteasomal degradation: BAF155 and BAF170 limit expression of BAF57. Mol Cell Biol. 2005;25:9016–27. doi: 10.1128/MCB.25.20.9016-9027.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curtis BJ, Zraly CB, Marenda DR, Dingwall AK. Histone lysine demethylases function as co-repressors of SWI/SNF remodeling activities during Drosophila wing development. Dev Biol. 2011;350:534–47. doi: 10.1016/j.ydbio.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 8.Decristofaro MF, Betz BL, Rorie CJ, Reisman DN, Wang W, Weissman BE. Characterization of SWI/SNF protein expression in human breast cancer cell lines and other malignancies. J Cell Physiol. 2001;186:136–45. doi: 10.1002/1097-4652(200101)186:1<136::AID-JCP1010>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 9.DelBove J, Rosson G, Strobeck M, Chen J, Archer TK, Wang W, et al. Identification of a core member of the SWI/SNF complex, BAF155/SMARCC1, as a human tumor suppressor gene. Epigenetics. 2011;6:1444–53. doi: 10.4161/epi.6.12.18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung I, Sohn DH, Choi J, Kim JM, Jeon S, Seol JH, et al. SRG3/mBAF155 stabilizes the SWI/SNF-like BAF complex by blocking CHFR mediated ubiquitination and degradation of its major components. Biochemical and biophysical research communications. 2012;418:512–7. doi: 10.1016/j.bbrc.2012.01.057. [DOI] [PubMed] [Google Scholar]
- 11.Moshkin YM, Mohrmann L, van Ijcken WF, Verrijzer CP. Functional differentiation of SWI/SNF remodelers in transcription and cell cycle control. Molecular and Cellular Biology. 2007;27:651–61. doi: 10.1128/MCB.01257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zraly CB, Middleton FA, Dingwall AK. Hormone-response genes are direct in vivo regulatory targets of Brahma (SWI/SNF) complex function. THE JOURNAL OF BIOLOGICAL CHEMISTRY. 2006;281:35305–15. doi: 10.1074/jbc.M607806200. [DOI] [PubMed] [Google Scholar]
- 13.Ho L, Ronan JL, Wu J, Staahl BT, Chen L, Kuo A, et al. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proceedings of the National Academy of Sciences. 2009;106:5181–6. doi: 10.1073/pnas.0812889106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kadoch C, Crabtree GR. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell. 2013;153:71–85. doi: 10.1016/j.cell.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, et al. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron. 2007;55:201–15. doi: 10.1016/j.neuron.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chai J, Charboneau AL, Betz BL, Weissman BE. Loss of the hSNF5 gene concomitantly inactivates p21CIP/WAF1 and p16INK4a activity associated with replicative senescence in A204 rhabdoid tumor cells. Cancer Res. 2005;65:10192–8. doi: 10.1158/0008-5472.CAN-05-1896. [DOI] [PubMed] [Google Scholar]
- 17.Weissman BE, Saxon PJ, Pasquale SR, Jones GR, Geiser AG, Stanbridge EJ. Introduction of a normal human chromosome 11 into a Wilms’ tumor cell line controls its tumorigenic expression. Science. 1987;236:175–80. doi: 10.1126/science.3031816. [DOI] [PubMed] [Google Scholar]
- 18.Weissman BE, Stanbridge EJ. Characterization of ouabain-resistant, hypoxanthine guanine phosphoribosyl transferase-deficient human cells and their usefulness as a general method for the production of human cell hybrids. Cytogenet Cell Genet. 1980;28:227–39. doi: 10.1159/000131536. [DOI] [PubMed] [Google Scholar]
- 19.Reincke BS, Rosson GB, Oswald BW, Wright CF. INI1 expression induces cell cycle arrest and markers of senescence in malignant rhabdoid tumor cells. J Cell Physiol. 2003;194:303–13. doi: 10.1002/jcp.10201. [DOI] [PubMed] [Google Scholar]
- 20.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:2509–14. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaeser MD, Aslanian A, Dong MQ, Yates JR, 3rd, Emerson BM. BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. The Journal of biological chemistry. 2008;283:32254–63. doi: 10.1074/jbc.M806061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuwahara Y, Charboneau A, Knudsen ES, Weissman BE. Reexpression of hSNF5 in malignant rhabdoid tumor cell lines causes cell cycle arrest through a p21(CIP1/WAF1)-dependent mechanism. Cancer Res. 2010;70:1854–65. doi: 10.1158/0008-5472.CAN-09-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeCristofaro MF, Betz BL, Wang W, Weissman BE. Alteration of hSNF5/INI1/BAF47 detected in rhabdoid cell lines and primary rhabdomyosarcomas but not Wilms’ tumors. Oncogene. 1999;18:7559–65. doi: 10.1038/sj.onc.1203168. [DOI] [PubMed] [Google Scholar]
- 24.Hast BE, Goldfarb D, Mulvaney KM, Hast MA, Siesser PF, Yan F, et al. Proteomic analysis of ubiquitin ligase KEAP1 reveals associated proteins that inhibit NRF2 ubiquitination. Cancer Res. 2013;73:2199–210. doi: 10.1158/0008-5472.CAN-12-4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature biotechnology. 2008;26:1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 26.Xu K, Ma H, McCown TJ, Verma IM, Kafri T. Generation of a stable cell line producing high-titer self-inactivating lentiviral vectors. Mol Ther. 2001;3:97–104. doi: 10.1006/mthe.2000.0238. [DOI] [PubMed] [Google Scholar]
- 27.Jagani Z, Mora-Blanco EL, Sansam CG, McKenna ES, Wilson B, Chen D, et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nature Medicine. 2010;16:1429–33. doi: 10.1038/nm.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamamichi N, Yamamichi-Nishina M, Mizutani T, Watanabe H, Minoguchi S, Kobayashi N, et al. The Brm gene suppressed at the post-transcriptional level in various human cell lines is inducible by transient HDAC inhibitor treatment, which exhibits antioncogenic potential. Oncogene. 2005;24:5471–81. doi: 10.1038/sj.onc.1208716. [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Rao S, Chu J, Shen X, Levasseur DN, Theunissen TW, et al. A protein interaction network for pluripotency of embryonic stem cells. Nature. 2006;444:364–8. doi: 10.1038/nature05284. [DOI] [PubMed] [Google Scholar]
- 30.Wang W, Cote J, Xue Y, Zhou S, Khavari PA, Biggar SR, et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. Embo J. 1996;15:5370–82. [PMC free article] [PubMed] [Google Scholar]
- 31.Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, et al. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3665–70. doi: 10.1073/pnas.1019736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asher G, Reuven N, Shaul Y. 20S proteasomes and protein degradation “by default”. Bioessays. 2006;28:844–9. doi: 10.1002/bies.20447. [DOI] [PubMed] [Google Scholar]
- 33.Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8:397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 34.Sohn DH, Lee KY, Lee C, Oh J, Chung H, Jeon SH, et al. SRG3 interacts directly with the major components of the SWI/SNF chromatin remodeling complex and protects them from proteasomal degradation. J Biol Chem. 2007;282:10614–24. doi: 10.1074/jbc.M610563200. [DOI] [PubMed] [Google Scholar]
- 35.Yan Z, Cui K, Murray DM, Ling C, Xue Y, Gerstein A, et al. PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes & Development. 2005;19:1662–7. doi: 10.1101/gad.1323805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen SM, Chastain PD, 2nd, Rosson GB, Groh BS, Weissman BE, Kaufman DG, et al. BRG1 co-localizes with DNA replication factors and is required for efficient replication fork progression. Nucleic Acids Res. 2010;38:6906–19. doi: 10.1093/nar/gkq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reyes JC, Barra J, Muchardt C, Camus A, Babinet C, Yaniv M. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2a) EMBO J. 1998;17:6979–91. doi: 10.1093/emboj/17.23.6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Sansam CG, Thom CS, Metzger D, Evans JA, Nguyen PT, et al. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Research. 2009;69:8094–101. doi: 10.1158/0008-5472.CAN-09-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuwahara Y, Wei D, Durand J, Weissman BE. SNF5 reexpression in malignant rhabdoid tumors regulates transcription of target genes by recruitment of SWI/SNF complexes and RNAPII to the transcription start site of their promoters. Mol Cancer Res. 2013;11:251–60. doi: 10.1158/1541-7786.MCR-12-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown SA, Imbalzano AN, Kingston RE. Activator-dependent regulation of transcriptional pausing on nucleosomal templates. Genes & Development. 1996;10:1479–90. doi: 10.1101/gad.10.12.1479. [DOI] [PubMed] [Google Scholar]
- 41.Neely KE, Hassan AH, Wallberg AE, Steger DJ, Cairns BR, Wright AP, et al. Activation domain-mediated targeting of the SWI/SNF complex to promoters stimulates transcription from nucleosome arrays. Molecular cell. 1999;4:649–55. doi: 10.1016/s1097-2765(00)80216-6. [DOI] [PubMed] [Google Scholar]
- 42.Zraly CB, Dingwall AK. The chromatin remodeling and mRNA splicing functions of the Brahma (SWI/SNF) complex are mediated by the SNR1/SNF5 regulatory subunit. Nucleic acids research. 2012;40:5975–87. doi: 10.1093/nar/gks288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yan Z. PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes & Development. 2005;19:1662–7. doi: 10.1101/gad.1323805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bourgo RJ, Siddiqui H, Fox S, Solomon D, Sansam CG, Yaniv M, et al. SWI/SNF deficiency results in aberrant chromatin organization, mitotic failure, and diminished proliferative capacity. Molecular biology of the cell. 2009;20:3192–9. doi: 10.1091/mbc.E08-12-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guidi CJ, Mudhasani R, Hoover K, Koff A, Leav I, Imbalzano AN, et al. Functional interaction of the retinoblastoma and ini1/snf5 tumor suppressors in cell growth and pituitary tumorigenesis. Cancer Res. 2006;66:8076–82. doi: 10.1158/0008-5472.CAN-06-1451. [DOI] [PubMed] [Google Scholar]
- 46.Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. The Journal of clinical investigation. 2012;122:2983–8. doi: 10.1172/JCI64400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Betz BL, Strobeck MW, Reisman DN, Knudsen ES, Weissman BE. Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene. 2002;21:5193–203. doi: 10.1038/sj.onc.1205706. [DOI] [PubMed] [Google Scholar]
- 48.Isakoff MS, Sansam CG, Tamayo P, Subramanian A, Evans JA, Fillmore CM, et al. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:17745–50. doi: 10.1073/pnas.0509014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Isakoff MS, Sansam CG, Tamayo P, Subramanian A, Evans JA, Fillmore CM, et al. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:17745–50. doi: 10.1073/pnas.0509014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morozov A, Lee SJ, Zhang ZK, Cimica V, Zagzag D, Kalpana GV. INI1 induces interferon signaling and spindle checkpoint in rhabdoid tumors. Clin Cancer Res. 2007;13:4721–30. doi: 10.1158/1078-0432.CCR-07-0054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.