

Abstract
Energy expenditure is determined by metabolic rate and diet-induced thermogenesis. Normally, energy expenditure increases due to neural mechanisms that sense plasma levels of ingested nutrients/hormones and reflexively increase sympathetic nerve activity (SNA). Here, we investigated neural mechanisms of glucose-driven sympathetic activation by determining contributions of neuronal activity in the hypothalamic paraventricular nucleus (PVN) and activation of corticotropin-releasing factor (CRF) receptors in the rostral ventrolateral medulla (RVLM). Glucose was infused intravenously (150 mg/kg, 10 min) in male rats to raise plasma glucose concentration to a physiological postprandial level. In conscious rats, glucose infusion activated CRF-containing PVN neurons and TH-containing RVLM neurons, as indexed by c-Fos immunofluorescence. In α-chloralose/urethane-anesthetized rats, glucose infusion increased lumbar and splanchnic SNA, which was nearly prevented by prior RVLM injection of the CRF receptor antagonist astressin (10 pmol/50 nl). This cannot be attributed to a nonspecific effect, as sciatic afferent stimulation increased SNA and ABP equivalently in astressin- and aCSF-injected rats. Glucose-stimulated sympathoexcitation was largely reversed during inhibition of PVN neuronal activity with the GABA-A receptor agonist muscimol (100 pmol/50 nl). The effects of astressin to prevent glucose-stimulated sympathetic activation appear to be specific to interruption of PVN drive to RVLM because RVLM injection of astressin prior to glucose infusion effectively prevented SNA from rising and prevented any fall of SNA in response to acute PVN inhibition with muscimol. These findings suggest that activation of SNA, and thus energy expenditure, by glucose is initiated by activation of CRF receptors in RVLM by descending inputs from PVN.
Keywords: corticotropin releasing factor, sympathetic nerve activity, rostral ventrolateral medulla
decreasing the risk of obesity-related metabolic (e.g., type 2 diabetes) and cardiovascular diseases (hypertension, atherosclerosis, and stroke) is often achieved by weight loss. Normal body weight is maintained by a balance between food intake and energy expenditure, and even small perturbations in this balance can produce large changes in body composition over time. Therefore, it is imperative to understand the neural circuits and synaptic mechanisms that control energy expenditure. Daily energy expenditure is determined by three major components: resting metabolic rate, exercise-induced increases in metabolic rate, and diet-induced thermogenesis (37). Among sedentary individuals, resting metabolic rate is the major determinant of daily energy expenditure and is maintained by a combination of sympathetic nerve activity (SNA), cortisol levels, and thyroid hormones (41, 42). Many features of the neural circuitry that drive energy expenditure, especially through activation of SNA, remain incompletely understood. Food intake stimulates energy expenditure through neural mechanisms that sense the elevation of nutritive factors such as glucose and insulin. Glucose infusion in humans and rodents increases SNA (17, 26, 38), and previous studies have shown that this effect is blunted during obesity (18, 33) but can be reestablished with weight loss (34). The neural circuits mediating this response to glucose have not been examined extensively. Indeed, the majority of studies have focused on neural targets and pathways activated by glucose and insulin using hyperinsulinemic euglycemic clamps, which mimic the abnormal hormonal profile of insulin resistance such as occurs in metabolic syndrome (3, 40). Such studies have identified important roles for the arcuate nucleus (ARC) (5, 22), hypothalamic paraventricular nucleus (PVN) (5, 40),and rostral ventrolateral medulla (RVLM) (3).
With regard to control of SNA, neurons of the PVN and RVLM play pivotal roles. PVN neurons contribute to a multitude of sympathoexcitatory states (10, 12, 15), have major projections to the RVLM (28, 32), and are activated by glucose (9). RVLM neurons comprise the major vasomotor center of the hindbrain and as a result are best known for their role in regulating arterial blood pressure (ABP) (12). Activity of RVLM neurons is controlled by a variety of neurotransmitters (11, 12, 15, 16), including the 41-amino acid neuropeptide corticotropin-releasing factor (CRF) (23).
Although CRF is expressed in a number of limbic regions of the central nervous system, it is most densely localized to parvocellular neurons of the PVN. CRFergic neurons of the PVN are best known for stimulating adrenocorticotropic hormone (ACTH) release from the anterior pituitary via activation of type 1 CRF receptors. CRF also colocalizes with melanocortin receptors in the PVN that mediate sympathoexcitation by hyperinsulinemia (24, 40), which is consistent with evidence that PVN is the principle source of CRF-immunoreactive terminals in the RVLM (23). Of special interest is evidence that ∼30% of all RVLM-projecting PVN (PVN-RVLM) neurons are reported to express CRF (16). Moreover, bilateral RVLM injections of CRF acutely increase ABP (23), an observation consistent with CRF driving sympathoexcitatory RVLM neuronal discharge. From the preceding observations, we formed the hypothesis that increases in plasma glucose will not only increase SNA but will do so through a mechanism involving activation of CRF-containing PVN neurons as well as CRF receptors in the RVLM.
MATERIALS AND METHODS
Ethical Assurance
All experimental procedures conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio.
Animals
Studies used male Sprague-Dawley rats (350–450 g; Charles River Laboratories) that were housed in a temperature-controlled room (22 ± 1°C) with a 14:10-h light-dark cycle. Rats consumed standard chow and had ad libitum access to deionized drinking water. Rats used for c-Fos immunostaining studies were housed singly for ∼10 days prior to use. Rats used for acute brain injection and nerve recording studies were housed two to three per cage.
Glucose Infusions and Measurement of Blood Glucose
Because plasma glucose concentration increases in healthy humans by 30–40 mg/dl following consumption of an average meal (7, 20), initial experiments were performed to establish parameters of intravenous (iv) glucose infusion (concentration and rate) that would produce such a physiological increase in plasma glucose. Rats were anesthetized by intraperitoneal injection of a mixture of urethane (750 mg/kg) and α-chloralose (75 mg/kg), and catheters (PE-50 tubing) were implanted in a femoral artery and vein. After stabilizing for >1 h, rats received a 10-min iv infusion of a 100 mg/ml solution of glucose in normal saline. Blood glucose was measured at baseline and every 15 min thereafter from a drop of arterial blood using a commercial glucometer (Freestyle Lite; Abbott Laboratories).
Induction of c-Fos in Conscious Rats
Rats were anesthetized with 2–3% isoflurane (in oxygen), and a catheter (PE-50 tubing) was placed in a femoral vein. Catheters were tunneled subcutaneously to exit between the scapulae and sutured in place. Catheters were flushed daily with sterile heparinized saline (40 U/ml). Postoperatively, each rat received daily injections of penicillin G (30,000 U/100 g body wt sc) and buprenorphine (0.05 mg/kg sc) for 3 days after surgery. Thereafter, rats were handled daily for 7 days to habituate them to experimental procedures used for c-Fos induction. Rats (n = 3–7/group) were randomly assigned to receive saline or glucose infusion through the femoral vein catheter while conscious. An infusion pump was used to deliver a predetermined iv glucose load of 150 mg/kg or an equal volume of saline (∼0.6 ml) over 10 min. Rats were anesthetized (5% isoflurane) 2 h later and perfused transcardially with 200 ml of 0.1 M phosphate-buffered saline (PBS) followed by 200 ml of 4% paraformaldehyde (PFA) in 0.1 M PBS. Brains were removed, postfixed in 4% PFA at 4°C for 24 h, and transferred to 30% sucrose in 0.1 M PBS for ≥2 days. Brain tissue that included PVN and RVLM was sectioned using a sliding microtome at 30 μM. Sections were placed in three vials of polyvinyl-pyrrolidone (PVP-40) cryoprotectant and stored at −20°C.
Immunohistochemistry
Detection of c-Fos induced among PVN and RVLM neurons was performed as described previously (11, 27, 32). Briefly, tissue sections from saline- and glucose-infused rats were removed from cryoprotectant, rinsed in 0.1 M PBS, incubated in 0.5% sodium borohydride in PBS for 30 min, and then incubated for 2 h in PBS containing 3% donkey serum and 0.25% Triton-X 100. Sections were then rinsed and incubated for 72 h with a polyclonal rabbit anti-rat c-Fos antibody (1:10,000; Millipore) at 4°C. After thorough rinsing, sections were incubated for 2 h in biotinylated donkey anti-rabbit secondary antibody (1:250; Vector Laboratories). To visualize immunoreactivity, sections were incubated for 10 min with a streptavidin-Alexa Fluor 594 conjugate (1:250; Invitrogen).
For detection of c-Fos among CRF and tyrosine hydroxylase (TH)-containing neurons, alternate sections of c-Fos-stained tissue were incubated for 72 h with a polyclonal guinea pig anti-rat CRF antibody (1:1,000; Bachem) or monoclonal mouse anti-rat TH antibody (1:1,000; Millipore), respectively. Sections were then rinsed and incubated for 2 h in a FITC-conjugated donkey anti-guinea pig secondary antibody (1:250; Millipore) or Alexa Fluor 488 donkey anti-mouse IgG antibody (1:250; Millipore). Sections were mounted on gelatin-coated slides, air-dried for 1–2 days in the dark, and coverslipped with ProLong Gold to preserve fluorescence.
Tissue sections stained for c-Fos alone or for c-Fos and CRF or TH were examined with an Olympus IX50 microscope under appropriate fluorescence illumination. The PVN in coronal sections was identified by the presence of the fornix and optic chiasm/tracts. The RVLM was identified by the presence of the inferior olive and nucleus ambiguous. Images of PVN and RVLM immunofluorescence were captured with a Spot digital camera (Diagnostic Instruments). ImageJ software (http://rsbweb.nih.gov/ij/) was used to merge images.
In Vivo Studies in Anesthetized Rats
Surgical procedures.
Rats were anesthetized by intraperitoneal injection of a urethane (750 mg/kg) and α-chloralose (75 mg/kg) mixture. Catheters (PE-50 tubing) were implanted in a femoral artery and vein to record ABP and administer drugs, respectively. Lumbar and splanchnic SNA were recorded as described previously (1, 2, 13, 29). Animals were artificially ventilated with oxygen-enriched room air and end-tidal CO2 was monitored and maintained between 4.5 and 5%. Rats were paralyzed with gallamine triethiodide (20 mg/ml, 0.25 ml/h iv) to avoid movement artifacts in SNA recordings. An adequate depth of anesthesia was determined by lack of a limb withdrawal reflex to noxious pinching of the foot prior to paralysis. Thereafter, adequacy of anesthesia was determined by lack of a pressor or sympathoexcitatory response to noxious foot pinch. Supplemental anesthesia (10% of initial dose) was given as needed. Body temperature was maintained at 37 ± 0.5°C. Recorded variables were allowed to stabilize for ∼1 h after surgery before an experiment began.
Brain site-specific nanoinjections.
For injections into RVLM, rats were placed in a stereotaxic head frame with the incisor bar positioned 11 mm below the interaural line. A single-barreled glass pipette (tip outer diameter: 20–40 μm) angled 20° rostrally was lowered into the left RVLM at the following coordinates referenced to calamus scriptorius: 1.8 mm lateral, 1.8 mm rostral, and 2.8 mm ventral. Initially, l-glutamate (1 nmol/50 nl) was injected into the RVLM at three different sites separated by 300 μm in the rostral-caudal plane to identify the site that produced the largest increase in ABP; subsequent injections were performed at these coordinates.
For injections into PVN, rats were placed in a stereotaxic head frame, and the skull was leveled between bregma and lambda. A midline craniotomy was performed, and a single-barreled glass pipette was lowered into the PVN at the following coordinates referenced to bregma: 1.8 mm caudal, 7.4–7.6 mm ventral, and 0.4–0.5 mm lateral. Drugs and vehicle were injected over an ∼30-s period using a pneumatic pump in a volume of 50 nl/site. Injection volumes were determined by calibrated meniscus movement. Injection sites were marked with rhodamine beads added to the injectate solution in a final concentration of 0.2%. At the conclusion of the experiments, brains were removed, postfixed in 4% paraformaldehyde for 3–6 days, cryoprotected in 30% sucrose-PBS, and sectioned at 50 μm on a sliding microtome.
Experimental protocols.
To determine the contribution of RVLM CRF receptors in mediating glucose-stimulated sympathoexcitation, separate groups of rats were prepared as described above. Initial experiments determined a dose of the nonselective CRF receptor antagonist astressin (American Peptides) that was capable of blocking SNA responses to the 41-amino acid CRF peptide (25 pmol/50 nl; American Peptides). CRF was unilaterally injected into the RVLM of four rats. Responses of SNA and ABP were compared before and after various doses of astressin. In separate groups of rats, either artificial cerebrospinal fluid (aCSF) or the effective dose of astressin, which was determined to be 10 pmol/50 nl, was bilaterally microinjected into the RVLM 5 min prior to the start of glucose infusion. Variables were recorded for a total of 45 min.
To assess the role of PVN neuronal activity in glucose-stimulated increases of SNA, aCSF was injected bilaterally into RVLM 5 min prior to the start of glucose infusion, and 30 min thereafter the GABA-A receptor agonist muscimol (100 pmol/50 nl) was bilaterally injected into the PVN to inhibit neuronal discharge. Variables were then recorded for an additional 30 min. To affirm that blockade of CRF receptors in the RVLM was sufficient to prevent PVN-mediated support of glucose-stimulated SNA, the CRF receptor antagonist astressin was injected bilaterally into RVLM 5 min prior to glucose infusion, and PVN neuronal activity was inhibited 30 min later by bilateral injection of muscimol.
Sciatic nerve stimulation.
To determine whether astressin attenuates sympathetic responses to other excitatory stimuli mediated by the RVLM, a separate group of rats was prepared as described above. The sciatic afferent nerve was isolated and stimulated electrically (1 ms, 500 μA, 5-s train) over a range of frequencies (1, 2, 5, 10, 20, and 40 Hz) following bilateral RVLM microinjection of aCSF, astressin (25 pmol/50 nl), or the ionotropic glutamate receptor antagonist kynurenic acid (KYN; 5 nmol/50 nl).
Data Analysis
Neuronal nuclei stained for c-Fos were counted in a single representative section from the rostral, middle, and caudal portions of the PVN and RVLM of each rat, as described previously (11, 27, 32). To insure consistency and to guard against possible counting errors introduced by costaining for CRF or TH, counts from PVN and RVLM sections stained for c-Fos only were compared with counts from alternate sections double-labeled for c-Fos and CRF or TH. For functional studies, responses of integrated SNA are expressed either as a percent of baseline or percent change from baseline, as indicated in each figure. In all cases, SNA responses were quantified after subtraction of background noise, which was determined as the signal remaining 5–10 min after treatment with the ganglionic blocker hexamethonium (30 mg/kg iv). For all recorded variables, the average of a 2-min data segment at baseline was compared with a similar average determined 15, 30, and 45 min after each RVLM or PVN injection. For sciatic stimulation, a 2-min baseline was compared with a 5-s peak response. Summary data were analyzed by one- or two-way ANOVA with repeated measurements as appropriate. Post hoc tests were performed with independent or paired t-tests with a layered Bonferroni correction. A critical value of P < 0.05 was deemed statistically significant. Data are expressed as means ± SE.
RESULTS
Effects of Glucose Infusion on Plasma Glucose
Experiments initially determined a concentration of glucose and an infusion rate that produced a physiological postprandial increase (+30 to 40 mg/dl) of plasma glucose. As shown in Table 1 (n = 6/group), infusion of 150 mg/kg glucose over 10 min produced a significant (P < 0.05), physiologically relevant rise in plasma glucose within 15 to 30 min. Within 45 min of infusion onset, plasma glucose returned to baseline. As expected, saline infusion had no effect on plasma glucose at any time point.
Table 1.
Effect of glucose infusion (iv) on plasma glucose concentration (mg/dl)
| Infusate | Baseline | 15 Min | 30 Min | 45 Min |
|---|---|---|---|---|
| Glucose | 95 ± 6 | 126 ± 9*# | 116 ± 8*# | 94 ± 6 |
| Saline | 93 ± 8 | 75 ± 9 | 83 ± 8 | 86 ± 10 |
Values are means ± SE; n = 6.
P < 0.05 vs. baseline;
P < 0.05 vs. saline.
Glucose Infusion Elevates SNA
To investigate neural mechanisms of glucose-stimulated increases in energy expenditure, we next confirmed that our predetermined physiological increase in plasma glucose caused the expected increase in SNA. Figure 1A shows representative responses to a 10-min infusion of saline (top) or glucose (bottom). Summary data in Fig. 1B (n = 6/group) show that saline had no effect on recorded variables, whereas glucose significantly (P < 0.05) increased lumbar and splanchnic SNA without changing mean arterial pressure (MAP).
Fig. 1.

Acute effects of glucose infusion on lumbar sympathetic nerve activity (SNA), splanchnic SNA, and arterial blood pressure (ABP). A: representative traces of lumbar SNA, splanchnic SNA, and ABP during intravenous (iv) infusion (black bars) of saline (top) or glucose (bottom) for 10 min. Note that glucose but not saline promptly increased lumbar and splanchnic SNA. As expected, neither glucose nor saline altered ABP. B: corresponding summary data (n = 6) showing that glucose-stimulated increases of SNA reached statistical significance within 15 min and continued increasing thereafter. The period of saline/glucose infusion is indicated in each graph by the black horizontal bar along the x-axis. *P < 0.05 vs. saline. MAP, mean arterial pressure.
Glucose Infusion Activates CRF-Containing PVN Neurons and TH-Containing RVLM Neurons
A major goal of this study was to identify circuit elements activated during glucose-stimulated increases of SNA/energy expenditure. Having determined that our acute glucose infusion increased both lumbar and splanchnic SNA, we next sought to investigate involvement of CRF neurons in the hypothalamic PVN. To do so, glucose or saline was infused in conscious rats, and c-Fos staining was used as a marker of activated neurons. Figure 2A shows representative PVN sections from a saline- (left) and glucose-infused (right) rat. As expected, most CRF immunoreactive neurons were located in the medial and ventrolateral regions of PVN, with some staining also in the dorsal cap and lateral parvocellular regions. A few CRF-positive neurons were also scattered in the central magnocellular region (Fig. 2A, image 1). In addition, the number of nuclei stained for c-Fos was significantly greater in glucose- than in saline-infused rats (Fig. 2A, image 2). In glucose-infused rats, stained nuclei were most often located in the medial and ventrolateral parvocellular regions. Merged images of CRF and c-Fos immunofluorescence are shown in Fig. 2A, image 3, with higher resolution images in Fig. 2A, image 4, which revealed little fluorescence overlap in tissue from the saline-infused rat (left) compared with that of the glucose-infused rat (right) where numerous c-Fos (red) and CRF (green) double-labeling can be seen. The latter indicates that glucose activated numerous CRF-containing PVN neurons, especially in the medial portion of the ventrolateral region. Summary data (n = 3–7/group) in Fig. 2B demonstrate that glucose infusion activated a significant population of CRF-containing PVN neurons. Not only was the average number of c-Fos-activated PVN neurons/section significantly greater (P < 0.05) in glucose than in saline-infused rats (Fig. 2B, left), glucose infusion also caused a significantly greater (P < 0.05) increase in the average number of CRF neurons/section that coexpressed c-Fos (Fig. 2B, right).
Fig. 2.
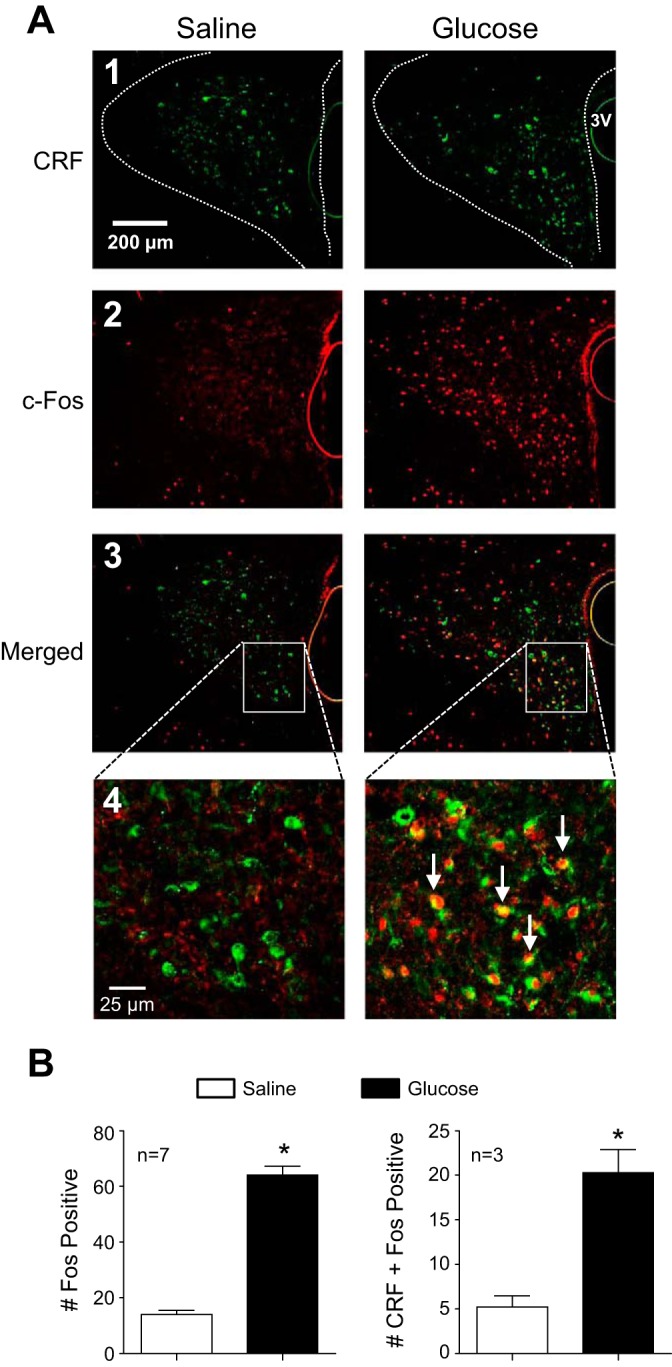
Effect of glucose infusion on c-Fos expression in corticotropin-releasing factor (CRF) neurons of the paraventricular nucleus (PVN). A: representative PVN sections from a saline-infused (left) and glucose-infused (right) rat showing neurons immunoreactive for CRF (image 1), c-Fos (image 2), and a merged image (image 3). In a higher-magnification merged image (image 4), numerous double-labeled neurons (arrows) can be seen only in tissue from the glucose-infused rat. B: summary data from saline- (open bar) and glucose-infused (black bar) rats (n = 3–7) revealed a significantly greater average no. of c-Fos-positive nuclei per PVN section in the glucose-infused group compared with saline-infused controls (left). The average no. of CRF-containing neurons that were colabeled with c-Fos immunoreactivity was also increased significantly by glucose infusion (right). *P < 0.05 vs. saline.
Because RVLM neurons are a major sympathoexcitatory target of the PVN (28) that have already been implicated in glucose-driven sympathetic activation (3), we next investigated whether TH-containing RVLM neurons were activated by acute glucose infusion. Figure 3A shows representative RVLM images from a saline- (left) and glucose-infused (right) rat. As expected, TH-expressing neurons were found throughout the RVLM (Fig. 3A, image 1). The number of fos-positive nuclei was significantly greater in glucose- than in saline-infused rats (Fig. 3A, image 2). Merged images of TH and c-Fos are shown in Fig. 3A, image 3, with higher resolution images in Fig. 3A, image 4, which showed negligible overlap in saline-infused (left) compared with glucose-fused rats (right). Summary data in Fig. 3B show that glucose infusion activated a significantly greater portion of TH-expressing RVLM neurons.
Fig. 3.
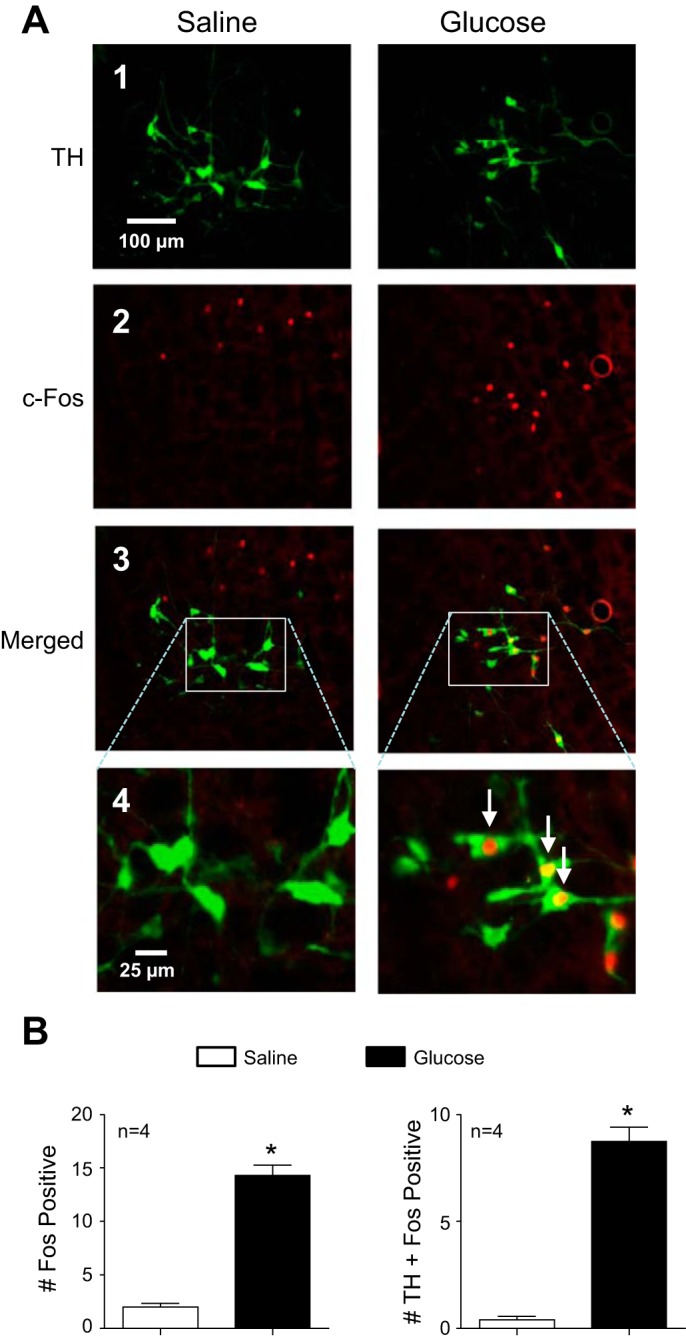
Effect of glucose infusion on c-Fos expression in tyrosine hydroxylase (TH) neurons of rostral ventrolateral medulla (RVLM). A: representative RVLM sections from a saline-infused (left) and glucose-infused (right) rat showing neurons immunoreactive for TH (image 1), c-Fos (image 2), and a merged image (image 3). In a higher-magnification merged image (image 4), numerous double-labeled neurons (arrows) can be seen only in tissue from the glucose-infused rat. B: summary data from saline- (open bar) and glucose-infused (black bar) rats (n = 4) revealed a significantly greater average no. of c-Fos positive nuclei per RVLM section in the glucose-infused group compared with saline infused controls (left). The average no. of TH-containing neurons that were colabeled with c-Fos immunoreactivity was also increased significantly by glucose infusion (right). *P < 0.05 vs. saline.
Astressin Prevents Glucose-Induced Sympathoexcitation
Because glucose infusion increased SNA (Fig. 1) and the number of c-Fos positive CRF neurons in PVN (Fig. 2), we next tested involvement of CRF receptor activation in glucose-stimulated sympathoexcitation. Efforts focused on CRF receptors in the RVLM, as c-fos was also elevated in TH neurons (Fig. 3). First, we determined a dose of the CRF receptor antagonist astressin that was capable of blocking responses to bilateral RVLM injection of CRF (n = 4). In Fig. 4A, a representative response to RVLM CRF shows that both lumbar SNA and MAP were acutely increased, as expected. Whereas RVLM injection of a 10-pmol dose of astressin alone had no effect on recorded variables, the response to subsequent injection of CRF was clearly blunted. Summary data in Fig. 4B indicate that whereas RVLM CRF significantly increased lumbar SNA and MAP (P < 0.05) prior to the injection of astressin, responses were significantly blunted 10–15 min thereafter (P < 0.05) and remained blunted for ≥60 min (data not shown).
Fig. 4.
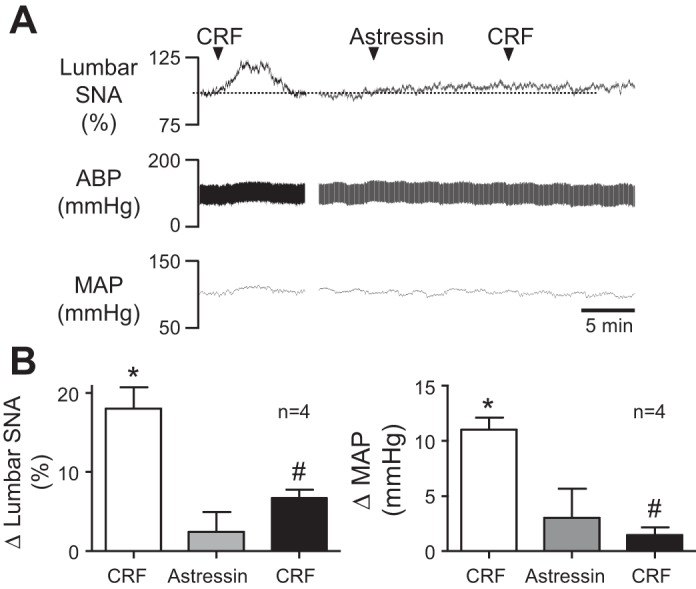
Effect of RVLM CRF receptor blockade on CRF-induced sympathoexcitation. A: representative responses of lumbar SNA, ABP, and MAP to RVLM injection of CRF before (left) and after (right) RVLM injection of the CRF antagonist astressin. Note that astressin nearly prevented sympathoexcitatory and pressor responses to CRF. B: corresponding summary data (n = 4) showing that RVLM CRF (25 pmol/50 nl) significantly (P < 0.05) increased lumbar SNA (left) and MAP (right) relative to baseline. Whereas astressin (10 pmol/50 nl) alone had no effect on either recorded variable, it significantly (P < 0.05) attenuated responses to subsequent injection of CRF. *P < 0.05 vs. baseline; #P < 0.05 vs. CRF before astressin.
To determine whether RVLM CRF receptors are necessary for glucose-induced sympathoexcitation, aCSF or the dose of astressin (10 pmol/50 nl) that blocked responses to CRF was bilaterally microinjected into RVLM prior to glucose infusion (n = 5/group). Figure 5A shows representative responses to acute glucose infusion following RVLM injection of aCSF (top) or astressin (bottom). Summary data in Fig. 5B reveal that increases in lumbar and splanchnic SNA were significantly blunted (P < 0.05) following RVLM astressin compared with aCSF.
Fig. 5.

Effect of RVLM CRF receptor blockade on glucose-induced sympathoexcitation. A: representative examples of lumbar SNA, splanchnic SNA, and ABP during a 10-min iv glucose infusion in which artificial cerebrospinal fluid (aCSF; top) or astressin (bottom) was bilaterally microinjected into the RVLM 5 min before the start of iv glucose infusion. B: corresponding summary data showing that RVLM pretreatment with saline or astressin had no effect on baseline SNA or MAP. In contrast, astressin significantly blunted only glucose-stimulated increases of SNA. C: schematic drawings of 2 rostrocaudal coronal planes through the RVLM showing the location of aCSF (gray; left) and astressin (black; right) injection sites. Note that injection sites were similarly placed bilaterally but are shown unilaterally for clarity. The period of glucose infusion is indicated in each part by the black horizontal bar along the x-axis. *P < 0.05 vs. aCSF.
Figure 5C shows the location of aCSF (gray) and astressin (black) injections into RVLM. Injection sites were located 0 to 600 μm caudal to the caudal pole of the facial nucleus and were centered within a triangular region bordered dorsally by nucleus ambiguus, medially by the inferior olive, and laterally by the spinal trigeminal nucleus.
To determine whether astressin produced a nonspecific effect on RVLM neurons by decreasing excitability or interfering with glutamate receptor activity, the sciatic nerve afferent, which is well established to be mediated by RVLM ionotropic glutamate receptors (3), was stimulated across a range of frequencies. As shown in Fig. 6A, sciatic stimulation at 5 Hz produced similar increases in lumbar SNA and ABP following aCSF and astressin RVLM microinjection. Responses were not statistically different between groups across the range of frequencies (data not shown). In contrast, KYN completely abrogated the response. Summary data are shown in Fig. 6B. Sites of aCSF, astressin, and KYN microinjection into the RVLM did not differ from those shown in Fig. 5C.
Fig. 6.
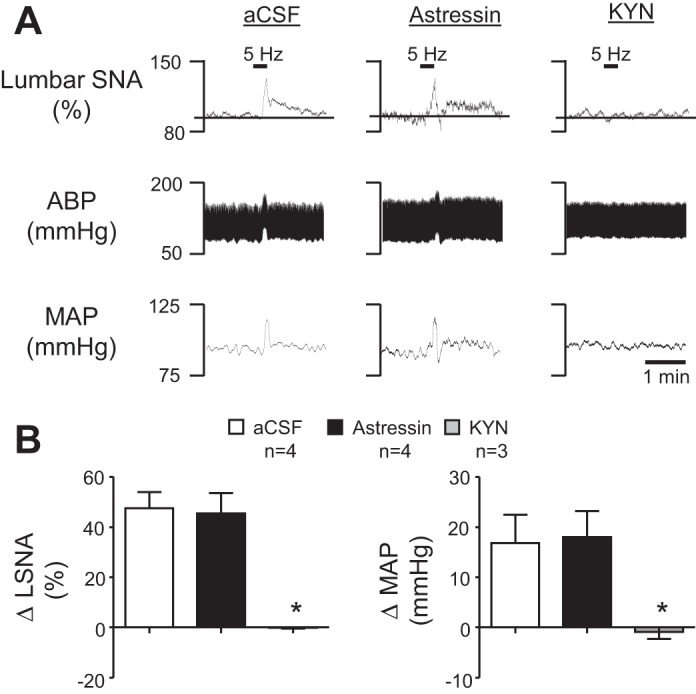
Effect of CRF receptor blockade on the sympathoexcitatory response to sciatic afferent stimulation. A: representative examples of lumbar SNA, ABP, and MAP following aCSF, astressin, or KYN microinjection into the RVLM in response to electrical stimulation of the sciatic nerve at 5 Hz. B: summary data of the change in lumbar SNA and MAP following sciatic nerve stimulation. There was no significant difference between aCSF- and astressin-treated rats, although KYN significantly blunted the response. *P < 0.05 vs. aCSF.
Activity of RVLM CRF Receptors is Required for PVN-Driven Sympathetic Activation by Glucose
Because glucose infusion activated CRF-containing PVN neurons and because RVLM astressin effectively prevented glucose-stimulated sympathoexcitation, we next investigated the role of PVN neuronal activity in glucose-induced sympathoexcitation and whether activity of CRF receptors in RVLM was required for PVN-driven support of glucose-stimulated SNA. Figure 7A shows representative responses from rats that received RVLM injections of aCSF (top) or astressin (bottom) prior to iv glucose infusion. Effects of PVN inhibition with muscimol 30 min later are also shown. Consistent with data in Fig. 5, summary data in Fig. 7B (n = 5/group) show that RVLM astressin nearly prevented glucose-stimulated increases in lumbar and splanchnic SNA compared with RVLM aCSF (P < 0.05). Among rats that first received RVLM injections of aCSF, glucose infusion caused the expected significant increases in SNA and MAP (P < 0.05). Inhibition of PVN 30 min later caused elevated lumbar SNA, splanchnic SNA, and MAP to fall toward baseline (P < 0.05). In contrast, prior RVLM injection of astressin effectively prevented glucose-stimulated SNA and MAP responses, and PVN inhibition with muscimol also failed to significantly alter ongoing SNA or MAP.
Fig. 7.

Effect of RVLM CRF receptor blockade on PVN-driven sympathetic activation by glucose. A: representative responses of lumbar SNA, splanchnic SNA, and ABP to bilateral RVLM injection of aCSF (top) or astressin (bottom) prior to iv glucose infusion and PVN injection of muscimol. B: corresponding summary data showing that PVN inhibition with muscimol restored glucose-stimulated lumbar SNA and splanchnic SNA while also reducing MAP in rats pretreated with aCSF in the RVLM. In contrast, PVN inhibition had no effect when glucose responses were prevented by prior RVLM injection of astressin. C: schematic drawings of 3 rostrocaudal coronal planes through the PVN showing the location of injected muscimol in rats infused with saline (gray; left) and glucose (black; right). The period of glucose infusion is indicated in each part by the black horizontal bar along the x-axis. *P < 0.05 vs. saline; ‡P < 0.05 vs. 30 min.
Figure 7C shows the location of muscimol injections into PVN of saline- (gray) and glucose-infused (black) rats. Injection sites were typically centered on the rostral-caudal midpoint of PVN, indicating that diffusional spread of muscimol would have likely encompassed the central magnocellular region along with most of the dorsal cap, ventrolateral, and periventricular parvocellular areas. Rhodamine beads did not enter the third ventricle or spread significantly to areas surrounding PVN. Sites of aCSF and astressin injection into RVLM did not differ from those shown in Fig. 5C.
DISCUSSION
Meal ingestion normally stimulates SNA, which in turn increases energy expenditure. Obesity and other metabolic diseases appear to disrupt normal coupling of energy intake with energy expenditure (27), which underscores the importance of elucidating neural mechanisms of nutrient-stimulated energy expenditure. In this study, we found, as expected, that acute glucose infusions increased both lumbar and splanchnic SNA and caused significant c-Fos activation of CRF-containing PVN neurons. Major downstream targets of PVN neurons are sympathoexcitatory neurons of the RVLM (11, 12, 28, 32), and unsurprisingly, c-fos was also increased in TH-expressing RVLM neurons. Evidence in the literature indicates that CRF delivered directly into RVLM acutely raises blood pressure (23). We found that RVLM CRF receptor activation also acutely activated lumbar SNA, indicating that sympathoexcitation likely mediates the pressor response to RVLM CRF. A major finding of this study is that RVLM CRF receptor blockade nearly prevented glucose-stimulated sympathoexcitation. This is important, because we also determined that glucose-induced sympathoexcitation was effectively reversed by acute chemical inhibition of the PVN. Additional findings indicate that blockade of CRF receptors in the RVLM prior to glucose infusion effectively prevented both the glucose-stimulated rise of SNA and the sympathoinhibitory response to PVN inhibition. Collectively, these findings strongly indicate that glucose activates CRF-containing PVN neurons and that recruitment of these neurons leads to activation of CRF receptors in the RVLM to increase lumbar and splanchnic SNA. Therefore, we postulate that activation of a CRFergic PVN/RLVM pathway is likely to play a significant role in glucose-stimulated energy expenditure.
In healthy individuals, meal consumption increases blood glucose concentration and thereby stimulates pancreatic secretion of insulin, which promotes cellular glucose disposal (6). As a result, postprandial blood glucose levels normally rise by an average of ∼30–50 mg/dl, depending on meal content (6, 7). To mimic physiological increases in energy expenditure, we first established a glucose infusion that would increase plasma glucose to a normal postprandial level within 30 min postinfusion. Under the current experimental conditions, this was achieved by a 10-min intravenous infusion of 150 mg glucose/kg body wt. Glucose infusions are known to increase muscle SNA in humans (38), and hyperinsulinemic euglycemic clamps in rodents have been reported to likewise increase lumbar SNA with little increase in renal SNA (3, 22, 40). Here, we reported that acutely elevated plasma glucose increased lumbar as well as splanchnic SNA. It is not possible to discern the extent to which these sympathoexcitatory responses were due to a direct action of glucose or to an indirect action of glucose-stimulated secretion of peptide hormones such as insulin, cholecystokinin, or glucagon-like peptide. This is the case because cerebrospinal fluid levels were not measured and because SNA began to rise and continued rising throughout the period when plasma glucose and likely other peptide hormones were also elevated. In addition, future studies will ascertain how short-term elevations in plasma glucose result in a prolonged sympathetic response that continues even after glucose levels return to baseline.
Here, we found that glucose-stimulated sympathoexcitation was accompanied by c-Fos activation of both CRF-containing PVN neurons and TH-containing RVLM neurons. Glucose-stimulated c-Fos was observed prominently in parvocellular areas of PVN known to contain hypophysiotropic and sympathetic neurons (32) and C1 neurons of the RVLM that control sympathetic output (12). Although our study did not differentiate between PVN neurons that project to RVLM vs. elsewhere, recent evidence indicates that ∼30% of CRF-expressing PVN neurons project to the RVLM (16). To our knowledge, this is the first use of double-labeling immunohistochemistry to demonstrate that an acute physiological increase of plasma glucose activates both populations of neurons. Although c-Fos was also expressed in the central magnocellular region of PVN, we did not investigate its colocalization with oxytocin or vasopressin containing neurosecretory neurons.
Little is known about the ability of CRF to modulate SNA, and only one study to date has investigated its role in the RVLM (23). Of note is that blockade of CRF receptors with astressin had no acute effect on the prevailing level of SNA. This suggests that RVLM CRF receptors lack functional tone under euglycemic conditions. Although CRF is best known for regulating release of adrenocorticotropin (ACTH) from the anterior pituitary (36), anatomic and functional studies have also linked CRF to the central melanocortin system (14, 19, 21, 25). This is potentially important, because a recent study demonstrated that PVN melanocortin receptor blockade reverses the sympathoexcitatory response to a hyperinsulinemic euglycemic clamp (40). Given the likelihood that both plasma glucose and insulin were elevated in the present study, it is important to reconcile these results with those of the present study. Whereas both studies implicate the PVN as an upstream driver of the RVLM, they differ in that ours focused on the role of CRF receptors in the RVLM and theirs focused on the role of glutamatergic excitation. At present, the mechanism of CRF action in the RVLM is unknown, although evidence in the literature is consistent with possibilities ranging from excitation to inhibition (4, 8, 25, 30, 35). Given the evidence that CRF receptor activation can facilitate glutamatergic synaptic activity (30), it is possible that CRF released in the RVLM interacts with glutamate to mediate sympathoexcitation. Given that sciatic afferent nerve stimulation was unaffected by RVLM astressin, it is unlikely that astressin caused a nonspecific dampening of RVLM excitability. Alternatively, CRF has also been reported to have neuronal inhibitory actions (4, 8, 35). Such actions in the RVLM would suggest that CRF might drive SNA by causing disinhibition of RVLM. In this regard, it is noteworthy that RVLM neurons normally operate with significant GABAergic inhibition. Collectively, our results and those published previously (40) suggest a possible model of nutrient-driven sympathetic activation in which elevated plasma glucose and/or insulin coactivate glutamatergic and CRFergic PVN neurons. In the RVLM, glutamatergic inputs would likely be necessary to insure depolarization of membrane potential to spike threshold. At the same time, activation of CRF receptors would enhance the efficacy of excitatory neurotransmission by diminishing the tonic level of GABAergic inhibition. Additional studies are clearly needed to validate such a model.
In addition to understanding integrative processing within the downstream RVLM, studies are also needed to determine upstream mechanisms of glucose/insulin-induced sympathetic activation and the first-order neurons responding to such changes in nutritive factors. Both glucose- and insulin-responsive neurons are expressed in regions of the hypothalamus, with major clusters located within the ARC, a brain region essential for nutrient/hormone-stimulated control of energy expenditure (39) and one known to mediate insulin-induced sympathoexcitation (22). Whereas insulin has been found to inhibit ARC neurons (31), glucose-excited ARC neurons have been reported to overlap with ARC neurons that are excited by α-melanocyte-stimulating hormone and inhibited by neuropeptide Y (39). These observations suggest that glucose could act, at least partially, through a direct excitatory mechanism separate from insulin. How these ARC responses would filter through to recruit PVN neurons or whether glucose can act directly on PVN neurons remains to be fully explored.
Of note in the present findings is that PVN inhibition failed to lower SNA in glucose-infused rats pretreated with RVLM astressin. This was not entirely unexpected given that RVLM astressin also effectively prevented glucose from initiating sympathetic activation. Despite PVN inhibition failing to reduce SNA in astressin-injected glucose-infused rats, we observed that it nevertheless did cause blood pressure to fall. The reason for this is presently unknown, but a possible explanation is that glucose could have stimulated SNA to vascular beds/tissues other than those we recorded (lumbar and splanchnic). If so, then PVN inhibition might have reduced these sympathetic activities, thereby reducing vascular resistance and allowing blood pressure to fall. This explanation implies that RVLM CRF receptors would play little or no role in regulating SNA to those “other” tissues.
In summary, the results of the present study provide new and potentially important insight into neurochemical mechanisms and neural pathways activated during glucose-stimulated increases in energy expenditure. As a novel energy-regulating neurotransmitter in the RVLM, CRF could play a significant role in maintaining/stimulating metabolic rate in healthy individuals. Additional studies are needed to expand our understanding of CRF actions in RVLM and to determine whether altered CRF signaling contributes to uncoupling of energy intake and energy expenditure associated with metabolic diseases such as type 2 diabetes and obesity.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute (NHLBI) Grants P01-HL-088052 and R01-HL-102310 (G. M. Toney). M. E. Bardgett was supported by NHLBI Grant T32-HL-07446.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.E.B., A.L.S., and G.M.T. conception and design of research; M.E.B. and A.L.S. performed experiments; M.E.B. analyzed data; M.E.B. interpreted results of experiments; M.E.B. prepared figures; M.E.B. drafted manuscript; M.E.B. and G.M.T. edited and revised manuscript; M.E.B., A.L.S., and G.M.T. approved final version of manuscript.
REFERENCES
- 1.Bardgett ME, Chen QH, Guo Q, Calderon AS, Andrade MA, Toney GM. Coping with dehydration: sympathetic activation and regulation of glutamatergic transmission in the hypothalamic PVN. Am J Physiol Regul Integr Comp Physiol 306: R804–R813, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bardgett ME, Holbein WW, Herrera-Rosales M, Toney GM. Ang II-salt hypertension depends on neuronal activity in the hypothalamic paraventricular nucleus but not on local actions of tumor necrosis factor-alpha. Hypertension 63: 527–534, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bardgett ME, McCarthy JJ, Stocker SD. Glutamatergic receptor activation in the rostral ventrolateral medulla mediates the sympathoexcitatory response to hyperinsulinemia. Hypertension 55: 284–290, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beckstead MJ, Gantz SC, Ford CP, Stenzel-Poore MP, Phillips PE, Mark GP, Williams JT. CRF enhancement of GIRK channel-mediated transmission in dopamine neurons. Neuropsychopharmacology 34: 1926–1935, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cassaglia PA, Hermes SM, Aicher SA, Brooks VL. Insulin acts in the arcuate nucleus to increase lumbar sympathetic nerve activity and baroreflex function in rats. J Physiol 589: 1643–1662, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coulston AM, Liu GC, Reaven GM. Plasma glucose, insulin and lipid responses to high-carbohydrate low-fat diets in normal humans. Metabolism 32: 52–56, 1983. [DOI] [PubMed] [Google Scholar]
- 7.Crapo PA, Reaven G, Olefsky J. Postprandial plasma-glucose and -insulin responses to different complex carbohydrates. Diabetes 26: 1178–1183, 1977. [DOI] [PubMed] [Google Scholar]
- 8.Dabrowska J, Hazra R, Guo JD, Dewitt S, Rainnie DG. Central CRF neurons are not created equal: phenotypic differences in CRF-containing neurons of the rat paraventricular hypothalamus and the bed nucleus of the stria terminalis. Front Neurosci 7: 156, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunn-Meynell AA, Govek E, Levin BE. Intracarotid glucose selectively increases Fos-like immunoreactivity in paraventricular, ventromedial and dorsomedial nuclei neurons. Brain Res 748: 100–106, 1997. [DOI] [PubMed] [Google Scholar]
- 10.Freeman KL, Brooks VL. AT1 and glutamatergic receptors in paraventricular nucleus support blood pressure during water deprivation. Am J Physiol Regul Integr Comp Physiol 292: R1675–R1682, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Gabor A, Leenen FH. Central neuromodulatory pathways regulating sympathetic activity in hypertension. J Appl Physiol (1985) 113: 1294–1303, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci 7: 335–346, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Holbein WW, Toney GM. Sympathetic network drive during water deprivation does not increase respiratory or cardiac rhythmic sympathetic nerve activity. J Appl Physiol (1985) 114: 1689–1696, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hwang BH, Guntz JM. Downregulation of corticotropin-releasing factor mRNA, but not vasopressin mRNA, in the paraventricular hypothalamic nucleus of rats following nutritional stress. Brain Res Bull 43: 509–514, 1997. [DOI] [PubMed] [Google Scholar]
- 15.Kenney MJ, Weiss ML, Haywood JR. The paraventricular nucleus: an important component of the central neurocircuitry regulating sympathetic nerve outflow. Acta Physiol Scand 177: 7–15, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Lee SK, Ryu PD, Lee SY. Differential distributions of neuropeptides in hypothalamic paraventricular nucleus neurons projecting to the rostral ventrolateral medulla in the rat. Neurosci Lett 556: 160–165, 2013. [DOI] [PubMed] [Google Scholar]
- 17.Levin BE, Dunn-Meynell AA, Routh VH. Brain glucose sensing and body energy homeostasis: role in obesity and diabetes. Am J Physiol Regul Integr Comp Physiol 276: R1223–R1231, 1999. [DOI] [PubMed] [Google Scholar]
- 18.Levin BE, Govek EK, Dunn-Meynell AA. Reduced glucose-induced neuronal activation in the hypothalamus of diet-induced obese rats. Brain Res 808: 317–319, 1998. [DOI] [PubMed] [Google Scholar]
- 19.Liposits Z, Sievers L, Paull WK. Neuropeptide-Y and ACTH-immunoreactive innervation of corticotropin releasing factor (CRF)-synthesizing neurons in the hypothalamus of the rat. An immunocytochemical analysis at the light and electron microscopic levels. Histochemistry 88: 227–234, 1988. [DOI] [PubMed] [Google Scholar]
- 20.Liu GC, Coulston AM, Reaven GM. Effect of high-carbohydrate-low-fat diets on plasma glucose, insulin and lipid responses in hypertriglyceridemic humans. Metabolism 32: 750–753, 1983. [DOI] [PubMed] [Google Scholar]
- 21.Lu XY, Barsh GS, Akil H, Watson SJ. Interaction between alpha-melanocyte-stimulating hormone and corticotropin-releasing hormone in the regulation of feeding and hypothalamo-pituitary-adrenal responses. J Neurosci 23: 7863–7872, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luckett BS, Frielle JL, Wolfgang L, Stocker SD. Arcuate nucleus injection of an anti-insulin affibody prevents the sympathetic response to insulin. Am J Physiol Heart Circ Physiol 304: H1538–H1546, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milner TA, Reis DJ, Pickel VM, Aicher SA, Giuliano R. Ultrastructural localization and afferent sources of corticotropin-releasing factor in the rat rostral ventrolateral medulla: implications for central cardiovascular regulation. J Comp Neurol 333: 151–167, 1993. [DOI] [PubMed] [Google Scholar]
- 24.Rahmouni K, Haynes WG, Morgan DA, Mark AL. Role of melanocortin-4 receptors in mediating renal sympathoactivation to leptin and insulin. J Neurosci 23: 5998–6004, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richard D, Lin Q, Timofeeva E. The corticotropin-releasing factor family of peptides and CRF receptors: their roles in the regulation of energy balance. Eur J Pharmacol 440: 189–197, 2002. [DOI] [PubMed] [Google Scholar]
- 26.Rowe JW, Young JB, Minaker KL, Stevens AL, Pallotta J, Landsberg L. Effect of insulin and glucose infusions on sympathetic nervous system activity in normal man. Diabetes 30: 219–225, 1981. [DOI] [PubMed] [Google Scholar]
- 27.Sandoval D, Cota D, Seeley RJ. The integrative role of CNS fuel-sensing mechanisms in energy balance and glucose regulation. Annu Rev Physiol 70: 513–535, 2008. [DOI] [PubMed] [Google Scholar]
- 28.Sawchenko PE, Swanson LW. Immunohistochemical identification of neurons in the paraventricular nucleus of the hypothalamus that project to the medulla or to the spinal cord in the rat. J Comp Neurol 205: 260–272, 1982. [DOI] [PubMed] [Google Scholar]
- 29.Sharpe AL, Andrade MA, Herrera-Rosales M, Britton SL, Koch LG, Toney GM. Rats selectively bred for differences in aerobic capacity have similar hypertensive responses to chronic intermittent hypoxia. Am J Physiol Heart Circ Physiol 305: H403–H409, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silberman Y, Winder DG. Corticotropin releasing factor and catecholamines enhance glutamatergic neurotransmission in the lateral subdivision of the central amygdala. Neuropharmacology 70: 316–323, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spanswick D, Smith MA, Mirshamsi S, Routh VH, Ashford ML. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat Neurosci 3: 757–758, 2000. [DOI] [PubMed] [Google Scholar]
- 32.Stocker SD, Cunningham JT, Toney GM. Water deprivation increases Fos immunoreactivity in PVN autonomic neurons with projections to the spinal cord and rostral ventrolateral medulla. Am J Physiol Regul Integr Comp Physiol 287: R1172–R1183, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Straznicky NE, Lambert GW, Masuo K, Dawood T, Eikelis N, Nestel PJ, McGrane MT, Mariani JA, Socratous F, Chopra R, Esler MD, Schlaich MP, Lambert EA. Blunted sympathetic neural response to oral glucose in obese subjects with the insulin-resistant metabolic syndrome. Am J Clin Nutr 89: 27–36, 2009. [DOI] [PubMed] [Google Scholar]
- 34.Straznicky NE, Lambert GW, McGrane MT, Masuo K, Dawood T, Nestel PJ, Eikelis N, Schlaich MP, Esler MD, Socratous F, Chopra R, Lambert EA. Weight loss may reverse blunted sympathetic neural responsiveness to glucose ingestion in obese subjects with metabolic syndrome. Diabetes 58: 1126–1132, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tao J, Hildebrand ME, Liao P, Liang MC, Tan G, Li S, Snutch TP, Soong TW. Activation of corticotropin-releasing factor receptor 1 selectively inhibits CaV3.2 T-type calcium channels. Mol Pharmacol 73: 1596–1609, 2008. [DOI] [PubMed] [Google Scholar]
- 36.Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science 213: 1394–1397, 1981. [DOI] [PubMed] [Google Scholar]
- 37.Van Gaal LF, Vansant GA, De Leeuw IH. Factors determining energy expenditure during very-low-calorie diets. Am J Clin Nutr 56: 224S–229S, 1992. [DOI] [PubMed] [Google Scholar]
- 38.Vollenweider P, Tappy L, Randin D, Schneiter P, Jequier E, Nicod P, Scherrer U. Differential effects of hyperinsulinemia and carbohydrate metabolism on sympathetic nerve activity and muscle blood flow in humans. J Clin Invest 92: 147–154, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang R, Liu X, Hentges ST, Dunn-Meynell AA, Levin BE, Wang W, Routh VH. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes 53: 1959–1965, 2004. [DOI] [PubMed] [Google Scholar]
- 40.Ward KR, Bardgett JF, Wolfgang L, Stocker SD. Sympathetic response to insulin is mediated by melanocortin 3/4 receptors in the hypothalamic paraventricular nucleus. Hypertension 57: 435–441, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Young JB, Landsberg L. Stimulation of the sympathetic nervous system during sucrose feeding. Nature 269: 615–617, 1977. [DOI] [PubMed] [Google Scholar]
- 42.Young JB, Landsberg L. Suppression of sympathetic nervous system during fasting. Science 196: 1473–1475, 1977. [DOI] [PubMed] [Google Scholar]