

Abstract
Inappropriate glucagon secretion contributes to hyperglycemia in inflammatory disease. Previous work implicates the proinflammatory cytokine interleukin-6 (IL-6) in glucagon secretion. IL-6-KO mice have a blunted glucagon response to lipopolysaccharide (LPS) that is restored by intravenous replacement of IL-6. Given that IL-6 has previously been demonstrated to have a transcriptional (i.e., slow) effect on glucagon secretion from islets, we hypothesized that the rapid increase in glucagon following LPS occurred by a faster mechanism, such as by action within the brain. Using chronically catheterized conscious mice, we have demonstrated that central IL-6 stimulates glucagon secretion uniquely in the presence of an accompanying stressor (hypoglycemia or LPS). Contrary to our hypothesis, however, we found that IL-6 amplifies glucagon secretion in two ways; IL-6 not only stimulates glucagon secretion via the brain but also by direct action on islets. Interestingly, IL-6 augments glucagon secretion from both sites only in the presence of an accompanying stressor (such as epinephrine). Given that both adrenergic tone and plasma IL-6 are elevated in multiple inflammatory diseases, the interactions of the IL-6 and catecholaminergic signaling pathways in regulating GCG secretion may contribute to our present understanding of these diseases.
Keywords: glucagon, inflammation, interleukin-6, hypoglycemia, endotoxemia
inflammatory disease causes profound alterations in glucose homeostasis and accompanying insulin resistance. Exquisite control of insulin (INS) and glucagon (GCG) secretion from the pancreas maintains euglycemia in healthy individuals (56). However, this balance is altered in inflammatory disease. The bulk of glucoregulatory research has focused on the INS side of the story for some years, whereas less research has focused on therapies to attenuate GCG secretion or action. Demonstrating the importance of GCG, hyperglycemia associated with INS deficiency does not manifest in the absence of GCG-dependent signaling in rodent models of diabetes (52). Moreover, in models of type 2 diabetes, inhibition of GCG also improves glucose homeostasis (44). Although diabetes and acute or chronic inflammation are vastly different in etiology and pathology, they have some similarities; hyperglycemia is associated with either a rise in absolute levels of GCG or in the ratio of GCG to INS as well as in proinflammatory cytokines. Moreover, improving glycemic control improves patient morbidity and mortality (20, 28, 54). Therefore, understanding the mechanisms that cause inappropriate GCG secretion is critically important for developing treatment strategies to improve glucose homeostasis in inflammatory disease.
Previous work has demonstrated a role of the proinflammatory cytokine interleukin-6 (IL-6) in modulating GCG secretion. IL-6 is a cytokine with a variety of physiological roles. IL-6 acts in adipose tissue to stimulate lipolysis and inhibit insulin action on glucose transport and lipogenesis. In muscle, IL-6 stimulates fat oxidation and glucose uptake. Furthermore, IL-6 stimulates glucose production from liver and inhibits insulin action on glycogen synthesis (42). IL-6 has also been well established as both a pyrogen (via hypothalamus-pituitary-adrenal axis activation) and a stimulator of the acute-phase protein response via the liver. Plasma IL-6 is elevated in physiological and pathophysiological settings where GCG is also elevated: exercise (36), diabetes (4), and inflammatory stress (19). Furthermore, IL-6 has become implicated in metabolic disease as a predictive factor in the development of type 2 diabetes (45). Prior work from our laboratory demonstrated that IL-6-deficient (IL-6-KO) mice have a blunted GCG response to acute inflammation [lipopolysaccharide (LPS)] compared with their wild-type (WT) littermates. The GCG response is completely rescued by intravenous replacement of IL-6 (51). In those studies, other proinflammatory cytokines (e.g., TNFα and IL1-β) increased normally following LPS administration, suggesting a specific role for IL-6 in modulating the acute effect on GCG secretion. IL-6 may also have longer-acting effects on α-cells; human islets incubated for 4 or 24 h in the presence of IL-6 exhibited increased GCG secretion and proglucagon expression compared with untreated controls (13). The GCG response that we observed in our studies was much more rapid. Therefore, knowing that IL-6 can cross the blood-brain barrier (3) as well as be produced within the brain (15) and that IL-6 receptors are expressed rather ubiquitously throughout the brain (53), we hypothesized that the rapid GCG secretory response to LPS was due to direct actions within the brain, which in turn augmented autonomic drive to the islet.
Our previous data demonstrated that IL-6 acutely modulates GCG secretion in vivo during acute inflammation. Herein, we tested whether IL-6 exerts its actions via the brain, islets, or both. We demonstrated that central IL-6 augments GCG secretion, but it does so only in the presence of an accompanying stressor, be it acute inflammation or hypoglycemia. These data suggest a potent modulatory role for this cytokine. Contrary to our hypothesis, IL-6 also amplifies acute GCG secretion from isolated islets in the presence of an accompanying stressor. These data provide insight into the dual role of IL-6 in amplifying neural drive to the pancreas and increasing the effectiveness of adrenergic signaling at the islet to stimulate GCG secretion.
RESEARCH DESIGN AND METHODS
Animal Care and Husbandry
All procedures performed were approved by the Vanderbilt University Animal Care and Use Committee. Male IL-6−/− (IL-6-KO) mice on a C57Bl/6 background (B6.129S2-Il6tm1Kopf/J; Jackson Laboratories, Bar Harbor, ME), WT (C57Bl/6) mice from our in-house colony (originally from Jackson Laboratories), or mice (C57Bl/6 background) expressing tandem-dimer red fluorescent protein (tdRFP) under control of the proglucagon promoter (24) were used. Experiments were performed at 12–14 wk of age. IL-6-KO mice at this age are not obese and have not yet developed complications characterized in older mice (55). Furthermore, we have demonstrated in IL-6-KO mice that intravenous administration of IL-6 rescues the GCG response to peripheral LPS, demonstrating that these mice respond normally to LPS when the normal LPS-induced increase in circulating IL-6 is present (51). Mice were separated postoperatively and maintained in microisolator cages on a 12:12-h light-dark cycle with free access to food and water. Mice were handled extensively on a daily basis following surgery to minimize stress on the day of an experiment.
Surgical Procedures
One week prior to catheterization surgery, mice were anesthetized using isoflurane and connected to a Stoelting (Kiel, WI) stereotaxic frame for cannulation. Mice underwent lateral ventricle cannulation (coordinates: 0.6 mm posterior to bregma, 1.5 mm lateral to midline, 1.4 mm below the surface of the skull) with a Plastics One guide cannula (Roanoke, VA) and FujiCEM glass ionomer cement (Instech Laboratories, Plymouth Meeting, PA) to secure cannula placement. A Plastics One dummy cannula was subsequently screwed in place. Successful cannulation was assessed following the experiment by cryosection or by using methylene blue dye injection.
The surgical procedures utilized to implant chronic catheters were as described previously (2, 31). Briefly, mice were anesthetized using isoflurane. The left common carotid artery and right jugular vein were catheterized for sampling and infusions, respectively. The free ends of the catheters were subcutaneously tunneled to the back of the neck, where the loose ends of the catheters were attached via stainless-steel connectors to polyethylene tubing (0.043 in outer diameter), which were exteriorized and sealed with stainless-steel plugs. Animals were housed individually after surgery, and body weight was recorded daily. The lines were flushed every other day with ∼50 μl of saline containing 200 U/ml heparin and 5 mg/ml ampicillin.
In Vivo Metabolic Experiments
Experiments were performed following an ∼5-day postoperative recovery period to see whether mice were within 10% of presurgery weight. Conscious, unrestrained mice were fasted for 5 h. Approximately 2 h prior to the experiment, animals were connected to a dual channel stainless-steel swivel (Instech Laboratories) to allow simultaneous jugular vein infusion and sampling of arterial blood as well as microinfusion into the lateral ventricle. An infusion of donor red blood cells (washed with 10% heparinized saline) was begun 1 h prior to the experiment and continued for the duration of the study to replace blood removed during the study. The hyperinsulinemic hypoglycemic clamp and LPS studies utilized IL-6-KO mice (Jackson Laboratories) that were both catheterized and cannulated, whereas the ganglionic blockade and sympathetic/parasympathetic antagonist studies utilized WT mice that were catheterized only. At the end of all studies, mice were euthanized via a lethal dose of pentobarbital sodium (iv).
Central IL-6 and peripheral lipopolysaccharide.
At t = −10 min, IL-6-KO mice were given a microinjection of 2 μl of saline (vehicle control) or 200 ng of recombinant mouse IL-6 (100 ng/μl IL-6 at 1 μl/min for 2 min; Biomyx, San Diego, CA) via the lateral ventricle cannula. This dose of IL-6 was used based on our prior work in which the concentration of plasma IL-6 in response to LPS was assessed (51), taking into account brain fluid volume, and on historic experiments (55). It should be noted that this dose was comparatively much lower than other intracerebroventricular (icv) doses that have been administered to mice historically (46). At t = 0 min, either saline or 1 mg/kg LPS (Sigma-Aldrich, St. Louis, MO) dissolved in saline was given as a bolus via the jugular vein. Arterial blood was sampled throughout the study to measure blood glucose and plasma GCG and insulin.
Sympathetic/parasympathetic blockade and peripheral LPS.
At t = −10 min, WT mice were given either 1) phentolamine and propranolol (2 mg/kg priming bolus for both, followed by a 0.02 mg·kg−1·min−1 iv infusion for the duration of the study; Sigma-Aldrich) to block α- and β-adrenergic receptors, respectively, 2) atropine (1 mg/kg prime, followed by 0.01 mg·kg−1·min−1 iv infusion for the duration the study; Sigma-Aldrich) to block muscarinic cholinergic receptors, or 3) saline. At t = 0 min, 1 mg/kg LPS was given as a bolus via the jugular vein. Arterial blood was sampled throughout the study to monitor blood glucose and plasma GCG, insulin, and IL-6.
Ganglionic blockade and peripheral LPS.
At t = −30 min, WT mice were given a bolus of 12 mg/kg chlorisondamine diiodide (Sigma-Aldrich) or saline as a control. At t = 0 min, 1 μg/g LPS was given as a bolus via the jugular vein. Since chlorisondamine decreases blood pressure, the carotid artery catheter was connected to a blood pressure monitor (via a Y-connector at the carotid artery catheter) to ensure efficacy of the chlorisondamine bolus; successful ganglionic blockade reduced mean arterial pressure by ∼50 mmHg. Arterial blood was sampled throughout the study to measure the blood glucose and plasma GCG, insulin, and IL-6 response.
Central IL-6 and hyperinsulinemic hypoglycemic clamp.
At t = −10 min, IL-6-KO mice were given a 2-μl microbolus of either bacteriostatic saline (vehicle control) or IL-6 (as described above) into the lateral ventricle cannula. At t = 0 min, a constant jugular vein infusion of INS (10 mU·kg−1·min−1) was begun, and a variable amount of glucose (20% dextrose) was administered to target or “clamp” the blood glucose at ∼60 mg/dl. Arterial blood was sampled throughout the study to assess blood glucose and plasma GCG, insulin, and catecholamines.
Islet Isolation and Perifusion
Islets from IL-6-KO mice were isolated as described previously (8). The ability of IL-6 to modulate GCG secretion was examined in the dynamic cell perifusion system (8). One-hundred islet equivalents were placed in a chamber and washed with baseline medium (5.6 mM glucose; Sigma-Aldrich) for 30 min prior to the experiment. Islets were perfused with multiple GCG secretagogues, preceded each time by a washout period with 5.6 mM glucose-containing buffer. Secretagogues were in the presence or absence of 200 ng/ml recombinant mouse IL-6 (Biomyx). Effluent fractions were collected using an automatic fraction collector. The GCG content of each fraction was subsequently assessed.
Imaging Calcium Activity in Islets
Islets were isolated and imaged from mice expressing tdRFP under control of the proglucagon promoter, as described previously (24). Islets were incubated with 5 μM Fluo-4 AM for 30 min at 2.8 mM glucose. After washing, islets were placed in a microfluidic device (39) and allowed to equilibrate on the microscope stage for 15 min in the same solution. Islets were exposed to low glucose (1.7 mM), low glucose plus epinephrine (1 μM; Sigma-Aldrich), low glucose plus IL-6 (200 ng/ml), or low glucose plus IL-6 plus epinephrine. Fluo-4 was excited at 488 nm and detected between 495 and 545 nm. Red α-cells were localized by recording tdRFP fluorescence excited by 561 nm. Time courses were collected with an LSM780 (Carl Zeiss) using a Fluar ×40/1.3NA lens and a 2 Airy unit pinhole. Mean intensities were normalized to the first five frames of data 8 min following reagent change.
Plasma/Sample Analysis
Immunoreactive INS and GCG were assayed using a Linco Rat Radioimmunoassay kit (Linco Research, St. Charles, MO), using a double antibody method. Catecholamines were assessed using HPLC with electrochemical detection following alumina extraction and subsequent elution (1, 26). Plasma IL-6 was assessed using ELISA (Millipore, Billerica, MA).
Statistical Analysis
All data are presented as means ± SE. All in vivo data were analyzed using two-way repeated-measures ANOVA, followed by Bonferroni post hoc test as appropriate. Perifusion data and calcium imaging data were analyzed using Student's t-test.
RESULTS
Central IL-6 Enhances GCG Secretion During Acute Inflammation in IL-6-KO Mice
To determine whether central IL-6 signaling modulates GCG secretion, chronically catheterized, cannulated IL-6-KO mice were microinjected with IL-6 or vehicle (Veh) via the lateral ventricle. Because acute inflammation is a potent stimulator of GCG secretion, separate cohorts of mice were exposed to intravenous LPS (1 mg/kg iv) to determine whether central IL-6 could augment LPS-stimulated GCG secretion. IL-6-KO mice were used to avoid endogenous production of IL-6 that might confound interpretation of these studies.
IL-6-KO mice experienced no change in plasma GCG in response to IL-6 microinjection alone (54 ± 11 vs. 49 ± 5 pg/ml at 120 min, Veh vs. IL-6 injection; Fig. 1A). In contrast, central IL-6 microinjection nearly doubled circulating GCG by 120 min when mice were administered LPS (85 ± 14 vs. 143 ± 12 pg/ml, Veh vs. IL-6 injection). LPS administration elevated plasma glucose levels slightly throughout the period of study (Fig. 1B), but central IL-6 did not further modulate the glucose response. There were no differences observed between groups in plasma INS prior to or 2 h following either LPS/saline injection (Fig. 1C). IL-6 did not escape the central nervous system, as circulating IL-6 concentrations were undetectable following central administration. Consistent with the data found here in IL-6-KO mice, in a small cohort of WT mice, central IL-6 injection alone did not enhance GCG secretion at any time point observed (t = 0 min, 56 ± 6 vs. 64 ± 6; t = 30 min, 64 ± 14 vs. 59 ± 6; t = 60 min, 50 ± 0 vs. 68 ± 7 pg/ml; Veh vs. IL-6 injection). These results provide evidence in IL-6-KO mice that IL-6 acutely modulates GCG secretion via the brain, but interestingly, it does so only in the presence of an accompanying stressor such as LPS.
Fig. 1.
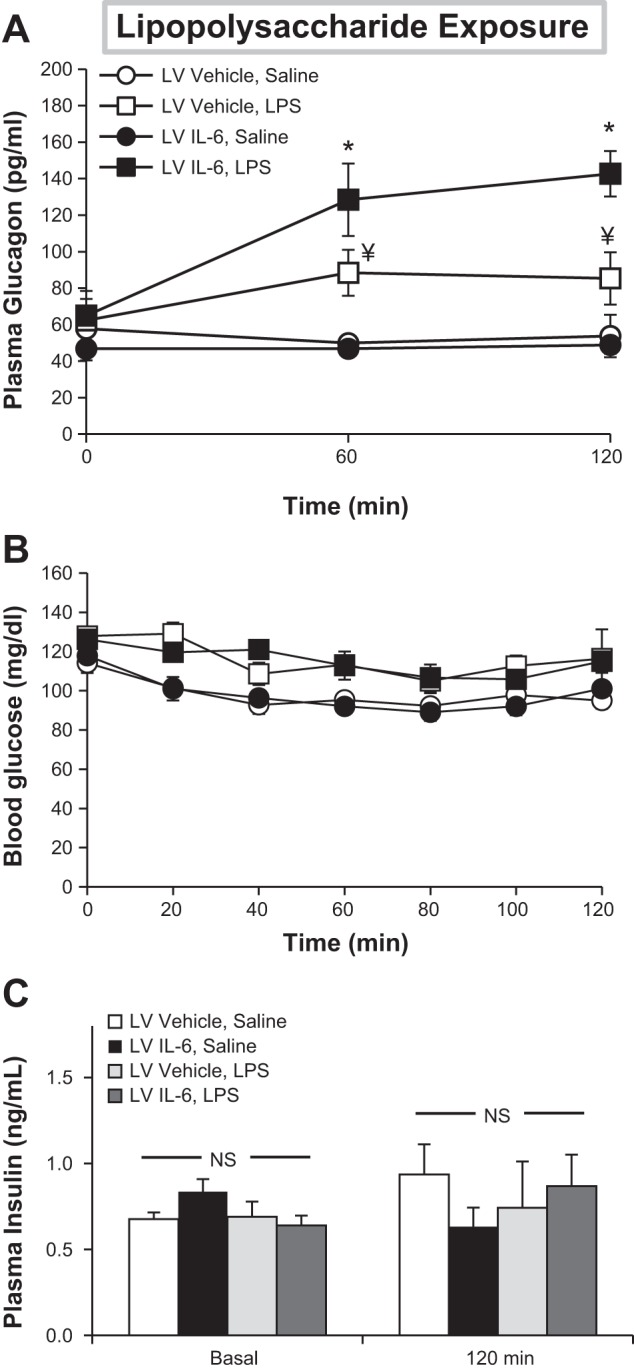
Central IL-6 augments the glucagon (GCG) response to endotoxin. Chronically catheterized, cannulated IL-6-deficient (IL-6-KO) mice were administered either IL-6 or vehicle (Veh) into the lateral ventricle (LV) prior to iv injection of lipopolysaccharide (LPS) or saline. Plasma glucagon (A), blood glucose (B), and insulin (C) were assessed over time; n = 5–7 mice/group. *P < 0.05 relative to both LV Veh, saline and LV Veh, and LPS treatments. ¥P < 0.05 relative to LV Veh and saline. NS, not significant.
The Sympathetic and Parasympathetic Nervous Systems Drive LPS-Stimulated GCG in WT Mice
Although the data support the involvement of the autonomic nervous system (ANS) in the GCG response to hypoglycemia (16, 17, 48), the role of the ANS in the GCG response to LPS is yet to be elucidated. Activation of both the sympathetic and parasympathetic nervous system can stimulate GCG secretion. Therefore, understanding whether one branch predominates in the response to LPS could inform our grasp of how central IL-6 signaling influences autonomic drive. To determine the role of each branch of the ANS in the GCG response to LPS, chronically catheterized, 5-h-fasted WT male mice were pretreated with pharmacological antagonists of either the sympathetic nervous system or parasympathetic nervous system, followed by exposure to LPS (1 mg/kg iv). To target the sympathetic nervous system (SNS), combined α- and β-adrenergic receptor blockade with phentolamine and propranolol, respectively, was employed. The parasympathetic nervous system (PNS) was targeted using muscarinic cholinergic receptor blockade with atropine.
Pharmacological blockade of the SNS but not PNS resulted in a rapid fall in blood glucose in response to LPS administration (Fig. 2A). Interestingly, however, the LPS-induced glucagon response was blunted following both sympathetic blockade (SB) and parasympathetic blockade (PB) (129 ± 4 vs. 59 ± 8 vs. 74 ± 6 pg/ml at 120 min; control vs. SB vs. PB; Fig. 2B). No differences in terminal plasma INS were observed (Fig. 2C). Terminal plasma was also assessed for IL-6 levels. Neither adrenergic blockade nor cholinergic blockade had an effect on terminal plasma IL-6 compared with controls (Fig. 2D). Despite the hypoglycemic (and thus further GCG-stimulating) environment in mice with SB, plasma GCG levels were blunted even greater than mice with PB. This suggests that although both branches of the ANS mediate the GCG secretory response to LPS, the SNS plays the predominant role.
Fig. 2.

Both the sympathetic and parasympathetic nervous systems are involved in the GCG response to LPS. Chronically catheterized wild-type (WT) mice were administered (iv) parasympathetic blocker (PB), sympathetic blockers (SB), or saline (control). Mice were then administered LPS. Blood glucose (A) and plasma GCG (B) were assessed over time. Terminal plasma insulin (C) and IL-6 (D) were also assessed; n = 6 mice/group. *P < 0.05 relative to control; ¥P < 0.05 relative to PB.
Neural Drive is Necessary for the GCG Response to LPS in WT Mice
To further determine the role of neural drive in the GCG response to LPS, chronically catheterized, 5-h-fasted WT mice were exposed to ganglionic blocker (12 mg/kg iv chlorisondamine) or saline control and subsequently treated with LPS (1 mg/kg).
No significant difference in blood glucose levels was observed throughout the study between treatments (Fig. 3A). Mice treated with ganglionic blocker (GB) failed to increase GCG in response to LPS (Fig. 3B), with the effect most pronounced at the end of the study (126 ± 14 vs. 51 ± 7 pg/ml; control vs. GB). GB suppressed plasma GCG even prior to administration of LPS (t = 0, 68 ± 15 vs. 46 ± 5 pg/ml; control vs. GB), supporting the role of the ANS in modulating GCG levels, even in the nonstimulated resting state. Interestingly, GB also blunted the LPS-induced rise in plasma IL-6 (175 ± 25 vs. 37 ± 11 ng/ml; control vs. GB) (Fig. 3C); this was not observed with either sympathetic or parasympathetic blockade alone (Fig. 2D). However, the IL-6 response was not abolished by ganglionic blockade because IL-6 levels are <0.007 ng/ml in the absence of LPS. These results suggest that neural drive is needed not only for the GCG response but also for a robust IL-6 response to LPS.
Fig. 3.
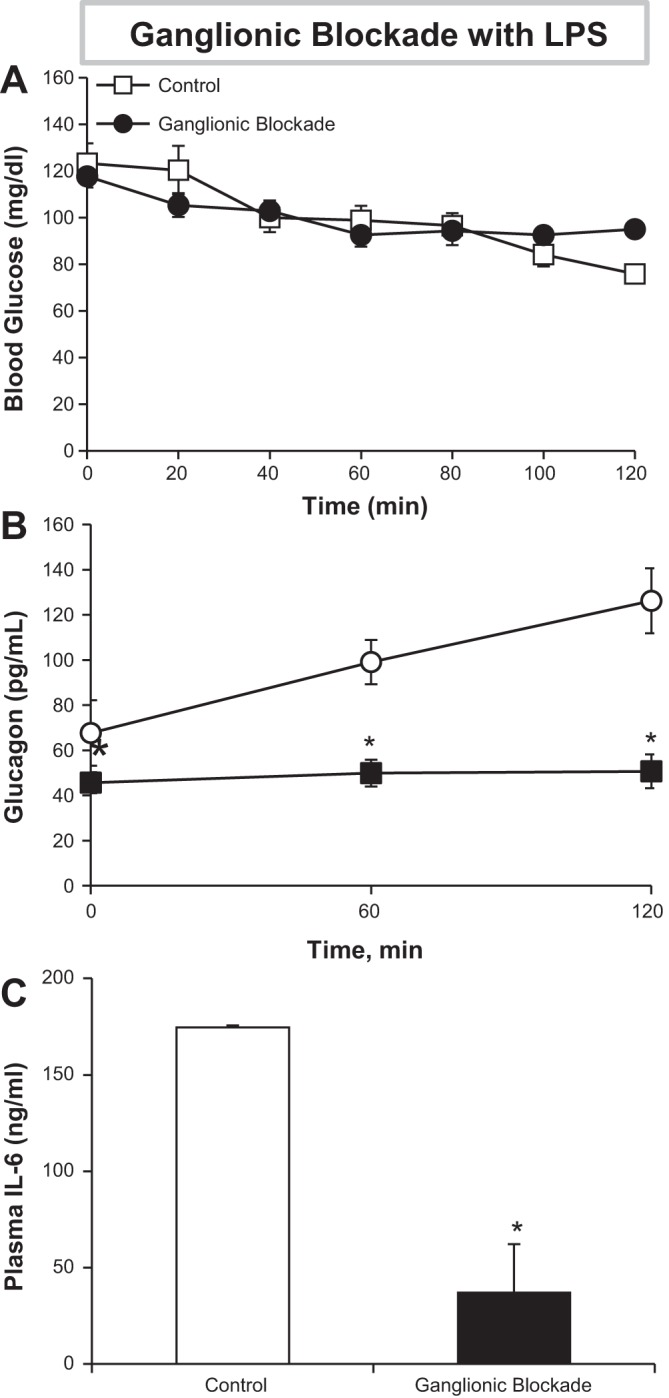
Autonomic drive is necessary for the GCG response to endotoxin. Chronically catheterized WT mice were given a jugular bolus of a ganglionic blocker (GB) or saline (control). Mice were subsequently administered LPS. A and B: blood glucose (A) and plasma GCG (B) were assessed over time. C: terminal plasma IL-6 was also assessed; n = 6 mice/group. *P < 0.05 relative to control.
Central IL-6 Augments the GCG Response to Hypoglycemia in IL-6-KO Mice
Prior work indicates the importance of the CNS for a full counterregulatory (GCG, epinephrine) response to hypoglycemia (47). Knowing that plasma IL-6 levels rise during hypoglycemia (12), we sought to determine whether central IL-6 could modulate GCG secretion in response to this alternative stressor. Chronically catheterized, cannulated IL-6-KO mice underwent hyperinsulinemic hypoglycemic clamps.
The rate of fall in blood glucose and the absolute glucose levels during hypoglycemia were well-matched between Veh and IL-6-microinjected mice, allowing for proper comparison of the counterregulatory response (Fig. 4A). By 20 min of hypoglycemia, the target glucose was reached and was maintained throughout the clamp period. No differences were observed in baseline GCG (Fig. 4B), but when IL-6 was administered centrally, there was a significant increase in plasma GCG (t = 30 min) in mice treated with central IL-6 (95 ± 15 vs. 137 ± 17 pg/ml; Veh vs. IL-6). This trend was maintained throughout the clamp study. Plasma INS concentrations were similar at baseline and rose similarly during the clamp (Fig. 4C). Catecholamine levels were not different between treatment groups (Fig. 4, D and E). Thus, centrally administered IL-6 enhances the GCG response to INS-induced hypoglycemia without amplifying circulating epinephrine and norepinephrine. These data provide evidence that IL-6 augments GCG secretion during multiple stressors.
Fig. 4.

Central IL-6 augments the GCG response to hypoglycemia. Chronically catheterized, cannulated IL-6-KO mice were given either IL-6 or Veh into the LV prior to onset of insulin-induced hypoglycemia (10 mU·kg−1·min−1). Blood glucose (A) and plasma GCG (B), insulin (C), epinephrine (Epi; D), and norepinephrine (Norepi; E) were also assessed; n = 6–7 mice/group. *P < 0.05 relative to vehicle.
IL-6 Enhances Epinephrine-Stimulated GCG Secretion from IL-6-KO Islets
Although studies have examined longer-term transcription-related effects of IL-6 on GCG secretion, none have yet characterized the acute action of the cytokine at the islets. A dynamic islet perifusion system was used to determine whether IL-6 acutely modulates GCG secretion from the pancreas. Islets isolated from IL-6-KO mice were used to avoid the contribution of endogenous IL-6 to the studies. Islets were perfused with media containing 200 ng/ml IL-6 or vehicle and known GCG secretagogues.
IL-6 had no effect on GCG secretion in low-glucose (1.7 mM) media (Fig. 5A). Surprisingly, however, in the presence of low glucose plus 1 μM epinephrine, IL-6 enhanced the GCG response (765 ± 76 vs. 1,017 ± 55 pg glucagon/islet equivalents), suggesting a potential interaction between the IL-6 and catecholamine-signaling pathways to increase GCG secretion. IL-6 did not enhance arginine-stimulated GCG secretion. Since insulin can inhibit GCG secretion, the concentration of glucose in the perfusate was kept low in these studies to inhibit insulin release; none was detectable for all treatments. To verify that IL-6-KO islets have a similar GCG-secreting capacity as WT islets, a separate experiment was conducted using the same secretagogues and concentrations (in the absence of IL-6). We found that GCG secretion was similar in both WT and IL-6-KO islets in response to secretagogues (data not shown). These data demonstrate that IL-6 modulates GCG secretion from islets in the presence of adrenergic (epinephrine) signaling, but not independently.
Fig. 5.

IL-6 enhances the Epi-stimulated GCG response via a calcium-independent mechanism. A: IL-6-KO islets were isolated and perfused in a cell perifusion system with 1.7 mM glucose either alone (Lo) or with 1 μM Epi or 20 mM arginine (Arg). One-half of the islets received concurrent IL-6 exposure (200 ng/ml). Fractions were collected over time and analyzed for GCG (AUC presented). B and C: islets isolated from GCG-tdRFP mice were incubated with the calcium indicator Fluo-4 and placed in a microfluidic device. Islets were first incubated with 1.7 mM glucose and subsequently switched to media containing 1 μM Epi, 200 ng/ml IL-6, or both. Calcium oscillations were observed using confocal microscopy. Normalized Fluo-4 intensity (B) and representative traces (C) are presented. *P < 0.05 relative to control buffer; **P < 0.005; ***P < 0.0005.
Synergy Between IL-6 and Epinephrine Signaling is Calcium Independent
To determine whether IL-6 may augment epinephrine-stimulated GCG secretion via a global intracellular Ca2+ ([Ca2+]i)-dependent mechanism, islets were isolated from mice with tdRFP-expressing α-cells (24) and subsequently loaded with the calcium (Ca2+) indicator dye Fluo-4. Using a microfluidic device (38), Fluo-4 intensity was monitored over time in islets from four mice treated first with 1.7 mM glucose and subsequently switched into 1.7 mM glucose buffer containing IL-6 (200 ng/ml), epinephrine (1 μM), or both.
Epinephrine induced a 2.22 ± 0.09-fold increase (P < 0.0001) relative to 1.7 mM glucose alone (Fig. 5B). IL-6 alone did not alter [Ca2+]i (1.89 ± 0.04-fold over glucose alone). Surprisingly, cotreatment of islets with IL-6 and epinephrine blunted the epinephrine-induced increase in [Ca2+]i (1.5 ± 0.09-fold over 1.7 mM glucose, P = 0.0008). Therefore, IL-6 is likely exerting its effect on GCG secretion in a calcium-independent manner.
DISCUSSION
The results presented herein demonstrate that IL-6 enhances GCG secretion via at least two distinct yet complimentary mechanisms in IL-6-KO mice. GCG is often elevated in inflammatory disease despite accompanying hyperglycemia. Furthermore, inflammatory diseases often associate with autonomic dysfunction. These studies support a synergistic link between IL-6 signaling and autonomic drive to enhance GCG secretion. The hyperglucagonemia observed in certain disease states may result from IL-6-mediated effects on autonomic function. Indeed, acute IL-6 delivery 1) enhances neural drive to islets and 2) increases the effectiveness of epinephrine at islets to augment GCG secretion.
In previous studies, the vascular infusion of IL-6 into IL-6-KO mice completely restored the GCG response to LPS (51). We hypothesized that part of the rapid GCG secretory response was via action of IL-6 in the brain. Indeed, infusing IL-6 into the lateral ventricle (LV) of IL-6-KO mice enhanced LPS-induced GCG secretion by nearly twofold (a physiologically significant change). Plasma IL-6 levels were undetectable following these studies, indicating that central IL-6 alone (i.e., in the absence of vascular IL-6) was sufficient to augment GCG secretion in response to LPS. Furthermore, we found in a separate cohort of WT mice that central injection of IL-6 alone did not stimulate GCG secretion, consistent with our data in KO mice. These data support our hypothesis that IL-6 mediates the cross-talk between the brain and pancreas during inflammation. These data are consistent with work indicating that IL-6 injected into the LV of the anesthetized rat (anesthesia is a stressor) increases splenic autonomic nerve activity (18). Others have demonstrated that proinflammatory cytokines in the brain acutely increase sympathetic tone in renal failure-induced hypertension (57). Although this work does not directly address the regulation of autonomic drive by IL-6, our GCG secretion data lead us to speculate that during settings of stress, IL-6 augments autonomic tone. Unfortunately, catecholamine samples were compromised in the LPS experiments. However, based on our previous work, peripheral IL-6 does not augment circulating epinephrine or norepinephrine during inflammatory stress (51). Thus, the central effects of IL-6 are likely via direct changes in autonomic tone to the pancreas and do not augment adrenal catecholamine secretion. Further studies to directly measure autonomic tone to the pancreas are needed to corroborate this hypothesis.
Other groups have found using in vitro or ex vivo models that IL-6 can also regulate islet β-cell function (14, 41, 43). Although no differences in terminal insulin were observed between any of the cohorts, it is possible that central IL-6 modulated an early insulin response to LPS, which was not tested in these studies. An early rise in insulin would explain why elevated GCG would not increase glucose levels following LPS. In our hands, we have found that there is a short, transient (∼30 min) increase in insulin following initial exposure to the selected dose of LPS. Central IL-6 may enhance this transient elevation in insulin. However, we would anticipate that GCG levels would actually be reduced in IL-6-treated mice, given that insulin is a negative regulator of GCG secretion. Nonetheless, it is possible that central IL-6 also augmented an early insulin response to LPS.
Our studies point to a dominant role of the sympathetic nervous system in the GCG response to LPS. Sympathetic blockade almost completely blunted the GCG response to LPS in WT mice, whereas parasympathetic blockade did so to a lesser degree. Sympathetic blockade also decreased blood glucose levels compared with control and parasympathetic blockade. Hypoglycemia is a potent stimulator of GCG secretion. Despite having both LPS and hypoglycemia on board, sympathetic blockade still suppressed GCG to a greater degree than parasympathetic blockade. Whereas blood glucose levels fell with sympathetic blockade, blood glucose levels following LPS treatment were similar in control, parasympathetic, and ganglionic blocker-treated mice. Propranolol crosses the blood-brain barrier, whereas phentolamine, atropine, and chlorisondamine do not (34). Thus, propranolol delivery may have resulted in off-target effects in the brain. Alternatively, plasma catecholamines may have increased and thus kept blood glucose levels from dropping in mice treated with parasympathetic blockade (atropine). Unfortunately, the catecholamine samples were compromised for these LPS studies, and this remains a point of speculation. These data lead us to hypothesize that centrally administered IL-6 may augment sympathetic drive to the pancreas in response to LPS.
We also found that abolishing total neural drive (ganglionic blockade) completely blunted the GCG response to LPS in WT mice. In fact, ganglionic blockade lowered basal GCG levels prior to LPS treatment, demonstrating that autonomic tone to the pancreas sustains GCG secretion even in the resting state. Interestingly, the plasma IL-6 response to LPS was blunted in mice with ganglionic blockade. This contrasts with mice treated with sympathetic or parasympathetic blockade, where plasma IL-6 was elevated similarly to control-treated mice. Other investigators have found that increased adrenergic (sympathetic) and cholinergic (parasympathetic) tone can increase local production of IL-6 (10, 29, 37). Since we found that both branches contribute to LPS-stimulated GCG secretion, and both are thus activated in response to LPS, the fact that targeting only one branch did not decrease plasma IL-6 levels is not surprising. These data are consistent with previous work demonstrating that vagotomy impaired the proinflammatory cytokine mRNA response to LPS in the brain and in the periphery; this was associated with decreased LPS-induced sleep (58). Cumulatively, the data lead us to hypothesize that an afferent signal(s) following LPS administration leads to elevated proinflammatory cytokines in the brain, which in turn enhances neural drive and amplifies the production of proinflammatory cytokines in the periphery.
Hypoglycemia is an alternative setting of both increased plasma IL-6 (12) and increased GCG secretion. Increases in GCG and autonomic nervous system activation work to limit the nadir in blood glucose levels during episodes of hypoglycemia. Our studies utilized IL-6-KO mice to determine whether central IL-6 signaling alone was sufficient to enhance GCG secretion in response to insulin-induced hypoglycemia. Intracerebroventricular delivery of IL-6 enhanced the rapid rise in GCG secretion. Furthermore, no significant differences in plasma epinephrine were detected during hypoglycemia with IL-6 administration. These data imply that IL-6 triggers GCG secretion independent of adrenal function; this is consistent with the observation we made previously in the presence of LPS (51). These results are consistent with other studies that suggest that separate neural pathways may stimulate glucagon and epinephrine secretion in response to glucoprivation (35, 50). Collectively, these data indicate that central IL-6 augments GCG secretion not only in response to LPS but also in response to insulin-induced hypoglycemia. Further studies are required to delineate the specific site(s) of action and mechanism whereby central IL-6 enhances autonomic drive to the pancreas in these stressed states.
IL-6 not only modified neural drive but also acutely modulated GCG secretion in isolated islets in the presence of epinephrine. Ellingsgaard et al. (13) demonstrated that human islets incubated with IL-6 undergo a slow-onset increase in GCG production; by 4 h there is a significant increase in proglucagon gene expression, and by 24 h there is an increase in secreted GCG. Our islet perifusion experiments demonstrated that IL-6 had no acute effect on islets perfused with low glucose alone or with arginine, but IL-6 amplified epinephrine-stimulated GCG secretion. These data suggest that the adrenergic signaling pathway may synergize with IL-6 signaling to augment GCG secretion. This relationship may be extrapolated to glucose-sensing regions of the brain. Szepietowski et al. (47) showed that modulating adrenergic signaling in the glucose-sensing ventromedial hypothalamus (VMH) modulates the GCG response to insulin-induced hypoglycemia in rats. Moreover, endogenous production of norepinephrine in this region increases during hypoglycemia (5). Since IL-6 receptors are expressed throughout the brain (53), adrenergic and IL-6 signaling may synergize not only in the islet but also in the glucose-sensing regions of the brain.
Since calcium can modulate GCG secretion, we determined whether IL-6 could enhance epinephrine-stimulated calcium signaling. Surprisingly, IL-6 actually reduced the epinephrine-stimulated increase in calcium oscillations while not altering basal calcium oscillations. These data suggest that epinephrine and IL-6 synergize to increase GCG secretion through a calcium-independent mechanism. Both IL-6 and catecholaminergic signaling (Gq-coupled) may converge at the level of the MAPK/ERK signaling pathway. Moreover, ERK has been implicated as a positive effector of GCG secretion (11). IL-6 and catecholaminergic signals may operate through the ERK signaling pathway to increase GCG production. Further studies are needed to address this possibility.
Both our in vivo and ex vivo data suggest that IL-6 does not by itself acutely modulate GCG secretion. It is only in the presence of an additional stressor that IL-6 augments GCG secretion. Both LPS and hypoglycemia are associated with increased autonomic tone. Our islet perifusion data used epinephrine as a surrogate for elevated adrenergic tone at the islet. It was only with epinephrine that we saw the ability of IL-6 to amplify GCG secretion. What occurs during this shift in autonomic tone that allows for IL-6 to presumably amplify neural drive to the pancreas and increase GCG secretion is yet to be elucidated.
A great body of work in a critical glucose-sensing region of the brain, the VMH (49), points to a potential mechanism by which central IL-6 may enhance GCG secretion in settings of stress. Local induction of glucopenia in this region results in increased systemic levels of GCG and epinephrine (7), and delivery of glucose in the VMH during systemic glucopenia prevents the release of these hormones (6). Increasing adrenergic signaling in the VMH increases the GCG response to hypoglycemia, whereas blocking adrenergic signaling decreases the response (47). Furthermore, norepinephrine levels increase in the VMH during hypoglycemia (5), and norepinephrine can increase local production of IL-6 (27, 32). These data, when combined with our findings that catecholaminergic signaling and IL-6-signaling pathways synergize in islets, lead us to speculate that a similar synergy may also happen within the VMH and potentially other glucoregulatory brain regions. IL-6 receptors are abundant throughout the hypothalamus, including within the VMH (53). In a separate cohort of mice, we also found that administration of IL-6 to the lateral ventricle activates phosphorylation of STAT3 (an index of IL-6 signaling) in regions surrounding the ventricles, including the VMH (data not presented). Furthermore, pathophysiological settings such as endotoxemia or physiological settings such as hypoglycemia are also associated with elevated adrenergic tone and elevated IL-6. Therefore, it is possible that IL-6 signaling in the brain may synergize with catecholaminergic signaling to amplify the GCG response in each of these settings.
Clinical research has demonstrated that targeting the IL-6 pathway is of benefit to diabetic patients. Tocilizumab is a humanized IL-6 receptor (IL6Rα) antibody used clinically to treat rheumatoid arthritis (RA). When administered to diabetics with RA, tocilizumab reduced Hb A1c in type 2 diabetics compared with those without diabetes, who showed no changes in Hb A1c (33). Although this particular study is complicated by the concurrent RA of these patients, targeting the IL-6 pathway clearly shows promise in lowering Hb A1c levels. We speculate that tocilizumab may suppress GCG secretion and thus contribute to the improvement of Hb A1c levels in these patients. Reducing GCG production via IL-6 antagonism may represent an additional treatment option in diabetics with hyperglucagonemia. Because chronic levels of circulating IL-6 can also contribute to reduced insulin sensitivity in adipose, muscle, and liver (9, 21–23, 30, 40), reducing IL-6 signaling could have benefits beyond controlling GCG levels.
In summary, our data support IL-6 as an important driver of GCG secretion. IL-6 exerts its actions via at least two mechanisms; IL-6 1) acts centrally to enhance GCG secretion in response to both inflammation and insulin-induced hypoglycemia and 2) enhances epinephrine-stimulated GCG secretion in the islet. Thus, IL-6 may modulate GCG secretion in vivo by augmenting neural drive to the pancreas and amplifying the effects of circulating epinephrine or neural drive directly at the pancreas. However, future work is needed to demonstrate that both effects are happening simultaneously in vivo as well as to determine whether varying concentrations of IL-6 will yield similar results. IL-6 is elevated in multiple disease states, many of which associate with autonomic dysfunction. These findings implicate IL-6 in contributing to the hyperglucagonemia that exists in multiple inflammatory settings. The refore, the IL-6 signaling pathway may represent a novel target in the treatment of glucoregulatory disorders that are also characterized by autonomic dysfunction.
GRANTS
This work was supported by National Institutes of Health Grants DK-043748 and DK-078188(principal investigator: O. P. McGuinness). Special thanks to the Mouse Metabolic Phenotyping Center (DK-59637) and the Islet Procurement & Analysis Core (DK-020593) and Hormone Assay and Analytical Services Core (DK-059637 and DK-020593). T. M. Barnes was supported by T32-DK-07563. O. P. McGuinness had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
DISCLOSURES
The authors have no conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
T.M.B. and M.B. conception and design of research; T.M.B., Y.F.O., A.D.E., A.D.L., C.M.M., and A.G.C. performed experiments; T.M.B. and M.B. analyzed data; T.M.B., Y.F.O., A.D.E., M.B., D.W.P., and O.P.M. interpreted results of experiments; T.M.B. prepared figures; T.M.B. drafted manuscript; T.M.B., Y.F.O., A.D.E., and O.P.M. edited and revised manuscript; T.M.B., Y.F.O., and O.P.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Paul Gresch (Vanderbilt University) for help in teaching cannulation surgery techniques to T. M. Barnes. We also thank Lawrence McLean House II (UT Medical) and Travis Cyphert (Vanderbilt University) for helping with blood collection at the end of the LPS studies. Finally, we thank David Wasserman and Clinton Hasenour (Vanderbilt University) for help in editing the manuscript.
REFERENCES
- 1.Anton AH, Sayre DF. A study of the factors affecting the aluminum oxide-trihydroxyindole procedure for the analysis of catecholamines. J Pharmacol Exp Ther 138: 360–375, 1962. [PubMed] [Google Scholar]
- 2.Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 55: 390–397, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Banks WA, Kastin AJ, Gutierrez EG. Penetration of interleukin-6 across the murine blood-brain barrier. Neurosci Lett 179: 53–56, 1994. [DOI] [PubMed] [Google Scholar]
- 4.Bastard JP, Maachi M, Van Nhieu JT, Jardel C, Bruckert E, Grimaldi A, Robert JJ, Capeau J, Hainque B. Adipose tissue IL-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro. J Clin Endocrinol Metab 87: 2084–2089, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Beverly JL, De Vries MG, Bouman SD, Arseneau LM. Noradrenergic and GABAergic systems in the medial hypothalamus are activated during hypoglycemia. Am J Physiol Regul Integr Comp Physiol 280: R563–R569, 2001. [DOI] [PubMed] [Google Scholar]
- 6.Borg MA, Sherwin RS, Borg WP, Tamborlane WV, Shulman GI. Local ventromedial hypothalamus glucose perfusion blocks counterregulation during systemic hypoglycemia in awake rats. J Clin Invest 99: 361–365, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borg WP, Sherwin RS, During MJ, Borg MA, Shulman GI. Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes 44: 180–184, 1995. [DOI] [PubMed] [Google Scholar]
- 8.Brissova M, Shostak A, Shiota M, Wiebe PO, Poffenberger G, Kantz J, Chen Z, Carr C, Jerome WG, Chen J, Baldwin HS, Nicholson W, Bader DM, Jetton T, Gannon M, Powers AC. Pancreatic islet production of vascular endothelial growth factor-a is essential for islet vascularization, revascularization, and function. Diabetes 55: 2974–2985, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 11: 183–190, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen C, Du J, Feng W, Song Y, Lu Z, Xu M, Li Z, Zhang Y. β-Adrenergic receptors stimulate interleukin-6 production through Epac-dependent activation of PKCδ/p38 MAPK signalling in neonatal mouse cardiac fibroblasts. Br J Pharmacol 166: 676–688, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chuang JC, Sakata I, Kohno D, Perello M, Osborne-Lawrence S, Repa JJ, Zigman JM. Ghrelin directly stimulates glucagon secretion from pancreatic alpha-cells. Mol Endocrinol 25: 1600–1611, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dotson S, Freeman R, Failing HJ, Adler GK. Hypoglycemia increases serum interleukin-6 levels in healthy men and women. Diabetes Care 31: 1222–1223, 2008. [DOI] [PubMed] [Google Scholar]
- 13.Ellingsgaard H, Ehses JA, Hammar EB, Van Lommel L, Quintens R, Martens G, Kerr-Conte J, Pattou F, Berney T, Pipeleers D, Halban PA, Schuit FC, Donath MY. Interleukin-6 regulates pancreatic alpha-cell mass expansion. Proc Natl Acad Sci USA 105: 13163–13168, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, Eppler E, Bouzakri K, Wueest S, Muller YD, Hansen AM, Reinecke M, Konrad D, Gassmann M, Reimann F, Halban PA, Gromada J, Drucker DJ, Gribble FM, Ehses JA, Donath MY. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med 17: 1481–1489, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci 8: 1254–1266, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Havel PJ, Mundinger TO, Taborsky GJ., Jr Pancreatic sympathetic nerves contribute to increased glucagon secretion during severe hypoglycemia in dogs. Am J Physiol Endocrinol Metab 270: E20–E26, 1996. [DOI] [PubMed] [Google Scholar]
- 17.Havel PJ, Veith RC, Dunning BE, Taborsky GJ., Jr Role for autonomic nervous system to increase pancreatic glucagon secretion during marked insulin-induced hypoglycemia in dogs. Diabetes 40: 1107–1114, 1991. [DOI] [PubMed] [Google Scholar]
- 18.Helwig BG, Craig RA, Fels RJ, Blecha F, Kenney MJ. Central nervous system administration of interleukin-6 produces splenic sympathoexcitation. Auton Neurosci 141: 104–111, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirano T. Interleukin 6 in autoimmune and inflammatory diseases: a personal memoir. Proc Jpn Acad Ser B Phys Biol Sci 86: 717–730, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang G, Zhang BB. Glucagon and regulation of glucose metabolism. Am J Physiol Endocrinol Metab 284: E671–E678, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Klover PJ, Clementi AH, Mooney RA. Interleukin-6 depletion selectively improves hepatic insulin action in obesity. Endocrinology 146: 3417–3427, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Klover PJ, Zimmers TA, Koniaris LG, Mooney RA. Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes 52: 2784–2789, 2003. [DOI] [PubMed] [Google Scholar]
- 23.Lagathu C, Bastard JP, Auclair M, Maachi M, Capeau J, Caron M. Chronic interleukin-6 (IL-6) treatment increased IL-6 secretion and induced insulin resistance in adipocyte: prevention by rosiglitazone. Biochem Biophys Res Commun 311: 372–379, 2003. [DOI] [PubMed] [Google Scholar]
- 24.Le Marchand SJ, Piston DW. Glucose suppression of glucagon secretion: metabolic and calcium responses from alpha-cells in intact mouse pancreatic islets. J Biol Chem 285: 14389–14398, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macdonald IA, Lake DM. An improved technique for extracting catecholamines from body fluids. J Neurosci Methods 13: 239–248, 1985. [DOI] [PubMed] [Google Scholar]
- 27.Maimone D, Cioni C, Rosa S, Macchia G, Aloisi F, Annunziata P. Norepinephrine and vasoactive intestinal peptide induce IL-6 secretion by astrocytes: synergism with IL-1 beta and TNF alpha. J Neuroimmunol 47: 73–81, 1993. [DOI] [PubMed] [Google Scholar]
- 28.McGuinness OP. Defective glucose homeostasis during infection. Annu Rev Nutr 25: 9–35, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Mohamed-Ali V, Flower L, Sethi J, Hotamisligil G, Gray R, Humphries SE, York DA, Pinkney J. beta-Adrenergic regulation of IL-6 release from adipose tissue: in vivo and in vitro studies. J Clin Endocrinol Metab 86: 5864–5869, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Nieto-Vazquez I, Fernandez-Veledo S, de Alvaro C, Lorenzo M. Dual role of interleukin-6 in regulating insulin sensitivity in murine skeletal muscle. Diabetes 57: 3211–3221, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Niswender KD, Shiota M, Postic C, Cherrington AD, Magnuson MA. Effects of increased glucokinase gene copy number on glucose homeostasis and hepatic glucose metabolism. J Biol Chem 272: 22570–22575, 1997. [DOI] [PubMed] [Google Scholar]
- 32.Norris JG, Benveniste EN. Interleukin-6 production by astrocytes: induction by the neurotransmitter norepinephrine. J Neuroimmunol 45: 137–145, 1993. [DOI] [PubMed] [Google Scholar]
- 33.Ogata A, Morishima A, Hirano T, Hishitani Y, Hagihara K, Shima Y, Narazaki M, Tanaka T. Improvement of HbA1c during treatment with humanised anti-interleukin 6 receptor antibody, tocilizumab. Ann Rheum Dis 70: 1164–1165, 2011. [DOI] [PubMed] [Google Scholar]
- 34.Olesen J, Hougard K, Hertz M. Isoproterenol and propranolol: ability to cross the blood-brain barrier and effects on cerebral circulation in man. Stroke 9: 344–349, 1978. [DOI] [PubMed] [Google Scholar]
- 35.Paranjape SA, Chan O, Zhu W, Horblitt AM, McNay EC, Cresswell JA, Bogan JS, McCrimmon RJ, Sherwin RS. Influence of insulin in the ventromedial hypothalamus on pancreatic glucagon secretion in vivo. Diabetes 59: 1521–1527, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pedersen BK, Steensberg A, Schjerling P. Exercise and interleukin-6. Curr Opin Hematol 8: 137–141, 2001. [DOI] [PubMed] [Google Scholar]
- 37.Perez DM, Papay RS, Shi T. alpha1-Adrenergic receptor stimulates interleukin-6 expression and secretion through both mRNA stability and transcriptional regulation: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB. Mol Pharmacol 76: 144–152, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rocheleau JV, Walker GM, Head WS, McGuinness OP, Piston DW. Microfluidic glucose stimulation reveals limited coordination of intracellular Ca2+ activity oscillations in pancreatic islets. Proc Natl Acad Sci USA 101: 12899–12903, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rocheleau JV, Walker GM, Head WS, McGuinness OP, Piston DW. Microfluidic glucose stimulation reveals limited coordination of intracellular Ca2+ activity oscillations in pancreatic islets. Proc Natl Acad Sci USA 101: 12899–12903, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem 278: 45777–45784, 2003. [DOI] [PubMed] [Google Scholar]
- 41.Sandler S, Bendtzen K, Eizirik DL, Welsh M. Interleukin-6 affects insulin secretion and glucose metabolism of rat pancreatic islets in vitro. Endocrinology 126: 1288–1294, 1990. [DOI] [PubMed] [Google Scholar]
- 42.Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes 51: 3391–3399, 2002. [DOI] [PubMed] [Google Scholar]
- 43.Shimizu H, Ohtani K, Kato Y, Mori M. Interleukin-6 increases insulin secretion and preproinsulin mRNA expression via Ca2+-dependent mechanism. J Endocrinol 166: 121–126, 2000. [DOI] [PubMed] [Google Scholar]
- 44.Sorensen H, Brand CL, Neschen S, Holst JJ, Fosgerau K, Nishimura E, Shulman GI. Immunoneutralization of endogenous glucagon reduces hepatic glucose output and improves long-term glycemic control in diabetic ob/ob mice. Diabetes 55: 2843–2848, 2006. [DOI] [PubMed] [Google Scholar]
- 45.Spranger J, Kroke A, Mohlig M, Hoffmann K, Bergmann MM, Ristow M, Boeing H, Pfeiffer AF. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes 52: 812–817, 2003. [DOI] [PubMed] [Google Scholar]
- 46.Sukoff Rizzo SJ, Neal SJ, Hughes ZA, Beyna M, Rosenzweig-Lipson S, Moss SJ, Brandon NJ. Evidence for sustained elevation of IL-6 in the CNS as a key contributor of depressive-like phenotypes. Transl Psychiatry 2: e199, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szepietowska B, Zhu W, Chan O, Horblitt A, Dziura J, Sherwin RS. Modulation of beta-adrenergic receptors in the ventromedial hypothalamus influences counterregulatory responses to hypoglycemia. Diabetes 60: 3154–3158, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taborsky GJ, Jr., Mundinger TO. Minireview: The role of the autonomic nervous system in mediating the glucagon response to hypoglycemia. Endocrinology 153: 1055–1062, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thorens B. Brain glucose sensing and neural regulation of insulin and glucagon secretion. Diabetes Obes Metab 13, Suppl 1: 82–88, 2011. [DOI] [PubMed] [Google Scholar]
- 50.Tong Q, Ye C, McCrimmon RJ, Dhillon H, Choi B, Kramer MD, Yu J, Yang Z, Christiansen LM, Lee CE, Choi CS, Zigman JM, Shulman GI, Sherwin RS, Elmquist JK, Lowell BB. Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab 5: 383–393, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tweedell A, Mulligan KX, Martel JE, Chueh FY, Santomango T, McGuinness OP. Metabolic response to endotoxin in vivo in the conscious mouse: role of interleukin-6. Metabolism 60: 92–98, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest 122: 4–12, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vallieres L, Rivest S. Interleukin-6 is a needed proinflammatory cytokine in the prolonged neural activity and transcriptional activation of corticotropin-releasing factor during endotoxemia. Endocrinology 140: 3890–3903, 1999. [DOI] [PubMed] [Google Scholar]
- 54.van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R. Intensive insulin therapy in critically ill patients. N Engl J Med 345: 1359–1367, 2001. [DOI] [PubMed] [Google Scholar]
- 55.Wallenius V, Wallenius K, Ahren B, Rudling M, Carlsten H, Dickson SL, Ohlsson C, Jansson JO. Interleukin-6-deficient mice develop mature-onset obesity. Nat Med 8: 75–79, 2002. [DOI] [PubMed] [Google Scholar]
- 56.Wasserman DH. Four grams of glucose. Am J Physiol Endocrinol Metab 296: E11–E21, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ye S, Mozayeni P, Gamburd M, Zhong H, Campese VM. Interleukin-1β and neurogenic control of blood pressure in normal rats and rats with chronic renal failure. Am J Physiol Heart Circ Physiol 279: H2786–H2796, 2000. [DOI] [PubMed] [Google Scholar]
- 58.Zielinski MR, Dunbrasky DL, Taishi P, Souza G, Krueger JM. Vagotomy attenuates brain cytokines and sleep induced by peripherally administered tumor necrosis factor-α and lipopolysaccharide in mice. Sleep 36: 1227–1238, 1238A, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]