

Abstract
Cancer deaths are primarily caused by distant metastases, rather than by primary tumor growth; however, the role of smoking in metastasis remains unclear. We demonstrated previously that endothelial cell platelet-activating factor (PAF) production results in enhanced inflammatory cell recruitment to the lung. We propose that endothelial cell PAF accumulation plays a role in cancer cell migration to distal locations. We used cigarette smoke extract (CSE) to inhibit the activity of endothelial cell PAF acetylhydrolase (PAF-AH), which hydrolyzes and inactivates PAF, and determined whether this results in increased endothelial cell PAF accumulation and breast cancer adherence. Incubation of human lung microvascular endothelial cells (HMVEC-L) with CSE resulted in a significant inhibition of PAF-AH activity that was accompanied by increased PAF production and adherence of highly invasive MDA-MB-231 breast cancer cells. Pretreatment of HMVEC-L with (S)-bromoenol lactone to inhibit calcium-independent phospholipase A2β (iPLA2β, which initiates endothelial cell PAF production) prior to CSE exposure resulted in complete inhibition of MDA-MB-231 cell adherence. Similarly, pretreatment of MDA-MB-231 cells with the PAF receptor antagonist Ginkgo biloba resulted in inhibition of adherence to the endothelium. Immunoblot analysis indicated an increase in MDA-MB-231 cell PAF receptor expression with CSE exposure. Taken together, our data indicate that CSE exposure increases endothelial cell PAF production, resulting in enhanced adherence of tumor cells to the endothelium. Our in vitro data indicate that increased tumor cell adherence would lead to enhanced metastasis formation in smokers. Potential therapeutic targets include endothelial cell iPLA2β or the tumor cell PAF receptor.
Keywords: platelet-activating factor, cigarette smoke, lung endothelium, breast cancer, metastasis
cancer continues to be a major cause of mortality worldwide, with 90% of all cancers stemming from environmental and lifestyle factors (1, 3, 11). Almost one-third of all cancer deaths are a direct result of smoking (1, 25). According to the Centers for Disease Control, 18% of adults in the United States (42.1 million) continue to smoke, despite the known risks. Smoking has been linked to various cancers, and recently the link has been established with breast cancer (1). Several cohort studies have described the link between patients with breast cancer and long-term smoking history (1). Recently, a cohort study of American Cancer Society Cancer Preventative Studies II reported a higher rate of new breast cancer cases in patients with a history of smoking than in those who had never smoked. Because the underlying mechanisms involved in cigarette smoke-induced cancer risk, as well as contribution to progression and metastasis, remain unknown, this disease becomes very difficult to manage.
An important step in metastasis is the process by which tumor cells arrest in circulation and attach to the endothelial wall to execute extravasation into the distant tissue (6). In the setting of metastasis, many studies have characterized the role of adhesion molecules in tumor cell extravasation and endothelial cell attachment. However, the role of platelet-activating factor (PAF) and its receptor (PAF-R) has been less studied. PAF, which has been implicated in tumor cell growth, angiogenesis, and metastasis, could potentially play a role in the setting of smoking and cancer (4, 5, 9, 18).
Endothelial cell PAF production is increased during inflammatory or hypersensitivity responses and is instrumental in mediating endothelial cell-inflammatory cell interactions (15, 17, 32). PAF is a highly potent membrane phospholipid-derived metabolite that is tightly regulated within cells. PAF is synthesized by endothelial cells in response to stimulation by a variety of agonists. The synthesis of endothelial cell PAF occurs via the remodeling pathway that begins with activation of phospholipase A2 (PLA2) (14, 17, 19). We demonstrated previously that the majority of endothelial cell PLA2 activity that is responsible for PAF production is calcium-independent PLA2β (iPLA2β) (22, 23). The biological activity of PAF can be rapidly terminated by PAF acetylhydrolases (PAF-AH), a family of unique iPLA2 enzymes that hydrolyze the acetyl group at the sn-2 position of PAF to generate biologically inactive lyso-PAF and acetate (17). Inhibition of production and enhanced degradation of PAF have been proposed as anti-inflammatory targets. In a previous study we demonstrated that the iPLA2 inhibitor methyl arachidonyl fluorophosphonate (MAFP) inhibits endothelial cell PAF-AH, resulting in enhanced PAF accumulation and polymorphonuclear leukocyte adherence (13, 30). Pretreatment of endothelial cells with bromoenol lactone (BEL), an inhibitor of iPLA2 activity and PAF production, abolishes MAFP-induced PAF accumulation (30). More recently, we demonstrated that cigarette smoke extract (CSE) inhibits endothelial cell PAF-AH activity, resulting in PAF accumulation (24).
Smoking has been attributed to development of lung cancer and has recently been associated with increased breast cancer risk (12, 16, 25, 27). However, the effects of smoking on metastasis, particularly attachment of tumor cells to the endothelium, remain unknown (1). One of the most common sites of breast cancer metastasis is the lung (31). Therefore, in this study we determined whether CSE increases adherence of breast cancer cells to the lung endothelium. We utilized various breast cancer cell lines, including low-invasive hormone-positive (MCF-7), moderately invasive triple-negative (MDA-MB-468), highly invasive triple-negative (MDA-MB-231), and normal mammary epithelial (MCF-10A) cells. We determined that breast tumor cell adherence could be modulated by inhibition of endothelial cell iPLA2 activity or pretreatment of tumor cells with the PAF-R antagonist ginkgolide B. Recruitment of circulating breast cancer cells to the lung via PAF-AH inhibition may contribute to the increased risk of metastases, particularly in smokers. Although these data represent a mechanism for increased metastasis in breast cancer patients who smoke, this mechanism of enhanced metastasis is likely translational to other metastatic cancers.
MATERIALS AND METHODS
MDA-MB-231, MDA-468, MCF-7, and MCF-10A cells were obtained from American Type Culture Collection (Manassas, VA), and human lung microvascular endothelial cells (HMVEC-L) were obtained from Lonza (Walkersville, MD). CSE was obtained from Murty Pharmaceuticals (Lexington, KY), and [3H]acetic acid, sodium salt and hexadecyl-2-acetyl-sn-glyceryl-2-phosphorylcholine-1-O-[acetyl-3H(N)] were purchased from PerkinElmer (Boston, MA). All other chemicals were obtained from Sigma Chemical (St. Louis, MO).
Cell culture.
MDA-MB-231, MDA-468, MCF-7, and MCF-10A cells were grown to confluence in Dulbecco's modified Eagle's medium with 10% fetal bovine serum and penicillin-streptomycin-amphotericin. HMVEC-L were grown to confluence in EGM-2MV medium (Lonza) supplemented with 5% fetal bovine serum. Cells were incubated at 37°C in an atmosphere of 95% O2-5% CO2. Cells were subcultured using 0.05% trypsin-EDTA and passaged in a 1:3 ratio. HMVEC-L or MDA-MB-231 cells were incubated in certain experiments with CSE (20 μg/ml), the iPLA2 inhibitor BEL (5 μM), or the PAF-AH inhibitor MAFP (5 μM) for 1–18 h.
Measurement of PAF production.
HMVEC-L PAF production was measured using an ELISA kit (Biotang, Waltham, MA). Endothelial cell monolayers were washed with ice-cold Dulbecco's phosphate-buffered saline and frozen at −20°C. After two freeze-thaw cycles, aliquots of the suspension were added to microtiter plates with a biotin-conjugated polyclonal antibody specific for PAF. PAF content in samples was determined spectrophotometrically at 450 nm using a Synergy 2 microplate reader (Biotek, Winooski, VT).
PAF-AH activity.
HMVEC-L grown to confluence in 35-mm dishes were removed from the tissue culture plate in 1.2 mM Ca2+-HEPES buffer and sonicated on ice. Cellular protein (25 μg) was incubated with 0.1 mM [acetyl-3H]PAF (10 mCi/mmol) for 30 min at 37°C. The reaction was stopped by addition of 50 μl of 10 M acetic acid and 1.5 ml of 0.1 M sodium acetate. The reaction mixture was passed through a C18 gel cartridge (Baker Chemical, Phillipsburg, NJ) to isolate released [3H]acetic acid, and radioactivity was measured using a liquid scintillation counter.
Adherence of breast cancer cells to HMVEC-L.
MDA-MB-231 or MCF-7 cells were labeled with calcein-AM (4 μg/ml; Alexis Biochemicals, Lausen, Switzerland) for 45 min at 37°C. After they were washed three times, 2 × 106 cells were layered onto confluent HMVEC-L monolayers. Medium and unbound cells were removed and discarded. Adherent breast cancer cells and endothelial cells were washed with Dulbecco's phosphate-buffered saline and lysed with 1 ml of 0.2% Triton X-100. Samples were sonicated (550 Sonic Dismembrator, Fisher Scientific, Pittsburgh, PA) for 10 s. The amount of calcein-AM fluorescence was measured using a Synergy 2 microplate reader at an excitation wavelength of 485 nm and emission wavelength of 530 nm. The percentage of MDA-MB-231 cell adherence was calculated from the amount of calcein-AM fluorescence measured in 2 × 106 cells.
Immunoblot analysis.
Cells were suspended in lysis buffer containing 20 mM HEPES (pH 7.6), 250 mM sucrose, 2 mM dithiothreitol, 2 mM EDTA, 2 mM EGTA, 10 mM β-glycerophosphate, 1 mM sodium orthovanadate, 2 mM phenylmethylsulfonyl fluoride, 20 μg/ml leupeptin, 10 μg/ml aprotinin, and 5 μg/ml pepstatin A. Cells were sonicated on ice and centrifuged at 20,000 g at 4°C for 20 min to remove cellular debris and nuclei. Cytosolic protein was separated by SDS-PAGE and electrophoretically transferred to nitrocellulose membranes (Bio-Rad, Richmond, CA). The blocked nitrocellulose membrane was incubated with primary antibodies to PAF-R and horseradish peroxidase-conjugated secondary antibodies. Regions of antibody binding were detected using enhanced chemiluminescence (Amersham, Arlington Heights, IL) after exposure to film (Hyperfilm, Amersham). Equal loading was verified by immunoblot analysis for actin.
RESULTS
Inhibition of PAF-AH activity and PAF accumulation in HMVEC-L.
HMVEC-L were incubated with CSE (20 μg/ml), BEL (5 μM), or MAFP (5 μM) for 1 or 18 h, and PAF-AH activity was measured (Fig. 1). After 1 h, PAF-AH activity was significantly inhibited by CSE and MAFP but unaffected by the iPLA2 and subsequent PAF inhibitor BEL. At 18 h, PAF-AH activity was inhibited by >90% by MAFP and CSE but remained unaffected by BEL.
Fig. 1.
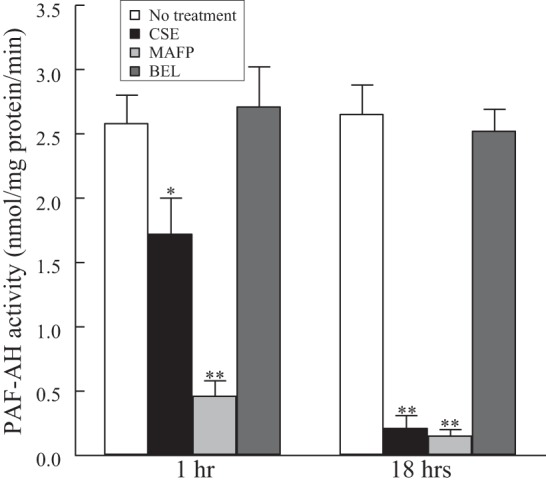
Platelet-activating factor (PAF) acetylhydrolase (PAF-AH) activity in human lung microvascular endothelial cells (HMVEC-L) incubated with cigarette smoke extract (CSE, 20 μg/ml), methyl arachidonyl fluorophosphonate (MAFP, 5 μM), or bromoenol lactone (BEL, 5 μM) for 1 or 18 h. Values are means ± SE for 4 separate cell cultures. *P < 0.05; **P < 0.01 vs. no treatment at each time point.
The inhibition of PAF-AH activity with MAFP or CSE resulted in the subsequent increase in HMVEC-L PAF accumulation after 18 h (Fig. 2). PAF accumulation was diminished when HMVEC-L were incubated with the iPLA2 inhibitor BEL (Fig. 2). HMVEC-L pretreated with BEL (5 μM, 1 h) prior to incubation with CSE (20 μg/ml, 18 h) demonstrated no increase in PAF accumulation. Thus, increased PAF accumulation as a result of PAF-AH inhibition can be prevented by blocking iPLA2 activity and, hence, PAF production in HMVEC-L.
Fig. 2.
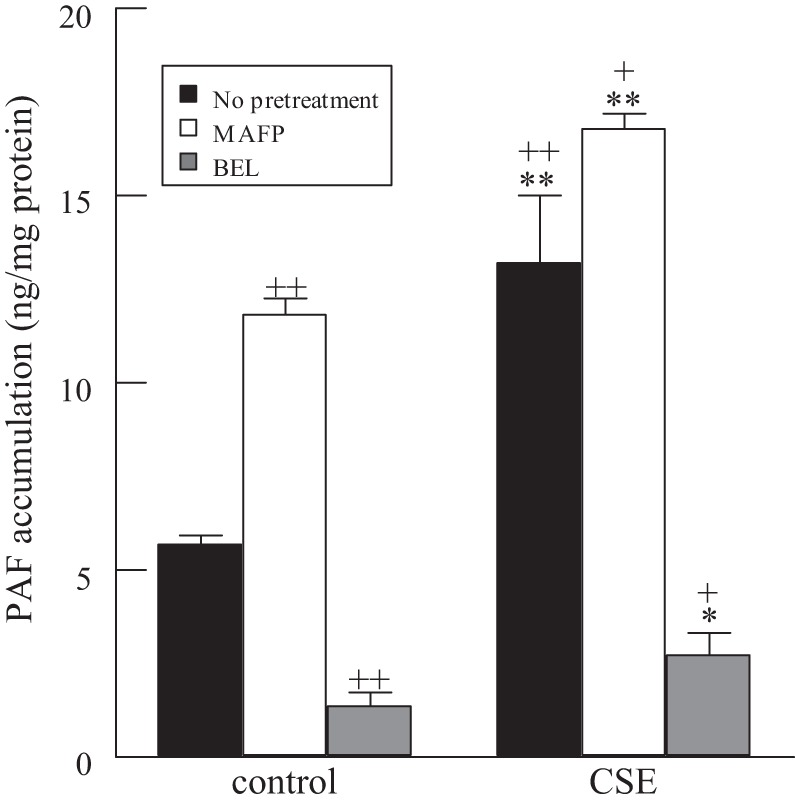
PAF accumulation in HMVEC-L incubated with CSE (20 μg/ml, 18 h) in the presence or absence of MAFP (5 μM, 30 min prior to CSE addition) or BEL (5 μM, 30 min prior to CSE addition). Values are means ± SE for 4 separate cell cultures. *P < 0.05; **P < 0.01 vs. corresponding room air control. +P < 0.05, ++P < 0.01, presence vs. absence of calcium-independent phospholipase A2 (iPLA2) inhibitor.
Increased HMVEC-L PAF accumulation is associated with an increase in human breast cancer cell adherence.
To investigate whether the CSE-induced increase in HMVEC-L PAF accumulation resulted in increased adherence of breast cancer cells, we utilized highly invasive triple-negative (MDA-MB-231) and hormone-positive noninvasive (MCF-7) breast cancer cells and measured adherence to lung endothelial cells that had been incubated with CSE for up to 24 h (Fig. 3). At the indicated time of CSE exposure, breast cancer cells were added to the HMVEC-L monolayer and incubated for 30 min. We observed a significant increase in MCF-7 and MDA-MB-231 cell adherence to HMVEC-L as CSE incubation time increased. However, the CSE-dependent increase in adherence was higher in the aggressive breast cancer cell line MDA-MB-231 than in the MCF-7 cell line at each time point (Fig. 3).
Fig. 3.
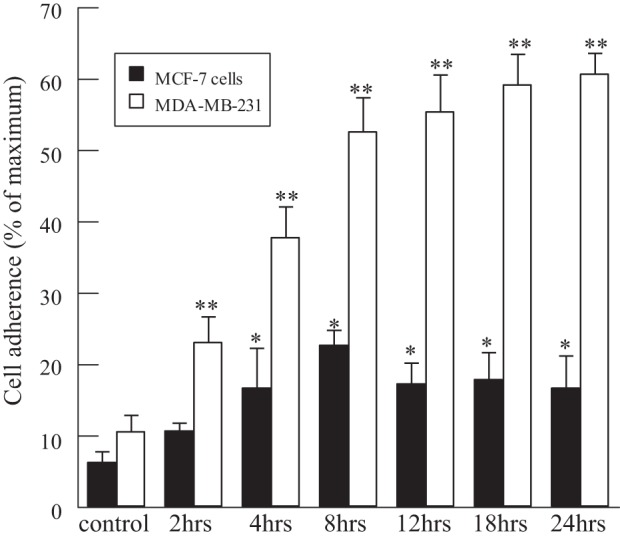
Breast cancer cell adherence to HMVEC-L monolayers incubated with CSE (20 μg/ml) for 2–24 h. MDA-MB-231 and MCF-7 cells were labeled with calcein-AM and added to CSE-treated HMVEC-L for 30 min, and adherence was measured by calcein-AM fluorescence. Values are means ± SE for 4 separate cell culture experiments. *P < 0.05; **P < 0.01 vs. control.
PAF-R expression of breast tumor cells is increased with CSE exposure.
After the observation that exposure of HMVEC-L to CSE resulted in increased MDA-MB-231 cell adherence, we determined whether CSE had an effect on PAF-R expression on the breast tumor cell surface. We incubated MDA-MB-231, MDA-MB-468, and MCF-7 breast tumor cells plus the mammary epithelial cell line MCF-10A with CSE (20 μg/ml) for up to 48 h (Fig. 4). PAF-R expression was not detected in MCF-10A cells but was present in each breast cancer cell line tested. When PAF-R expression was normalized to actin, we observed a significant increase in relative expression when MDA-MB-231 cells were exposed to CSE (Fig. 4B, bottom). In contrast, we observed minimal change in PAF-R expression in either MDA-MB-468 or MCF-7 cells in response to CSE (Fig. 4B, top). Incubation with 5 μM MAFP for 1 h resulted in a fourfold increase in normalized PAF-R expression, but no significant change was observed in MCF-7 or MDA-MB-468 cells.
Fig. 4.

PAF receptor (PAF-R) expression in CSE-exposed breast cell lines. B: representative immunoblots of PAF-R expression in MCF-7, MDA-MB-468, MDA-MB-231, and MCF-10A cell lines exposed to CSE (20 μg/ml, 6–48 h) or MAFP (5 μM, 18 h). A: densitometry of immunoblots shows significantly increased PAF-R expression in triple-negative highly metastatic MDA-MB-231 cells and minimal change in PAF-R expression in MCF-7 and MDA-MB-468 cells. Values are means ± SE for 4 separate cell culture experiments. *P < 0.05; **P < 0.01 vs. control.
MDA-MB-231 cells were exposed to CSE for 24 h prior to addition to HMVEC-L. As shown in Fig. 5, in breast cancer cells exposed to CSE, adherence to HMVEC-L was increased under control conditions and at each time point of CSE exposure compared with adherence of MDA-MB-231 cells incubated in medium alone.
Fig. 5.
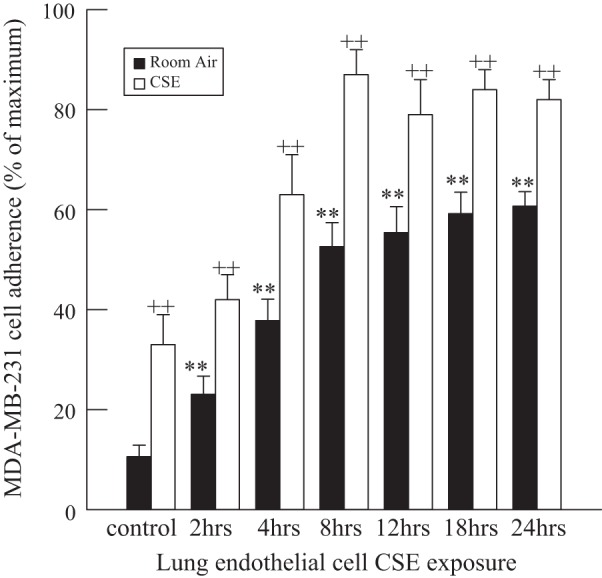
MDA-MB-231 cell adherence to HMVEC-L monolayers incubated with CSE (20 μg/ml) for 2–24 h. MDA-MB-231 cells were exposed to CSE in medium or room air, labeled with calcein-AM, and added to HMVEC-L for 30 min, and adherence was measured by calcein-AM fluorescence. Adherence to HMVEC-L was significantly increased in cells incubated with CSE compared with cells in room air. Values are means ± SE for 4 separate cell culture experiments. **P < 0.01 vs. control. ++P < 0.01 vs. room air.
MDA-MB-231 cell adherence to HMVEC-L can be modified using PAF and PAF-R antagonists.
To mitigate breast cancer cell adherence to lung endothelial cells mediated by CSE, we pretreated HMVEC-L with the iPLA2β-selective inhibitor (S)-BEL (5 μM) for 10 min prior to CSE exposure. We measured adherence of MDA-MB-231 cells to HMVEC-L and observed a significant inhibition of MDA-MB-231 cell adherence to HMVEC-L pretreated with (S)-BEL prior to CSE exposure (Fig. 6). Additionally, pretreatment of MDA-MB-231 breast tumor cells with 10 μM ginkgolide B, a PAF-R antagonist, for 10 min resulted in almost complete inhibition of tumor cell adherence to CSE-exposed human HMVEC-L (Fig. 6).
Fig. 6.
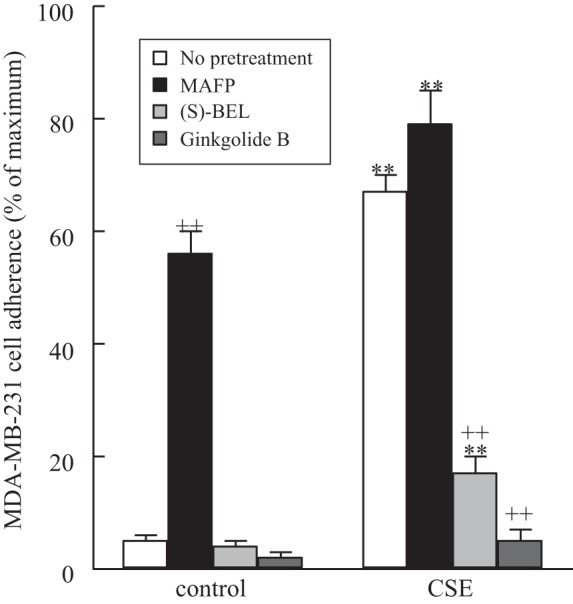
MDA-MB-231 cell adherence to HMVEC-L monolayers incubated with CSE (20 μg/ml, 18 h). MDA-MB-231 cells were labeled with calcein-AM and added to HMVEC-L for 30 min, and adherence was measured by calcein-AM fluorescence. HMVEC-L were pretreated with MAFP (5 μM, 30 min) or (S)-BEL (5 μM, 30 min) prior to CSE exposure. In select experiments, MDA-MB-231 cells were pretreated with the PAF-R antagonist ginkgolide B (10 μM, 30 min) prior to addition to the HMVEC-L monolayers. Values are means ± SE for 4 separate cell culture experiments. **P < 0.01 vs. corresponding treatment in the absence of CSE (control). ++P < 0.01 vs. no pretreatment.
DISCUSSION
In women, breast cancer is the second most commonly diagnosed cancer (after nonmelanoma skin cancer) and a leading cause of cancer mortality (26). The association between breast cancer and smoking has been controversial. Several cohort studies published in the 1990s and early 2000s stated that the association was “unclear” (1, 20, 28).
More recently, studies from 2005 to the present have supported a causal association between active smoking and elevated breast cancer risk (12), resulting in release of a statement by the Centers for Disease Control in 2014 that breast cancer patients who smoke are more likely than nonsmokers to die from the disease (1). The mechanism by which cigarette smoke can contribute to metastasis, especially to common sites such as the lung, remains unknown.
Cigarette smoking is associated with increased morbidity and mortality and remains a prevalent lifestyle choice, with >18% of all adults in the United States being classified as smokers (1). Despite declines during the past three decades, cigarette smoking among adults in the United States remains widespread, with 42.1 million smokers, and year-to-year decreases in prevalence have been seen only intermittently in recent years (1, 2). There are ∼7,000 chemicals in cigarettes, and ≥70 of them are carcinogenic, i.e., capable of damaging cellular DNA and initiating tumorigenesis (1). Components of cigarette smoke have been found in the DNA of epithelial cells of breast milk, as well as nipple aspirate, of female smokers, demonstrating that cigarette components access breast tissue (21, 29). Other studies have identified increased motility and epithelial-to-mesenchymal transition in breast tumor cells exposed to cigarette smoke components (8). Data to this point support plausible mechanisms by which cigarette smoke could lead to breast cancer development and enhance progression, yet a detailed mechanistic model remains undiscovered. However, in consideration of the fact that 90% of all cancer deaths are caused by metastasis, rather than by growth of the primary tumor, a greater emphasis should be placed on understanding the mechanism by which cigarette smoke can increase metastasis in the setting of cancer (6).
Metastatic disease is responsible for ∼90% of all cancer deaths and is difficult to manage. Despite the clinical importance of metastasis, it continues to be the least understood component of cancer pathogenesis (7). The steps of metastasis include primary tumor cell intravasation into the circulation, transit and arrest at distant sites, and subsequent attachment and extravasation into distant tissues (6). Metastatic cancers are often organ-specific in their spread, such as metastatic breast cancer, which commonly metastasizes to the lung (7). Many groups have characterized the role of adhesion molecules in metastasis, yet PAF has never been closely examined. PAF was first suggested to play a role in cigarette smoke exposure-induced injury when it was determined that PAF concentrations were higher in plasma from tobacco smokers than in plasma from nonsmokers (10). We propose that inhibition of PAF-AH activity in smokers leads to enhanced endothelial cell surface expression of PAF and increased tumor cell adherence and transmigration. This mechanism of cigarette smoke-enhanced tumor adherence may explain the increased risk for lung metastasis in breast cancer patients. As shown in the data presented here, cigarette smoke exposure to HMVEC-L led to inhibition of PAF-AH activity and subsequent increases in PAF accumulation and adherence of breast cancer cells (Figs. 1–3). Breast cancer cell adherence to CSE-exposed HMVEC-L was maximal in MDA-MB-231 cells that were exposed to CSE prior to addition to the endothelium. We propose that this is primarily due to increased expression of the PAF-R, as shown in Fig. 4. When invasive breast cancer cells are treated with the PAF-R antagonist ginkgolide B, their adherence to lung endothelial cells is markedly inhibited (Fig. 6). In addition, when lung endothelial cells are treated with the iPLA2β inhibitor (S)-BEL to prevent PAF accumulation, breast cancer cell adherence is decreased. Thus we suggest that inhibiting lung endothelial cell PLA2 activity to reduce PAF production or blocking the PAF-R on circulating breast cancer cells may be an effective treatment for metastatic disease.
Our data provide two new mechanisms by which tumor cell adherence to lung endothelium can be inhibited: blocking PAF accumulation in the endothelium or blocking PAF-R on the circulating tumor cell. These two mechanisms could potentially inhibit metastasis, which is responsible for the majority of cancer deaths. In accordance, the highly metastatic triple-negative cell line MDA-MB-231 exhibited much greater adherence to lung endothelial cells than the low invasive hormone-positive cell line MCF-7. When exposed to CSE, the MDA-MB-231 cell line shows a significant increase in PAF-R expression; however, nonmetastatic cell lines show only minimal increases. These novel data suggest that cigarette smoke-mediated mechanisms of metastasis may be particularly relevant in triple-negative metastatic breast cancers.
GRANTS
This work was supported in part by the Saint Louis University Presidents Research Fund Award (to J. McHowat).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.E.K., J.O.M., and J.M. are responsible for conception and design of the research; S.E.K., J.O.M., and J.M. performed the experiments; S.E.K., J.O.M., and J.M. analyzed data; S.E.K., J.O.M., and J.M. interpreted the results of the experiments; S.E.K., J.O.M., and J.M. prepared the figures; S.E.K., J.O.M., and J.M. drafted the manuscript; S.E.K., J.O.M., and J.M. edited and revised the manuscript; S.E.K., J.O.M., and J.M. approved the final version of the manuscript.
REFERENCES
- 1.Anonymous. The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General. Rockville, MD: Department of Health and Human Services, Public Health Service, Office of the Surgeon General, 2014. [Google Scholar]
- 2.Agaku IT, King BA, Dube SR. Current cigarette smoking among adults—United States, 2005–2012. MMWR Morb Mortal Wkly Rep 63: 29–34, 2014. [PMC free article] [PubMed] [Google Scholar]
- 3.Anand P, Kunnumakkara AB, Kunnumakara AB, Sundaram C, Harikumar KB, Tharakan ST, Lai OS, Sung B, Aggarwal BB. Cancer is a preventable disease that requires major lifestyle changes. Pharm Res 25: 2097–2116, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bussolati B, Biancone L, Cassoni P, Russo S, Rola-Pleszczynski M, Montrucchio G, Camussi G. PAF produced by human breast cancer cells promotes migration and proliferation of tumor cells and neo-angiogenesis. Am J Pathol 157: 1713–1725, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bussolino F, Arese M, Montrucchio G, Barra L, Primo L, Benelli R, Sanavio F, Aglietta M, Ghigo D, Rola-Pleszczynski MR. Platelet activating factor produced in vitro by Kaposi's sarcoma cells induces and sustains in vivo angiogenesis. J Clin Invest 96: 940–952, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science 331: 1559–1564, 2011. [DOI] [PubMed] [Google Scholar]
- 7.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2: 563–572, 2002. [DOI] [PubMed] [Google Scholar]
- 8.Dasgupta P, Rizwani W, Pillai S, Kinkade R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D, Haura E, Chellappan S. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer 124: 36–45, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Im SY, Ko HM, Kim JW, Lee HK, Ha TY, Lee HB, Oh SJ, Bai S, Chung KC, Lee YB, Kang HS, Chun SB. Augmentation of tumor metastasis by platelet-activating factor. Cancer Res 56: 2662–2665, 1996. [PubMed] [Google Scholar]
- 10.Imaizumi T. Intravascular release of a platelet-activating factor-like lipid (PAF-LL) induced by cigarette smoking. Lipids 26: 1269–1273, 1991. [DOI] [PubMed] [Google Scholar]
- 11.Jha P, Peto R. Global effects of smoking, of quitting, and of taxing tobacco. N Engl J Med 370: 60–68, 2014. [DOI] [PubMed] [Google Scholar]
- 12.Johnson KC, Miller AB, Collishaw NE, Palmer JR, Hammond SK, Salmon AG, Cantor KP, Miller MD, Boyd NF, Millar J, Turcotte F. Active smoking and secondhand smoke increase breast cancer risk: the report of the Canadian Expert Panel on Tobacco Smoke and Breast Cancer Risk (2009). Tob Control 20: e2, 2011. [DOI] [PubMed] [Google Scholar]
- 13.Kell PJ, Creer MH, Crown KN, Wirsig K, McHowat J. Inhibition of platelet-activating factor (PAF) acetylhydrolase by methyl arachidonyl fluorophosphonate potentiates PAF synthesis in thrombin-stimulated human coronary artery endothelial cells. J Pharmacol Exp Ther 307: 1163–1170, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Lee T, Lenihan DJ, Malone B, Roddy LL, Wasserman SI. Increased biosynthesis of platelet-activating factor in activated human eosinophils. J Biol Chem 259: 5526–5530, 1984. [PubMed] [Google Scholar]
- 15.Levi R, Burke JA, Guo ZG, Hattori Y, Hoppens CM, McManus LM, Hanahan DJ, Pinckard RN. Acetyl glyceryl ether phosphorylcholine (AGEPC). A putative mediator of cardiac anaphylaxis in the guinea pig. Circ Res 54: 117–124, 1984. [DOI] [PubMed] [Google Scholar]
- 16.Luo J, Margolis KL, Wactawski-Wende J, Horn K, Messina C, Stefanick ML, Tindle HA, Tong E, Rohan TE. Association of active and passive smoking with risk of breast cancer among postmenopausal women: a prospective cohort study. BMJ 342: d1016, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montrucchio G, Alloatti G, Camussi G. Role of platelet-activating factor in cardiovascular pathophysiology. Physiol Rev 80: 1669–1699, 2000. [DOI] [PubMed] [Google Scholar]
- 18.Montrucchio G, Sapino A, Bussolati B, Ghisolfi G, Rizea-Savu S, Silvestro L, Lupia E, Camussi G. Potential angiogenic role of platelet-activating factor in human breast cancer. Am J Pathol 153: 1589–1596, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ninio E, Mencia-Huerta JM, Heymans F, Benveniste J. Biosynthesis of platelet-activating factor. I. Evidence for an acetyl-transferase activity in murine macrophages. Biochim Biophys Acta 710: 23–31, 1982. [DOI] [PubMed] [Google Scholar]
- 20.Palmer JR, Rosenberg L. Cigarette smoking and the risk of breast cancer. Epidemiol Rev 15: 145–156, 1993. [DOI] [PubMed] [Google Scholar]
- 21.Petrakis NL, Gruenke LD, Beelen TC, Castagnoli N, Craig JC. Nicotine in breast fluid of nonlactating women. Science 199: 303–305, 1978. [DOI] [PubMed] [Google Scholar]
- 22.Rastogi P, McHowat J. Inhibition of calcium-independent phospholipase A2 prevents inflammatory mediator production in pulmonary microvascular endothelium. Respir Physiol Neurobiol 165: 167–174, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma J, Turk J, McHowat J. Endothelial cell prostaglandin I2 and platelet-activating factor production are markedly attenuated in the calcium-independent phospholipase A2β knockout mouse. Biochemistry 49: 5473–5481, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma J, Young DM, Marentette JO, Rastogi P, Turk J, McHowat J. Lung endothelial cell platelet-activating factor production and inflammatory cell adherence are increased in response to cigarette smoke component exposure. Am J Physiol Lung Cell Mol Physiol 302: L47–L55, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shopland DR. Tobacco use and its contribution to early cancer mortality with a special emphasis on cigarette smoking. Environ Health Perspect 103 Suppl 8: 131–142, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 63: 11–30, 2013. [DOI] [PubMed] [Google Scholar]
- 27.Stein CJ, Colditz GA. Modifiable risk factors for cancer. Br J Cancer 90: 299–303, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Terry PD, Rohan TE. Cigarette smoking and the risk of breast cancer in women: a review of the literature. Cancer Epidemiol Biomarkers Prev 11: 953–971, 2002. [PubMed] [Google Scholar]
- 29.Thompson PA, DeMarini DM, Kadlubar FF, McClure GY, Brooks LR, Green BL, Fares MY, Stone A, Josephy PD, Ambrosone CB. Evidence for the presence of mutagenic arylamines in human breast milk and DNA adducts in exfoliated breast ductal epithelial cells. Environ Mol Mutagen 39: 134–142, 2002. [DOI] [PubMed] [Google Scholar]
- 30.Vinson SM, Rickard A, Ryerse JS, McHowat J. Neutrophil adherence to bladder microvascular endothelial cells following platelet-activating factor acetylhydrolase inhibition. J Pharmacol Exp Ther 314: 1241–1247, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Weigelt B, Peterse JL, van't Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer 5: 591–602, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Whatley RE, Zimmerman GA, McIntyre TM, Taylor R, Prescott SM. Production of platelet-activating factor by endothelial cells. Semin Thromb Hemost 13: 445–453, 1987. [DOI] [PubMed] [Google Scholar]