

Abstract
Insulin acts within the central nervous system to regulate food intake and sympathetic nerve activity (SNA). Strong evidence indicates that glucocorticoids impair insulin-mediated glucose uptake and food intake. However, few data are available regarding whether glucocorticoids also modulate the sympathoexcitatory response to insulin. Therefore, the present study first confirmed that chronic administration of glucocorticoids attenuated insulin-induced increases in SNA and then investigated whether these effects were attributed to deficits in central insulin-mediated responses. Male Sprague-Dawley rats were given access to water or a drinking solution of the glucocorticoid agonist dexamethasone (0.3 μg/ml) for 7 days. A hyperinsulinemic-euglycemic clamp significantly increased lumbar SNA in control rats. This response was significantly attenuated in rats given access to dexamethasone for 7, but not 1, days. Similarly, injection of insulin into the lateral ventricle or locally within the arcuate nucleus (ARC) significantly increased lumbar SNA in control rats but this response was absent in rats given access to dexamethasone. The lack of a sympathetic response to insulin cannot be attributed to a generalized depression of sympathetic function or inactivation of ARC neurons as electrical activation of sciatic afferents or ARC injection of gabazine, respectively, produced similar increases in SNA between control and dexamethasone-treated rats. Western blot analysis indicates insulin produced similar activation of Akt Ser473 and rpS6 Ser240/244 in the ventral hypothalamus of control and dexamethasone-treated rats. Collectively, these findings suggest that dexamethasone attenuates the sympathoexcitatory actions of insulin through a disruption of ARC neuronal function downstream of Akt or mammalian target of rapamycin (mTOR) signaling.
Keywords: sympathetic nerve activity, arcuate nucleus, dexamethasone, Akt, mTOR
the peptide hormone insulin is released from the pancreas and plays a pivotal role in the regulation of glucose homeostasis, body weight, and autonomic function. These effects depend, at least in part, on the ability of insulin to act within the central nervous system to inhibit food intake (Begg and Woods 2012; Obici et al. 2002), alter hepatic glucose production (Kalsbeek et al. 2010), elevate sympathetic nerve activity (SNA) (Anderson et al. 1991; Luckett et al. 2013; Morgan et al. 1993; Muntzel et al. 1994a; Ward et al. 2011), and alter baroreflex function (Pricher et al. 2008; Young et al. 2010). Accumulating evidence indicates that insulin activates the phosphatidylinositol 3-kinase (PI3K)-Akt-signaling pathway within neurons of the hypothalamic arcuate nucleus (ARC) to modulate the activity of several hypothalamic pathways. First, ARC neurons express a high abundance of insulin receptors (Marks et al. 1990; Werther et al. 1987), and insulin injection directly into the ARC increases lumbar SNA and alters baroreflex function (Cassaglia et al. 2011; Luckett et al. 2013). Second, ARC pretreatment with an anti-insulin affibody prevents or attenuates the sympathoexcitatory response to ARC or intracerebroventricular (icv) injection of insulin and a hyperinsulinemic-euglycemic clamp (Luckett et al. 2013). Third, acute icv injection of insulin increases PI3K activity and activates Akt in the ventromedial hypothalamus (Niswender et al. 2003; Rahmouni et al. 2004). In vitro patch-clamp recordings also suggest insulin alters the activity of ARC neurons through a PI3K-dependent mechanism (Hill et al. 2008; Qiu et al. 2014; Williams et al. 2010). Furthermore, inhibition of PI3K-Akt signaling prevents the insulin-induced lumbar sympathoexcitatory response (Rahmouni et al. 2004) and decrease in food intake (Niswender et al. 2003).
Chronic exposure to glucocorticoids leads to insulin resistance accompanied by metabolic dysregulation and impairment of the primary insulin-signaling pathways (i.e., IRS/PI3K-mTOR and MAPK) in several tissues including liver, adipose tissue, and muscle (Geer et al. 2014; Ruzzin et al. 2005; Shah et al. 2000). Within skeletal muscle, glucocorticoids contribute to muscle wasting at least in part via inhibition of signaling through PI3K/Akt to the mammalian target of rapamycin complex I (mTORC1) (Ruzzin et al. 2005; Shah et al. 2000). Interestingly, glucocorticoid receptors are also highly expressed within several hypothalamic regions including the ARC and paraventricular nucleus (PVN) (Aronsson et al. 1988; McEwen et al. 1986; Morimoto et al. 1996). The icv infusion of the glucocorticoid agonist dexamethasone increases food intake and body weight (Mori et al. 2009) and impairs glucose uptake in peripheral tissues (Norman et al. 2004). Moreover, local infusion of dexamethasone into ARC induces hepatic insulin resistance during a hyperinsulinemic-euglycemic clamp in rats (Yi et al. 2012). Shimizu et al. (2010) reported that glucocorticoids may also impair hypothalamic-signaling pathways as 24-h dexamethasone treatment decreased the phosphorylation of the mTORC1 substrate p70S6K1 and its downstream substrate ribosomal protein S6 (rpS6) in hypothalamic organotypic cultures. Collectively, these findings suggest glucocorticoids may interfere with the central actions of insulin through a disruption of PI3K-Akt and mTORC1 signaling.
Despite the ability of glucocorticoids to act centrally and affect glucose homeostasis and body weight, only one study (Scherrer et al. 1993) has investigated whether glucocorticoids directly modulate the sympathoexcitatory effects of insulin. These authors reported that 48-h treatment with dexamethasone eliminated the increase in muscle SNA to a hyperinsulinemic-euglycemic clamp in humans (Scherrer et al. 1993). Therefore, the initial purpose of this study was to confirm that glucocorticoid administration attenuated the sympathoexcitatory response to a hyperinsulinemic-euglycemic clamp in rodents. Although a previous report indicates that dexamethasone interferes with insulin-mediated transport across the blood-brain-barrier (Baura et al. 1996), subsequent experiments were designed to test the hypothesis that dexamethasone attenuates the centrally mediated insulin responses by icv injection of insulin or local injection of insulin into the ARC nucleus. A final set of experiments examined whether dexamethasone attenuated insulin-stimulated increases in Akt Ser473 and rpS6 Ser240/244 of the ventral hypothalamus since prior evidence suggests that the sympathoexcitatory response to insulin depends on PI3K-Akt signaling in the hypothalamus (Rahmouni et al. 2004) and observations in other tissues suggest that glucocorticoids interfere with insulin-mediated actions through a disruption in PI3K-Akt and mTORC1 signaling.
MATERIALS AND METHODS
Animals.
All of the experimental procedures conform to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the Pennsylvania State College of Medicine. Male Sprague-Dawley rats (initial body weight: 275–325 g; Charles River Laboratories) were singly housed in a temperature-controlled room (22–23°C) with a 12:12-h light-dark cycle. Rats were fed standard chow (Harlan Teklad 2018) and given access to deionized water or a dexamethasone solution (0.3 μg/ml) for either 1 or 7 days. The dexamethasone solution was replaced daily and diluted from a stock solution (2 mg/ml; Bimeda). Food and water intake were measured daily. All experiments were conducted in the ab libitum fed state.
Sympathetic nerve recordings.
Animals were anesthetized with isoflurane (2–3% in 100% O2) and instrumented with a femoral arterial catheter for arterial blood pressure (ABP) measurement, a double-lumen catheter in the femoral vein for intravenous infusions, and a brachial arterial catheter for blood sampling. The lumbar and renal sympathetic nerves were isolated through a ventral midline and a retroperitoneal incision as described previously (Bardgett et al. 2010; Simmonds et al. 2014; Ward et al. 2011). After a tracheotomy, animals were artificially ventilated. End-tidal CO2 was maintained between 4 and 4.5% by adjusting respiratory rate (60–80 breaths/min) or tidal volume (1 ml/100 g body wt). Body temperature was measured through a rectal probe and maintained at 37 ± 0.5°C by a water-circulating blanket. After completion of all surgical procedures, isoflurane anesthesia was replaced by α-chloralose (50 mg/kg body wt bolus followed by a continuous infusion of 25 mg·kg−1·h−1 iv). The level of anesthesia was assessed by the lack of withdrawal reflex to a foot pinch. Variables were stabilized for >60 min before experiments began. Background noise was assessed at the end of experiments after ganglionic blockade with hexamathonium (30 mg/kg iv) or physical crushing of the nerve.
Experiment 1–hyperinsulinemic-euglycemic clamp.
The first set of experiments was performed to confirm that dexamethasone attenuates the sympathoexcitatory response to insulin in rodents. Hyperinsulinemic-euglycemic clamps were performed as described previously (Bardgett et al. 2010; Ward et al. 2011) in rats ingesting water or dexamethasone for 1 or 7 days. Briefly, insulin (3.75 mU·kg−1·min−1) and a 50% dextrose solution (0.1–2.0 ml/h iv) were infused for 120 min. This dose of insulin has been reported previously in our laboratory to produce plasma insulin levels equivalent to those of rats fed a moderately high-fat diet for 13 wk and those of obese Zucker rats (Bardgett et al. 2010). In the current experiments, plasma insulin levels were measured from blood samples (0.2 ml) collected from the arterial catheter at −10, 60, and 120 min. Samples were centrifuged (10,000 g, 1 min) and analyzed by ELISA (Millipore, Billerica, MA). Blood glucose was measured from a drop of arterial blood every 10 min using a standard glucometer (One Touch Ultra). The dextrose infusion rate was experimentally adjusted to maintain euglycemia. To test whether dexamethasone attenuates sympathetic responses to stimuli other than insulin, the sciatic afferent nerve was isolated and stimulated electrically (1 ms, 500 μA, 5-s train) over a range of frequencies (1, 2, 5, 10, and 20 Hz) while animals were paralyzed with gallamine triethiodide (25 mg/kg iv).
Experiment 2–icv injection of insulin.
To establish that dexamethasone attenuates centrally mediated actions of insulin, SNA responses were measured in response to lateral ventricle injection of insulin. Initially, chronic brain cannulas targeted at the lateral ventricle were implanted in naïve rats as described previously (Bardgett et al. 2010; Stocker et al. 2003). Cannula placement was verified by a positive drinking response (>3 ml) to angiotensin II injection (20 ng/2 μl). Then, animals were given access to water or dexamethasone solution for 7 days and prepared for SNA and ABP recordings as described above. Insulin (50 mU/2 μl) was injected into the lateral ventricle, and responses were measured for an additional 120 min. Blood glucose was measured every 30 min. At the end of the experiment, cannula placement was verified again by the presence of dye in the third and fourth ventricles after injection of Evan's blue dye (1%, 2 μl) through the cannula.
Experiment 3–arcuate microinjection.
A third set of experiments was performed to determine whether the central actions of insulin were attenuated by dexamethasone in the ARC nucleus. Rats were given access to water or dexamethasone for 7 days and then prepared for ABP and SNA recordings as described above. Rats were placed into a stereotaxic head frame. A small craniotomy was performed to gain access to the cortex overlying the arcuate nucleus. After variables stabilized for 20 min, gabazine (1 mM, 20 nl) was injected unilaterally into the arcuate nucleus using single-barrel micropipettes (20 μm OD) as described previously (Luckett et al. 2013). Once variables returned to baseline values for 20 min, insulin (4 μU, 30 nl) was injected bilaterally using a glass pipette lowered into each arcuate nucleus. Variables were recorded for an additional 2 h. Drug solutions contained 0.2% rhodadmine of FITC-labeled microspheres. At the end of experiments, animals were perfused transcardially with 4% paraformaldehyde. Brains were harvested, postfixed in 4% paraformaldehyde, sectioned at 100 μm using a vibratome, and counterstained as described previously (Luckett et al. 2013). Data are reported for those animals that displayed labeled microspheres within the boundaries of the ARC as described previously (Luckett et al. 2013).
Experiment 4–Western blot analysis.
To assess the activation of the PI3K/Akt pathway in response to insulin following glucocorticoid treatment, rats were first instrumented with chronic brain cannulas targeted at the lateral ventricle. Placement was verified by a drinking response to angiotensin II as described above. Animals were then given access to water or dexamethasone solution for 7 days and acclimated to the ventricular injection procedure each day. On day 7, rats received an injection of artificial cerebrospinal fluid (aCSF; 2 μl) or insulin (50 mU/2 μl). At 30 min after injection, rats were decapitated and a block of the ventral hypothalamus was isolated underneath a microscope and immediately frozen in liquid nitrogen. The block of tissue was defined laterally by the fornix, dorsally by the fornix, rostrally by the optic chiasm, and caudally by mammillary bodies. Samples were stored at −80°C.
Hypothalamic samples (1.5–13 mg) were homogenized using a glass mortar and pestle in 7 vol of ice-cold buffer consisting of the following: 50 mM HEPES pH 7.8, 137 mM NaCl, 2 mM EDTA, 1 mM magnesium chloride, 1 mM sodium orthovanadate, 1% NP-40, and 1 cOmplete Mini EDTA-free Protease Inhibitor Cocktail Tablet (Roche Applied Science) per 10 ml of buffer. Protein concentration was quantified using Bio-Rad Protein Assay Dye reagent concentrate (Bio-Rad, Hercules, CA), and SDS-PAGE was carried out using equal amounts of total protein (10 μg) per sample. The membranes were incubated overnight at 4°C with primary antibody (1:1000; Cell Signaling, Beverly, MA) including AKT, p-AKT Ser473, ribosomal protein S6, ribosomal protein S6 Ser240/244, mTOR Ser2481, and mTOR. The FluorChem M Multifluor System (ProteinSimple, San Jose, CA) was used for visualization following exposure to ECL reagent (Thermo Scientific, Leesport, PA), and images were analyzed using AlphaView (ProteinSimple) and ImageJ software (NIH).
Statistical analysis.
All data are expressed as means ± SE. Changes in SNA were calculated by subtracting background noise after physical crushing the nerve. When applicable, data were averaged over 5-min bins and compared with multiple 5-min baseline periods (before infusion or injection). Sciatic nerve responses were compared using peak 1-s responses to 30-s baseline segment. All data were analyzed by one- or two-way ANOVA with repeated measures when appropriate. All post hoc tests were performed using independent or paired t-tests with a layered Bonferroni correction. P < 0.05 were considered statistically significant.
RESULTS
Table 1 summarizes characteristics of rats given access to water or dexamethasone solution for 7 days pooled from all experiments. Although there was no difference in baseline body weight between groups, there was a significant difference at 7 days as rats with access to dexamethasone did not increase body weight and ingested ∼20% less food and water than controls. There were no differences in baseline renal SNA or mean ABP. Lumbar SNA tended to be higher in dexamethasone-treated rats, but this effect was not statistically different (P = 0.06, two-tailed). Heart rate was significantly higher in rats given access to dexamethasone for 7 days vs. water.
Table 1.
Characteristics of rats given access to water or dexamethasone solution for 7 days
| Control Group | 7-Day Dexamethasone | |
|---|---|---|
| n | 34 | 36 |
| Initial body weight, g | 320 ± 7 | 301 ± 8 |
| Final body weight, g | 372 ± 12 | 298 ± 7* |
| Average daily food intake, g | 29 ± 1 | 23 ± 1* |
| Average daily fluid intake, ml | 35 ± 1 | 27 ± 1* |
| Mean ABP, mmHg | 116 ± 4 (18) | 123 ± 5 (21) |
| Heart rate, beats/min | 363 ± 9 (18) | 395 ± 13 (21)* |
| Lumbar SNA, μV | 1.51 ± 0.37 (18) | 3.31 ± 0.91 (21) |
| Renal SNA, μV | 5.76 ± 2.07 (18) | 4.66 ± 0.87 (21) |
Values are means ± SE. Numbers in parentheses represents the number of animals in the respective group for the cardiovascular measurements. ABP, arterial blood pressure; SNA, sympathetic nerve activity.
P < 0.05 vs. control group.
Chronic dexamethasone treatment attenuates sympathoexcitatory response to a hyperinsulinemic-euglycemic clamp.
To confirm a previous study in humans (Scherrer et al. 1993) that dexamethasone attenuates the sympathoexcitatory response to insulin, rats were given access to water or dexamethasone solution for 1 or 7 days and then infused with insulin iv under euglycemic conditions. As previously reported (Bardgett et al. 2010; Luckett et al. 2013; Ward et al. 2011), a hyperinsulinemic-euglycemic clamp significantly increased lumbar SNA but did not alter mean ABP in control rats (Fig. 1). In contrast, the lumbar sympathoexcitatory response was significantly attenuated in rats drinking the dexamethasone solution for 7 days but not after 1 day (Fig. 1). Despite significantly higher plasma insulin levels in rats drinking dexamethasone for 7 days, the glucose infusion rate to maintain euglycemia was significantly less in rats drinking dexamethasone for 7 days vs. control rats or rats drinking dexamethasone for 1 day. Heart rate did not change from baseline values (control: 355 ± 12 beats/min; 1 day: 375 ± 15 beats/min; 7 day: 365 ± 10 beats/min; P > 0.3 from overall ANOVA). In addition, a hyperinsulinemic-euglycemic clamp did not alter renal SNA in any group (data not shown).
Fig. 1.

Example of arterial blood pressure (ABP), mean ABP (grey line), integrated lumbar sympathetic nerve activity (SNA), and 1-s segments of raw lumbar SNA of rats drinking water (A) or dexamethasone (B; Dex) for 7 days and then infused with insulin plus dextrose. C: means ± SE of mean ABP, lumbar SNA, blood glucose, glucose infusion rate, and plasma insulin concentrations. *P < 0.05, control vs. 7-day dexamethasone. #P < 0.05, 1-day vs. 7-day dexamethasone.
To test whether dexamethasone treatment produced a nonspecific attenuation of sympathetic reflexes, the sciatic nerve afferents were stimulated electrically across a range of frequencies. As illustrated in Fig. 2, electrical activation of sciatic nerve afferents produced frequency-dependent increases in mean ABP, lumbar SNA, and renal SNA within both groups. However, the responses were not statistically different between rats drinking water vs. dexamethasone.
Fig. 2.
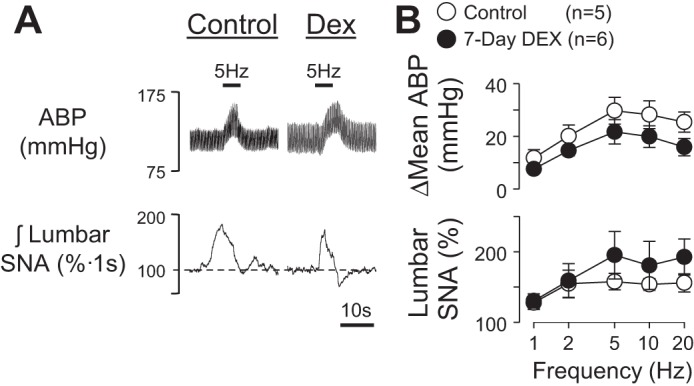
A: example of ABP and integrated lumbar SNA of rats drinking water or dexamethasone for 7 days in response to electrical stimulation of the sciatic nerve. B: means ± SE of Δmean ABP and lumbar SNA. There were no statistically significant differences between control vs. dexamethasone-treated rats.
Dexamethasone treatment attenuates sympathoexcitatory response to icv injection of insulin.
To directly test whether dexamethasone attenuated the central sympathoexcitatory action of insulin independent of blood-brain-barrier transport, insulin was injected into the lateral ventricle of rats drinking either water or dexamethasone for 7 days. As previously reported (Luckett et al. 2013; Muntzel et al. 1994b; Ward et al. 2011), icv injection of insulin in control rats significantly increased lumbar SNA without a change in mean ABP (Fig. 3). In marked contrast, the sympathoexcitatory response to icv insulin in dexamethasone-treated rats was significantly less at 90 and 120 min (Fig. 3C). The icv insulin did not alter blood glucose (Fig. 3) or heart rate (control: 410 ± 14 beats/min vs. 7-day dexamethasone: 417 ± 16 beats/min).
Fig. 3.

Example of ABP, mean ABP (grey line), integrated lumbar SNA, and 1-s segments of raw lumbar SNA of rats drinking (A) water or dexamethasone (B) for 7 days and infused icv with insulin (50 mU, 2 μl). C: means ± SE of mean ABP, lumbar SNA, and blood glucose. *P < 0.05, control vs. dexamethasone.
Dexamethasone treatment attenuates sympathoexcitatory response to arcuate nucleus injection of insulin but not gabazine.
As previous studies indicate that insulin acts on ARC neurons to increase SNA (Cassaglia et al. 2011; Luckett et al. 2013), a third series of experiments were performed to directly assess the extent by which dexamethasone attenuates the actions of insulin in the ARC. As expected (Cassaglia et al. 2011; Luckett et al. 2013), bilateral injection of insulin into the ARC of control rats significantly increased lumbar SNA but did not alter mean ABP (Fig. 4A) or renal SNA (data not shown). In marked contrast, lumbar SNA did not change after ARC injection of insulin in rats drinking dexamethasone for 7 days (Fig. 4A). Blood glucose (control: 71 ± 3 mg/dl; 7-day dexamethasone: 75 ± 2 mg/dl) and heart rate (control: 377 ± 15 beats/min; 7-day dexamethasone: 385 ± 13 beats/min) did not change after ARC injection of insulin in either group.
Fig. 4.

Data are means ± SE of mean ABP and integrated lumbar SNA during bilateral injection of insulin (4 μU, 30 nl; A) or unilateral injection of the GABAA receptor antagonist gabazine (1 mM, 20 nl; B) into the arcuate nucleus of rats drinking water or dexamethasone for 7 days. C: schematic illustration of arcuate injection sites. DMH, dorsomedial hypothalamus; VMH, ventromedial hypothalamus; ARC, arcuate nucleus.
To test whether dexamethasone treatment produced a general inactivation of ARC neurons, the same animals received a unilateral injection of gabazine. As reported previously (Kawabe et al. 2012; Luckett et al. 2013), injection of gabazine into the ARC significantly increased lumbar SNA and mean ABP (Fig. 4B). The magnitude of these responses was not different between rats drinking water or dexamethasone for 7 days. The location of injection sites was also not different between the groups (Fig. 4C).
Dexamethasone treatment did not interfere with central insulin signaling.
To investigate whether dexamethasone interferes with central insulin signaling, a final set of experiments analyzed the expression of several proteins in the ventral hypothalamus after the central injection of insulin. As expected, icv insulin in control rats significantly increased the expression of Akt Ser473 (Fig. 5A) and rpS6 Ser240/244 (Fig. 5B). However, icv insulin also increased the expression of Akt Ser473 (Fig. 5A) and rpS6 Ser240/244 in dexamethasone-treated rats (Fig. 5, A and B), and the levels of Akt Ser473 and rpS6 Ser240/244 after insulin treatment did not differ between control and dexamethasone-treated rats. Autophosphorylation of mTOR on residue Ser2481 was not different between treatment groups before normalization. However, levels of total mTOR protein were increased in all groups compared with control animals injected with aCSF. Therefore, when mTOR Ser2481 is expressed relative to total mTOR, significant decreases in this autophosphorylation site were detected in both control animals injected with insulin and dexamethasone-treated animals injected with aCSF (Fig. 5C).
Fig. 5.

Effects of dexamethasone and/or insulin on Akt and mammalian target of rapamycin complex I (mTORC1)-related signaling in the ventromedial hypothalamus of rats. Protein expression of Akt Ser473 (A), ribosomal protein S6 Ser240/244 (B), and mTORSer2481 (C) was quantified following treatment with either vehicle [artificial cerebrospinal fluid (aCSF); n = 8], dexamethasone (n = 7), insulin (n = 8), or the combination of dexamethasone and insulin (n = 8). Vertical lines separating samples from each condition have been drawn denoting where intervening lanes have been removed; however, samples for all 4 groups were run on same gel. All data are expressed relative to the vehicle treatment condition. Values are means ± SE. *P < 0.05 vs. aCSF; ^P < 0.05 vs. Dex.
DISCUSSION
Despite evidence that glucocorticoids act centrally to modulate glucose homeostasis and food intake (Mori et al. 2009; Norman et al. 2004; Shimizu et al. 2010; Yi et al. 2012), only one prior study directly assessed whether glucocorticoids altered the sympathetic response to insulin (Scherrer et al. 1993). The initial set of experiments in this study confirmed this observation in rodents as 7, but not 1, days of dexamethasone administration attenuated the increase in lumbar SNA during a hyperinsulinemic-euglycemic clamp. Subsequent experiments were then designed to determine whether glucocorticoids act centrally to disrupt insulin-mediated responses in SNA and PI3K-Akt/mTORC1 signaling. This series of experiments provide several novel observations including: 1) dexamethasone attenuates the increase in lumbar SNA after icv injection of insulin, 2) ARC injection of insulin fails to increase lumbar SNA in dexamethasone-treated rats, 3) dexamethasone does not affect the sympathetic responses to electrical activation of sciatic afferents or ARC injection of gabazine, and 4) dexamethasone does not affect the insulin-induced activation of Akt or rpS6 in the ventral hypothalamus. Collectively, these findings suggest glucocorticoids act centrally within the ARC nucleus to attenuate the sympathoexcitatory actions of insulin through a disruption of ARC neuronal function that is independent of Akt or mTORC1 signaling.
A number of studies have documented that glucocorticoids impair insulin signaling and function (Ruzzin et al. 2005; Shah et al. 2000; Yi et al. 2012). To our knowledge, only one prior study examined whether the administration of glucocorticoids attenuates the sympathoexcitatory response to insulin (Scherrer et al. 1993). Scherrer et al. reported that 48-h administration of dexamethasone abolished the increase in muscle SNA and calf vasodilation during a hyperinsulinemic-euglycemic-clamp. In contrast, dexamethasone did not affect the sympathetic and pressor responses to a Valsalva maneuver or cold pressor test. The present findings confirm these observations as dexamethasone administration for 7 days significantly attenuated the lumbar sympathoexcitatory response to a hyperinsulinemic-euglycemic clamp. The smaller sympathetic response was independent of changes in plasma insulin as dexamethasone-treated rats had significantly higher plasma insulin levels at 60 and 120 min compared with control rats (although the change in plasma insulin concentration from baseline levels was not different between groups). Again, these findings cannot be attributed to a general depression of all sympathetic circuits or reflexes, as electrical activation of sciatic nerve afferents produced similar increases in SNA and ABP between control and dexamethasone-treated rats. Therefore, these findings confirm the earlier study of Scherrer et al. (1993) and indicate that chronic dexamethasone treatment attenuates the sympathoexcitatory actions of insulin in rodents.
Accumulating evidence indicates that glucocorticoids act centrally to impact glucose homeostasis and body weight (Mori et al. 2009; Norman et al. 2004; Shimizu et al. 2010; Yi et al. 2012). A major goal of the present study was to establish whether the smaller sympathetic response to a hyperinsulinemic-euglycemic clamp in dexamethasone-treated animals (or humans) was attributable to a disruption of centrally mediated insulin responses. In the present study, injection of insulin into the lateral ventricle increased lumbar SNA in control rats, but this effect was significantly attenuated in rats treated with dexamethasone. Additionally, direct injection of insulin into the ARC increased lumbar SNA in control but not dexamethasone-treated rats. In marked contrast, ARC injection of the GABAA receptor antagonist gabazine produced similar increases in SNA between control and dexamethasone-treated rats. Collectively, these findings suggest that dexamethasone induces deficits in centrally mediated actions of insulin within ARC neurons. These findings are consistent with previous reports that glucocorticoids attenuate centrally mediated effects of insulin on hepatic glucose production (Yi et al. 2012). It is also noteworthy that dexamethasone has been reported to decrease insulin transport from plasma to the cerebrospinal fluid (Baura et al. 1996), and the decreased transport of insulin may contribute to the attenuated sympathetic response during a hyperinsulinemic-euglycemic clamp. The present findings demonstrate that glucocorticoids have an additional central effect to interfere with insulin-mediated responses.
Consistent with previous observations in our laboratory (Bardgett et al. 2010; Luckett et al. 2013; Ward et al. 2011), but in contrast to those of others (Morgan and Rahmouni 2010; Rahmouni et al. 2003, 2004), we did not observe an increase in renal SNA during a hyperinsulinic-euglycemic clamp or after ARC injection of insulin. The reason for the discrepancy is not clear. However, data from humans indicate that a hyperinsulinemic-euglycemic clamp increased muscle SNA but failed to alter renal norepinephrine spillover (Gudbjornsdottir et al. 1994). While it remains unclear why insulin would directly alter lumbar but not renal SNA, these data highlight the ability of the central nervous system to differentially control SNA in response to various stimuli.
The actions of insulin are mediated by PI3K/Akt associated activation of mTORC1 signaling in both peripheral tissues as well as the central nervous system (Inoki et al. 2002; Rahmouni et al. 2004; Sancak et al. 2007). Since previous reports in skeletal muscle indicate that glucocorticoids impair these same insulin-signaling pathways (Ruzzin et al. 2005; Shah et al. 2000), we hypothesized that the smaller sympathetic response to insulin would be attributed to a blunted activation of the PI3K/Akt and mTORC1 pathways. Instead, we found that phosphorylation of Akt, a protein downstream of PI3K, and rpS6, a protein phosphorylated by one of the mTORC1 primary substrates, p70S6K1, were unchanged by dexamethasone treatment under both basal and insulin-stimulated conditions. While these data may suggest that the sympathetic response to insulin does not depend on PI3K/Akt and mTORC1 pathways, Rahmouni et al. (2004) have reported that centrally administered insulin increases PI3K/Akt in the ventral hypothalamus and icv pretreatment with LY294002 or wortmannin prevented the insulin-induced increase in lumbar SNA. Therefore, a more plausible explanation is that dexamethasone disrupts insulin signaling downstream of PI3K/Akt or mTORC1. Since tissue punches were collected from the ventral hypothalamus, it is possible that changes specifically within the ARC or within subsets of ARC neuronal populations [neuropeptide Y (NPY) vs. proopiomelanocortin] were missed.
The lack of basal changes following dexamethasone treatment contrasts ex vivo work in rat hypothalamic organotypic cultures in which incubation with dexamethasone for 24-h significantly decreased phosphorylation of both p70S6K1 and rpS6 (Shimizu et al. 2010). A possible explanation for these differences, aside from the obvious methodological variations, is the influence of the animals' nutritional status (i.e., fed) and/or time of tissue collection following injection of insulin on PI3K/Akt/mTORC1 signaling. Previous work has shown maximal binding (and assumed activity) of IRS-1 to PI3K between 15 and 30 min after icv insulin in rats (Rahmouni et al. 2004), while in a separate study maximal phosphorylation of the mTORC1 substrates p70S6K1 Thr389 and rpS6 Ser240/244 occurred at 60–120 min after icv injection of leucine (Harlan et al. 2013) showing that PI3K and mTORC1 signaling may not always follow temporally. Irrespective of these methodological differences, our current work provides the first evidence that insulin-stimulated Akt and mTORC1 signaling is maintained in ARC following dexamethasone treatment.
Unexpectedly, changes in the phosphorylation of mTOR did not mimic those of Akt and rpS6 with the ARC following insulin and dexamethasone treatment and as such the physiological significance of the increase in total mTOR protein levels remains to be determined. Despite evidence that the phosphorylation of Ser2481 is enhanced by mTORC1-activating signals (i.e., amino acids, cellular energy), autophosphorylation of mTOR is not a necessary event in the activation of S6K1 and 4E-BP1 and is in agreement with the current findings (Peterson et al. 2000; Soliman et al. 2010). Evidently, the effects of short-term dexamethasone treatment on insulin-signaling pathways are differentially regulated in peripheral and central tissues while insulin-induced-signaling pathways within the central nervous system appear to be somewhat resistant to prolonged exposure to dexamethasone in contrast to the effects observed on SNA.
An alternative explanation for the findings is that glucocorticoids may attenuate the sympathoexcitatory response to insulin through activation of NPY-containing neurons in the ARC. First, glucocorticoid administration upregulates the expression of NPY in the ARC (Akabayashi et al. 1994; Yi et al. 2012). Second, NPY-positive ARC neurons densely innervate PVN (Broberger et al. 1999), and injection of NPY into the PVN decreases SNA and ABP (Cassaglia et al. 2014). In agreement, sympathetic and cardiovascular responses evoked from the ARC are partially mediated by NPY receptor activation in the PVN (Kawabe et al. 2012). Moreover, a recent study reported that local infusion of dexamethasone into the ARC attenuated insulin-mediated suppression of glucose production (Yi et al. 2012) and this effect was prevented by icv antagonism of NPY receptors (Yi et al. 2012). This evidence suggests that glucocorticoids may attenuate the sympathoexcitatory actions of insulin through the upregulation of NPY signaling; however, this hypothesis has not been tested.
In summary, the present study demonstrates that glucocorticoids attenuate the central sympathoexcitatory actions of insulin. Although we originally hypothesized that an attenuation of the sympathetic response by glucocorticoids would be associated with a blunted activation of PI3K/Akt or mTORC1 signaling, central insulin administration was found to robustly activate this pathway. These findings indicate that glucocorticoids have profound and widespread effects on metabolic function in both peripheral and central tissues, but the underlying mechanisms for these effects may be tissue or cell specific. Future studies will be needed to understand the cellular mechanisms within ARC and perhaps NPY neurons mediated by glucocorticoids.
GRANTS
This research was supported by NIH Grants HL-090826 (to S. D. Stocker), HL-113270 (to S. D. Stocker), GM-38032 (to C. H. Lang), and F32-AA-023422 (to J. L. Steiner); an American Heart Association Established Investigator Grant (to S. D. Stocker); and a Great Rivers Predoctoral Fellowship (to L. Wolfgang).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.L.S., M.E.B., L.W., C.H.L., and S.D.S. conception and design of research; J.L.S., M.E.B., L.W., and S.D.S. performed experiments; J.L.S., M.E.B., L.W., C.H.L., and S.D.S. analyzed data; J.L.S., M.E.B., L.W., C.H.L., and S.D.S. interpreted results of experiments; J.L.S. and S.D.S. prepared figures; J.L.S. and S.D.S. drafted manuscript; J.L.S., C.H.L., and S.D.S. edited and revised manuscript; J.L.S., M.E.B., L.W., C.H.L., and S.D.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Sarah Simmonds and Jennifer Lay for technical assistance.
REFERENCES
- Akabayashi A, Watanabe Y, Wahlestedt C, McEwen BS, Paez X, Leibowitz SF. Hypothalamic neuropeptide Y, its gene expression and receptor activity: relation to circulating corticosterone in adrenalectomized rats. Brain Res 665: 201–212, 1994. [DOI] [PubMed] [Google Scholar]
- Anderson EA, Hoffman RP, Balon TW, Sinkey CA, Mark AL. Hyperinsulinemia produces both sympathetic neural activation and vasodilation in normal humans. J Clin Invest 87: 2246–2252, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronsson M, Fuxe K, Dong Y, Agnati LF, Okret S, Gustafsson JA. Localization of glucocorticoid receptor mRNA in the male rat brain by in situ hybridization. Proc Natl Acad Sci USA 85: 9331–9335, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardgett ME, McCarthy JJ, Stocker SD. Glutamatergic receptor activation in the rostral ventrolateral medulla mediates the sympathoexcitatory response to hyperinsulinemia. Hypertension 55: 284–290, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baura GD, Foster DM, Kaiyala K, Porte D, Jr, Kahn SE, Schwartz MW. Insulin transport from plasma into the central nervous system is inhibited by dexamethasone in dogs. Diabetes 45: 86–90, 1996. [DOI] [PubMed] [Google Scholar]
- Begg DP, Woods SC. The central insulin system and energy balance. Handb Exp Pharmacol 111–129, 2012. [DOI] [PubMed] [Google Scholar]
- Broberger C, Visser TJ, Kuhar MJ, Hokfelt T. Neuropeptide Y innervation and neuropeptide-Y-Y1-receptor-expressing neurons in the paraventricular hypothalamic nucleus of the mouse. Neuroendocrinology 70: 295–305, 1999. [DOI] [PubMed] [Google Scholar]
- Cassaglia PA, Hermes SM, Aicher SA, Brooks VL. Insulin acts in the arcuate nucleus to increase lumbar sympathetic nerve activity and baroreflex function in rats. J Physiol 589: 1643–1662, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassaglia PA, Shi Z, Li B, Reis WL, Clute-Reinig NM, Stern JE, Brooks VL. Neuropeptide Y acts in the paraventricular nucleus to suppress sympathetic nerve activity and its baroreflex regulation. J Physiol 592: 1655–1675, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer EB, Islam J, Buettner C. Mechanisms of glucocorticoid-induced insulin resistance: focus on adipose tissue function and lipid metabolism. Endocrinol Metab Clin North Am 43: 75–102, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjornsdottir S, Friberg P, Elam M, Attvall S, Lonnroth P, Wallin BG. The effect of metformin and insulin on sympathetic nerve activity, norepinephrine spillover and blood pressure in obese, insulin resistant, normoglycemic, hypertensive men. Blood Press 3: 394–403, 1994. [DOI] [PubMed] [Google Scholar]
- Harlan SM, Guo DF, Morgan DA, Fernandes-Santos C, Rahmouni K. Hypothalamic mTORC1 signaling controls sympathetic nerve activity and arterial pressure and mediates leptin effects. Cell Metab 17: 599–606, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, Cowley MA, Cantley LC, Lowell BB, Elmquist JK. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest 118: 1796–1805, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648–657, 2002. [DOI] [PubMed] [Google Scholar]
- Kalsbeek A, Bruinstroop E, Yi CX, Klieverik LP, La Fleur SE, Fliers E. Hypothalamic control of energy metabolism via the autonomic nervous system. Ann NY Acad Sci 1212: 114–129, 2010. [DOI] [PubMed] [Google Scholar]
- Kawabe T, Kawabe K, Sapru HN. Cardiovascular responses to chemical stimulation of the hypothalamic arcuate nucleus in the rat: role of the hypothalamic paraventricular nucleus. PLoS One 7: e45180, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckett BS, Frielle JL, Wolfgang L, Stocker SD. Arcuate nucleus injection of an anti-insulin affibody prevents the sympathetic response to insulin. Am J Physiol Heart Circ Physiol 304: H1538–H1546, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks JL, Porte D, Jr, Stahl WL, Baskin DG. Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology 127: 3234–3236, 1990. [DOI] [PubMed] [Google Scholar]
- McEwen BS, De Kloet ER, Rostene W. Adrenal steroid receptors and actions in the nervous system. Physiol Rev 66: 1121–1188, 1986. [DOI] [PubMed] [Google Scholar]
- Morgan DA, Balon TW, Ginsberg BH, Mark AL. Nonuniform regional sympathetic nerve responses to hyperinsulinemia in rats. Am J Physiol Regul Integr Comp Physiol 264: R423–R427, 1993. [DOI] [PubMed] [Google Scholar]
- Morgan DA, Rahmouni K. Differential effects of insulin on sympathetic nerve activity in agouti obese mice. J Hypertens 28: 1913–1919, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H, Inoki K, Munzberg H, Opland D, Faouzi M, Villanueva EC, Ikenoue T, Kwiatkowski D, MacDougald OA, Myers MG, Jr, Guan KL. Critical role for hypothalamic mTOR activity in energy balance. Cell Metab 9: 362–374, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto M, Morita N, Ozawa H, Yokoyama K, Kawata M. Distribution of glucocorticoid receptor immunoreactivity and mRNA in the rat brain: an immunohistochemical and in situ hybridization study. Neurosci Res 26: 235–269, 1996. [DOI] [PubMed] [Google Scholar]
- Muntzel M, Beltz T, Mark AL, Johnson AK. Anteroventral third ventricle lesions abolish lumbar sympathetic responses to insulin. Hypertension 23: 1059–1062, 1994a. [DOI] [PubMed] [Google Scholar]
- Muntzel MS, Morgan DA, Mark AL, Johnson AK. Intracerebroventricular insulin produces nonuniform regional increases in sympathetic nerve activity. Am J Physiol Regul Integr Comp Physiol 267: R1350–R1355, 1994b. [DOI] [PubMed] [Google Scholar]
- Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG, Jr, Seeley RJ, Schwartz MW. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes 52: 227–231, 2003. [DOI] [PubMed] [Google Scholar]
- Norman AW, Mizwicki MT, Norman DP. Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat Rev Drug Discov 3: 27–41, 2004. [DOI] [PubMed] [Google Scholar]
- Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci 5: 566–572, 2002. [DOI] [PubMed] [Google Scholar]
- Peterson RT, Beal PA, Comb MJ, Schreiber SL. FKBP12-rapamycin-associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J Biol Chem 275: 7416–7423, 2000. [DOI] [PubMed] [Google Scholar]
- Pricher MP, Freeman KL, Brooks VL. Insulin in the brain increases gain of baroreflex control of heart rate and lumbar sympathetic nerve activity. Hypertension 51: 514–520, 2008. [DOI] [PubMed] [Google Scholar]
- Qiu J, Zhang C, Borgquist A, Nestor CC, Smith AW, Bosch MA, Ku S, Wagner EJ, Ronnekleiv OK, Kelly MJ. Insulin excites anorexigenic proopiomelanocortin neurons via activation of canonical transient receptor potential channels. Cell Metab 19: 682–693, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmouni K, Haynes WG, Morgan DA, Mark AL. Role of melanocortin-4 receptors in mediating renal sympathoactivation to leptin and insulin. J Neurosci 23: 5998–6004, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmouni K, Morgan DA, Morgan GM, Liu X, Sigmund CD, Mark AL, Haynes WG. Hypothalamic PI3K and MAPK differentially mediate regional sympathetic activation to insulin. J Clin Invest 114: 652–658, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzin J, Wagman AS, Jensen J. Glucocorticoid-induced insulin resistance in skeletal muscles: defects in insulin signalling and the effects of a selective glycogen synthase kinase-3 inhibitor. Diabetologia 48: 2119–2130, 2005. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25: 903–915, 2007. [DOI] [PubMed] [Google Scholar]
- Scherrer U, Vollenweider P, Randin D, Jequier E, Nicod P, Tappy L. Suppression of insulin-induced sympathetic activation and vasodilation by dexamethasone in humans. Circulation 88: 388–394, 1993. [DOI] [PubMed] [Google Scholar]
- Shah OJ, Anthony JC, Kimball SR, Jefferson LS. Glucocorticoids oppose translational control by leucine in skeletal muscle. Am J Physiol Endocrinol Metab 279: E1185–E1190, 2000. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Arima H, Ozawa Y, Watanabe M, Banno R, Sugimura Y, Ozaki N, Nagasaki H, Oiso Y. Glucocorticoids increase NPY gene expression in the arcuate nucleus by inhibiting mTOR signaling in rat hypothalamic organotypic cultures. Peptides 31: 145–149, 2010. [DOI] [PubMed] [Google Scholar]
- Simmonds SS, Lay J, Stocker SD. Dietary salt intake exaggerates sympathetic reflexes and increases blood pressure variability in normotensive rats. Hypertension 64: 583–589, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman GA, Acosta-Jaquez HA, Dunlop EA, Ekim B, Maj NE, Tee AR, Fingar DC. mTOR Ser-2481 autophosphorylation monitors mTORC-specific catalytic activity and clarifies rapamycin mechanism of action. J Biol Chem 285: 7866–7879, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker SD, Smith CA, Kimbrough CM, Stricker EM, Sved AF. Elevated dietary salt suppresses renin secretion but not thirst evoked by arterial hypotension in rats. Am J Physiol Regul Integr Comp Physiol 284: R1521–R1528, 2003. [DOI] [PubMed] [Google Scholar]
- Ward KR, Bardgett JF, Wolfgang L, Stocker SD. Sympathetic response to insulin is mediated by melanocortin 3/4 receptors in the hypothalamic paraventricular nucleus. Hypertension 57: 435–441, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werther GA, Hogg A, Oldfield BJ, McKinley MJ, Figdor R, Allen AM, Mendelsohn FA. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology 121: 1562–1570, 1987. [DOI] [PubMed] [Google Scholar]
- Williams KW, Margatho LO, Lee CE, Choi M, Lee S, Scott MM, Elias CF, Elmquist JK. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci 30: 2472–2479, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi CX, Foppen E, Abplanalp W, Gao Y, Alkemade A, la Fleur SE, Serlie MJ, Fliers E, Buijs RM, Tschop MH, Kalsbeek A. Glucocorticoid signaling in the arcuate nucleus modulates hepatic insulin sensitivity. Diabetes 61: 339–345, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CN, Deo SH, Chaudhary K, Thyfault JP, Fadel PJ. Insulin enhances the gain of arterial baroreflex control of muscle sympathetic nerve activity in humans. J Physiol 588: 3593–3603, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]