

Abstract
Local Ca2+ signals (Ca2+ sparks) play an important role in multiple cellular functions in airway smooth muscle cells (ASMCs). Protein kinase Cϵ is known to downregulate ASMC Ca2+ sparks and contraction; however, no complementary phosphatase has been shown to produce opposite effects. Here, we for the first time report that treatment with a specific calcineurin (CaN) autoinhibitory peptide (CAIP) to block CaN activity decreases, whereas application of nickel to activate CaN increases, Ca2+ sparks in both the presence and absence of extracellular Ca2+. Treatment with xestospogin-C to eliminate functional inositol 1,4,5-trisphosphate receptors does not prevent CAIP from inhibiting local Ca2+ signaling. However, high ryanodine treatment almost completely blocks spark formation and prevents the nickel-mediated increase in sparks. Unlike CAIP, the protein phosphatase 2A inhibitor endothall has no effect. Local Ca2+ signaling is lower in CaN catalytic subunit Aα gene knockout (CaN-Aα−/−) mouse ASMCs. The effects of CAIP and nickel are completely lost in CaN-Aα−/− ASMCs. Neither CAIP nor nickel produces an effect on Ca2+ sparks in type 1 ryanodine receptor heterozygous knockout (RyR1−/+) mouse ASMCs. However, their effects are not altered in RyR2−/+ or RyR3−/− mouse ASMCs. CaN inhibition decreases methacholine-induced contraction in isolated RyR1+/+ but not RyR1−/+ mouse tracheal rings. Supportively, muscarinic contractile responses are also reduced in CaN-Aα−/+ mouse tracheal rings. Taken together, these results provide novel evidence that CaN regulates ASMC Ca2+ sparks specifically through RyR1, which plays an important role in the control of Ca2+ signaling and contraction in ASMCs.
Keywords: local calcium signaling, contraction
asthma attacks, due to hypercontractile responses to nonspecific stimuli of airway smooth muscle cells (ASMCs) encircling the respiratory tree, pose a life-threatening risk to >300 million people worldwide on a daily basis. The pathological airway hyperresponsiveness (AHR) has long been thought to be associated with dysfunctional Ca2+-signaling pathways involved in the regulation of ASMC contraction.
Ca2+ signaling may occur in the form of local Ca2+ release events such as a Ca2+ spark, which is due to the synchronized opening of a cluster of ryanodine receptors (RyRs), the main intracellular Ca2+ release channels on the sarcoplasmic reticulum (SR) (6, 7). Ca2+ sparks have been observed in equine (44), guinea pig (51), porcine (32), mouse (26), and human ASMCs (10). These local Ca2+ events are known to regulate ASMC membrane potential through the activation of hyperpolarizing spontaneous transient outward currents (STOCs) or depolarizing spontaneous transient inward currents (STICs) (49). Ca2+ sparks can cause STOCs by opening big-conductance Ca2+-activated K+ (BK) channels that are located nearby RyRs in ASMCs, leading to K+ efflux, membrane hyperpolarization, and a decrease in the sensitivity of ASMCs to excitation (20, 22). Oppositely, Ca2+ sparks activate STICs due to a close association of RyRs with highly dense clusters of Ca2+-activated Cl− (CaCl) channels (1) that have now been identified as the transmembrane protein 16A CaCl channel (5, 36, 46). Of great clinical importance in determining the cause of asthmatic AHR, Ca2+ sparks have recently been shown to exist in human ASMCs (10) and to be upregulated in freshly isolated ASMCs from a sensitized murine model of asthma (41). Thus, Ca2+ sparks may play an important role in physiological and pathological functions in ASMCs.
We have reported that a direct activation of native M3 muscarinic receptors following exposure of the receptor agonist methacholine (MCh) or membrane depolarization results in activation of a Gq protein-coupled receptor-mediated signaling cascade, initiating the phospholipase C (PLC)-dependent breakdown of phosphatidylinositol 4,5-bisphosphate into diacylgycerol (DAG) and inositol 1,4,5-trisphosphate (IP3); IP3 activates IP3 receptors (IP3Rs), inducing intracellular Ca2+ release; this activates nearby RyR2 via a local Ca2+-induced Ca2+-release (CICR) mechanism, amplifying agonist-evoked, IP3R-mediated Ca2+ release and contraction in ASMCs (25). As a result of the MCh exposure or membrane depolarization, PLC also produces DAG, which activates protein kinase C (PKC). It has been shown that PKC downregulates Ca2+ sparks in vascular smooth muscle cells (SMCs) (3). Our study has shown that PKCϵ inhibits Ca2+ sparks specifically through RyR1 and MCh-induced contraction in ASMCs (24). Because a kinase is generally believed to regulate an ion channel in coordination with one or more complementary phosphatases, a protein phosphatase may promote RyR1-mediated local Ca2+ signals and contraction in ASMC. The serine/threonine protein phosphatase 2B (calcineurin, CaN) has been shown to regulate Ca2+ release in a C2C12 cell line derived from mouse skeletal muscle, indicating its role in the control of RyR1 activity (37). It is believed that CaN has a regulatory effect and potential physical interaction with intracellular Ca2+ release channels (4). CaN has also been shown to oppositely modulate PKC substrates like the transient receptor potential vanilloid-1 (TRPV1) channels in chorda tympani taste neurons (28), the myristoylated alanine-rich C kinase substrate (MARCKS), and growth-associated protein43 (GAP43) in the rat hippocampus (47) and L-type Ca2+ channels in vascular SMCs (30). Taken together, in the current study, we performed a series of pharmacological and genetic studies to test a novel hypothesis that CaN is a suitable phosphatase of interest in the regulation of local Ca2+ signaling and associated contraction in ASMCs, and the role of CaN may also be specifically mediated by RyR1.
MATERIALS AND METHODS
Cell isolation.
Freshly isolated mouse ASMCs were prepared as previously reported (24–26). In brief, adult male Swiss Webster mice were killed by an intraperitoneal injection of pentobarbital sodium according to the protocol approved by the Animal Care and Use Committee of Albany Medical College. The trachea was removed and kept in ice-cold physiological saline solution (PSS) containing (in mM): 125 NaCl, 5 KCl, 1 MgSO4, 10 glucose, 10 HEPES, and 1.8 CaCl2 (pH 7.4). The tissue was then cleaned of epithelium, cartilage, and connective tissue and then digested using a two-step enzymatic method. First the trachealis muscle tissue strip was incubated in low (0.1 mM)-Ca2+ PSS (37°C) containing (in mg/ml): 1 papain, 0.2 dithioerythritol, and 1 bovine serum albumin (BSA) for ∼12 min and then in low-Ca2+ PSS containing (in mg/ml): 1 dithiothreitol, 1 collagenase H, 1 collagenase II, and 1 BSA for ∼27 min. The tissue was washed four times for 5 min with 5 ml of low-Ca2+ PSS containing 1 mg/ml BSA and then gently triturated to release single cells for daily use (∼6 h) in high-Ca2+ PSS.
Animals.
Swiss Webster male mice at an age of 6–8 wk were purchased from Taconic Farms (Rensselaer, NY). RyR1 heterozygous, RyR2 heterozygous, and RyR3 homozygous gene knockout mice (RyR1−/+, RyR2−/+, and RyR3−/−) of C57/BL6 background were originally provided by Dr. Takeshima at Kyoto University Graduate School of Pharmaceutical Sciences (Kyoto, Japan) and bred to produce RyR1−/+, RyR2−/+, and RyR3−/− mice and their corresponding wild-type (RyR1+/+, RyR2+/+, and RyR3+/+) animals, as described in our previous publications (24–26). C57/BL6-based CaN catalytic subunit Aα homozygous knockout (CaN Aα−/−) mice were offered by Dr. J. L. Gooch at Emory University School of Medicine (Atlanta, GA) and bred to generate CaN Aα−/−, CaN Aα−/+, and CaN Aα+/+ mice, as we reported previously (39). RyR1−/− mice die at or just before birth; RyR2−/− mice are embryonically lethal; and RyR1−/+, RyR2−/+, RyR3−/−, CaN-Aα−/−, as well as CaN-Aα−/+ mice show normal growth, viability, fertility, and behaviors and do not have noticeable mortality or morbidity. However, CaN Aα KO mice are rarely born and have only been provided by Dr. Zhang and Dr. Wu at the National Institute of Neurological Disorders and Stroke (NINDS, Bethesda, MD). Thus, male RyR1−/+, RyR2−/+, RyR3−/−, CaN Aα−/−, CaN Aα−/+, and their wild-type mice at an age of 6–10 wk were used in experiments.
Ca2+ imaging.
Spontaneous local Ca2+ signaling (Ca2+ sparks) was measured in freshly isolated ASMCs using an LSM510 laser-scanning confocal microscope (Carl Zeiss) (26). Cells were loaded with fluo 4-AM (2.5 μM) in 1.8 mM Ca2+ PSS at room temperature for 30 min and then perfused with PSS to rinse away excess fluo 4-AM. Images were taken using a line-scanning mode with a Zeiss ×40 oil immersion objective. Fluo 4 was excited at 488 nm, and emitted fluorescence was measured at 505 nm. The confocal pinhole was set at 1 airy unit to provide a spatial resolution of 0.9 μm in the x-y axis. Each line-scan image was taken every 1.9 ms. The original line-scan recording times before and after application of agents were set at 5 s to obtain a number of Ca2+ sparks and minimize laser toxicity.
Muscle contraction.
Muscle contraction in isolated tracheal rings was measured using the organ bath technique, as described previously (44), with isometric transducers (Harvard Apparatus, South Natick, MA) and a PowerLab/4SP recording system (AD Instruments, Colorado Springs, CO). Tracheas were quickly removed from mice and transferred into ice-cold 1.8 mM PSS. After the connective tissue and epithelia were removed, tracheal rings were mounted vertically in 2-ml organ bath chambers containing Krebs solution (in mM): 110 NaCl, 3.4 KCl, 2.4 CaCl2, 0.8 MgSO4, 25.8 NaHCO3, 1.2 KH2PO4, and 5.6 glucose (equilibrated with 20% O2, 5% CO2, and 75% N2, pH 7.4) at 37°C. The resting tension was set at 500 mg. During a 60-min equilibration period, rings were washed every 20 min and restretched to 500 mg. The muscarinic receptor agonist MCh was given to induce muscle contraction.
Reagents.
CaN autoinhibitory peptide (CAIP), xestospongin-C (Xes-C), and phorbol 12-myristate,13-acetate were purchased from Calbiochem. Fluo 4-AM was purchased from Molecular Probes. These chemicals were dissolved in water or dimethyl sulfoxide with a final concentration ≤0.1%. Nickel chloride, MCh, and other remaining reagents were purchased from Sigma. The Calcineurin Cellular Activity Assay Kit was purchased from Enzolifesciences (catalog no. BML-AK816–0001). Pharmacological agents were delivered on the cell through a glass pipette connected to a Picospritzer III pressure controller (Parker Instrumentation) or directly added by pipette to the organ tissue bath solution. All experiments were conducted at room temperature (∼22°C). Data were obtained from a minimum of three different animals.
Statistical analysis.
All of the data are presented as means ± SE. Statistical comparisons in the same cells before and after treatment with pharmacological agents were performed using the paired Student's t-test and normalized to control. Statistical comparisons between different mouse genotypes were performed using an unpaired Student's t-test. Statistical analysis of isolated tracheal rings before and after treatment was done using a paired Student's t-test. A statistical comparison between contractile responses in different mouse genotypes was performed using an unpaired Student's t-test. Differences with a P value <0.05 were considered statistically significant.
RESULTS
Specific inhibition of CaN decreases ASMC local Ca2+ signaling.
Specific inhibition of CaN was achieved by treatment with the synthetic CAIP (20 μM). Ca2+ sparks were first recorded in freshly isolated ASMCs before treatment as control. After that, cells were exposed to CAIP, which was delivered by a pressure injection via a micropippette connected to a Picospritzer III system, as described in our previous publication (43). Following exposure of CAIP for 8 min, Ca2+ sparks were recorded again. Due to the cell, animal, and day-to-day variation, the frequencies and amplitudes of Ca2+ sparks were normalized to those before treatment with CAIP (control) and analyzed using a paired Student's t-test to determine their statistically significant differences. As shown in Fig. 1A, CAIP treatment reduced local Ca2+ signaling, decreasing the spontaneous Ca2+ spark frequency by 58 ± 8% (n = 11, P < 0.05; Fig. 1B). However, the amplitude of Ca2+ sparks was not significantly changed. In control experiments, we found that a pressure injection of normal bath solution for 8 min had no effect on either Ca2+ spark frequency or amplitude in eight cells tested.
Fig. 1.
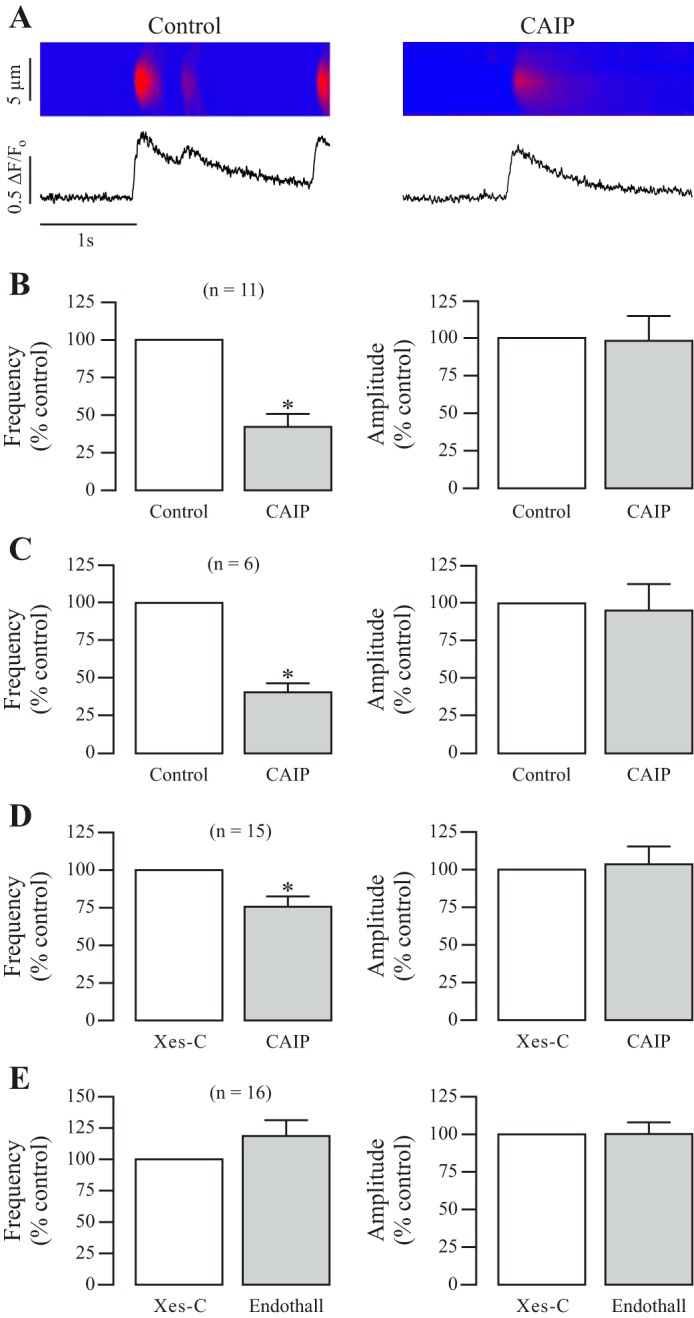
Specific inhibition of calcineurin (CaN) decreases local Ca2+ signaling in mouse airway smooth muscle cells (ASMCs). A: original recordings exhibit local Ca2+ signals (sparks) in a mouse ASMC before (control) and after treatment with a specific CaN autoinhibitory peptide (CAIP, 20 μM) for 8 min. Ca2+ sparks were recorded using a Zeiss LSM510 laser-scanning confocal microscope with a line-scanning mode. B: bar graphs summarize the effects of CAIP on the frequency and amplitude of Ca2+ sparks. Nos. in parentheses indicate the no. of individual cells tested from a minimum of three mice. *P < 0.05 compared with control (before application of CAIP). C: effect of CAIP treatment on Ca2+ sparks in ASMCs in the absence of extracellular Ca2+ (nominally free Ca2+ PSS supplemented with 0.5 mM EGTA). D: effects of CAIP on the frequency and amplitude of Ca2+ sparks in ASMCs pretreated with xestongin-C (Xes-C, 10 μM) for 8 min to functionally inhibit inositol 1,4,5-trisphosphate receptors (IP3Rs). E: effects of the protein phosphatase 2A endothall (1 μM) for 8 min on local Ca2+ signaling in cells pretreated with Xes-C (10 μM) for 8 min.
After seeing this, we wanted to verify that the effect of CAIP on local Ca2+ signaling was due to intracellular Ca2+ release, rather than extracellular Ca2+ influx. Thus, we repeated the initial experiments with cells bathed in nominally free Ca2+ PSS supplemented with 0.5 mM EGTA to scavenge extracellular Ca2+. Highlighted in Fig. 1C, we saw that CAIP decreased local Ca2+ signaling by over 50% in the absence of extracellular Ca2+ with no effect on amplitude, similar to those in the presence of extracellular Ca2+. These results suggest that CaN promotes Ca2+ sparks through intracellular Ca2+ release pathways.
We have previously shown that IP3Rs promote local Ca2+ release events in ASMCs through a local CICR mechanism (24–26), and so the action of a phosphatase, which increases Ca2+ sparks, could function through the IP3R-mediated CICR process. To exclude this possible complication, we tested the effects of CaN inhibition (via CAIP) on Ca2+ sparks in the absence of functional IP3Rs. In the current study, the functional elimination of IP3Rs was obtained by pretreating cells with Xes-C at 10 μM for 8 min, as we described previously (26). Ca2+ sparks were measured in cells pretreated with Xes-C as control; then, the cells were treated with CAIP (20 μM) for 8 min, and Ca2+ sparks were recorded. Figure 1D shows that CAIP still decreased local Ca2+ signals even in cells pretreated with Xes-C, signifying that CaN might upregulate local Ca2+ signaling by regulating RyRs in ASMCs.
To confirm the specific effect of CaN in general, we tested whether inhibition of protein phosphatase 2A (PP2A) might have a similar effect. In Fig. 1E, cells were pretreated with Xes-C to functionally inhibit IP3Rs, and then endothall was applied at 1 μM for 8 min to inhibit PP2A. Endothall had no effect on Ca2+ spark frequency or amplitude, suggesting a specific and novel role for the serine/threonine protein phosphatase CaN in promoting local Ca2+ signaling in ASMCs through its effect on RyRs.
Pharmacological activation of CaN increases local Ca2+ signaling in ASMCs.
Because CaN inhibition decreases local Ca2+ signaling, we next examined whether CaN activation might lead to an increase in local Ca2+ signals. The activation of CaN was achieved by treatment with nickel, which has been shown to activate purified CaN from bovine brain and bacteria (18, 33, 34). In contrast to CAIP, application of nickel at 500 μM for 5 min greatly increased the mean frequency of Ca2+ sparks by 110 ± 25% (n = 13; P < 0.05; Fig. 2A), whereas the mean amplitude of Ca2+ sparks was unaltered.
Fig. 2.
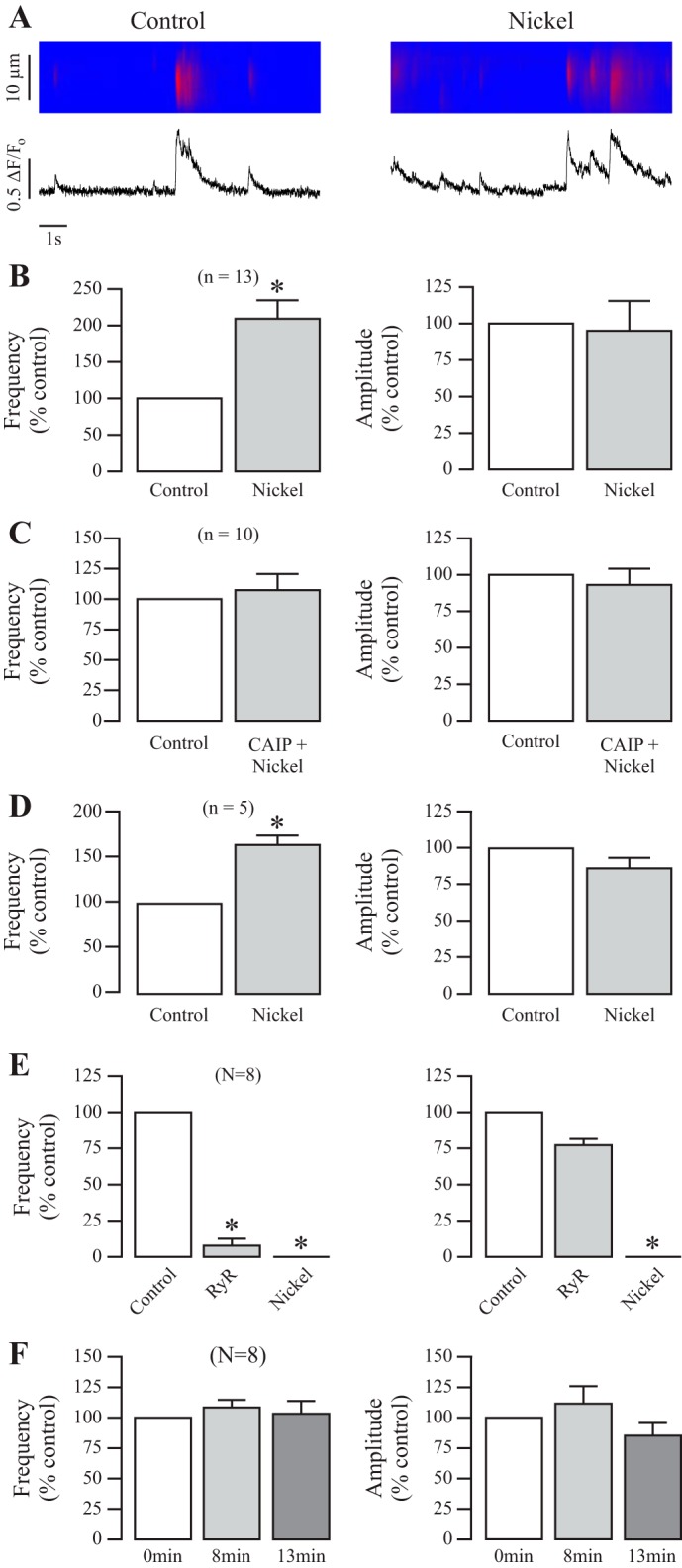
Pharmacological activation of CaN increases local Ca2+ signaling in ASMCs. A: original recordings exhibit local Ca2+ signals (sparks) in a mouse ASMC before (control) and after treatment with nickel (500 μM) for 5 min. B: bar graphs summarize the effects of nickel on the frequency and amplitude of Ca2+ sparks. Nos. in parentheses indicate the no. of individual cells tested from a minimum of three mice. *P < 0.05 compared with control (before application of nickel). C: effects of CAIP (20 μM) for 8 min followed by nickel (500 μM) for 5 min on the frequency and amplitude of ASMC Ca2+ sparks. D: effect of nickel treatment on Ca2+ sparks in ASMCs in the absence of extracellular Ca2+ (nominally free Ca2+ PSS supplemented with 0.5 mM EGTA). E: effect of ryanodine (100 μM) for 7 min and then nickel on the frequency and amplitude of ASMC Ca2+ sparks. F: effect of 1.8 mM Ca2+ PSS for 8 and 13 min on the frequency and amplitude of local Ca2+ signaling in mouse ASMCs.
Our subsequent experiment was to test whether nickel could increase sparks by interacting with CaN following treatment with CAIP. In these experiments, Ca2+ sparks were recorded in cells before and after the successive treatment of CAIP (20 μM) for 8 min followed by nickel (500 μM) for 5 min (13 min total between scans). Our results in Fig. 2C show no change in local Ca2+ signaling before and after the dual treatment, signifying that nickel was able to reverse the expected decrease in sparks following CAIP (seen in Fig. 1), restoring local Ca2+ signaling to control levels and further supporting the claim that nickel may increase Ca2+ sparks by activating CaN. We double checked the effect of nickel on Ca2+ sparks in the absence of extracellular Ca2+ and found that nickel was still able to significantly increase local Ca2+ signals even in the absence of extracellular Ca2+ (Fig. 2D).
Subsequently, we wanted to verify that RyR-mediated Ca2+ release is indeed responsible for Ca2+ sparks and that RyRs are required for the CaN-based regulation of local Ca2+ signaling. We treated spontaneously sparking mouse ASMCs with ryanodine (100 μM) for 7 min, followed by nickel (500 μM) for 5 min. As summarized in Fig. 2E, a high concentration of ryanodine treatment almost completely blocked all sparks in all cells, as was also seen in a previous report (26), and currently ryanodine treatment prevented nickel from generating any further Ca2+ release in the form of sparks. These results illustrate that RyRs are indeed responsible for Ca2+ spark formation and that CaN requires them to regulate sparks.
As a control experiment, to confirm that the time between scans for various treatments has no effect on sparking cells, we analyzed sparks before and after 8 and 13 min of 1.8 mM Ca2+ PSS. As summarized in Fig. 2F, there was no difference in spark frequency or amplitude throughout a 13-min timespan with saline treatment. Together, our studies provide clear evidence that CaN may upregulate local Ca2+ signaling in ASMCs.
Genetic inhibition of CaN decreases local Ca2+ signaling in ASMCs.
To further verify this role for CaN, we tested whether genetic inhibition of CaN changed spontaneous local Ca2+ signaling. We used CaN-Aα−/− mice because the CaN-Aα domain has been shown to play an important role in controlling the activity of CaCl channels and ATP-sensitive K+ channels in SMCs (15, 31). Freshly isolated ASMCs were prepared in parallel, from tracheal tissue obtained from CaN-Aα+/+ and CaN-Aα−/− mice provided by Dr. Zhang and Dr. Wu at the NINDS. Ca2+ sparks were measured in both cell types, and their frequency and amplitudes were compared using an unpaired Student's t-test. Figure 3A shows example recordings of spontaneous local Ca2+ signaling in a CaN-Aα+/+ mouse ASMC vs. CaN-Aα−/−. As summarized in Fig. 3B, the mean frequency of Ca2+ sparks was 0.0409 ± 0.00419 sparks·μm−1·s−1 in CaN-Aα−/− cells and 0.0552 ± 0.00755 sparks·μm−1·s−1 in CaN-Aα+/+, whereas the amplitude was similar in both cell types, providing evidence that CaN-Aα−/− impairs ASMC local Ca2+ signaling.
Fig. 3.
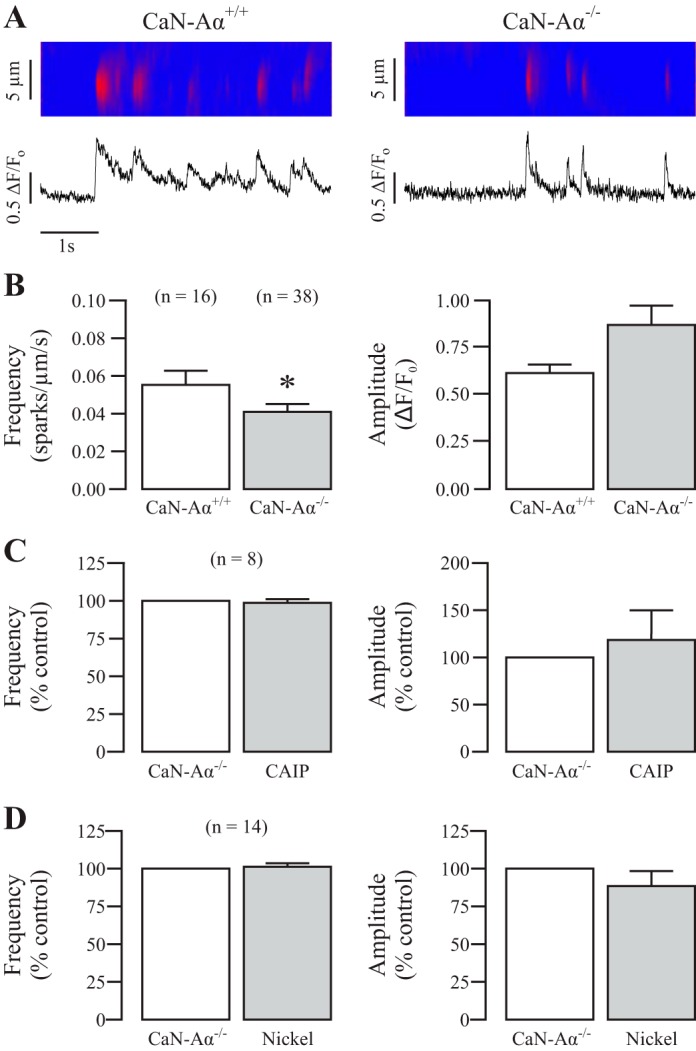
Genetic inhibition of CaN decreases spontaneous local Ca2+ and prevents the effects of pharmacological inhibition and activation. A: original recordings exhibit local Ca2+ signals (sparks) in a CaN-Aα wild-type (WT) mouse compared with a CaN-Aα−/− mouse ASMC. B: bar graphs present the mean frequency and amplitude of Ca2+ sparks from CaN-Aα WT and CaN-Aα−/− mouse ASMCs. Nos. in parentheses indicate the no. of individual cells tested from a minimum of three mice. *P < 0.05 comparing CaN-Aα WT and CaN-Aα−/−. C: effect of CAIP (20 μM) for 8 min on Ca2+ sparks in CaN-Aα−/− mouse ASMCs. D: effect of nickel on the frequency and amplitude of local Ca2+ signaling in CaN-Aα−/− mouse ASMCs.
With access to tissue from these knockout animals, we were able to verify the results of our pharmacological experiments using CAIP or nickel. As seen in Fig. 3C, treatment with the specific synthetic peptide CAIP, to inhibit CaN, did not affect Ca2+ sparks in CaN-Aα−/− ASMCs. Similarly, application of nickel to activate CaN did not produce an effect in CaN-Aα−/− cells either (Fig. 3D). Collectively, the effects of CAIP and nickel on local Ca2+ signaling in ASMCs result from their specific inhibition and activation of CaN, respectively, and local Ca2+ signaling is reduced in CaN-Aα−/− ASMCs.
RyR1 gene deletion blocks the role of CaN in the regulation of local Ca2+ signaling in ASMCs.
Because we have just outlined the role for the serine/threonine protein phosphatase CaN in regulating local Ca2+ signaling, we questioned if it works through RyR1. First, RyR1 heterozygous gene deletion (RyR1−/+) mice were used because RyR1 homozygous knockout mice (RyR1−/−) die before or at birth; second, local Ca2+ signaling is significantly different in embryonic vs. adult ASMCs; and third, the effects of PKCϵ are fully blocked in RyR1−/+ ASMCs (24, 25). Here, we found that treatment with CAIP was without effect on local Ca2+ signals in RyR1−/+ ASMCs (Fig. 4, A and B), whereas it significantly decreased spark frequency in RyR1+/+ cells (Fig. 4C).
Fig. 4.
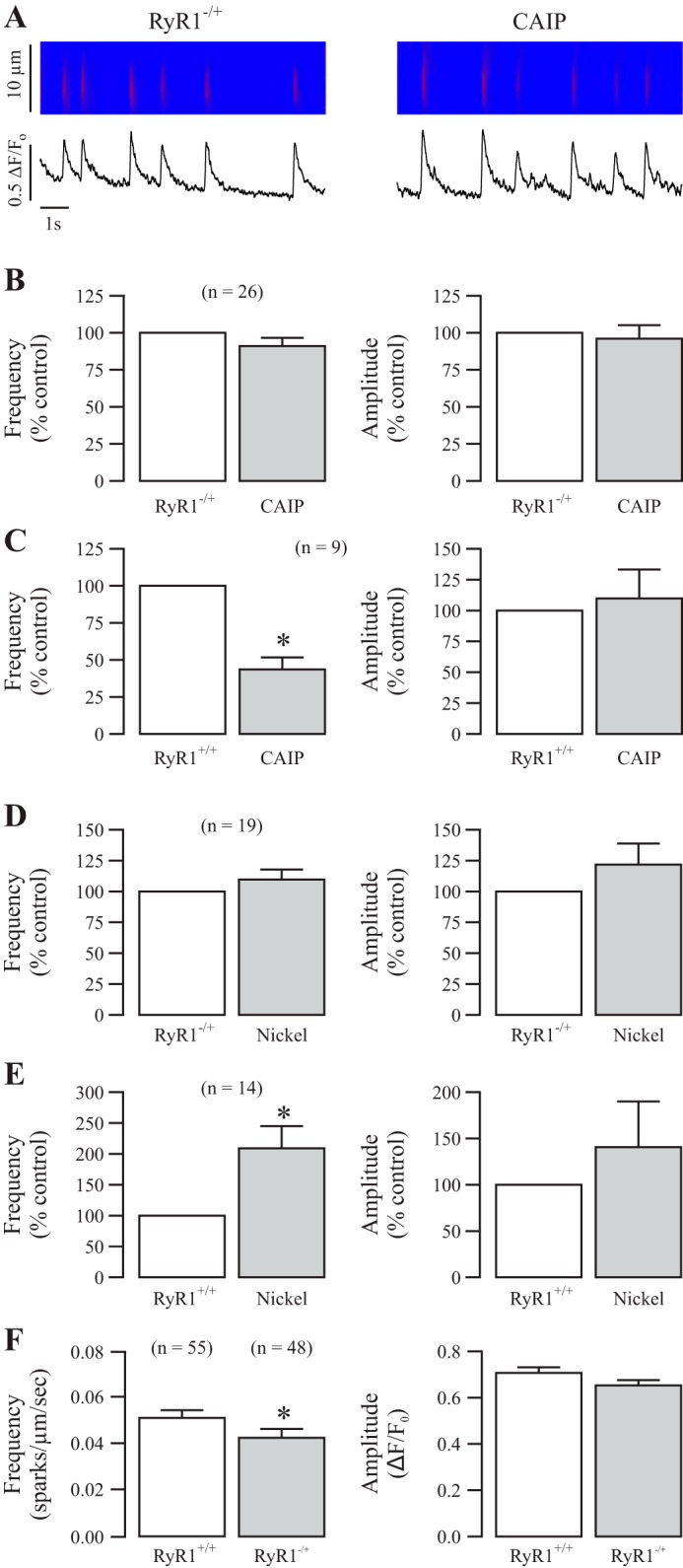
Ryanodine receptor-1 (RyR1) gene deletion blocks the role of CaN in the regulation of local Ca2+ signaling. A: original recordings exhibit local Ca2+ signals (sparks) in a RyR1−/+ mouse ASMC before (control) and after treatment with CAIP (20 μM) for 8 min. B: bar graphs summarize the effects of CAIP on the frequency and amplitude of Ca2+ sparks in RyR1−/+. Nos. in parentheses indicate the no. of individual cells tested from a minimum of three mice. *P < 0.05 compared with control (before application of CAIP). C: effect of CAIP on the frequency and amplitude of Ca2+ sparks in RyR1+/+. D: effect of nickel (500 μM) for 5 min on the frequency and amplitude of Ca2+ sparks in RyR1−/+. E: effect of nickel on the frequency and amplitude of Ca2+ sparks in RyR1+/+. F: bar graphs represent the mean frequency and amplitude of local Ca2+ signals from RyR1+/+ and RyR1−/+ mouse ASMCs.
After that, we tested the effect of CaN activation in RyR1−/+ ASMCs. Application of nickel to activate CaN did not alter the frequency and amplitude of Ca2+ sparks in RyR1−/+ cells (Fig. 4D), although it greatly increased Ca2+ spark frequency in RyR1+/+ cells (Fig. 4E), paralleling the results of Fig. 2, A and B.
To further examine the potentially important role of RyR1 in mediating CaN-based regulation of ASMC local Ca2+ signaling, we sought to determine if there is a difference in basal local Ca2+ signaling between RyR1+/+ and RyR1−/+ ASMCs. We prepared freshly isolated ASMCs from both RyR1+/+ and RyR1−/+ mice in parallel and imaged spontaneously sparking cells from both populations comparing their frequency and amplitude with an unpaired t-test. The results summarized in Fig. 4F reveal that local Ca2+ signaling is significantly higher in RyR1+/+ (0.05122 ± 0.00339 sparks·μm−1·s−1) vs. RyR1−/+ (0.04266 ± 0.00383) mice. Although the amplitudes are not significantly different, the P value for an unpaired t-test approached ∼0.06, suggesting that the amplitude of sparks might also be lower in RyR1−/+. Altogether, our results reveal that CaN upregulates local Ca2+ signals through its specific effect on RyR1 in ASMCs and that RyR1 plays a critical role in the regulation of local Ca2+ signaling.
CaN regulates local Ca2+ signals independent of RyR2 or RyR3.
Next, we examined whether RyR2 or RyR3 might also mediate the role of CaN in controlling the activity of local Ca2+ signaling. Different from the results seen in RyR1−/+ ASMCs (Fig. 4), RyR2 heterozygous gene deletion (RyR2−/+) did not block the effect of CaN inhibition or activation through the application of CAIP or nickel, respectively. Figure 5, A and B, reveals that CAIP resulted in a significant decrease in the activity of Ca2+ sparks in RyR2−/+, whereas Fig. 5C shows that nickel significantly increased local Ca2+ signals in RyR2−/+ mouse ASMCs.
Fig. 5.
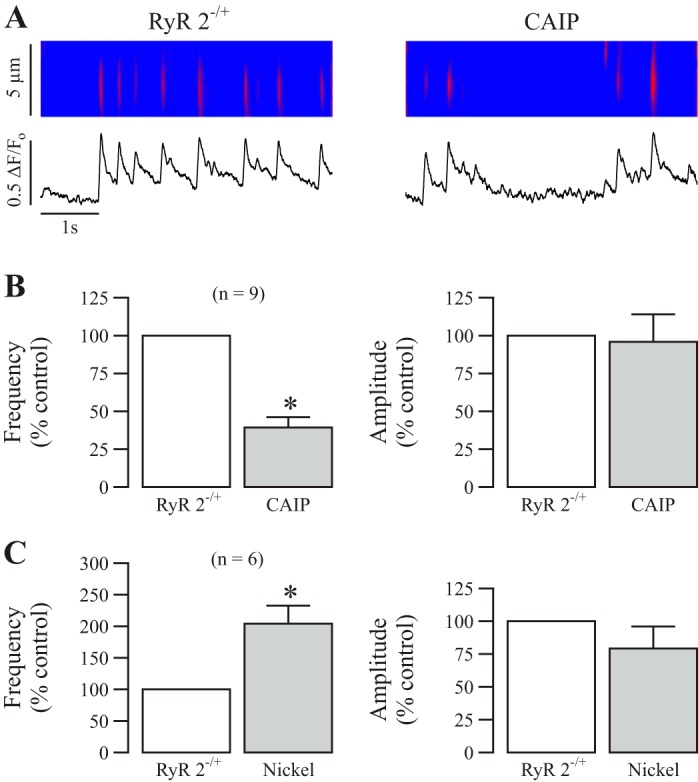
CaN regulates local Ca2+ signals independent of RyR type 2. A: original recordings exhibit local Ca2+ signals (sparks) in a RyR2−/+ mouse ASMC before (control) and after treatment with CAIP (20 μM) for 8 min. B: bar graphs summarize the effects of CAIP on the frequency and amplitude of Ca2+ sparks in RyR2−/+. Nos. in parentheses indicate the no. of individual cells tested from a minimum of three mice. *P < 0.05 compared with control (before CAIP). C: effects of nickel (500 μM) for 5 min on the frequency and amplitude of Ca2+ sparks in RyR2−/+.
Similar to RyR2−/+, RyR3−/− gene knockout was unable to prevent CaN inhibition and activation from decreasing and increasing the activity of Ca2+ sparks, respectively. In Fig. 6, A and B, application of CAIP significantly decreased spark frequency in RyR3−/− ASMCs (n = 12; P < 0.05), whereas in Fig. 6C nickel significantly increased spark frequency in RyR3−/− cells (n = 21; P < 0.05). These data reinforce the view that CaN upregulates local Ca2+ signaling in ASMCs through its specific effect on RyR1.
Fig. 6.

CaN regulates local Ca2+ signals independent of RyR type 3. A: original recordings exhibit local Ca2+ signals (sparks) in a RyR3−/− mouse ASMC before (control) and after treatment with CAIP (20 μM) for 8 min. B: bar graphs summarize the effect of CAIP on the frequency and amplitude of Ca2+ sparks in RyR3−/−. Nos. in parentheses indicate the no. of individual cells tested from a minimum of three mice. *P < 0.05 compared with control (before CAIP). C: effects of nickel (500 μM) for 5 min on the frequency and amplitude of Ca2+ sparks in RyR3−/−.
CaN inhibition regulates muscarinic contraction in airway muscle through RyR1.
Having seen the novel role for CaN in upregulating local Ca2+ signaling through RyR1, we hypothesized that inhibiting CaN might affect the physiological contractile response of airway muscle to the classical muscarinic receptor agonist MCh. In Fig. 7A we tested the contraction of isolated tracheal rings to MCh before and after the addition of saline (control) or CAIP (20 μM) for 45 min. Application of MCh (10 μM) resulted in repeated contractile responses with similar amplitudes in isolated tracheal rings. In contrast, tracheal rings pretreated with CAIP exhibited a decreased contractile response.
Fig. 7.
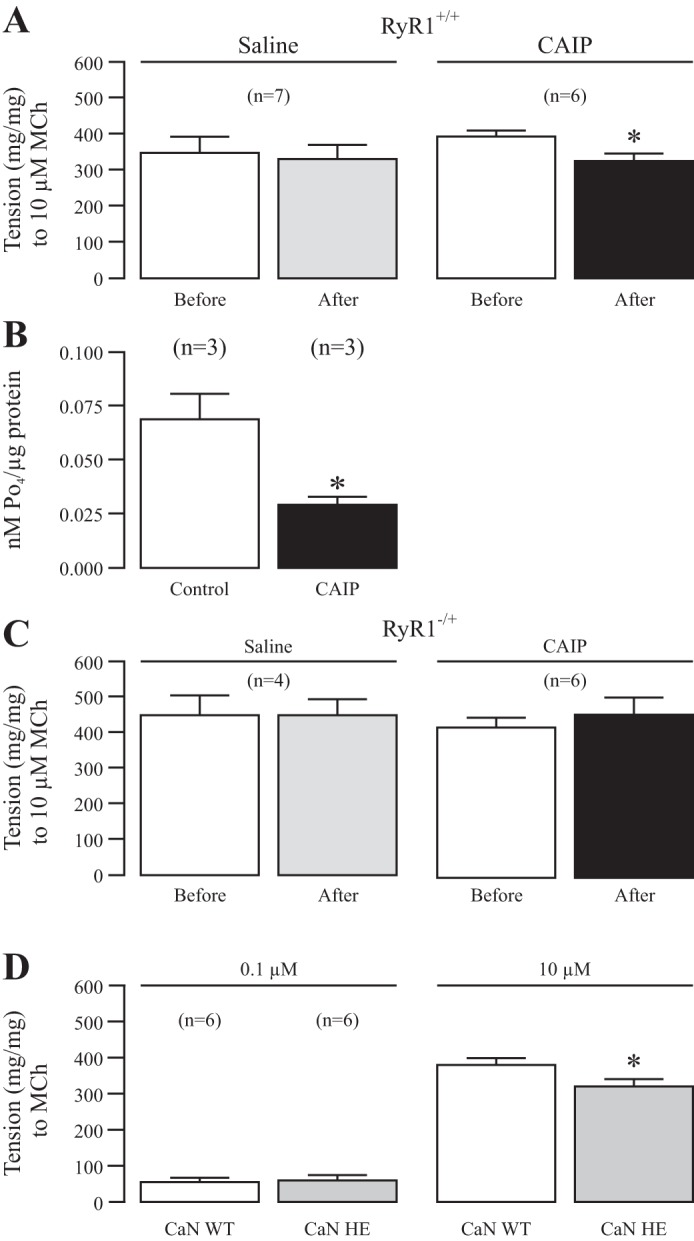
CaN inhibition regulates muscarinic contraction in airway muscle through RyR1. A: muscle contraction induced by the muscarinic receptor agonist methacholine at 10 μM was measured in isolated tracheal rings from RyR1+/+ mice before and after treatment with saline or CAIP (20 μM) for 45 min. The results are expressed as mg tension/mg of tissue. Nos. in parentheses indicate the no. of individual rings tested from a minimum of three mice. *P < 0.05 compared with before saline or CAIP. B: CAIP inhibits the activity of CaN in tracheal ring tissue. CaN activity was inhibited in tracheal ring tissues incubated with CAIP at 20 μM for 45 min vs. saline as measured through the CaN Cellular Activity Assay Kit (Enzolifesciences: catalog no. BML-AK816-0001). Nos. in parentheses denote the no. of times the kit was run with four mouse airway tissue strips being used each time. *P < 0.05 for CAIP vs. control. C: methacholine (10 μM)-induced contraction in isolated tracheal rings from RyR−/+ mice before and after saline or CAIP (20 μM) for 45 min. D: methacholine (0.1 and 10 μM) induced contraction in isolated tracheal rings from CaN WT and heterozygous gene deletion mice.
To verify the effect of CAIP in airway smooth muscle tissues, we incubated the isolated tracheal rings with CAIP at 20 μM or saline and then measured the activity of CaN in tracheal tissues with a Calcineurin Cellular Assay Kit (Calbiochem). The results are shown in Fig. 7B, in which CaN activity was significantly decreased in tracheal tissues after incubation of CAIP. Thus, CAIP can steadily enter ASMCs in tracheal tissues to produce an inhibitory effect on Ca2+ sparks and contraction.
Furthermore, we wanted to see if RyR1 heterozygous gene deletion could block the effects of CAIP on MCh-induced contraction. In Fig. 7C, we repeated the same experiments in Fig. 7A but with tracheal rings taken from RyR1−/+ mice. The results of our work show no change in contraction with saline or CAIP in RyR1−/+, supporting the hypothesis that CaN upregulates local Ca2+ signaling and contraction through RyR1 in airway smooth muscle.
Enhancing our understanding of the physiological contractile role of CaN, we assessed the effect of genetic inhibition of CaN on MCh-induced contraction in isolated tracheal rings. Previous studies have revealed that CaN-Aα−/+ mice have decreased protein expression and activity (12, 13); thus, we thought it suitable to compare MCh-evoked contractile responses in CaN-Aα+/+ and CaN-Aα−/+ mouse tracheal rings. As seen in Fig. 7D, CaN-Aα−/+ rings have a decreased contractile response to 10 μM MCh vs. CaN-Aα+/+ tissues, supporting our novel hypothesis that CaN regulates ASMC contraction through RyR1-based local Ca2+ signaling.
DISCUSSION
Ca2+ sparks, localized transient Ca2+ release events due to the coordinated opening of a cluster of RyRs in a functional Ca2+ release unit (CRU) on the SR, are known to regulate excitation-contraction coupling, membrane excitability, neurotransmitter release and secretion, cell proliferation and migration, and gene expression in a variety of cell types (23, 29). We and other investigators have shown the presence of Ca2+ sparks in equine, guinea pig, porcine, and mouse ASMCs (23), making them an active area of respiratory research. Within ASMCs, these localized Ca2+ release events directly mediate a basal tone of contraction, with cell relaxation and contraction following a decrease or increase in their activity, respectively. These local Ca2+ signals are also able to regulate ASMC membrane potential (23). RyRs on the SR can localize with other local cytosolic or membrane-bound proteins/channels, structurally forming membrane-membrane nanojunctions and functionally communicating using Ca2+ concentration as a currency (42). It has been generally accepted that RyRs may highly colocalize with ClCa and BK channels in ASMCs. By this unique micromachinery, RyR-mediated Ca2+ sparks readily activate these two channels to generate STICs and STOCs, respectively. STICs lead to membrane depolarization, LTCC activation, and extracellular Ca2+ influx; in contrast, STOCs result in membrane hyperpolarization and LTCC inhibition, thereby inhibiting extracellular Ca2+ influx (23, 29). However, at resting membrane potential, Ca2+ sparks preferentially activate STICs, whereas at more positive membrane potential STOCs are initiated (19). STICs have been shown to be involved in ASMC contraction (16) and asthmatic AHR (48). In human ASMCs, some believe the spark-STOC relationship underlies relaxation of ASMCs following activation of bitter taste receptors (10). Nevertheless, previous reports have shown that ASMC contraction is not significantly affected by LTCC blockers in isolated animal ASMCs and tissues, and clinical studies indicate that LTCC blockers are ineffective in the treatment of airway smooth muscle contraction and asthma (9, 14, 38). Our recent research demonstrates that, under physiological conditions, membrane depolarization causes a direct activation of Gq protein-coupled M3 muscarinic receptors, leading to intracellular Ca2+ release and contraction without the involvement of LTCCs in ASMCs (25), signifying the especially important role of intracellular Ca2+ release for ASMC contraction. Of great clinical importance in the context of the aforementioned information is the fact that local Ca2+ signals have been shown to be significantly upregulated in a mouse model of asthma (41). These findings lead us to believe that an increase in local Ca2+ signals may perturb the dynamic balance of STICs and STOCs, forming a mechanistic setting for the exaggerated AHR seen in asthma. Thus, new regulators of ASMC local Ca2+ signaling, once defined, may become novel drug targets for the treatment of asthma.
Recent work from our laboratory has shown that PKCϵ downregulates local Ca2+ signaling through its specific effect on RyR1 as well as inhibiting the contractile response of ASMCs to MCh (23). Because a kinase regulates the activity of its substrate in coordination with one or more phosphatases by causing phosphorylation and dephosphorylation, we wondered whether and which phosphatase is involved in the regulation of local Ca2+ signals in ASMCs. Protein phosphatase 2B/CaN (CaN) is known to regulate RyR-mediated Ca2+ release in a C2C12 cell line (37). Moreover, this serine/threonine protein phosphatase, opposite to the serine/threonine protein kinase PKC, controls the activity of TRPV1 channels (28) GAP43, MARCKS (47), and LTCCs in vascular SMCs (30). Therefore, we proposed a novel hypothesis that CaN may upregulate local Ca2+ signaling and contraction in ASMCs. In support of this view, we have found that specific inhibition of CaN with CAIP reduces, whereas activation of CaN with nickel increases, local Ca2+ signaling (Figs. 1A, 1B, 2A, and 2B). The inhibitory effect of CaN in C2C12 cells is thought to depend on CaN, RyR, and the 12-kDa FK-506-binding protein (FKBP 12) being associated in a trimeric complex (37); however, within ASMCs, FKBP 12 does not bind to RyRs (44), providing support that CaN regulates ASMC RyR activity through a novel regulatory mechanism. As shown in Fig. 1C, a loss of extracellular Ca2+ influx (under nominally free extracellular Ca2+ conditions with EGTA) has no effects on the decrease in Ca2+ sparks due to CAIP; similarly, the effect of CaN activation with nickel is not affected either (Fig. 2D), suggesting that extracellular Ca2+ is not necessary for the regulation of Ca2+ sparks by CaN. Within a murine model of asthma, ASMC local Ca2+ signaling is increased (41); it is unknown how this increase is maintained, and it might seem reasonable to suggest that the canonical transient receptor potential-3 channel, which mediates extracellular Ca2+ influx and has increased expression and activity in asthma (45), may play a role in maintaining this signaling. We have further reinforced the specific role for the protein phosphatase CaN by finding out that inhibition of PP2A with endothall does not affect local Ca2+ signals (Fig. 1E), unlike in cardiac myocytes where PP2A was shown to increase Ca2+ sparks (40). RyRs are essential for spark formation; however, IP3Rs cross talk with RyRs, promoting Ca2+ spark formation through a local CICR process (25, 26). In the present study, we have revealed that CaN is able to modulate Ca2+ sparks in the absence of functional IP3Rs (Fig. 1D) but not in the absence of functional RyRs (Figs. 2E, 4A, 4B, and 4D). These results suggest that CaN regulates RyRs and then local Ca2+ signaling in ASMCs.
More importantly, local Ca2+ signaling is decreased in CaN-Aα−/− ASMCs (Fig. 3), and both CAIP and nickel fail to affect Ca2+ sparks in the knockout cells. These results indicate that CAIP and nickel are specifically targeting CaN, providing clear evidence for the novel role of CaN in upregulating ASMC local Ca2+ signaling.
Our earlier study (24) has demonstrated a specific role for RyR1 in mediating the downregulation of local Ca2+ signals by PKCϵ, leading us to hypothesize that CaN may function through RyR1. Local Ca2+ signaling is significantly different between embryonic and adult ASMCs, suggesting that different regulatory mechanisms are in play (24). Furthermore, within a given SMC there are multiple spark-generating sites where individual Ca2+ spark currents have been measured over a wide range, suggesting the involvement of any number from 1 to 50 RyRs in a single CRU (50) with each CRU made up of a heterogeneous mix of RyR1 and RyR2 (but perhaps not RyR3). As we reported previously (24), the effect of PKCϵ on Ca2+ sparks is completely blocked in RyR1 heterozygous deletion (RyR1−/+) ASMCs, which is similar to that in RyR1 homozygous gene deletion (RyR1−/−) cells. With all these considerations, we have used RyR1−/+ mice to test our current hypothesis. Our data reveal a specific role for RyR1 in mediating the CaN-based regulation of Ca2+ sparks since the effects of chemical inhibition and activation of CaN are lost in RyR1−/+ ASMCs (Fig. 4). As a consequence, RyR1 full expression is required for the CaN-based regulation of local Ca2+ signaling in ASMCs. In support, RyR1 is also required for the Ca2+ spark formation in skeletal muscle cells (35), depolarization-induced Ca2+ spark in cultured portal vein (8), and embryonic bladder SMCs (11). RyR1−/+ ASMCs have a lower level of local Ca2+ signaling, further indicating the specific importance of RyR1 in this cell type. However, all three known RyR subtypes (RyR1, RyR2, and RyR3) are expressed in ASMCs (23). However, in support of the specific role for RyR1 in mediating CaN-based regulation of Ca2+ sparks, we have found that the effects of CaN inhibition and activation are not blocked in RyR2−/+ or RyR3−/− ASMCs (Figs. 5 and 6). The role of RyR2 in Ca2+ spark signaling has been extensively studied in the heart, but we know the least about the role of RyR3 in the generation of Ca2+ sparks in SMCs. RyR3 gene knockout or knockdown does not alter local Ca2+ signaling in bladder and portal vein SMCs (8, 17); yet, it has been reported that the frequency of STOCs is augmented in RyR3−/− cerebral artery myocytes (27). This result has been interpreted as evidence for the inhibitory role of RyR3 in the development of Ca2+ sparks, since STOCs are generally thought to be activated by Ca2+ sparks. However, the frequency of Ca2+ sparks in RyR3−/− cerebral artery myocytes is not significantly increased. Furthermore, we have shown that resting and depolarization-induced Ca2+ sparks are not prevented in RyR3−/− ASMCs (25) and that the downregulation of local Ca2+ signaling by PKCϵ is not blocked in RyR3−/− cells either (24). These findings, taken together, clearly point out that CaN regulates local Ca2+ signaling specifically through RyR1, but not RyR2 or RyR3.
As previously stated, Ca2+ sparks play an essential role in mediating ASMC contractile tone as well as regulating ASMC excitation-contraction coupling through the activation of STICs and STOCs. For the first time, we have shown how CaN promotes airway muscle contraction in response to muscarinic stimulation through RyR1-mediated local Ca2+ signaling. CaN inhibition via CAIP decreases MCh-induced contraction in RyR1+/+ rings (Fig. 7A), but not in RyR1−/+ rings (Fig. 7C). In support, a similar smaller muscarinic contractile response has been observed in CaN-Aα−/+, relative to CaN-Aα+/+, mouse tracheal rings. Opposite of PKCϵ's ability to downregulate local Ca2+ signaling and contraction as reported in our previous publication (24), the studies herein have shown how CaN may play an important role in physiological contractile responses by regulating the activity of RyR1-mediated ASMC Ca2+ sparks.
RyR is steadily regulated by phosphorylation, redox modifications from reactive oxygen/nitrogen species, and a variety of small proteins and ions, with the majority of its posttranslational modifications occurring on the large cytoplasmic domain; moreover, RyR1 activity in skeletal myocytes is also regulated by disease-causing mutations and the proteins such as calsequestrin, triadin, and junctin located within the SR (2, 21). All of these unique characteristics make RyRs a hub for integrating a known and unknown number of signaling pathways that use Ca2+ as an intracellular second messenger. In conclusion, we have for the first time shown how CaN upregulates local Ca2+ signaling and contraction in ASMCs through RyR1. The importance of this physiological regulatory mechanism in disease has yet to be determined; however, RyR-generated Ca2+ sparks are a local Ca2+-signaling mechanism conserved across mammalian species, including humans (10), and has been shown to be increased in a mouse model of asthma (41). Further work is needed to extend these novel findings in human tissue samples and to determine the potentially important role of CaN and RyR1 in asthmatic AHR.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant R01-HL-071000 (Y.-X. Wang), an American Heart Association Established Investigator Award 0340160N (Y.-X. Wang), as well as Scientist Development Grants 0730242N (Q.-H. Liu) and 0630236N (Y.-M. Zheng).
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the authors.
AUTHOR CONTRIBUTIONS
C.P.S., Q.-H.L., Y.-M.Z., and Y.-X.W. conception and design of research; C.P.S., Q.-H.L., Y.-M.Z., V.R.Y., Z.Z., L.-G.W., and Y.-X.W. performed experiments; C.P.S., Q.-H.L., Y.-M.Z., and V.R.Y. analyzed data; C.P.S., Q.-H.L., Y.-M.Z., V.R.Y., Z.Z., L.-G.W., and Y.-X.W. interpreted results of experiments; C.P.S., Q.-H.L., Y.-M.Z., V.R.Y., and Y.-X.W. prepared Figs.; C.P.S., Q.-H.L., Y.-M.Z., V.R.Y., and Y.-X.W. drafted manuscript; C.P.S., Q.-H.L., Y.-M.Z., V.R.Y., Z.Z., L.-G.W., and Y.-X.W. edited and revised manuscript; C.P.S., Q.-H.L., Y.-M.Z., V.R.Y., Z.Z., L.-G.W., and Y.-X.W. approved final version of manuscript.
REFERENCES
- 1.Bao R, Lifshitz LM, Tuft RA, Bellve K, Fogarty KE, ZhuGe R. A close association of RyRs with highly dense clusters of Ca2+-activated Cl- channels underlies the activation of STICs by Ca2+ sparks in mouse airway smooth muscle. J Gen Physiol 132: 145–160, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beard NA, Wei L, Dulhunty AF. Control of muscle ryanodine receptor calcium release channels by proteins in the sarcoplasmic reticulum lumen. Clin Exp Pharmacol Physiol 36: 340–345, 2009. [DOI] [PubMed] [Google Scholar]
- 3.Bonev AD, Jaggar JH, Rubart M, Nelson MT. Activators of protein kinase C decrease Ca2+ spark frequency in smooth muscle cells from cerebral arteries. Am J Physiol Cell Physiol 273: C2090–C2095, 1997. [DOI] [PubMed] [Google Scholar]
- 4.Bultynck G, Vermassen E, Szlufcik K, De Smet P, Fissore RA, Callewaert G, Missiaen L, De Smedt H, Parys JB. Calcineurin and intracellular Ca2+-release channels: regulation or association? Biochem Biophys Res Commun 311: 1181–1193, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322: 590–594, 2008. [DOI] [PubMed] [Google Scholar]
- 6.Cheng H, Lederer WJ. Calcium sparks. Physiol Rev 88: 1491–1545, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science 262: 740–744, 1993. [DOI] [PubMed] [Google Scholar]
- 8.Coussin F, Macrez N, Morel JL, Mironneau J. Requirement of ryanodine receptor subtypes 1 and 2 for Ca(2+)-induced Ca(2+) release in vascular myocytes. J Biol Chem 275: 9596–9603, 2000. [DOI] [PubMed] [Google Scholar]
- 9.Daniel EE, Jury J, Serio R, Jager LP. Role of depolarization and calcium in contractions of canine trachealis from endogenous or exogenous acetylcholine. Can J Physiol Pharmacol 69: 518–525, 1991. [DOI] [PubMed] [Google Scholar]
- 10.Deshpande DA, Wang WC, McIlmoyle EL, Robinett KS, Schillinger RM, An SS, Sham JS, Liggett SB. Bitter taste receptors on airway smooth muscle bronchodilate by localized calcium signaling and reverse obstruction. Nat Med 16: 1299–1304, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fritz N, Morel JL, Jeyakumar LH, Fleischer S, Allen PD, Mironneau J, Macrez N. RyR1-specific requirement for depolarization-induced Ca2+ sparks in urinary bladder smooth muscle. J Cell Sci 120: 3784–3791, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Gooch JL, Guler RL, Barnes JL, Toro JJ. Loss of calcineurin Aalpha results in altered trafficking of AQP2 and in nephrogenic diabetes insipidus. J Cell Sci 119: 2468–2476, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Gooch JL, Toro JJ, Guler RL, Barnes JL. Calcineurin A-alpha but not A-beta is required for normal kidney development and function. Am J Pathol 165: 1755–1765, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gordon EH, Wong SC, Klaustermeyer WB. Comparison of nifedipine with a new calcium channel blocker, flordipine, in exercise-induced asthma. J Asthma 24: 261–265, 1987. [DOI] [PubMed] [Google Scholar]
- 15.Greenwood IA, Ledoux J, Sanguinetti A, Perrino BA, Leblanc N. Calcineurin Aalpha but not Abeta augments ICl(Ca) in rabbit pulmonary artery smooth muscle cells. J Biol Chem 279: 38830–38837, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Huang F, Zhang H, Wu M, Yang H, Kudo M, Peters CJ, Woodruff PG, Solberg OD, Donne ML, Huang X, Sheppard D, Fahy JV, Wolters PJ, Hogan BL, Finkbeiner WE, Li M, Jan YN, Jan LY, Rock JR. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc Natl Acad Sci USA 109: 16354–16359, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ji G, Feldman ME, Greene KS, Sorrentino V, Xin HB, Kotlikoff MI. RYR2 proteins contribute to the formation of Ca(2+) sparks in smooth muscle. J Gen Physiol 123: 377–386, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King MM, Huang CY. Activation of calcineurin by nickel ions. Biochem Biophys Res Commun 114: 955–961, 1983. [DOI] [PubMed] [Google Scholar]
- 19.Kotlikoff MI, Wang YX. Calcium release and calcium-activated chloride channels in airway smooth muscle cells. Am J Respir Crit Care Med 158: S109–S114, 1998. [DOI] [PubMed] [Google Scholar]
- 20.Kume H. Large-conductance calcium-activated potassium channels. In: Calcium Signaling In Airway Smooth Muscle Cell, edited by Wang Y-X. Switzerland: Springer, 2014, p. 49–83. [Google Scholar]
- 21.Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harbor Perspect Biol 2: a003996, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lifshitz LM, Carmichael JD, Lai FA, Sorrentino V, Bellve K, Fogarty KE, ZhuGe R. Spatial organization of RYRs and BK channels underlying the activation of STOCs by Ca(2+) sparks in airway myocytes. J Gen Physiol 138: 195–209, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu QH, Savoia C, Wang YX, Zheng YM. Local calcium signaling in airway smooth muscle cells. In: Calcium Signaling In Airway Smooth Muscle Cells, edited by Wang Y-X. Switzerland: Springer, 2014, p. 107–124. [Google Scholar]
- 24.Liu QH, Zheng YM, Korde AS, Li XQ, Ma J, Takeshima H, Wang YX. Protein kinase C-epsilon regulates local calcium signaling in airway smooth muscle cells. Am J Respir Cell Mol Biol 40: 663–671, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu QH, Zheng YM, Korde AS, Yadav VR, Rathore R, Wess J, Wang YX. Membrane depolarization causes a direct activation of G protein-coupled receptors leading to local Ca2+ release in smooth muscle. Proc Natl Acad Sci USA 106: 11418–11423, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu QH, Zheng YM, Wang YX. Two distinct signaling pathways for regulation of spontaneous local Ca2+ release by phospholipase C in airway smooth muscle cells. Pflügers Arch 453: 531–541, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Lohn M, Jessner W, Furstenau M, Wellner M, Sorrentino V, Haller H, Luft FC, Gollasch M. Regulation of calcium sparks and spontaneous transient outward currents by RyR3 in arterial vascular smooth muscle cells. Circ Res 89: 1051–1057, 2001. [DOI] [PubMed] [Google Scholar]
- 28.Lyall V, Phan TH, Mummalaneni S, Melone P, Mahavadi S, Murthy KS, DeSimone JA. Regulation of the benzamil-insensitive salt taste receptor by intracellular Ca2+, protein kinase C, and calcineurin. J Neurophysiol 102: 1591–1605, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mei L, Zheng YM, Wang YX. Ryanodine and inositol triphosphate receptors/Ca2+ release channels in airway smooth muscle cells. In: Calcium Signaling In Airway Smooth Muscle Cells, edited by Wang Y.-X. Switzerland: Springer, 2014, p. 1–20. [Google Scholar]
- 30.Navedo MF, Amberg GC, Nieves M, Molkentin JD, Santana LF. Mechanisms underlying heterogeneous Ca2+ sparklet activity in arterial smooth muscle. J Gen Physiol 127: 611–622, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orie NN, Thomas AM, Perrino BA, Tinker A, Clapp LH. Ca2+/calcineurin regulation of cloned vascular K ATP channels: crosstalk with the protein kinase A pathway. Br J Pharmacol 157: 554–564, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pabelick CM, Prakash YS, Kannan MS, Sieck GC. Spatial and temporal aspects of calcium sparks in porcine tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 277: L1018–L1025, 1999. [DOI] [PubMed] [Google Scholar]
- 33.Pallen CJ, Wang JH. Stoichiometry and dynamic interaction of metal ion activators with calcineurin phosphatase. J Biol Chem 261: 16115–16120, 1986. [PubMed] [Google Scholar]
- 34.Ping L, Ke Z, Benqiong X, Qun W. Effect of metal ions on the activity of the catalytic domain of calcineurin. Biometals 17: 157–165, 2004. [DOI] [PubMed] [Google Scholar]
- 35.Schneider MF, Ward CW. Initiation and termination of calcium sparks in skeletal muscle. Front Biosci 7: d1212–d1222, 2002. [DOI] [PubMed] [Google Scholar]
- 36.Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 134: 1019–1029, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shin DW, Pan Z, Bandyopadhyay A, Bhat MB, Kim DH, Ma J. Ca(2+)-dependent interaction between FKBP12 and calcineurin regulates activity of the Ca(2+) release channel in skeletal muscle. Biophys J 83: 2539–2549, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sly PD, Olinsky A, Landau LI. Does nifedipine affect the diurnal variation of asthma in children? Pediatr Pulmonol 2: 206–210, 1986. [DOI] [PubMed] [Google Scholar]
- 39.Sun T, Wu XS, Xu J, McNeil BD, Pang ZP, Yang W, Bai L, Qadri S, Molkentin JD, Yue DT, Wu LG. The role of calcium/calmodulin-activated calcineurin in rapid and slow endocytosis at central synapses. J Neurosci 30: 11838–11847, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Terentyev D, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Gyorke S. Protein phosphatases decrease sarcoplasmic reticulum calcium content by stimulating calcium release in cardiac myocytes. J Physiol 552: 109–118, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tuo QR, Ma YF, Chen W, Luo XJ, Shen J, Guo D, Zheng YM, Wang YX, Ji G, Liu QH. Reactive oxygen species induce a Ca(2+)-spark increase in sensitized murine airway smooth muscle cells. Biochem Biophys Res Commun 434: 498–502, 2013. [DOI] [PubMed] [Google Scholar]
- 42.van Breemen C, Fameli N, Evans AM. Pan-junctional sarcoplasmic reticulum in vascular smooth muscle: nanospace Ca2+ transport for site- and function-specific Ca2+ signalling. J Physiol 591: 2043–2054, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang YX, Kotlikoff MI. Inactivation of calcium-activated chloride channels in smooth muscle by calcium/calmodulin-dependent protein kinase. Proc Natl Acad Sci USA 94: 14918–14923, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang YX, Zheng YM, Mei QB, Wang QS, Collier ML, Fleischer S, Xin HB, Kotlikoff BP. MIFK12.6 and cADPR regulation of Ca2+ release in smooth muscle cells. Am J Physiol Cell Physiol 286: C538–C546, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Xiao JH, Zheng YM, Liao B, Wang YX. Functional role of canonical transient receptor potential 1 and canonical transient receptor potential 3 in normal and asthmatic airway smooth muscle cells. Am J Respir Cell Mol Biol 43: 17–25, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455: 1210–1215, 2008. [DOI] [PubMed] [Google Scholar]
- 47.Yi H, Kim SH, Park HG, Yu HS, Kim YS. The effect of systemic injection of cyclosporin A on the phosphorylation of the PKC substrates MARCKS and GAP43 in the rat hippocampus. Neurosci Lett 497: 17–21, 2011. [DOI] [PubMed] [Google Scholar]
- 48.Zhang CH, Li Y, Zhao W, Lifshitz LM, Li H, Harfe BD, Zhu MS, Zhuge R. The transmembrane protein 16A Ca2+-activated Cl- channel in airway smooth muscle contributes to airway hyperresponsiveness. Am J Respir Crit Care Med 187: 374–381, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhuge R, Bao R, Fogarty KE, Lifshitz LM. Ca2+ sparks act as potent regulators of excitation-contraction coupling in airway smooth muscle. J Biol Chem 285: 2203–2210, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhuge R, Fogarty KE, Tuft RA, Lifshitz LM, Sayar K, Walsh JV., Jr Dynamics of signaling between Ca(2+) sparks and Ca(2+)- activated K(+) channels studied with a novel image-based method for direct intracellular measurement of ryanodine receptor Ca(2+) current. J Gen Physiol 116: 845–864, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhuge R, Sims SM, Tuft RA, Fogarty KE, Walsh JV., Jr Ca2+ sparks activate K+ and Cl- channels, resulting in spontaneous transient currents in guinea-pig tracheal myocytes. J Physiol 3: 711–718, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]