

Abstract
G protein-coupled receptors are the most pervasive signaling superfamily in the body and act as receptors to endogenous agonists and drugs. For β-agonist-mediated bronchodilation, the receptor-G protein-effector network consists of the β2-adrenergic receptor (β2AR), Gs, and adenylyl cyclase, expressed on airway smooth muscle (ASM). Using ASM-targeted transgenesis, we previously explored which of these three early signaling elements represents a limiting factor, or bottleneck, in transmission of the signal from agonist binding to ASM relaxation. Here we overexpressed Gαs in transgenic mice and found that agonist-promoted relaxation of airways was enhanced in direct proportion to the level of Gαs expression. Contraction of ASM from acetylcholine was not affected in Gαs transgenic mice, nor was relaxation by bitter taste receptors. Furthermore, agonist-promoted (but not basal) cAMP production in ASM cells from Gαs-transgenic mice was enhanced compared with ASM from nontransgenic littermates. Agonist-promoted inhibition of platelet-derived growth factor-stimulated ASM proliferation was also enhanced in Gαs mouse ASM. The enhanced maximal β-agonist response was of similar magnitude for relaxation, cAMP production, and growth inhibition. Taken together, it appears that a limiting factor in β-agonist responsiveness in ASM is the expression level of Gαs. Gene therapy or pharmacological means of increasing Gαs (or its coupling efficiency to β2AR) thus represent an interface for development of novel therapeutic agents for improvement of β-agonist therapy.
Keywords: bronchodilation, relaxation, stoichiometry, β-agonist
β2-adrenergic receptors (β2AR) on airway smooth muscle (ASM) are the targets for β-agonist bronchodilators, utilized in the treatment of obstructive lung diseases such as asthma and chronic obstructive pulmonary disease (26). The effectiveness of these agents is subject to modification by the disease state (5, 12, 17, 20), prior agonist exposure causing desensitization (19), and genetic variation (14, 15). β2AR carry out signal transduction by binding to and dissociating the heterotrimeric G protein Gs, with the Gαs subunit activating adenylyl cyclase, which catalyzes conversion of ATP to the second messenger cAMP, which evokes ASM relaxation via PKA-dependent and -independent mechanisms (2, 4). However, it is unclear which of these three major components in these early events, β2AR, Gs, or adenylyl cyclase, represents the limiting factor in physiological signaling. Ascertaining which element represents the bottleneck for β-agonist-mediated bronchodilation might identify an interface for novel therapeutics. In cardiomyocytes, which primarily express β1AR, it has been shown that the expression level of adenylyl cyclase represents such a bottleneck that limits signaling from receptor to cAMP production and cardiac contractility (9, 10). Indeed, gene therapy trials are underway to increase cardiac adenylyl cyclase expression in patients with chronic heart failure, with the goal of enhancing inotropy (trial NCT00787059). We previously overexpressed or knocked down several of these elements and other β-agonist signal transduction components in mice to ascertain whether, in ASM, such a limiting element exists, potentially leading to therapeutic approaches to increase that component and enhance β-agonist effectiveness. These have included the β2AR itself (22), G protein-coupled receptor kinase (GRK) (28), and PKA function (28), Gαi (21), and adenylyl cyclase (29). In the current work we have performed ASM-targeted transgenesis of Gαs in mice to ascertain the biochemical and physiological effects of increasing this component.
METHODS
Generation of transgenic mice.
These studies were approved by the University of Maryland School of Medicine Institutional Animal Care and Use Committee. FVB/N mice (Taconic, Hudson, NY) were used for transgenesis, as previously described (28). The long form of human Gαs (GNAS) was subcloned into the XhoI site of the smooth muscle α-actin promoter SMP8 (8) and confirmed by sequencing. We previously showed the effectiveness and specificity of this promoter for targeting genes to ASM (22). Isolation and purification of a NotI-digested fragment provided material for injection to generate F0 founder mice. Genomic DNA derived from tail clips was utilized for screening by PCR using transgene-specific primers. Positive founders were mated to nontransgenic (NTG) littermates to maintain heterozygous colonies. Typically, F3–F6 mice were studied. From the cultured ASM cell lines (passages 3–8), quantitative RT-PCR was carried out using primers for Gαs, thereby providing net (endogenous and transgene-based) mRNA quantitation. These same cells were utilized in immunoblots of whole cell lysates to determine Gαs and GAPDH expression using primary antibodies obtained from Santa Cruz Biotechnology (Dallas, TX) and Millipore (Billerica, MA), respectively. Blots were processed using enhanced chemiluminescence (GE Healthcare, Cleveland, OH), and signal intensities were quantitated using a ChemiDoc imager (Bio-Rad, Hercules, CA).
Airway physiology.
Ex vivo studies of tracheal rings were carried out in 14- to 16-wk-old mice, as described previously (7). After CO2 narcosis, tracheae were rapidly removed and cut into 4-mm cross sections, which were placed between the two wires of a lateral isometric myograph. Rings were maintained in Krebs buffer at 37°C with 95% O2 aeration for the duration of the study. Rings were studied in groups of four. After 15 min of incubation, the rings were passively stretched to 5 mN, and the force was monitored for an additional 30 min to ensure stabilization. For each ring, a dose-response study with acetylcholine (100 nM–1 mM) was performed to ascertain contractile properties. To determine the relaxation response to the β-agonist (−)isoproterenol, rings were contracted with acetylcholine using the ED70 dose, and then increasing concentrations (1 nM–1 μM) of (−)isoproterenol were added to the bath. Recordings were made on a continuous basis, and the response was typically stable by 2 min after addition of the agonist. β-Agonist response data are presented as raw force (in mN) or normalized to percentage of the acetylcholine force.
Establishment of ASM cell lines.
Freshly excised tracheae were cut in a longitudinal manner, and 2- to 3-mm cross sections were placed lumen-side-down in polystyrene culture dishes. The medium for this initial proliferation was Dulbecco's modified Eagle's medium (DMEM) with 20% fetal calf serum and 100 μg/ml gentamicin, 1% penicillin, 1% streptomycin, and 0.5 μg/ml amphotericin B. After 7 days of incubation at 37°C in a 5% air-95% CO2 atmosphere, the trachea and surrounding cells were adherent and the medium was changed to DMEM with 10% fetal calf serum and 50 μg/ml gentamicin, 1% penicillin, 1% streptomycin, and 0.25 μg/ml amphotericin B. Subsequently, the tracheae were removed, the plates were washed with PBS, and fresh medium with antibiotics was added, and the cells were grown to confluence. Subsequent culture conditions for later passages did not include amphotericin B. The cells had typical morphology and staining characteristics of ASM, as previously documented (22). Cells from passages 3–8 were utilized in these studies.
cAMP and cell proliferation assays.
ASM cells were transferred to 48-well plates (∼5 × 104 cells/well) and, after attachment, maintained in Krebs-Ringer phosphate buffer in the absence of serum for the cAMP assay. They were pretreated with 100 μM isobutylmethylxanthine for 30 min at 37°C. Then ascorbic acid (100 μM) alone or ascorbic acid with 10 μM (−)isoproterenol was added, and the plate was gently swirled and incubated at 37°C for 30 min. The reaction was stopped by addition of a hypotonic buffer, and the plate was frozen at −80°C. cAMP was measured using a fluorescent competitive immunoassay (Catch Point, Molecular Devices, Sunnyvale, CA) and read on a FlexStation III plate reader (Molecular Devices). Data are represented as picomoles of cAMP per milligram of whole cell lysate. For the proliferation assay, cells were seeded in 96-well plates in DMEM with 10% fetal calf serum for 24 h, serum-starved for 24 h, and then treated with medium alone, platelet-derived growth factor (PDGF, 25 ng/ml), or PDGF + 10 μM (−)isoproterenol for 24 h. Cells were trypsinized and counted in triplicate with an automated cell counter (Z1 Coulter, Beckman Coulter, Brea, CA) and presented as cells per milliliter.
Statistics.
For the physiological studies with dose-response data, results were fit to sigmoidal curves with variable slopes using the program PRISM (Graph Pad, La Jolla, CA). The maximal responses for each experiment as derived from these curves (rather than the responses to only the maximal dose) were compared between lines or conditions. In addition, curves were compared by two-way ANOVA to ascertain differences across the range of agonists. Results from these studies and the biochemical studies were compared by two-way t-tests, with significance imparted when P < 0.05. Results are shown as means ± SE.
RESULTS
Human Gαs cDNA was cloned into the smooth muscle α-actin promoter SMP8 (Fig. 1A). A digested fragment was utilized for injection to generate founder transgenic mice with use of methods previously described. Tail-derived genomic DNA was screened by a transgene-specific PCR (Fig. 1B), and three founders that passed the transgene to progeny were identified. These are designated as TG1, TG6, and TG8. RT-PCR was performed on RNA isolated from cultured ASM cells. For the aforementioned mouse lines, Gαs expression was 54,000 ± 1,200, 43,000 ± 3,000, and 290 ± 21 fold, respectively, over endogenous Gαs in NTG mice (Fig. 1B). Gαs protein expression from cultured ASM was ∼28% greater in TG1 and ∼10% greater in TG6 than NTG and not different from NTG in TG8 (Fig. 1B).
Fig. 1.
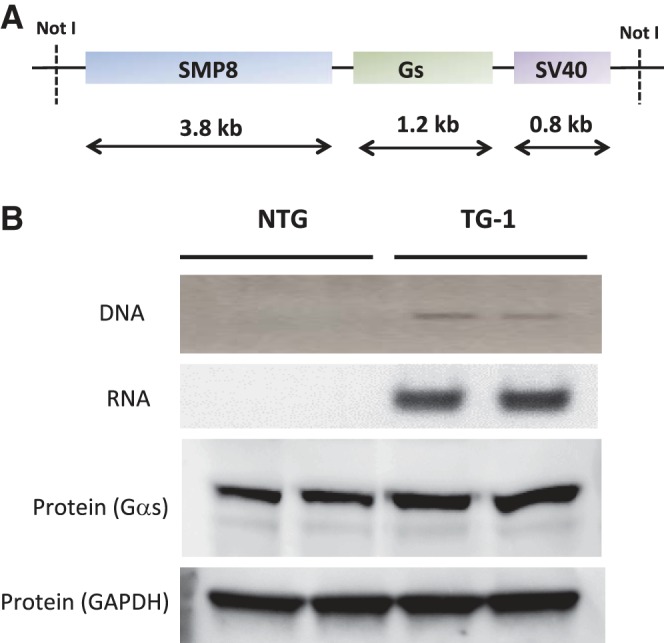
Generation of transgenic mice overexpressing Gαs. A: diagram of the DNA construct engineered for oocyte injection. SMP8, smooth muscle α-actin promoter; SV40, simian virus 40. B: PCR screen of tail-clip DNA using 1 primer from Gαs and 1 primer from the SV40 portion of the transgene (top) and expression of Gαs by RT-PCR from cultured airway smooth muscle (ASM) cells from transgenic (TG1) and nontransgenic (NTG) littermates (bottom).
Initial experiments were performed with TG1, the highest-expressing line. Airway rings were studied for their contractile and relaxation responses. A baseline passive stretch (tension) of 5 mN was applied to each ring, and data are shown as the change from that baseline. As seen in Fig. 2A, the contractile response to acetylcholine was not different between TG1 and NTG. For relaxation studies, rings were first contracted with acetylcholine, then increasing concentrations of isoproterenol were added to the bath (Fig. 2B). The maximal relaxation response to this β-agonist was ∼37% greater in TG1 than NTG (33 ± 1.4% vs. 24 ± 1.3%, P < 0.01, n = 4). In contrast, the relaxation response to the bitter taste receptor agonist quinine was identical between the two mouse lines (Fig. 2C). Similar studies were performed with the TG6 and TG8 lines. The intermediate-Gαs-expressing line (TG6) showed a modest and statistically significant increase in the relaxation response to isoproterenol compared with NTG (Fig. 3A), while the lowest-expressing line (TG8) showed no difference in isoproterenol-mediated airway relaxation compared with NTG (Fig. 3B).
Fig. 2.

Gαs overexpression in ASM enhances relaxation of airways from TG1 mice. A: contraction response to acetylcholine (ACh) was unaltered in TG1 airways. B: extent of relaxation of acetylcholine-contracted rings to the β-agonist isoproterenol (Iso) was greater in TG1 airways overexpressing Gαs than in NTG littermates. C: relaxation response to the bitter taste receptor agonist quinine (Qui) was not altered in TG1 mice. Values are means ± SE; n = 4 mice in each group. *P < 0.01 vs. NTG.
Fig. 3.

Relaxation responses to isoproterenol in TG6 and TG8 mice. A: extent of relaxation in TG6 mice, which had an intermediate level of Gαs ASM protein expression, compared with TG1 and TG8 mice, was greater than in NTG littermates. B: TG8 mouse airways, which had no detectable increase in ASM Gαs protein expression, showed no differences in the relaxation response to isoproterenol compared with NTG. Values are means ± SE; n = 4 mice in each group. *P < 0.05 vs. NTG.
The improvement in airway relaxation with increasing abundance of Gαs indicates that the expression level of Gαs limits the bronchodilating function of β2AR. If this is so, one would expect to see enhanced agonist-stimulated cAMP in ASM cells from the Gαs transgenic mice that show the enhanced relaxation. The TG8 mouse line was lost, but we were able to establish cultured ASM cells from NTG, TG1, and TG6. Adhered cultured mouse ASM cells were treated with the phosphodiesterase inhibitor isobutylmethylxanthine and then exposed for 30 min to vehicle or 10 μM isoproterenol. cAMP levels for the three lines are shown in Fig. 4. None of the transgenic lines had higher basal cAMP levels than NTG. TG1 cells, derived from mice with the greatest enhancement of isoproterenol-mediated relaxation, showed ∼60% greater isoproterenol-stimulated cAMP levels (P < 0.01) than NTG (P > 0.05). The apparent increase in isoproterenol-stimulated cAMP levels produced from TG6 cells was not statistically different from NTG levels (P = 0.08; Fig. 4).
Fig. 4.

β-Agonist-promoted intracellular cAMP accumulation is increased in cultured TG1 ASM cells overexpressing Gαs. Adhered cells were treated with 100 μM ascorbic acid (basal) or 10 μM isoproterenol + ascorbic acid (Iso) for 30 min at 37°C. Values are means ± SE; n = 5 experiments. *P < 0.01 vs. NTG.
β-Agonists have been variably reported to decrease ASM cell proliferation (3). The extent of such a decrease is dependent on how the cells are stimulated, the degree of confluence, and the time course of observation. In our studies we stimulated proliferation with PDGF and ascertained the effect of this mitogen in the absence or presence of 10 μM isoproterenol after 24 h. As shown in Fig. 5, NTG ASM showed very little inhibition of PDGF-promoted proliferation by isoproterenol. However, TG1 ASM displayed a ∼85% inhibition of proliferation by isoproterenol. The apparent small inhibition of PDGF-stimulated proliferation by isoproterenol for the ASM from the intermediate-Gαs-expressing TG6 line was not statistically significant.
Fig. 5.

Inhibition of platelet-derived growth factor (PDGF)-promoted ASM cell proliferation by β-agonist is enhanced in TG1-derived ASM cells. Cells were serum-starved and treated with PDGF or PDGF + isoproterenol for 24 h. Cells were counted in a standard volume in triplicate using an automated cell counter. Values are means ± SE; n = 8 experiments. *P < 0.02 vs. NTG.
DISCUSSION
In the current work we have explored the intracellular signaling and physiological consequences of increasing the expression of Gαs in ASM using targeted transgenesis. Our goal over the last few years has been to understand the relationship between the immediate signaling elements in the β2AR pathway and β-agonist-mediated function in the airway. These elements, the β2AR, Gαs, and adenylyl cyclase, represent the earliest components that functionally interact in the classic pathway leading to increased intracellular cAMP and, ultimately, ASM relaxation. When set to the activated state by agonist binding, β2AR couple to the heterotrimeric G protein Gs, releasing Gαs, which activates adenylyl cyclase catalyzing the generation of cAMP. Additional elements are also important in these early events. As agonist activation persists, β2AR are phosphorylated by GRKs at specific residues. GRK-phosphorylated β2AR promotes binding of β-arrestins, which interdict between receptor and Gs, thus partially quenching the cAMP response. In ASM, we have shown that this event is rapid and, indeed, integrated into the initial response that is observed with agonist binding (28). β2AR also couple to Gαi, albeit in a less efficient manner than Gαs (6). This event appears to require PKA phosphorylation of the β2AR, as well as β-arrestin recruitment. Such coupling acts to depress adenylyl cyclase activation (27), since Gαi inhibits the enzyme and also promotes additional signaling, such as to Erk1/2 (6).
In previous studies we created transgenic mice that overexpressed the β2AR on ASM cells to ∼75-fold over endogenous levels of the protein (22). Surprisingly, the gain in agonist-promoted cAMP generation in these ASM cells was modest (<3-fold) at best and was certainly not in direct proportion to the increase in receptor protein expression. Moreover, maximal β-agonist-promoted relaxation in tracheal rings was not enhanced, but the response curves were left-shifted. We next overexpressed by approximately threefold adenylyl cyclase (type V, the predominant isoform in ASM cells) in the ASM of transgenic mice (29). Here we found that the increase in adenylyl cyclase caused a reciprocal increase in expression of Gαi, potentially in compensation from a sustained basal cAMP. When treated with pertussis toxin to block the inhibiting effect of increased Gαi, we found no enhancement of β-agonist-promoted relaxation in the adenylyl cyclase-overexpressing mice (29). We thus concluded that adenylyl cyclase was not the limiting factor, and, regardless, efforts to enhance the enzyme for therapeutic purposes would be restrained by the increase in Gαi.
We thus began to focus on Gαs as the potential limiting factor in β2AR signaling in ASM. Three transgenic lines were generated, and, despite substantial increases in mRNA levels, protein expression was modestly increased in two lines and not significantly different from endogenous Gαs in the other line. This discrepancy between mRNA levels and Gαs protein expression has been previously reported in mice with cardiomyocyte overexpression of Gαs and suggests posttranscriptional or posttranslational regulation (11). These low levels of transgene protein expression that we obtained in the current study were fortuitous, in that we indeed observed enhanced β-agonist-mediated ASM relaxation in the TG1 line, in which we observed a 28% increase in Gαs expression. Remarkably, the enhanced relaxation in these mice was 37%, suggesting a close relationship between the level of Gαs expression and β2AR physiological function. Consistent with this finding, β-agonist-promoted cAMP was ∼60% greater in cultured ASM cells from these Gαs transgenic mice than in NTG ASM. In the intermediate-expressing TG6 mice, there was a smaller enhancement of β-agonist-mediated ASM relaxation. We also observed that the modest Gαs overexpression resulted in an enhancement of β-agonist-mediated attenuation of cell proliferation induced by PDGF. This effect on proliferation was not observed in NTG littermates, although the phenomenon may be condition-dependent. Nevertheless, a decrease in ASM mass evoked by β-agonist would be considered a long-term positive therapeutic outcome (13, 16).
The results that we obtained here in ASM cells appear to be different from those in other cell types where Gαs expression has been increased. In the heart, such overexpression did not result in an increase in basal or agonist-promoted adenylyl cyclase activities (11). In HEK 293 cells, expression of a β2AR-Gαs fusion protein, which maintains a 1:1 stoichiometry, showed properties of constitutive activation (24). A similar finding was observed in Sf9 cells, with marked overexpression of β2AR and Gαs β1γ2 (1). However, in CHW-1102 cells, where β2AR were moderately overexpressed with Gαs, or in cells that were transfected to express the β2AR-Gαs fusion protein, only the latter cells showed an increase in basal adenylyl cyclase activities (25). Regardless, we found no increase in basal cAMP levels or baseline relaxation to suggest constitutive activity of β2AR in our transgenic mouse airways or ASM cells.
We conclude that the limiting element in the β2AR signal transduction pathway leading to ASM relaxation is the amount of Gαs. This is based on the very similar levels of increase in β-agonist-mediated airway relaxation, ASM cAMP accumulation, and inhibition of ASM proliferation in the Gαs transgenic mice, as well as the gene dose response observed in the three transgenic mouse lines. As indicated earlier, in our previous investigations of transgenic mice overexpressing other components, we did not observe this tight relationship between expression and gain (or loss) of function. Therapeutics acting to increase ASM expression of Gαs, such as modulators of transcription factors or Gαs turnover or Gαs gene therapy, could ultimately improve β-agonist efficacy as defined by bronchodilation and, potentially, decreased ASM mass. In a similar manner, increasing the efficiency of β2AR coupling to existing Gαs would also lead to a similar improvement. This could be accomplished with positive allosteric modulators (18), β-agonists that bias signaling away from GRK phosphorylation/β-arrestin binding (30), or small molecules that attenuate the GRK/β-arrestin effect (23). Interestingly, we have generated β2AR (GRK-negative) mice and found that the increase in ASM relaxation in these mice, which lack GRK-phosphorylatable β2AR (28), is the same level of magnitude as increasing expression of Gαs, confirming the notion that increasing Gαs expression, or the efficiency of its coupling, can accomplish the same physiological end point. Taken together, the results from the current study reveal that Gαs (or its interface with β2AR) is a nodal point for targeting new therapeutics for the treatment of obstructive lung disease.
GRANTS
This work was funded by National Heart, Lung, and Blood Institute Grants HL-045967, HL-071609, and HL-104119 and the Cracchiolo Family Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
W.C.H.W., S.S.A., B.C.-M., and S.B.L. are responsible for conception and design of the research; W.C.H.W., S.H.P., D.C.S., M.A.D., D.J.D., A.P., and S.B.L. performed the experiments; W.C.H.W., S.H.P., D.C.S., M.A.D., D.J.D., A.P., and S.B.L. analyzed the data; W.C.H.W., S.H.P., D.C.S., M.A.D., D.J.D., A.P., S.S.A., B.C.-M., and S.B.L. interpreted the results of the experiments; W.C.H.W., S.H.P., D.C.S., and S.B.L. prepared the figures; W.C.H.W. and S.B.L. drafted the manuscript; W.C.H.W., S.H.P., D.C.S., M.A.D., D.J.D., A.P., S.S.A., B.C.-M., and S.B.L. edited and revised the manuscript; W.C.H.W., S.H.P., D.C.S., M.A.D., D.J.D., A.P., S.S.A., B.C.-M., and S.B.L. approved the final version of the manuscript.
ACKNOWLEDGMENTS
The authors thank Charmaine Disimile for manuscript preparation.
Current affiliation of W. C. H. Wang: Division of Cardiovascular Sciences, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD.
REFERENCES
- 1.Azzi M, Pineyro G, Pontier S, Parent S, Ansanay H, Bouvier M. Allosteric effects of G protein overexpression on the binding of β-adrenergic ligands with distinct inverse efficacies. Mol Pharmacol 60: 999–1007, 2001. [DOI] [PubMed] [Google Scholar]
- 2.Barnes PJ. β-Adrenergic receptors and their regulation. Am J Respir Crit Care Med 152: 838–860, 1995. [DOI] [PubMed] [Google Scholar]
- 3.Billington CK, Ojo OO, Penn RB, Ito S. cAMP regulation of airway smooth muscle function. Pulm Pharmacol Ther 26: 112–120, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Billington CK, Penn RB. Signaling and regulation of G protein-coupled receptors in airway smooth muscle. Respir Res 4: 2–24, 2003. [PMC free article] [PubMed] [Google Scholar]
- 5.Chhabra J, Li YZ, Alkhouri H, Blake AE, Ge Q, Armour CL, Hughes JM. Histamine and tryptase modulate asthmatic airway smooth muscle GM-CSF and RANTES release. Eur Respir J 29: 861–870, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature 390: 88–91, 1997. [DOI] [PubMed] [Google Scholar]
- 7.Deshpande DA, Wang WC, McIlmoyle EL, Robinett KS, Schillinger RM, An SS, Sham JS, Liggett SB. Bitter taste receptors on airway smooth muscle bronchodilate by localized calcium signaling and reverse obstruction. Nat Med 16: 1299–1304, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foster DN, Min B, Foster LK, Stoflet ES, Sun S, Getz MJ, Strauch AR. Positive and negative cis-acting regulatory elements mediate expression of the mouse vascular smooth muscle α-actin gene. J Biol Chem 267: 11995–12003, 1992. [PubMed] [Google Scholar]
- 9.Gao M, Ping P, Post S, Insel PA, Tang R, Hammond HK. Increased expression of adenylylcyclase type VI proportionately increases β-adrenergic receptor-stimulated production of cAMP in neonatal rat cardiac myocytes. Proc Natl Acad Sci USA 95: 1038–1043, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao MH, Lai NC, Roth DM, Zhou J, Zhu J, Anzai T, Dalton N, Hammond HK. Adenylylcyclase increases responsiveness to catecholamine stimulation in transgenic mice. Circulation 99: 1618–1622, 1999. [DOI] [PubMed] [Google Scholar]
- 11.Gaudin C, Ishikawa Y, Wight DC, Maier M, Mahdavi V, Nadal-Ginard B, Wagner TE, Vatner DE, Homcy CJ. Overexpression of Gsα protein in the hearts of transgenic mice. J Clin Invest 95: 1676–1683, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldie RG, Spina D, Henry PJ, Lulich KM, Paterson JW. In vitro responsiveness of human asthmatic bronchus to carbachol, histamine, β-adrenoreceptor agonists and theophylline. Br J Clin Pharmacol 22: 669–676, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirst SJ, Martin JG, Bonacci JV, Chan V, Fixman ED, Hamid QA, Herszberg B, Lavoie JP, McVicker CG, Moir LM, Nguyen TT, Peng Q, Ramos-Barbon D, Stewart AG. Proliferative aspects of airway smooth muscle. J Allergy Clin Immunol 114: S2–S17, 2004. [DOI] [PubMed] [Google Scholar]
- 14.Israel E, Chinchilli VM, Ford JG, Boushey HA, Cherniack RM, Craig TJ, Deykin A, Fagan JK, Fahy JV, Fish J, Kraft M, Kunselman SJ, Lazarus SC, Lemanske RF, Liggett SB, Martin RJ, Mitra N, Peters SP, Silverman E, Sorkness CA, Szefler SJ, Wechsler ME, Weiss ST, Drazen JM. Use of regularly scheduled albuterol treatment in asthma: genotype-stratified, randomised, placebo-controlled cross-over trial. Lancet 364: 1505–1512, 2004. [DOI] [PubMed] [Google Scholar]
- 15.Israel E, Drazen JM, Liggett SB, Boushey HA, Cherniack RM, Chinchilli VM, Cooper DM, Fahy JV, Fish JE, Ford JG, Kraft M, Kunselman S, Lazarus SC, Lemanske RF, Martin RJ, McLean DE, Peters SP, Silverman EK, Sorkness CA, Szefler SJ, Weiss ST, Yandava CN. The effect of polymorphisms of the β2-adrenergic receptor on the response to regular use of albuterol in asthma. Am J Respir Crit Care Med 162: 75–80, 2000. [DOI] [PubMed] [Google Scholar]
- 16.Johnson PR, Roth M, Tamm M, Hughes M, Ge Q, King G, Burgess JK, Black JL. Airway smooth muscle cell proliferation is increased in asthma. Am J Respir Crit Care Med 164: 474–477, 2001. [DOI] [PubMed] [Google Scholar]
- 17.Jude JA, Panettieri RA, Jr, Walseth TF, Kannan MS. TNF-α regulation of CD38 expression in human airway smooth muscle: role of MAP kinases and NF-κB. Adv Exp Med Biol 691: 449–459, 2011. [DOI] [PubMed] [Google Scholar]
- 18.Keov P, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology 60: 24–35, 2011. [DOI] [PubMed] [Google Scholar]
- 19.Lipworth BJ. Airway subsensitivity with long-acting β2-agonists. Is there cause for concern? Drug Saf 16: 295–308, 1997. [DOI] [PubMed] [Google Scholar]
- 20.Mahn K, Hirst SJ, Ying S, Holt MR, Lavender P, Ojo OO, Siew L, Simcock DE, McVicker CG, Kanabar V, Snetkov VA, O'Connor BJ, Karner C, Cousins DJ, Macedo P, Chung KF, Corrigan CJ, Ward JP, Lee TH. Diminished sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) expression contributes to airway remodelling in bronchial asthma. Proc Natl Acad Sci USA 106: 10775–10780, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGraw DW, Elwing JM, Fogel KM, Wang WC, Glinka CB, Mihlbachler KA, Rothenberg ME, Liggett SB. Crosstalk between Gi and Gq/Gs pathways in airway smooth muscle regulates bronchial contractility and relaxation. J Clin Invest 117: 1391–1398, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGraw DW, Forbes SL, Witte DP, Fortner CN, Paul RJ, Liggett SB. Transgenic overexpression of β2-adrenergic receptors in airway smooth muscle alters myocyte function and ablates bronchial hyperreactivity. J Biol Chem 274: 32241–32247, 1999. [DOI] [PubMed] [Google Scholar]
- 23.O'Callaghan K, Kuliopulos A, Covic L. Turning receptors on and off with intracellular pepducins: new insights into G-protein-coupled receptor drug development. J Biol Chem 287: 12787–12796, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seifert R, Wenzel-Seifert K, Lee TW, Gether U, Sanders-Bush E, Kobilka BK. Different effects of Gsα splice variants on β2-adrenoreceptor-mediated signaling. J Biol Chem 273: 5109–5116, 1998. [PubMed] [Google Scholar]
- 25.Small KM, Forbes SL, Rahman FF, Liggett SB. Fusion of β2-adrenergic receptor to Gαs in mammalian cells: identification of a specific signal transduction species not characteristic of constitutive activation or precoupling. Biochemistry 39: 2815–2821, 2000. [DOI] [PubMed] [Google Scholar]
- 26.Tattersfield AE. Clinical studies of β-agonists in adults. In: β2-Agonists in Asthma Treatment (1st ed.), edited by Pauwels R, O'Byrne PM. New York: Dekker, 1997, p. 283–317. [Google Scholar]
- 27.Tepe NM, Liggett SB. Functional receptor coupling to Gi is a mechanism of agonist-promoted desensitization of the β2-adrenergic receptor. J Recept Signal Transduct Res 20: 75–85, 2000. [DOI] [PubMed] [Google Scholar]
- 28.Wang WC, Mihlbachler KA, Brunnett AC, Liggett SB. Targeted transgenesis reveals discrete attenuator functions of GRK and PKA in airway β2-adrenergic receptor physiologic signaling. Proc Natl Acad Sci USA 106: 15007–15012, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang WC, Schillinger RM, Malone MM, Liggett SB. Paradoxical attenuation of β2-AR function in airway smooth muscle by Gi-mediated counterregulation in transgenic mice overexpressing type 5 adenylyl cyclase. Am J Physiol Lung Cell Mol Physiol 300: L472–L478, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wisler JW, Xiao K, Thomsen AR, Lefkowitz RJ. Recent developments in biased agonism. Curr Opin Cell Biol 27: 18–24, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]