

Abstract
Although AMP-activated protein kinase (AMPK) is involved in regulating carbohydrate and lipid metabolism, activated AMPK also plays an anti-inflammatory role in many cell populations. However, despite the ability of AMPK activation to diminish the severity of inflammatory responses, previous studies have found that AMPK activity is diminished in LPS-treated neutrophils and also in lungs of mice with LPS-induced acute lung injury (ALI). Since GSK3β participates in regulating AMPK activity, we examined potential roles for GSK3β in modulating LPS-induced activation of neutrophils and macrophages and in influencing severity of ALI. We found that GSK3β-dependent phosphorylation of T479-AMPK was associated with pT172 dephosphorylation and inactivation of AMPK following TLR4 engagement. GSK3β inhibitors BIO (6-bromoindirubin-3′-oxime), SB216763, or siRNA knockdown of GSK3β, but not the PI3K/AKT inhibitor LY294002, prevented Thr172-AMPK dephosphorylation. Exposure to LPS resulted in rapid binding between IKKβ and AMPKα, and phosphorylation of S485-AMPK by IKKβ. These results suggest that IKKβ-dependent phosphorylation of S485-AMPK was an essential step in subsequent phosphorylation and inactivation AMPK by GSK3β. Inhibition of GSK3β activity delayed IκBα degradation and diminished expression of the proinflammatory TNF-α in LPS-stimulated neutrophils and macrophages. In vivo, inhibition of GSK3β decreased the severity of LPS-induced lung injury as assessed by development of pulmonary edema, production of TNF-α and MIP-2, and release of the alarmins HMGB1 and histone 3 in the lungs. These results show that inhibition of AMPK by GSK3β plays an important contributory role in enhancing LPS-induced inflammatory responses, including worsening the severity of ALI.
Keywords: GSK3β, AMPK, inflammation, IKK, acute lung injury
amp-activated protein kinase (AMPK) is a serine threonine protein kinase that participates in the regulation of cellular bioenergetics and redox status (16, 17, 37). Recent studies have also shown that AMPK activation has potent anti-inflammatory effects under in vitro and in vivo conditions (24, 47, 61). AMPK is a heterotrimer that includes regulatory β and γ subunits as well as an α catalytic subunit (30). Cellular stress, such as that induced by ischemia or hypoxia, is associated with increased AMP-to-ATP ratios and binding of AMP to the regulatory AMPKγ subunit (42). Crystal structure analysis of AMPK showed that binding of AMP and ADP to the AMPKγ subunit results in allosteric domain rearrangement (55, 56) that in turn allows for phosphorylation of Thr172-AMPKα by upstream kinases, including liver kinase B1 (LKB1) and calcium-calmodulin kinase kinase beta (CaMKKβ) (19, 54). Recent studies have shown that glycogen binding to the AMPK β subunit or direct oxidation of specific reactive cysteine thiols in the AMPKα and β subunits are also followed by Thr172 phosphorylation and activation of AMPK (32, 40, 62).
AMPK phosphorylation and associated activation is a reversible process, with deactivation of AMPK primarily being mediated by dephosphorylation of Thr172-AMPKα by protein phosphatase 2C (PP2Cα) (10). Previous studies have shown that AKT/protein kinase B or the combination of AKT and glycogen synthase kinase-3β (GSK3β) signaling can promote AMPK inactivation, particularly through inducing phosphorylation of Thr479 and Ser485 in the AMPKα subunit (21, 33, 51). Phosphorylation of Thr479 and Ser485 through AKT and GSK3β associated pathways was sufficient to trigger dephosphorylation of pThr172 and inactivation of AMPK by PP2Cα (51).
Although the primary actions of AMPK were initially associated with metabolic regulation of carbohydrate, lipid, and protein synthesis, AMPK has also been shown to have potent anti-inflammatory effects, including diminishing TLR4 associated NF-κB activation in neutrophils, macrophages, and endothelial cells (47, 61). In addition, enhanced AMPK activation reduced the severity of LPS-induced acute lung injury (61). The ability of AMPK to be phosphorylated and activated is diminished in LPS-stimulated neutrophils and macrophages as well as in the lungs of mice with acute lung injury (52, 57). However, the mechanisms responsible for the inhibition of AMPK activation during TLR4 proinflammatory responses have not been well characterized.
GSK3β is a serine threonine protein kinase involved in regulating signaling pathways associated with cell metabolism, differentiation, apoptosis, proliferation, and viability (9, 31). Although originally described as a regulator of glycogen synthase implicated in the development of cancer, diabetes, and obesity as well as neurodegenerative diseases (2, 22, 28, 29, 38, 46), GSK3β also has important role in modulating the duration and magnitude of inflammatory conditions (7, 22, 39). For example, exposure of LPS-treated monocytes to GSK3β inhibitors or small interfering RNA (siRNA)-mediated knockdown of GSK3β diminished proinflammatory cytokine production (39). Moreover, blockade of GSK3β activation through treatment of mice with the specific inhibitor SB216763 significantly reduced LPS-associated mortality (39). Inhibition of GSK3β also provided protection from bleomycin-induced lung injury, decreased diabetes-associated elevations in glucose levels, and diminished neuroinflammation (8, 14, 29, 36, 59). In addition to potentiating the release of proinflammatory mediators, GSK3β has also been implicated in suppressing the production of anti-inflammatory cytokines, including IL-10 (7, 22, 39).
Although neutrophils play essential roles in innate immune responses and microbial eradication, exaggerated and excessively prolonged activation of neutrophils is associated with dysregulated inflammatory processes that can lead to organ dysfunction, including acute lung injury (ALI). In preclinical models of ALI, sepsis, and liver or kidney injury, treatment with the AMPK activators metformin or 5-aminoimidazole-4-carboxamide-1-beta-d-ribonucleoside (AICAR) diminished the severity of organ dysfunction and reduced mortality (5, 6, 15, 43, 61). In addition to diminishing release of cytokines and other proinflammatory mediators (45, 47, 61), AMPK activation also appears to decrease inflammation through enhancing the ability of neutrophils and macrophages to ingest apoptotic cells, a process known as efferocytosis that plays a central role in the resolution of inflammation (3, 26). However, despite the presence of conditions known to participate in the activation of AMPK, no activation of AMPK was found in the lungs of mice with ALI, even though AMP-ATP ratios were increased and elevated amounts of reactive oxygen species were generated (52, 57), suggesting that counterregulatory mechanisms induced in inflammation inhibit AMPK activation.
Given the role of GSK3β in promoting inflammation and in regulating the metabolic functions of AMPK (22, 39, 51), we examined whether GSK3β-dependent inhibition of AMPK might contribute to neutrophil proinflammatory activation and the severity of LPS-induced acute lung injury. Our results show that inhibition of AMPK by GSK3β enhances LPS-induced inflammatory responses, including worsening the severity of ALI.
MATERIALS AND METHODS
Mice.
Male C57BL/6 mice were purchased from the National Cancer Institute-Frederick (Frederick, MD). Male mice, 8 to 10 wk of age, were used for experiments. The mice were kept on a 12-h light-dark cycle with free access to food and water. All experiments were conducted in accordance with protocols approved by the University of Alabama at Birmingham Animal Care and Use Committee.
Reagents and antibodies.
The GSK3β inhibitor BIO (6-bromoindirubin-3′-oxime) was purchased from R&D Systems (Minneapolis, MN). Second GSK3β inhibitors, SB216763, as well as the IKK1/2 inhibitor PS-1145 were obtained from Sigma-Aldrich (St. Louis, MO). LY294002 was purchased from Cell Signaling (Danvers, MA). Antibodies for phospho-Thr172-AMPK, total AMPK, phospho-Ser9-GSK3β, total GSK3β, phospho-Ser79-ACC, total ACC, IKKβ, and IκBα were purchased from Cell Signaling Technology. β-Actin antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). Custom antibody mixtures and negative selection columns for neutrophil isolation were obtained from Stem Cell Technologies (Vancouver, BC). Anti-phospho-Thr479-AMPK antibody was generated as previously described (51). Mouse monoclonal antibody to histone 3 was obtained from Abcam (Cambridge, MA) whereas anti-HMGB1 antibody was purchased from R&D Systems. Goat anti-mouse κ-HRP was purchase from Southern Biotech (Birmingham, AL).
Neutrophil and peritoneal macrophage isolation and culture.
Bone marrow neutrophils were isolated as described previously (45, 64, 65). Neutrophil purity was consistently greater than 97%, as determined by Wright-Giemsa-stained cytospin preparations. Neutrophils were cultured in RPMI 1640 medium containing 0.5% FBS and treated as indicated in the figure legends. Neutrophil viability under experimental conditions was determined by Trypan blue staining and was consistently greater than 95%. Peritoneal macrophages were isolated as described previously (3). Macrophages were elicited in 8- to 10-wk-old mice by use of Brewer thioglycollate. Cells were collected 5 days after intraperitoneal injection of thioglycollate. Macrophages were then cultured in 12-well plates (105 cells/well) in RPMI 1640 media containing 5% FBS at 37°C. Nonadherent cells were removed by washing cells after 60 min of incubation. Cells were incubated with RPMI 1640 medium with FBS 0.5% serum for 2 h before and during treatments.
RAW 264.7 macrophage culture.
Murine macrophage like RAW 264.7 cells were maintained in DMEM medium supplemented with FBS serum (8%)(26). Cells were incubated with DMEM medium with FBS 0.5% serum for 2 h before and during treatments.
Cytokine ELISA.
ELISA was used to measure cytokines in bronchoalveolar lavage (BAL) fluid. Levels of TNF-α and macrophage inflammatory protein (MIP)-2 were determined by using commercially available ELISA kits (R&D Systems) according to the manufacturer's instructions and as previously described (63, 66)
Western blot analysis.
Western blot analysis was performed as described previously (62). Each experiment was carried out two or more times with cell populations obtained from separate groups of mice. In selected experiments, BAL fluids (30 μl) were mixed with Laemmli sample buffer and boiled for 5 min followed by Western blot analysis.
Coimmunoprecipitation assays.
Pull-down assay was performed as previously described (4). In brief, cell lysates (500 μg/ml) were incubated with anti-AMPKα or IKKβ antibody (5 μg/ml) overnight at 4°C. Samples were then incubated with protein-A agarose (25 μl/sample) for an additional 2 h at 4°C. The protein-A agarose conjugates were washed and proteins released by incubation with Laemmli sample buffer for 10 min at 95°C. The amounts of IKKβ associated with AMPKα were determined by Western Blot analysis.
siRNA knockdown of GSK3β.
Murine macrophage-like RAW 264.7 cells were incubated with scrambled siRNA or siRNA specific to GSK3β as described previously (3, 50). Briefly, cells (3 × 105/well) in 24-well plates were incubated in Accell media with scrambled siRNA (2 μM) or siRNA (2 μM) to GSK3β for 72 h. Cells were treated as described in the figure legends and then subjected to Western blot analysis as indicated.
ALI model.
ALI was induced by intratracheal administration of 1 mg/kg LPS in 50 μl of PBS as previously described (61). With this model, ALI is characterized by neutrophil infiltration into the lung interstitium and airways, development of interstitial edema, and increased pulmonary proinflammatory cytokine production, with the greatest degree of injury being present 24 h after LPS exposure (61, 64). Of note, although intratracheal instillation of LPS is clinically relevant model for moderate lung injury, several additional conditions are implicated in development of acute respiratory distress syndrome (ARDS), including severe infection (sepsis), blood loss (trauma/hemorrhage), and mechanical ventilation. Briefly, mice were anesthetized with isoflurane and then suspended by their upper incisors on a 60° incline board. The tongue was gently extended and LPS or PBS solution deposited into the pharynx (60). In selected experiments, GSK3β inhibitor SB216763 (20 mg/kg) in 1 ml of DMSO/saline (1:40) or control vehicle (DMSO/saline 1:40) was injected intraperitoneally for 14 h and 2 h prior to LPS intratracheal instillation. Mice were euthanized 24 h after LPS administration. BALs were obtained by lavaging the lungs three times with 1 ml PBS.
Statistical analysis.
Multigroup comparisons were performed using one-way ANOVA with Tukey's post hoc test. Statistical significance was determined by the Student's t-test for comparisons between two groups. A value of P < 0.05 was considered significant. Analyses were performed on SPSS version 16.0 (IBM, Armonk, NY) for Windows (Microsoft, Redmond, WA).
RESULTS
GSK3β inhibits AMPK activation in LPS-stimulated neutrophils.
Enhanced activation of AMPK was previously shown to diminish the proinflammatory properties of neutrophils, macrophages, and other cell populations (18, 24, 47, 53, 57, 65), and might therefore be expected to occur after TLR4 engagement. However, exposure to LPS (0, 100, 300, or 1,000 ng/ml) for 60 min, or inclusion of LPS (300 ng/ml) for 0, 20, 40, or 60 min, resulted in dose- and time-dependent dephosphorylation pThr172-AMPK in bone marrow neutrophils (Fig. 1, A and B). Because GSK3β was implicated in both enhancing inflammation and inhibiting the actions of AMPK in metabolic regulation (51), we determined whether GSK3β inhibitors affect Thr172-AMPK phosphorylation following TLR4 engagement. As shown in Fig. 1, C and D, whereas exposure to LPS (300 ng/ml) for 60 min decreased the levels of phospho-Thr172-AMPK in neutrophils, incubation of neutrophils with the GSK3β inhibitor BIO (5 μM) for 60 min prevented AMPK dephosphorylation after LPS stimulation. Western blot analysis showed that culture of neutrophils with LPS resulted in phosphorylation of Thr479-AMPK, a specific GSK3β-dependent phosphorylation site that was recently implicated in the inhibition of AMPK activation (Fig. 1, C and D) (51). Pretreatment with GSK3β inhibitors BIO (5 μM) or SB216763 (30 μM) for 60 min significantly diminished the ability of LPS to stimulate neutrophil activation, including expression of TNF-α (Fig. 2A).
Fig. 1.

GSK3β participates in inhibiting AMP-activated protein kinase (AMPK) activation in LPS-stimulated neutrophils. A and B: representative Western blots and quantitative data show the amounts of pT172-AMPK and total AMPK obtained from LPS-treated neutrophils. Means ± SD (n = 3), *P < 0.05, **P < 0.01, compared with untreated cells. C and D: the GSK3β inhibitor BIO (6-bromoindirubin-3′-oxime) diminishes LPS-induced phosphorylation of T479-AMPK and dephosphorylation of pT172-AMPK. Bone marrow neutrophils were treated with BIO (5 μM) for 60 min and then cultured with LPS (300 ng/ml) for an additional 60 min. C: Western blots showing pT172-AMPK, pT479-AMPK, total AMPK, and β-actin. D: means ± SD (n = 3), *P < 0.05, **P < 0.01.
Fig. 2.
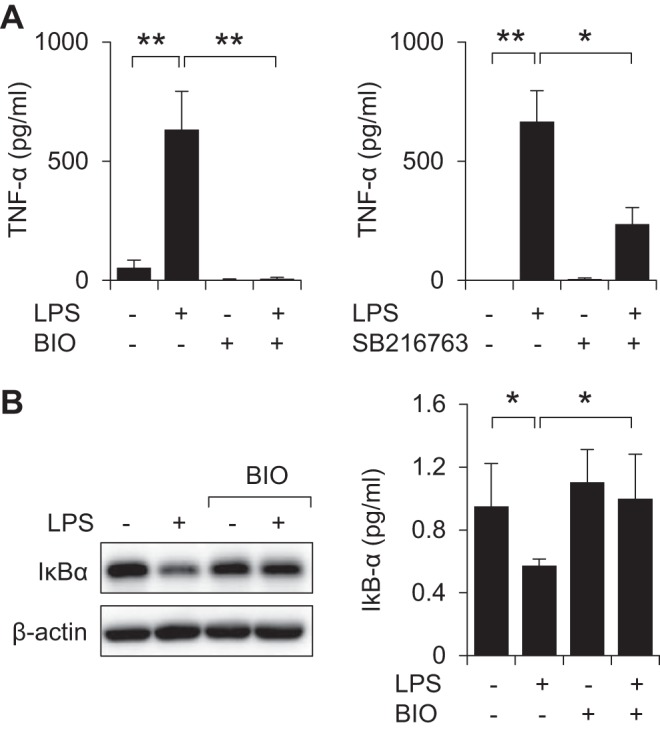
Inhibition of GSK3β diminishes LPS-induced neutrophil activation. A: bone marrow neutrophils were treated with BIO (0, or 5 μM) or SB216763 (30 μM) for 60 min followed by culture with or without LPS (300 ng/ml) for an additional 4 h. The amounts of TNF-α culture media were determined by ELISA. Means ± SD (n = 3), *P < 0.05, **P < 0.01. B: representative Western blots and quantitative data show the amount of IκBα in neutrophils treated with LPS in the presence or absence of BIO. The GSK3β inhibitor BIO (0 or 5 μM) was included in the cultures 60 min before the addition of LPS and then cells were cultured for an additional 60 min. Means ± SD (n = 3), *P < 0.05.
Previous studies indicated that a mechanism by which AMPK diminishes TLR4-induced activation of neutrophils was through decreasing degradation of IκBα, an essential step in initiating NF-κB translocation from cytosol to nucleus (61). As shown in Fig. 2B, incubation of neutrophils with the GSK3β inhibitor BIO (5 μM) for 60 min reduced IκBα degradation following LPS stimulation.
Inhibition of GSK3β prevents LPS-mediated Thr172-AMPK dephosphorylation and macrophage activation.
As shown in Fig. 3, A and B, rapid increases in levels of phospho-Thr479-AMPK were associated with time-dependent dephosphorylation of Thr172-AMPK in LPS (300 ng/ml)-treated peritoneal macrophages. Similarly, exposure to LPS (300 ng/ml) for 0, 5, 10, 20, or 30 min induced Thr172-AMPK dephosphorylation in RAW 264.7 cells (Fig. 3, C and D). Pretreatment of macrophages with the GSK3β inhibitor BIO (5 μM) for 60 min diminished the effects of LPS on AMPK dephosphorylation (Fig. 3, C and D). In addition to increased levels of phospho-Thr172-AMPK, GSK3β inhibition was also associated with enhanced phosphorylation of Ser79-acetyl-CoA carboxylase (ACC), a known downstream target of AMPK (Fig. 3C). As was seen with BIO, culture of macrophages with a second GSK3β inhibitor, SB216763, also resulted in inhibition of LPS-mediated Thr172-AMPK dephosphorylation (Fig. 3, E and F). Similar to inhibitors of GSK3β, siRNA-induced knockdown of GSK3β prevented dephosphorylation of pT172-AMPK in LPS-treated macrophages (Fig. 3, G and H). We also found that preincubation of RAW 264.7 macrophages with BIO (5 μM) or SB216763 (30 μM) for 60 min effectively diminished LPS induced TNF-α production (Fig. 4, A and B). IκBα degradation was also diminished in LPS-treated RAW 264.7 cells treated with BIO (Fig. 4C).
Fig. 3.

GSK3β contributes to dephosphorylation of pT172-AMPK in LPS-stimulated macrophages. A and B: peritoneal macrophages were treated with LPS (300 ng/ml) for the indicated times (0 to 60 min). Representative Western blots and quantitative data show fold changes of pT172-AMPK, pT479-AMPK, total AMPK, and β-actin. Means ± SD (n = 3), *P < 0.05, **P < 0.01, ***P < 0.001, compared with untreated cells. C and D: RAW 264.7 macrophages were pretreated with BIO (0 or 5 μM) for 60 min followed by incubation with LPS (300 ng/ml) for the indicated times. In E and F, cells were incubated with SB216763 (0, 5, 10, 20, or 30 μM) for 60 min and then with or without LPS (300 ng/ml) for an additional 60 min (results were obtained from 2 independent experiments). D and F: means ± SD (n = 3), *P < 0.05, **P < 0.01. G and H: representative Western blots and quantitative data show the amounts of pT172-AMPK in LPS (0 or 300 ng/ml)-treated [scrambled siRNA (scram)] or GSK3β-depleted RAW 264.7 cells. Means ± SD (n = 3), **P < 0.01, compared with untreated or treated with LPS.
Fig. 4.

GSK3β-dependent inhibition of AMPK enhances macrophage activation. A and B: RAW 264.7 macrophages were preincubated with BIO (0 or 5 μM) or SB216763 (0 or 30 μM) for 60 min followed by addition of LPS (0 or 10 ng/ml) to the cultures for 4 h. TNF-α levels were measured in culture media by ELISA (means ± SD, n = 3, ***P < 0.001). C: Western blot analysis and quantitative data (means ± SD, n = 3, **P < 0.01) show the amounts of IκBα obtained from macrophages that were treated with LPS (300 ng/ml). Cells were pretreated with or without BIO (5 μM) for 60 min before LPS exposure. D: TNF-α concentrations in the culture media were determined after treatment of RAW 264.7 macrophages or bone marrow neutrophils with LY294002 (0 or 10 μM) for 60 min followed by inclusion of LPS (0 or 10 ng/ml) in the cultures for an additional 4 h. Means ± SD (n = 3), ***P < 0.001; NS, not significant. E and F: Western blots and quantitative data show the levels of pT172-AMPK, pT479-AMPK or pS9-GSK3β, GSK3β, and β-actin in RAW 264.7 macrophages that were treated with LY294002 (0 or 10 μM) or BIO (0 or 5 μM) for 60 min followed by inclusion of LPS (0 or 300 ng/ml) in the cultures for an additional 60 min. Means ± SD (n = 3), *P < 0.05.
GSK3β-dependent inhibition of AMPK in LPS-treated neutrophils and macrophages is dependent on IKK1/2.
Engagement of TLR4 results in activation of downstream kinases responsible for initiation of the NF-κB signaling cascade, including IKK1/2 (13, 44). Unlike inhibition of PI3K/AKT (Fig. 4, D and E), inclusion of the IKK1/2 inhibitor PS1145 (10 μM) for 60 min effectively prevented dephosphorylation of Thr172AMPK in LPS-treated neutrophils and macrophages (Fig. 5, A and B), suggesting that IKK1/2 is involved in modulating AMPK activation following TLR4 engagement. Inclusion of PS1145 also diminished GSK3β-mediated inhibitory phosphorylation of Thr479-AMPK in LPS stimulated cells.
Fig. 5.

IKK and GSK3β participate in inhibiting AMPK phosphorylation in LPS-stimulated neutrophils and macrophages. A and B: bone marrow neutrophils (A) or RAW 264.7 macrophages (B) were treated with the IKK inhibitor PS1145 (0 or 10 μM) for 60 min followed by exposure to LPS (0 or 300 ng/ml) for 60 min. Western blots of pT172-AMPK, pT479-AMPK, AMPK, and β-actin and quantitative data are shown. Means ± SD (n = 3), *P < 0.05, **P < 0.01. C: Western blot analysis show the time-dependent increase in Ser485-AMPK phosphorylation in LPS (300 ng/ml)-treated RAW 264.7 cells. IKK inhibitor PS1141 (10 μM) was applied 60 min prior to LPS exposure. Means ± SD (n = 3), ***P < 0.001. D: RAW 264.7 cells were subsequently cultured with LY294002 (0 or 10 μM) for 60 min followed by inclusion of LPS (0 or 300 ng/ml) for an additional 60 min. Representative Western blots and quantitative are shown (means ± SD, n = 3). E: RAW 264.7 cells were cultured with or without LPS (300 ng/ml) for 30 min followed by pull-down assay with anti-AMPKα or anti-IKKβ antibody. Western blots show the amounts of AMPKα and IKKβ in cell lysates and after immunoprecipitation. Means ± SD (n = 3), *P < 0.05, **P < 0.01.
Previous studies have shown that the appearance of phospho-Ser485-AMPK is implicated in subsequent phosphorylation of Thr479-AMPK and inhibition of kinase activity by GSK3β (21, 33, 51). As shown in Fig. 5C, robust phosphorylation of Ser485-AMPK was evidenced after exposure of RAW 264.7 cells to LPS (300 ng/ml) for 30 or 60 min. Moreover, pretreatment with IKK1/2 inhibitor PS1141 (10 μM), but not PI3K/ATK inhibitor LY294002 (10 μM), effectively diminished the ability of LPS to induce phosphorylation of Ser485-AMPK (Fig. 5, C and D). Furthermore, results obtained from immunoprecipitation assay shows a rapid binding between IKKβ and AMPKα1, particularly after exposure of macrophages to LPS (300 ng/ml) for 30 min (Fig. 5E). These results suggest that IKKβ participated in phosphorylation of Ser485-AMPK, which than promoted inhibitory phosphorylation of Thr479-AMPK by GSK3β.
Inhibition of GSK3β diminishes the severity of LPS-induced ALI.
Our in vitro results indicate that GSK3β is involved in regulating AMPK activation and the production of proinflammatory mediators, including the cytokine TNF-α, in TLR4-stimulated neutrophils and macrophages. Given the anti-inflammatory actions of AMPK and importance of these cell populations in the development and perpetuation of inflammatory conditions, we next explored whether modulation of GSK3β can affect the development of ALI, a condition in which activated neutrophils and macrophages play central roles (1, 3, 61). In these experiments, mice were given the GSK3β inhibitor SB216763 (20 mg/kg ip) or vehicle (saline/DMSO 1:40 ip) 2 h before intratracheal instillation of saline (control) or LPS (2 mg/kg it).
As shown in Fig. 6, treatment with SB216763 diminished the severity of LPS-mediated ALI. In particular, decreased lung wet-to-dry ratios, indicative of less severe interstitial pulmonary edema, were present in mice that received the GSK3β inhibitor, compared with control mice (Fig. 6A). Compared with control LPS-exposed mice, administration of SB216763 resulted in diminished numbers of BAL neutrophils (Fig. 6B). Significant decreases in TNF-α and MIP-2 and total amount of proteins were also found in BALs of mice treated with SB216763 (Fig. 6C). Significant increases in the levels of the danger-associated molecular patterns (DAMPs) histone 3 and HMGB1 were found in BALs of LPS-exposed mice. Treatment of LPS-treated mice with SB216763 diminished levels of both HMGB1 and H3 in BALs compared with controls (Fig. 6D). Whereas phosphorylation of Thr172-AMPK and Ser79-ACC was diminished in lung homogenates from LPS-treated mice, phosphorylation of Thr479-AMPK increased after LPS exposure (Fig. 6, E and F). Such inhibition of AMPK activation in the lungs following LPS treatment was prevented by administration of SB216763. In particular, increased levels of pThr172-AMPK and pSer79-ACC, as well as diminished pThr479-AMPK phosphorylation, were present in the lungs of LPS-exposed mice after treatment with the GSK3β inhibitor SB216763.
Fig. 6.

Inhibition of GSK3β decreases the severity of LPS-induced acute lung injury. Mice were given SB216763 (20 mg/kg ip) 14 h and 2 h before intratracheal instillation of saline or LPS (2 mg/kg). A, B, and C show increase in lung wet-to-dry ratios, numbers of bronchoalveolar lavage (BAL) neutrophils, and TNF-α, MIP-2, and total protein concentrations in BAL fluids obtained 24 h after LPS administration. In A, fold increase above values present in mice receiving saline alone are shown (means ± SD, n = 5, *P < 0.05). In B, mean ± SE (n = 5), *P < 0.05, ***P < 0.001 compared with LPS alone, whereas in C, means ± SD (n = 5), *P < 0.05, ***P < 0.001. D: representative Western blots and quantitative data show the levels of HMGB1 and histone 3 (H3) in BAL fluids of control, LPS-treated, and LPS and SB216763-treated mice. Means ± SD (n = 6), *P < 0.05, ***P < 0.001. E and F: Western blots show the amounts of pT172-AMPK, pS79-ACC, pT479-AMPK, total AMPK, and β-actin in lung homogenates from control (saline)-treated mice or 24 h after LPS administration in mice given intratracheal LPS alone or LPS and SB216763 (given ip 14 and 2 h before LPS). Means ± SD (n = 3), *P < 0.05.
DISCUSSION
In this study, we have shown that GSK3β can modulate response to LPS-stimulated neutrophils and macrophages under both in vitro and in vivo conditions. In particular, inhibition of GSK3β with the specific inhibitor SB216763 diminished the severity of LPS-induced ALI. Although previous experiments (39) demonstrated the ability nuclear GSK3β to affect NF-κB transcriptional activity in monocytes, we demonstrated that GSK3β can also inhibit AMPK in LPS-treated neutrophils and macrophages. Given that AMPK has anti-inflammatory functions (35, 43, 61), our findings indicate that GSK3β activation may enhance inflammation through its effects on AMPK. This hypothesis was supported by experiments using the GSK3β inhibitors BIO and SB216763, which prevented GSK3β-mediated Thr172 dephosphorylation of AMPK, a step associated with AMPK inhibition. GSK3β inhibitors also diminished neutrophil and macrophage activation, including LPS-stimulated expression of TNF-α.
Our findings are consistent with a recent study that demonstrated the ability of GSK3β to inhibit AMPK, particularly in setting of serum-stimulated Thr172-AMPK dephosphorylation and inactivation (51). The ability of GSK3β to regulate the metabolic functions of AMPK appears to be a complex process that involves direct interaction between GSK3β and AMPK, leading to enhanced phosphorylation of Thr479-AMPK as well as phosphatase PP2Cα-dependent dephosphorylation of Thr172-AMPK. Previous studies have shown that AKT-mediated phosphorylation of Ser485-AMPK also facilitated GSK3β/PP2Cα-dependent Thr172 dephosphorylation (51). Although the PI3K/AKT pathway was shown to be involved in Ser485-AMPK phosphorylation in fibroblasts (21, 33), we found that inhibition of AKT had negligible effects on both Thr172-AMPK dephosphorylation and cytokine production in LPS-stimulated neutrophils and macrophages (Fig. 2 and 4). Engagement of TLR4 leads to activation of IKK1/2, which has a central role in inducing the phosphorylation and degradation of IκBα and translocation of NF-κB from the cytoplasm to the nucleus (13, 44). Unlike PI3K/AKT inhibition, which had only minimal effects on AMPK phosphorylation, inhibition of IKK1/2 effectively diminished LPS-mediated increase in pSer485-AMPK and also Thr172-AMPK dephosphorylation (Fig. 5). These results suggest that IKK1/2, but not PI3K/AKT, participates in the mechanism through which GSK3β inhibits AMPK activation in TLR4 stimulated neutrophils and macrophages.
Although AMPK has primarily been characterized as a major regulator of carbohydrate and lipid metabolism, AMPK also plays an important anti-inflammatory role in many cell populations (24, 34, 47, 61). The AMPK activators metformin and AICAR were shown to suppress production of nuclear factor NF-κB-dependent cytokines in TLR4-stimulated neutrophils (47, 61). Activation of AMPK was also implicated in enhanced phagocytosis and clearance of apoptotic cells, an important process in the resolution of inflammation (3, 26, 47). However, even though TLR4 engagement results in increased AMP to ATP ratios as well as enhanced reactive oxygen species formation, which are factors that should increase AMPK activation (20, 23, 48, 62), AMPK was not activated in the lungs of LPS-exposed mice or critically ill patients with ARDS. Recent studies demonstrated that engagement of TLR4 resulted in inhibition of AMPK in neutrophils, peritoneal macrophages, Raw 264.7 cells, and endothelial cells (25, 47, 52, 57, 58). Our results suggest that GSK3β-dependent phosphorylation of Thr479-AMPK followed by Thr172-AMPK dephosphorylation is an important mechanism contributing to AMPK inhibition in TLR4-stimulated cells.
Relatively little is known concerning mechanisms contributing to inhibition of AMPK activity. In addition to the ability of GSK3β to inhibit AMPK activation through PI3-K/AKT-dependent mechanisms (51), PKA-dependent phosphorylation of Ser173-AMPK has been shown to prevent AMPK activation through diminished phosphorylation of Thr172-AMPK (11). Recent studies, including results obtained from our laboratory, indicate that in addition to microbial associated mediators, such as LPS, host-derived proinflammatory factors can also modulate AMPK activation in macrophages. For example, we have found that high molecular group box 1 protein (HMGB1), a DAMP molecule, can directly bind to and inhibit AMPK upstream kinase LKB1 (52). Interferon-γ or human resistin has also been shown to inhibit AMPK activation (12, 27, 41, 49). Of note, whereas HMGB1 or human resistin had indirect effects on AMPK activity, our present studies suggest that subsequent engagement of LPS/TLR4, IKKβ-dependent phosphorylation of Ser485-AMPK and phosphorylation of Thr479-AMPK by GSK3β is a prompt mechanism of AMPK inactivation.
Our results indicate that GSK3β inhibition may be a feasible therapeutic approach to preserve the anti-inflammatory functions of AMPK and potentially improve the action of AMPK activators, such as metformin and AICAR, which have previously been shown to inhibit TLR2- and TLR4-induced activation of neutrophils and macrophages and diminish the severity of ALI. In these studies, GSK3β inhibition was able to diminish TLR4-associated activation of neutrophils and macrophages and also reduced LPS-induced ALI. Although there are concerns about involvement of GSK3β in regulating many signaling and metabolic pathways, we expect that combined approaches, by using pharmacological approaches to enhance AMPK activation while diminishing GSK3β-related inhibition of AMPK, may be beneficial in patients with ARDS and organ dysfunction. In addition, GSK3β inhibitors may be useful to prevent organ injury in other clinical settings where neutrophils play an important proinflammatory role.
GRANTS
This work was supported in part by National Institutes of Health Grants GM87748 and HL107585 to Jaroslaw W. Zmijewski.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
D.W.P., E.A., and J.W.Z. conception and design of research; D.W.P., S.J., and Y.L. performed experiments; D.W.P., S.J., G.P.S., K.I., E.A., and J.W.Z. analyzed data; D.W.P., S.J., K.I., E.A., and J.W.Z. Interpreted results of experiments; D.W.P., S.J., and J.W.Z. prepared figures; D.W.P., E.A., and J.W.Z. drafted manuscript; D.W.P., S.J., Y.L., K.I., E.A., and J.W.Z. approved final version of manuscript; E.A. and J.W.Z. edited and revised manuscript.
REFERENCES
- 1.Abraham E. Neutrophils and acute lung injury. Crit Care Med 31: S195–S199, 2003. [DOI] [PubMed] [Google Scholar]
- 2.Ali A, Hoeflich KP, Woodgett JR. Glycogen synthase kinase-3: properties, functions, and regulation. Chem Rev 101: 2527–2540, 2001. [DOI] [PubMed] [Google Scholar]
- 3.Bae HB, Zmijewski JW, Deshane JS, Tadie JM, Chaplin DD, Takashima S, Abraham E. AMP-activated protein kinase enhances the phagocytic ability of macrophages and neutrophils. FASEB J 25: 4358–4368, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banerjee S, Zmijewski JW, Lorne E, Liu G, Sha Y, Abraham E. Modulation of SCF beta-TrCP-dependent I kappaB alpha ubiquitination by hydrogen peroxide. J Biol Chem 285: 2665–2675, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergheim I, Luyendyk JP, Steele C, Russell GK, Guo L, Roth RA, Arteel GE. Metformin prevents endotoxin-induced liver injury after partial hepatectomy. J Pharmacol Exp Ther 316: 1053–1061, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Buler M, Aatsinki SM, Skoumal R, Komka Z, Toth M, Kerkela R, Georgiadi A, Kersten S, Hakkola J. Energy-sensing factors coactivator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1alpha) and AMP-activated protein kinase control expression of inflammatory mediators in liver: induction of interleukin 1 receptor antagonist. J Biol Chem 287: 1847–1860, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan MM, Cheung BK, Li JC, Chan LL, Lau AS. A role for glycogen synthase kinase-3 in antagonizing mycobacterial immune evasion by negatively regulating IL-10 induction. J Leukoc Biol 86: 283–291, 2009. [DOI] [PubMed] [Google Scholar]
- 8.Chuang DM. The antiapoptotic actions of mood stabilizers: molecular mechanisms and therapeutic potentials. Ann NY Acad Sci 1053: 195–204, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol 2: 769–776, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Davies SP, Helps NR, Cohen PT, Hardie DG. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett 377: 421–425, 1995. [DOI] [PubMed] [Google Scholar]
- 11.Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, Vaahtomeri K, Auchli Y, Rechsteiner H, Brunisholz RA, Viollet B, Makela TP, Wallimann T, Neumann D, Krek W. PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. EMBO J 29: 469–481, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frost RA, Nystrom GJ, Lang CH. Endotoxin and interferon-gamma inhibit translation in skeletal muscle cells by stimulating nitric oxide synthase activity. Shock 32: 416–426, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol 8: 837–848, 2008. [DOI] [PubMed] [Google Scholar]
- 14.Gurrieri C, Piazza F, Gnoato M, Montini B, Biasutto L, Gattazzo C, Brunetta E, Cabrelle A, Cinetto F, Niero R, Facco M, Garbisa S, Calabrese F, Semenzato G, Agostini C. 3-(2,4-Dichlorophenyl)-4-(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione (SB216763), a glycogen synthase kinase-3 inhibitor, displays therapeutic properties in a mouse model of pulmonary inflammation and fibrosis. J Pharmacol Exp Ther 332: 785–794, 2010. [DOI] [PubMed] [Google Scholar]
- 15.Hallows KR, Mount PF, Pastor-Soler NM, Power DA. Role of the energy sensor AMP-activated protein kinase in renal physiology and disease. Am J Physiol Renal Physiol 298: F1067–F1077, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251–262, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardie DG, Sakamoto K. AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology (Bethesda) 21: 48–60, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Hattori Y, Suzuki K, Hattori S, Kasai K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension 47: 1183–1188, 2006. [DOI] [PubMed] [Google Scholar]
- 19.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2: 9–19, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Horie T, Ono K, Nagao K, Nishi H, Kinoshita M, Kawamura T, Wada H, Shimatsu A, Kita T, Hasegawa K. Oxidative stress induces GLUT4 translocation by activation of PI3-K/Akt and dual AMPK kinase in cardiac myocytes. J Cell Physiol 215: 733–742, 2008. [DOI] [PubMed] [Google Scholar]
- 21.Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A, Schlattner U, Wallimann T, Carling D, Hue L, Rider MH. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem 281: 5335–5340, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT, Woodgett JR, Ivashkiv LB. IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity 24: 563–574, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Irrcher I, Ljubicic V, Hood DA. Interactions between ROS and AMP kinase activity in the regulation of PGC-1α transcription in skeletal muscle cells. Am J Physiol Cell Physiol 296: C116–C123, 2009. [DOI] [PubMed] [Google Scholar]
- 24.Jeong HW, Hsu KC, Lee JW, Ham M, Huh JY, Shin HJ, Kim WS, Kim JB. Berberine suppresses proinflammatory responses through AMPK activation in macrophages. Am J Physiol Endocrinol Metab 296: E955–E964, 2009. [DOI] [PubMed] [Google Scholar]
- 25.Ji G, Zhang Y, Yang Q, Cheng S, Hao J, Zhao X, Jiang Z. Genistein suppresses LPS-induced inflammatory response through inhibiting NF-kappaB following AMP kinase activation in RAW 264.7 macrophages. PLoS One 7: e53101, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang S, Park DW, Stigler WS, Creighton J, Ravi S, Darley-Usmar V, Zmijewski JW. Mitochondria and AMP-activated protein kinase-dependent mechanism of efferocytosis. J Biol Chem 288: 26013–26026, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang S, Park DW, Tadie JM, Gregoire M, Deshane J, Pittet JF, Abraham E, Zmijewski JW. Human resistin promotes neutrophil proinflammatory activation and neutrophil extracellular trap formation and increases severity of acute lung injury. J Immunol 192: 4795–4803, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 29: 95–102, 2004. [DOI] [PubMed] [Google Scholar]
- 29.Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res 32: 577–595, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1: 15–25, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Kaidanovich-Beilin O, Woodgett JR. GSK-3: functional insights from cell biology and animal models. Front Mol Neurosci 4: 40, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klaus A, Zorman S, Berthier A, Polge C, Ramirez S, Michelland S, Seve M, Vertommen D, Rider M, Lentze N, Auerbach D, Schlattner U. Glutathione S-transferases interact with AMP-activated protein kinase: evidence for S-glutathionylation and activation in vitro. PLoS One 8: e62497, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kovacic S, Soltys CL, Barr AJ, Shiojima I, Walsh K, Dyck JR. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem 278: 39422–39427, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Levine YC, Li GK, Michel T. Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells. Evidence for an AMPK → Rac1 → Akt → endothelial nitric-oxide synthase pathway. J Biol Chem 282: 20351–20364, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Liu TF, Brown CM, El Gazzar M, McPhail L, Millet P, Rao A, Vachharajani VT, Yoza BK, McCall CE. Fueling the flame: bioenergy couples metabolism and inflammation. J Leukoc Biol 92: 499–507, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lochhead PA, Coghlan M, Rice SQ, Sutherland C. Inhibition of GSK-3 selectively reduces glucose-6-phosphatase and phosphatase and phosphoenolpyruvate carboxykinase gene expression. Diabetes 50: 937–946, 2001. [DOI] [PubMed] [Google Scholar]
- 37.Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest 116: 1776–1783, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo J. Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett 273: 194–200, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol 6: 777–784, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McBride A, Ghilagaber S, Nikolaev A, Hardie DG. The glycogen-binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab 9: 23–34, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meares GP, Qin H, Liu Y, Holdbrooks AT, Benveniste EN. AMP-activated protein kinase restricts IFN-gamma signaling. J Immunol 190: 372–380, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michiels C. Physiological and pathological responses to hypoxia. Am J Pathol 164: 1875–1882, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493: 346–355, 2013. [DOI] [PubMed] [Google Scholar]
- 44.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol 12: 695–708, 2011. [DOI] [PubMed] [Google Scholar]
- 45.Park DW, Jiang S, Tadie JM, Stigler WS, Gao Y, Deshane J, Abraham E, Zmijewski JW. Activation of AMPK enhances neutrophil chemotaxis and bacterial killing. Mol Med 19: 387–398, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patel S, Woodgett J. Glycogen synthase kinase-3 and cancer: good cop, bad cop? Cancer Cell 14: 351–353, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sag D, Carling D, Stout RD, Suttles J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol 181: 8633–8641, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sandstrom ME, Zhang SJ, Bruton J, Silva JP, Reid MB, Westerblad H, Katz A. Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. J Physiol 575: 251–262, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sha Y, Zmijewski J, Xu Z, Abraham E. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol 180: 2531–2537, 2008. [DOI] [PubMed] [Google Scholar]
- 50.Steenport M, Khan KM, Du B, Barnhard SE, Dannenberg AJ, Falcone DJ. Matrix metalloproteinase (MMP)-1 and MMP-3 induce macrophage MMP-9: evidence for the role of TNF-alpha and cyclooxygenase-2. J Immunol 183: 8119–8127, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suzuki T, Bridges D, Nakada D, Skiniotis G, Morrison SJ, Lin JD, Saltiel AR, Inoki K. Inhibition of AMPK catabolic action by GSK3. Mol Cell 50: 407–419, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tadie JM, Bae HB, Deshane JS, Bell CP, Lazarowski ER, Chaplin DD, Thannickal VJ, Abraham E, Zmijewski JW. Toll-like receptor 4 engagement inhibits adenosine 5′-monophosphate-activated protein kinase activation through a high mobility group box 1 protein-dependent mechanism. Mol Med 18: 659–668, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsoyi K, Jang HJ, Nizamutdinova IT, Kim YM, Lee YS, Kim HJ, Seo HG, Lee JH, Chang KC. Metformin inhibits HMGB1 release in LPS-treated RAW 264.7 cells and increases survival rate of endotoxaemic mice. Br J Pharmacol 162: 1498–1508, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 13: 2004–2008, 2003. [DOI] [PubMed] [Google Scholar]
- 55.Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, Martin SR, Carling D, Gamblin SJ. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449: 496–500, 2007. [DOI] [PubMed] [Google Scholar]
- 56.Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, Gamblin SJ. Structure of mammalian AMPK and its regulation by ADP. Nature 472: 230–233, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xing J, Wang Q, Coughlan K, Viollet B, Moriasi C, Zou MH. Inhibition of AMP-activated protein kinase accentuates lipopolysaccharide-induced lung endothelial barrier dysfunction and lung injury in vivo. Am J Pathol 182: 1021–1030, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang Z, Kahn BB, Shi H, Xue BZ. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem 285: 19051–19059, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yuskaitis CJ, Jope RS. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell Signal 21: 264–273, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang M, Dong Y, Xu J, Xie Z, Wu Y, Song P, Guzman M, Wu J, Zou MH. Thromboxane receptor activates the AMP-activated protein kinase in vascular smooth muscle cells via hydrogen peroxide. Circ Res 102: 328–337, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao X, Zmijewski JW, Lorne E, Liu G, Park YJ, Tsuruta Y, Abraham E. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol 295: L497–L504, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zmijewski JW, Banerjee S, Bae H, Friggeri A, Lazarowski ER, Abraham E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J Biol Chem 285: 33154–33164, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zmijewski JW, Lorne E, Banerjee S, Abraham E. Participation of mitochondrial respiratory complex III in neutrophil activation and lung injury. Am J Physiol Lung Cell Mol Physiol 296: L624–L634, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zmijewski JW, Lorne E, Zhao X, Tsuruta Y, Sha Y, Liu G, Abraham E. Antiinflammatory effects of hydrogen peroxide in neutrophil activation and acute lung injury. Am J Respir Crit Care Med 179: 694–704, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zmijewski JW, Lorne E, Zhao X, Tsuruta Y, Sha Y, Liu G, Siegal GP, Abraham E. Mitochondrial respiratory complex I regulates neutrophil activation and severity of lung injury. Am J Respir Crit Care Med 178: 168–179, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zmijewski JW, Zhao X, Xu Z, Abraham E. Exposure to hydrogen peroxide diminishes NF-κB activation, IκB-α degradation, and proteasome activity in neutrophils. Am J Physiol Cell Physiol 293: C255–C266, 2007. [DOI] [PubMed] [Google Scholar]