

Abstract
β-Adrenergic receptor (β-AR) blockade is widely used to treat heart failure, since the adverse effects of chronic β-AR stimulation are central to the pathogenesis of this disease state. Transgenic (Tg) mice, where β-AR signaling is chronically enhanced by overexpression of cardiac β2-ARs, is a surrogate for this mechanism, since these mice develop cardiomyopathy as reflected by reduced left ventricular (LV) function, increased fibrosis, apoptosis, and myocyte hypertrophy. We hypothesized that disruption of type 5 adenylyl cyclase (AC5), which is in the β-AR signaling pathway in the heart, but exerts only a minor β-AR blocking effect, could prevent the cardiomyopathy in β2-AR Tg mice without the negative effects of full β-AR blockade. Accordingly, we mated β2-AR Tg mice with AC5 knockout (KO) mice. The β2-AR Tg × AC5 KO bigenic mice prevented the cardiomyopathy as reflected by improved LV ejection fraction, reduced apoptosis, fibrosis, and myocyte size and preserved exercise capacity. The rescue was not simply due to a β-blocking effect of AC5 KO, since neither baseline LV function nor the response to isoproterenol was diminished substantially compared with the negative inotropic effects of β-blockade. However, AC5 disruption in β2-AR Tg activates the antioxidant, manganese superoxide dismutase, an important mechanism protecting the heart from cardiomyopathy. These results indicate that disruption of AC5 prevents the cardiomyopathy induced by chronically enhanced β-AR signaling in mice with overexpressed β2-AR, potentially by enhancing resistance to oxidative stress and apoptosis, suggesting a novel, alternative approach to β-AR blockade.
Keywords: β-adrenergic receptor signaling, oxidative stress
after some initial controversy, it is now well accepted that chronic β-adrenergic receptor (β-AR) stimulation is deleterious and is involved in the pathogenesis of heart failure (13, 14, 21, 24, 34, 52, 60), and that β-AR blockade is an important adjunct to heart failure therapy (7, 43a). However, these drugs also reduce cardiac function, which sometimes cannot be tolerated in patients with heart failure (7, 29, 35). Accordingly, it would be desirable to have a therapeutic agent that inhibits β-AR signaling distal to the β-AR, and that does not have a significant negative inotropic effect. One candidate is inhibition of adenylyl cyclase (AC), which transduces β-AR stimulation to increase cAMP. There are two major isoforms of AC in the heart, AC types 5 and 6. Type 5 regulates a lesser fraction of AC and cAMP in the heart (roughly 25–30%) (30, 47), suggesting it would be less likely to exert a strong negative inotropic effect when blocked.
Therefore, the goal of the present investigation was to determine if inhibiting chronic β-AR stimulation, specifically by inhibiting only type 5 adenylyl cyclase (AC5), using an AC5 knockout (KO) model, would be useful in countering the adverse effects of chronic β-AR stimulation, i.e., the decrease in function and cardiomyopathy that develops (13, 14, 52, 60). Transgenic mice with cardiac overexpressed β2-AR (β2-AR Tg) were used as the model to elicit chronic β-AR stimulation, since they exhibit increased cardiac function at a young age but develop decreased cardiac function and cardiomyopathy as they age (13, 14, 52, 60). To accomplish this goal, β2-AR Tg mice were mated with AC5 KO mice and were examined for cardiac dysfunction and other manifestations of cardiomyopathy, e.g., cardiac fibrosis, apoptosis, and increased myocyte cross-sectional area, and compared with wild-type (WT) controls and β2-AR Tg mice as they aged to determine if the cardiomyopathy was prevented in the bigenic mice. We also examined if the AC5 KO would also prevent other clinical signs of cardiomyopathy, almost always observed in patients with heart disease, e.g., exercise intolerance (5, 8, 11, 23, 37). Because our hypothesis was that prevention of the cardiomyopathy would not be mediated by simply decreasing AC activity and cAMP, we also measured effects on oxidative stress as a potential mechanism based on our prior studies demonstrating manganese superoxide dismutase (MnSOD) involvement in longevity (64) and catecholamine stress (36) in AC5 KO.
MATERIALS AND METHODS
Animal models.
The development and characterization of mice with cardiac-specific overexpression of the β2-AR used in this study have been described previously (13, 44). Parent β2-AR mice were obtained from Jackson Laboratories. Parent AC5 KO mice used in this study have been described previously (48, 64). The mice with cardiac-specific overexpression of the β2-AR were mated with AC5 KO mice to generate littermate wild-type (WT), β2-AR, AC5 KO, and β2-AR × AC5 KO bigenic mice. Four groups of animals were studied (WT, AC5 KO, β2-AR Tg, and β2-AR Tg × AC5 KO bigenic mice) at 13–16 mo of age, at a time when cardiomyopathy develops in the β2-AR Tg mice. All protocols concerning animal use were approved by the Institutional Animal Care and Use Committee at the New Jersey Medical School. All of the investigations conformed to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health.
Echocardiography.
Transthoracic echocardiography was performed in 13- to 16-mo age-matched mice using an Acuson Sequoia with a 13-MHz transducer. Mice were anesthetized with 2.5% tribromoethanol (Avertin) injected intraperitoneally and placed on a warmed saline bag. Electrocardiographic leads were attached to each limb using needle electrodes (Grass Technologies). After a short-axis two-dimensional (2-D) image of the left ventricle (LV) was obtained at the level of the papillary muscles, a 2-D guided M-mode tracing crossing the anterior and posterior wall was recorded at a sweep speed of 200 mm/s. The following parameters were measured on the M-mode tracings using the leading-edge technique: LV internal dimensions of diastole and systole (LVIDd, LVIDs), LV external dimensions of diastole and systole, and wall thickness at diastole and systole. LV ejection fraction (LVEF) was calculated by the cubed method: LVEF = [(LVIDd)3 − (LVIDs)3]/(LVIDd)3.
Exercise capacity.
Mice were exercised on the treadmill. All mice were subjected to a practice trial 3 days before the experiment to adapt to the treadmill testing environment. Food was withdrawn at least 3 h before the exercise. At the time of the experiment, each mouse was placed on a treadmill at a constant 10° angle. The treadmill was started at 4 m/min, and the speed incrementally increased 2 m/min every 2 min until the mice reached exhaustion. Exhaustion was defined as spending time (10 s) on the electric stimulus platform without attempting to reengage the treadmill belt. The maximal running distance was calculated based on the time and speed of running.
Isoproterenol challenge.
Echocardiography was performed in WT and AC5 KO mice anesthetized with 2.5% tribromoethanol (290 mg/kg). For acute injection of isoproterenol (ISO), a PE-10 catheter was inserted in the right jugular vein, ISO was injected at the rate of 0.04 μg·kg−1·min−1 for 5 min, and LVEF fraction was measured. In WT mice, after recovery, 0.5 mg/kg propranolol was administered intravenously followed by LVEF measurement. ISO was given again 3 min after propranolol was infused followed by LVEF measurement.
Western blot analysis.
Total proteins were extracted from the LV of hearts as previously described (28, 64). Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. Immunoblotting was performed using polyclonal antibodies to MnSOD from Millipore and cleaved caspase-3 from Cell Signaling. Immunodetection was accomplished using a donkey anti-rabbit secondary antibody (1:2,000 dilution) and the enhanced chemiluminescence kit (Amersham Biosciences). Expression of these proteins was quantified by densitometry, and the data are presented as arbitrary units of density. GAPDH antibody Western blotting was used to verify equal protein loading of the blots.
Superoxide dismutase activity.
The superoxide dismutase (SOD) activity was measured by an SOD Assay kit from Cayman Chemicals (Ann Arbor, MI) following the manufacturer's instructions.
Histology.
The heart was excised and washed in cold PBS. A ring of LV tissue, cut at the level of the papillary muscles, was fixed in 10% buffered formalin, processed, and embedded in paraffin. Sections were cut 6-μm thick and deparaffinized. Images were obtained using an Olympus CCD video camera (DP 71; Olympus) attached to an Olympus microscope (Olympus BX 51) with a ×40 objective lens. Collagen volume percent was assessed using samples stained with picric sirius red. Myocyte cross-sectional area was determined on sections stained with rhodamine-labeled wheat germ agglutinin (1:250; Vector). Apoptosis was determined by using terminal deoxyribonucleotide transferase-mediated dUTP nick end-labeling, which detects apoptosis-induced DNA fragmentation by nick-end labeling of the fragmented DNA at the 3′-hydroxyl ends (Terminal Transferase, recombinant kit; Roche Diagnostics). The nuclei were costained with 4′,6-diamino-2-phenylindole. These techniques have all been used extensively (36, 50–52, 64). To further discriminate apoptosis in myocytes from smaller nonmyocytes, tissue sections (5 μm thickness) were costained with α-actinin for myocytes as previously described (18, 50, 51).
Statistical analysis.
All data were expressed as means ± SE. To compare two independent groups, we used Student's unpaired t-test. For a comparison of three or more groups, one-way ANOVA with Newman-Keuls post hoc test was used. P < 0.05 was taken as a minimal level of significance.
RESULTS
Cardiomyopathy in β2-AR Tg mice.
β2-AR Tg mice compared with WT developed cardiomyopathy as evidenced by decreased (P < 0.05) baseline LVEF (53 ± 3 vs. 63 ± 1%) (Fig. 1A), fractional shortening (FS) (23 ± 1 vs. 28 ± 1%), as well as increased LV end-diastolic and end-systolic diameter (Table 1). In addition, LV weight-to-tibial length (wt/TL) ratio, an index of LV hypertrophy, and lung wt/TL ratio, an index of LV decompensation, were also elevated in old β1-AR Tg (Table 1). Maximal running distance during treadmill exercise was significantly reduced in β2-AR Tg mice compared with WT mice (Fig. 1B). Furthermore, histological evidence of cardiomyopathy was also present, as reflected by increased levels, compared with WT, of myocardial fibrosis (18.1 ± 2.6 vs. 2.1 ± 0.9%, P < 0.05) and LV hypertrophy, as assessed by myocyte cross-sectional area (756 ± 57 vs. 477 ± 35 mm2) (Fig. 1, C and D). Apoptosis was also increased in the heart, P < 0.05, which occurred predominantly in nonmyocytes, and to a much lesser extent, in myocytes (Fig. 2A). Importantly, baseline levels of all these measurements in old AC5 KO were not different from WT, as exemplified by LVEF, which was 70 ± 2%, similar to the values in old WT shown above.
Fig. 1.

Type 5 adenylyl cyclase (AC5) knockout (KO) prevents β2-adrenergic receptor (β2-AR) cardiomyopathy. β2-AR transgenic (Tg) mice develop cardiomyopathy at the age of 13–15 mo. A: left ventricle ejection fraction (LVEF) was significantly depressed in old β2 Tg mice (n = 14) compared with wild-type mice (WT, n = 17). In contrast to the depressed left ventricle (LV) function observed in old β2 Tg mice, the old β2 Tg × AC5 KO bigenic mice (n = 30) exhibited normal cardiac function. B: exercise capacity as determined by maximal running distance was significantly reduced in β2 Tg mice (n = 8) compared with WT mice (n = 8). However, reduced exercise capacity was not observed in the β2 Tg × AC5 KO bigenic mice (n = 9). The representative images (left) and quantification (right) of fibrosis (C) and myocyte size (D) showed significantly higher levels in β2 Tg hearts compared with WT. In contrast to the β2 Tg mice, fibrosis and myocyte size in the bigenic mice were no longer different from WT; n = 4/group. The original magnification of hematoxylin and eosin (H&E) staining images for fibrosis was ×20. The original magnification of wheat germ agglutinin (WGA) staining images for myocyte size was ×40. Results are expressed as means ± SE. *P <0.05 by one-way ANOVA. WT and AC5 KO values were not significantly different, except for exercise capacity (n = 6 for AC5 KO), which was increased significantly, P <0.05, in AC5 KO.
Table 1.
LV function and pathology
| WT | AC5 KO | β2-AR Tg | β2-AR Tg × AC5 KO | |
|---|---|---|---|---|
| Echocardiographic results | ||||
| n | 17 | 11 | 14 | 12 |
| Age, mo | 16.2 | 15.1 | 15.6 | 15.1 |
| LVEF, % | 63 ± 1 | 68 ± 2* | 53 ± 3* | 70 ± 1† |
| Fraction shortening, % | 28 ± 1 | 32 ± 1* | 23 ± 1* | 33 ± 1† |
| End-diastolic diameter, mm | 4.0 ± 0.1 | 3.8 ± 0.1 | 4.5 ± 0.2* | 3.8 ± 0.1† |
| End-systolic diameter, mm | 2.9 ± 0.1 | 2.6 ± 0.1 | 3.5 ± 0.2* | 2.5 ± 0.1† |
| Pathology | ||||
| n | 10 | 11 | 9 | 12 |
| Body wt, g | 36 ± 1 | 29 ± 1* | 36 ± 2 | 30 ± 2† |
| LV wt, mg | 117 ± 7 | 96 ± 6* | 136 ± 6* | 95 ± 5† |
| Lung wt, mg | 171 ± 5 | 160 ± 5 | 196 ± 9* | 166 ± 6† |
| LV wt/body wt | 3.3 ± 0.2 | 3.4 ± 0.2 | 3.9 ± 0.3* | 3.2 ± 0.1† |
| Tibia length, mm | 18.3 ± 0.2 | 18.1 ± 0.1 | 18.4 ± 0.1 | 18.4 ± 0.2 |
| LV wt/TL | 6.1 ± 0.3 | 5.3 ± 0.3 | 7.4 ± 0.3* | 5.1 ± 0.2† |
| Lung wt/TL | 9.4 ± 0.3 | 8.9 ± 0.2 | 10.6 ± 0.5* | 9.0 ± 0.3† |
Values are means ± SE; n, no. of mice.
LV, left ventricle; WT, wild type; AC5, type 5 adenylyl cyclase; KO, knockout; β2-AR, β2-adrenergic receptor; LVEF, left ventricular ejection fraction.
P < 0.005 vs. WT;
P < 0.05 vs. β2 Tg.
Fig. 2.

AC5 disruption prevents apoptosis in β2-AR cardiomyopathy predominantly in nonmyocytes, and to a much lesser extent, in myocytes. A: quantification of terminal deoxyribonucleotide transferase-mediated dUTP nick end-labeling (TUNEL) assay in LV sections of old mice showed that nonmyocyte apoptosis was significantly rescued in β2 Tg × AC5 KO bigenic mice, n = 4/each group. B: representative images of the staining used to discriminate apoptosis in myocytes (bottom) from nonmyocytes (top). Myocytes were stained with sarcomeric α-actinin (red) and counterstained with TUNEL (green) and 4′,6-diamino-2-phenylindole (DAPI, blue). The original magnification of the images was × 40. C: the levels of cleaved caspase 3 were significantly higher in β2 Tg hearts compared with AC5 KO. In contrast to the β2 Tg mice, the bigenic mice were no longer different from WT; n = 4/group. Results are expressed as means ± SE. *P <0.05 by one-way ANOVA. WT and AC5 KO values were not significantly different.
Mating β2-AR Tg with AC5 KO mice prevents the cardiomyopathy in β2-AR Tg mice.
The cardiomyopathy was prevented in age-matched bigenic mice (β2-AR Tg × AC5 KO), as reflected by normal LVEF (70 ± 1%) and FS (33 ± 1%) (Fig. 1A). Increased LV end-diastolic and end-systolic diameter, as well as LV wt/TL ratio and Lung wt/TL ratio, were diminished in bigenic β2-AR Tg × AC5 KO mice (Table 1). Furthermore, apoptosis and fibrosis in the bigenic mice were no longer different from WT (Fig. 1, C and D, and Fig. 2, A and B). Reduced exercise capacity was also restored in the β2-AR Tg × AC5 KO bigenic mice (Fig. 1B).
The prevention of cardiomyopathy by AC5 disruption is not simply due to β-AR blockade.
Baseline LVEF was not decreased in AC5 KO mice compared with WT mice, whereas β-AR blockade with propranolol treatment reduced baseline LVEF significantly (P < 0.05, Fig. 3). The response to ISO challenge increased LVEF in WT and AC5 KO mice similarly, whereas β-AR blockade with propranolol fully abolished the increase in LVEF (Fig. 3). These data confirm that AC5 KO is not a β−blocker, and the prevention of cardiomyopathy by AC5 disruption is not simply due to β−blockade.
Fig. 3.
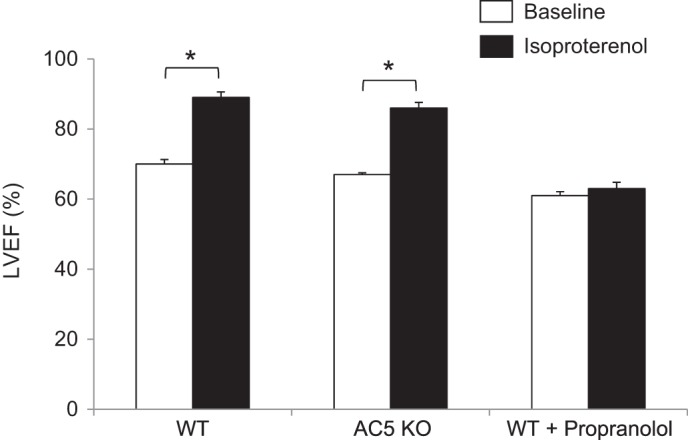
The effect of AC5 KO is not simply due to β−blockade. There is no decrease in baseline LVEF in AC5 KO mice, whereas β−blocker, propranolol, treatment reduced baseline LVEF. The response to isoproterenol (ISO) challenge is similar in WT (n = 6) and AC5KO (n = 4) mice. In contrast, the increase in LVEF with ISO is fully abolished by propranolol (n = 5). *P < 0.05 vs. WT by Student's t-test analysis.
Protection of cardiomyopathy in β2-AR Tg mice by inhibition of oxidative stress.
A major mechanism mediating the cardiac apoptosis and necrosis in cardiomyopathy is increased oxidative stress, and SOD is a major protective mechanism for oxidative stress. SOD activity and MnSOD levels were reduced in β2-AR Tg hearts but were rescued in the bigenic β2-AR Tg × AC5 KO hearts (Fig. 4), suggesting that this mechanisms is important in mediating the prevention of β2-AR cardiomyopathy by AC5 disruption.
Fig. 4.

Manganese superoxide dismutase (MnSOD) is activated in β2 Tg × AC5 KO bigenic mice. MnSOD expression levels determined by Western blotting (A) and superoxide dismutase (SOD) activity (B) were significantly downregulated in β2 Tg mice but preserved in β2 Tg × AC5 KO bigenic mice compared with WT mice; n = 4–7/group. Results are expressed as means ± SE. *P <0.05 by one-way ANOVA. MnSOD activity was increased significantly, P <0.05, in AC5 KO mice.
DISCUSSION
We demonstrated in this study that AC5 inhibition prevents the cardiomyopathy induced by chronic β-adrenergic receptor stimulation. The cardiomyopathy that developed as the mice aged was characterized by reduced cardiac function, increased cardiac apoptosis, fibrosis, and myocyte cross-sectional area, with reduced exercise tolerance. Except for reduced exercise tolerance, which has not been noted previously, prior studies also demonstrated the development of cardiomyopathy and increased mortality in β2-AR Tg mice (13, 52). Mortality was not examined in the current study because many mice had to be killed for the histological and biochemical studies, which reduced the power and statistical validity of a mortality analysis. A prior study indicated that the cause of the increased mortality in β2-AR Tg mice was due primarily to heart failure and potentially to arrhythmias, since ectopic beats were observed on the ECG (13). It is likely that the AC5 KO also protects against arrhythmias, since AC5 Tg mice are more susceptible to arrhythmias than AC5 KO or WT (65).
The potentially most clinically attractive feature of this prevention is that the older AC5 KO do not have reduced LV function at baseline, so adding AC5 inhibition to heart failure should not have the potential to reduce cardiac function further, yet the AC5 KO prevention of the adverse effects of chronic β-adrenergic receptor stimulation is still observed. One might predict that reducing AC activity will, by itself, be responsible for rescuing β-AR cardiomyopathy. However, the main source of AC activity in the heart is AC6, not AC5. We have demonstrated that AC5 KO reduces total AC activity in the heart by only 25–30% (30, 47). Our data in this study showing that AC5 KO did not act as a β-blocker also support the concept that the prevention of β2-AR Tg cardiomyopathy must involve a complex interaction of mechanisms, and is not simply due to the modest decrease in AC activity.
For example, there is considerable evidence supporting the concept that increased oxidative stress and apoptosis are involved in the pathogenesis of various types of cardiomyopathies (55), including dilated (9, 46), diabetic (57, 63), ischemic (3, 12), hypertensive (20), adriamycin-induced (39), and pressure overload (10, 31, 59) cardiomyopathies. Conversely, antioxidants, such as MnSOD, are able to protect the heart against apoptosis and development of cardiomyopathy (22, 32), whereas mice deficient in MnSOD die soon after birth with cardiomyopathy (22, 32, 40). Based on our previous findings that the protection against oxidative stress through activation of MnSOD is a powerful mechanism leading to longevity (64) as well as involved in resistance to cardiomyopathy induced by catecholamine stress in the AC5 KO mice (36), it is conceivable that resistance to oxidative stress could be an important mechanism in the protection against cell death and prevention of cardiomyopathy in β2-AR Tg × AC5 KO bigenic mice. Indeed, the β2-AR Tg mice showed increased necrosis, as reflected by myocardial fibrosis, and increased apoptosis when cardiomyopathy develops, and these Tg mice are less tolerant to oxidative stress, whereas fibrosis and apoptosis were normalized in β2-AR Tg × AC5 KO bigenic mice, and MnSOD was augmented in β2-AR Tg × AC5 KO mice, supporting the conclusion that prevention of oxidative stress in the AC5 KO mice plays a role in preventing the cardiomyopathy in β2-AR Tg. Moreover, we previously demonstrated that upregulation of MnSOD and resistance to oxidative stress in the AC5 KO heart are mediated by activation of MEK/ERK and SIRT1/FoxO3a pathways (36, 64). It is conceivable that MEK/ERK and SIRT1/FoxO3a-mediated activation of MnSOD is also involved in prevention of cardiomyopathy developed in β2 Tg mice.
Interestingly, the predominant fraction of the increased apoptosis in the cardiomyopathy and the decrease, when the cardiomyopathy was prevented by the AC5 KO mechanism, occurred in nonmyocytes. Whereas this might be understood, recognizing that over 70% of cells in the heart are nonmyocytes, it seems inconsistent with the currently held concept that apoptosis leads to cardiac dysfunction through reducing myocyte numbers (4, 19). However, the current results are consistent with our previous findings that the apoptosis occurs predominantly in nonmyocytes in several models of cardiomyopathy, including ischemic cardiomyopathy (50) and hypertrophic cardiomyopathy (18, 51). Based on our previous studies, the cell types of apoptotic nonmyocytes mainly include macrophages, neutrophils, fibroblasts, and endothelial cells (50, 51). The role of nonmyocyte apoptosis in the heart in mediating or preventing the cardiomyopathy is not clear, since the nonmyocytes are a double-edged sword. On the one hand, many of these cell types have been linked to myocyte cell damage (1, 17, 18, 33, 41, 42, 50), whereas on the other hand they have been linked to rescue (25, 38, 58, 61). For example, activation of both macrophages or myofibroblasts plays a beneficial role in tissue repair and the improvement of cardiac remodeling and function after myocardial infarction (38, 62).
Exercise intolerance is not only uniformly observed in patients with heart failure but is generally an early sign of cardiac dysfunction that precedes evidence of reduced baseline function at rest (2, 5, 26, 37, 53). However, exercise capacity is rarely examined in mouse models of cardiomyopathy, and has not been examined in transgenic mice, where β-AR signaling is chronically enhanced. However, it is known that, when β-AR signaling is enhanced in young mice, either genetically by increasing components of the β-AR signaling pathway (16) or pharmacologically (45), exercise performance is improved. AC5 KO mice exhibit enhanced exercise performance, whereas the older β2-AR Tg mice that developed cardiomyopathy had depressed exercise capacity, which was rescued in the bigenic mice. This finding is important because simply rescuing baseline cardiac function in patients with cardiomyopathy does not necessarily indicate that exercise tolerance will be normalized, leading to an improved quality of life.
Our current study supports the concept that inhibition of AC5 may be a novel approach to the treatment of cardiomyopathy/heart failure. β-Blockers are well-established drugs for treating heart failure (43a). However, because β-blockers can cause contractile dysfunction (15, 27, 54), some patients are intolerant to the administration of β-blockers (7, 29, 35), and the number of such patients who cannot benefit from β-blockers is, roughly estimated, to be over 1.3 million (43, 56). Inhibition of AC5 may be superior to inhibition of β-AR stimulation, since it does not decrease LV contractility (47–49, 64), and may lead to more effective β-AR desensitization (49). To demonstrate this point, we compared the extent to which the response to β-AR stimulation was blocked either by the AC5 KO or by a β-AR blocker propranolol. AC5 KO reduced the response to isoproterenol minimally, whereas propranolol completely abolished it. Thus, the development of a pharmacological AC5 inhibitor could be a useful alternative to β-blockers.
In summary, AC5 inhibition prevented the cardiomyopathy and depressed exercise tolerance induced by chronic β-adrenergic receptor stimulation. The prevention of cardiomyopathy by AC5 inhibition is not simply due, as might be thought, to reduced AC activity, thereby inhibiting sympathetic stimulation, as occurs with β-AR blockade, since AC activity is regulated to a minor extent by AC5 in the heart and consequently sympathetic stimulation was not blocked in the AC5 KO as it was with propranolol. Therefore, it is likely that there are distal mechanisms mediating the prevention, e.g., activation of SOD, which enhances resistance to oxidative stress through AC5 inhibition. Thus, AC5 inhibition may be a useful adjunct to β-AR blockade as a treatment for heart failure with the advantage of less inhibition of contractility.
GRANTS
This study was supported by National Institutes of Health Grants 5P01-AG-027211, 5R21-HL097264, 1R01-HL-102472, 5R01-HL-033107, 5T32-HL-069752, 5R01-HL-095888, 5P01-HL-069020, 5R01-HL-091781, R01-HL-106511, R01-HL-093481, and 1R01-HL-119464.
DISCLOSURES
The authors declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
Author contributions: L.Y. performed experiments; L.Y. analyzed data; L.Y., S.F.V., and D.E.V. interpreted results of experiments; L.Y. prepared figures; L.Y. and S.F.V. drafted manuscript; L.Y., S.F.V., and D.E.V. edited and revised manuscript; S.F.V. and D.E.V. conception and design of research; S.F.V. and D.E.V. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Drs. Chunbo Wang, Shumin Gao, Grace Lee, Yimin Tian, Hui Ge, Chujun Yuan, and Ronald Pachon for providing support.
REFERENCES
- 1.Adameova A, Dhalla NS. Role of microangiopathy in diabetic cardiomyopathy. Heart Fail Rev 19: 25–33, 2014. [DOI] [PubMed] [Google Scholar]
- 2.Azarbal F, Singh M, Finocchiaro G, Le VV, Schnittger I, Wang P, Myers J, Ashley E, Perez M. Exercise capacity and paroxysmal atrial fibrillation in patients with hypertrophic cardiomyopathy. Heart 100: 624–630, 2014. [DOI] [PubMed] [Google Scholar]
- 3.Bandyopadhyay D, Chattopadhyay A, Ghosh G, Datta AG. Oxidative stress-induced ischemic heart disease: protection by antioxidants. Curr Med Chem 11: 369–387, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Bernecker OY, Huq F, Heist EK, Podesser BK, Hajjar RJ. Apoptosis in heart failure and the senescent heart. Cardiovasc Toxicol 3: 183–190, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Borlaug BA. Mechanisms of exercise intolerance in heart failure with preserved ejection fraction. Circ J 78: 20–32, 2013. [DOI] [PubMed] [Google Scholar]
- 7.Bristow MR. β-Adrenergic receptor blockade in chronic heart failure. Circulation 101: 558–569, 2000. [DOI] [PubMed] [Google Scholar]
- 8.Brubaker PH. Exercise intolerance in congestive heart failure: a lesson in exercise physiology. J Cardiopulm Rehabil 17: 217–221, 1997. [DOI] [PubMed] [Google Scholar]
- 9.Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res 89: 279–286, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Condorelli G, Morisco C, Stassi G, Notte A, Farina F, Sgaramella G, de Rienzo A, Roncarati R, Trimarco B, Lembo G. Increased cardiomyocyte apoptosis and changes in proapoptotic and antiapoptotic genes bax and bcl-2 during left ventricular adaptations to chronic pressure overload in the rat. Circulation 99: 3071–3078, 1999. [DOI] [PubMed] [Google Scholar]
- 11.Conraads VM, Van Craenenbroeck EM, De Maeyer C, Van Berendoncks AM, Beckers PJ, Vrints CJ. Unraveling new mechanisms of exercise intolerance in chronic heart failure: role of exercise training. Heart Fail Rev 18: 65–77, 2013. [DOI] [PubMed] [Google Scholar]
- 12.Dhalla NS, Elmoselhi AB, Hata T, Makino N. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc Res 47: 446–456, 2000. [DOI] [PubMed] [Google Scholar]
- 13.Du XJ, Gao XM, Wang B, Jennings GL, Woodcock EA, Dart AM. Age-dependent cardiomyopathy and heart failure phenotype in mice overexpressing beta(2)-adrenergic receptors in the heart. Cardiovasc Res 48: 448–454, 2000. [DOI] [PubMed] [Google Scholar]
- 14.Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci USA 96: 7059–7064, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Epstein S, Robinson BF, Kahler RL, Braunwald E. Effects of beta-adrenergic blockade on the cardiac response to maximal and submaximal exercise in man. J Clin Invest 44: 1745–1753, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esposito G, Perrino C, Ozaki T, Takaoka H, Defer N, Petretta MP, De Angelis MC, Mao L, Hanoune J, Rockman HA, Chiariello M. Increased myocardial contractility and enhanced exercise function in transgenic mice overexpressing either adenylyl cyclase 5 or 8. Basic Res Cardiol 103: 22–30, 2008. [DOI] [PubMed] [Google Scholar]
- 17.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res 110: 159–173, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gelpi RJ, Park M, Gao S, Dhar S, Vatner DE, Vatner SF. Apoptosis in severe, compensated pressure overload predominates in nonmyocytes and is related to the hypertrophy but not function. Am J Physiol Heart Circ Physiol 300: H1062–H1068, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldspink DF, Burniston JG, Tan LB. Cardiomyocyte death and the ageing and failing heart. Exp Physiol 88: 447–458, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez A, Fortuno MA, Querejeta R, Ravassa S, Lopez B, Lopez N, Diez J. Cardiomyocyte apoptosis in hypertensive cardiomyopathy. Cardiovasc Res 59: 549–562, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Haber HL, Simek CL, Gimple LW, Bergin JD, Subbiah K, Jayaweera AR, Powers ER, Feldman MD. Why do patients with congestive heart failure tolerate the initiation of beta-blocker therapy? Circulation 88: 1610–1619, 1993. [DOI] [PubMed] [Google Scholar]
- 22.Hare JM. Oxidative stress and apoptosis in heart failure progression. Circ Res 89: 198–200, 2001. [PubMed] [Google Scholar]
- 23.Harrington D, Coats AJ. Mechanisms of exercise intolerance in congestive heart failure. Curr Opin Cardiol 12: 224–232, 1997. [DOI] [PubMed] [Google Scholar]
- 24.Hasking GJ, Esler MD, Jennings GL, Burton D, Johns JA, Korner PI. Norepinephrine spillover to plasma in patients with congestive heart failure: evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation 73: 615–621, 1986. [DOI] [PubMed] [Google Scholar]
- 25.Hayakawa K, Takemura G, Kanoh M, Li Y, Koda M, Kawase Y, Maruyama R, Okada H, Minatoguchi S, Fujiwara T, Fujiwara H. Inhibition of granulation tissue cell apoptosis during the subacute stage of myocardial infarction improves cardiac remodeling and dysfunction at the chronic stage. Circulation 108: 104–109, 2003. [DOI] [PubMed] [Google Scholar]
- 26.Haykowsky MJ, Brubaker PH, John JM, Stewart KP, Morgan TM, Kitzman DW. Determinants of exercise intolerance in elderly heart failure patients with preserved ejection fraction. J Am Coll Cardiol 58: 265–274, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higgins CB, Vatner SF, Franklin D, Braunwald E. Extent of regulation of the heart's contractile state in the conscious dog by alteration in the frequency of contraction. J Clin Invest 52: 1187–1194, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu CL, Chandra R, Ge H, Pain J, Yan L, Babu G, Depre C, Iwatsubo K, Ishikawa Y, Sadoshima J, Vatner SF, Vatner DE. Adenylyl cyclase type 5 protein expression during cardiac development and stress. Am J Physiol Heart Circ Physiol 297: H1776–H1782, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hunt SA. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure). J Am Coll Cardiol 46: e1–e82, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Iwatsubo K, Minamisawa S, Tsunematsu T, Nakagome M, Toya Y, Tomlinson JE, Umemura S, Scarborough RM, Levy DE, Ishikawa Y. Direct inhibition of type 5 adenylyl cyclase prevents myocardial apoptosis without functional deterioration. J Biol Chem 279: 40938–40945, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Jacob MH, Pontes MR, Araujo AS, Barp J, Irigoyen MC, Llesuy SF, Ribeiro MF, Bello-Klein A. Aortic-banding induces myocardial oxidative stress and changes in concentration and activity of antioxidants in male Wistar rats. Life Sci 79: 2187–2193, 2006. [DOI] [PubMed] [Google Scholar]
- 32.Kannan K, Jain SK. Oxidative stress and apoptosis. Pathophysiology 7: 153–163, 2000. [DOI] [PubMed] [Google Scholar]
- 33.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123: 594–604, 2011. [DOI] [PubMed] [Google Scholar]
- 34.Kiuchi K, Shannon RP, Komamura K, Cohen DJ, Bianchi C, Homcy CJ, Vatner SF, Vatner DE. Myocardial beta-adrenergic receptor function during the development of pacing-induced heart failure. J Clin Invest 91: 907–914, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ko DT, Hebert PR, Coffey CS, Curtis JP, Foody JM, Sedrakyan A, Krumholz HM. Adverse effects of beta-blocker therapy for patients with heart failure: a quantitative overview of randomized trials. Arch Intern Med 164: 1389–1394, 2004. [DOI] [PubMed] [Google Scholar]
- 36.Lai L, Yan L, Gao S, Hu CL, Ge H, Davidow A, Park M, Bravo C, Iwatsubo K, Ishikawa Y, Auwerx J, Sinclair DA, Vatner SF, Vatner DE. Type 5 adenylyl cyclase increases oxidative stress by transcriptional regulation of manganese superoxide dismutase via the SIRT1/FoxO3a pathway. Circulation 127: 1692–1701, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le VV, Perez MV, Wheeler MT, Myers J, Schnittger I, Ashley EA. Mechanisms of exercise intolerance in patients with hypertrophic cardiomyopathy. Am Heart J 158: e27–e34, 2009. [DOI] [PubMed] [Google Scholar]
- 38.Leor J, Rozen L, Zuloff-Shani A, Feinberg MS, Amsalem Y, Barbash IM, Kachel E, Holbova R, Mardor Y, Daniels D, Ocherashvilli A, Orenstein A, Danon D. Ex vivo activated human macrophages improve healing, remodeling, and function of the infarcted heart. Circulation 114: I94–I100, 2006. [DOI] [PubMed] [Google Scholar]
- 39.Li T, Danelisen I, Bello-Klein A, Singal PK. Effects of probucol on changes of antioxidant enzymes in adriamycin-induced cardiomyopathy in rats. Cardiovasc Res 46: 523–530, 2000. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 11: 376–381, 1995. [DOI] [PubMed] [Google Scholar]
- 41.Lindner D, Zietsch C, Tank J, Sossalla S, Fluschnik N, Hinrichs S, Maier L, Poller W, Blankenberg S, Schultheiss HP, Tschope C, Westermann D. Cardiac fibroblasts support cardiac inflammation in heart failure (Abstract). Basic Res Cardiol 109: 428, 2014. [DOI] [PubMed] [Google Scholar]
- 42.Litt MR, Jeremy RW, Weisman HF, Winkelstein JA, Becker LC. Neutrophil depletion limited to reperfusion reduces myocardial infarct size after 90 minutes of ischemia. Evidence for neutrophil-mediated reperfusion injury. Circulation 80: 1816–1827, 1989. [DOI] [PubMed] [Google Scholar]
- 43.Mannino DM, Homa DM, Akinbami LJ, Moorman JE, Gwynn C, Redd SC. Surveillance for asthma–United States, 1980–1999. MMWR Surveill Summ 51: 1–13, 2002. [PubMed] [Google Scholar]
- 43a.MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 353: 2001–2007, 1999. [PubMed] [Google Scholar]
- 44.Milano CA, Allen LF, Rockman HA, Dolber PC, McMinn TR, Chien KR, Johnson TD, Bond RA, Lefkowitz RJ. Enhanced myocardial function in transgenic mice overexpressing the beta 2-adrenergic receptor. Science 264: 582–586, 1994. [DOI] [PubMed] [Google Scholar]
- 45.Miura S, Kawanaka K, Kai Y, Tamura M, Goto M, Shiuchi T, Minokoshi Y, Ezaki O. An increase in murine skeletal muscle peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) mRNA in response to exercise is mediated by beta-adrenergic receptor activation. Endocrinology 148: 3441–3448, 2007. [DOI] [PubMed] [Google Scholar]
- 46.Narula J, Pandey P, Arbustini E, Haider N, Narula N, Kolodgie FD, Dal Bello B, Semigran MJ, Bielsa-Masdeu A, Dec GW, Israels S, Ballester M, Virmani R, Saxena S, Kharbanda S. Apoptosis in heart failure: release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proc Natl Acad Sci USA 96: 8144–8149, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okumura S, Kawabe J, Yatani A, Takagi G, Lee MC, Hong C, Liu J, Takagi I, Sadoshima J, Vatner DE, Vatner SF, Ishikawa Y. Type 5 adenylyl cyclase disruption alters not only sympathetic but also parasympathetic and calcium-mediated cardiac regulation. Circ Res 93: 364–371, 2003. [DOI] [PubMed] [Google Scholar]
- 48.Okumura S, Takagi G, Kawabe J, Yang G, Lee MC, Hong C, Liu J, Vatner DE, Sadoshima J, Vatner SF, Ishikawa Y. Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc Natl Acad Sci USA 100: 9986–9990, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okumura S, Vatner DE, Kurotani R, Bai Y, Gao S, Yuan Z, Iwatsubo K, Ulucan C, Kawabe J, Ghosh K, Vatner SF, Ishikawa Y. Disruption of type 5 adenylyl cyclase enhances desensitization of cyclic adenosine monophosphate signal and increases Akt signal with chronic catecholamine stress. Circulation 116: 1776–1783, 2007. [DOI] [PubMed] [Google Scholar]
- 50.Park M, Shen YT, Gaussin V, Heyndrickx GR, Bartunek J, Resuello RR, Natividad FF, Kitsis RN, Vatner DE, Vatner SF. Apoptosis predominates in nonmyocytes in heart failure. Am J Physiol Heart Circ Physiol 297: H785–H791, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park M, Vatner SF, Yan L, Gao S, Yoon S, Lee GJ, Xie LH, Kitsis RN, Vatner DE. Novel mechanisms for caspase inhibition protecting cardiac function with chronic pressure overload (Abstract). Basic Res Cardiol 108: 324, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peter PS, Brady JE, Yan L, Chen W, Engelhardt S, Wang Y, Sadoshima J, Vatner SF, Vatner DE. Inhibition of p38 alpha MAPK rescues cardiomyopathy induced by overexpressed beta 2-adrenergic receptor, but not beta 1-adrenergic receptor. J Clin Invest 117: 1335–1343, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pina IL, Apstein CS, Balady GJ, Belardinelli R, Chaitman BR, Duscha BD, Fletcher BJ, Fleg JL, Myers JN, Sullivan MJ. Exercise and heart failure: A statement from the American Heart Association Committee on exercise, rehabilitation, and prevention. Circulation 107: 1210–1225, 2003. [DOI] [PubMed] [Google Scholar]
- 54.Reiken S, Gaburjakova M, Gaburjakova J, He Kl KL, Prieto A, Becker E, Yi Gh GH, Wang J, Burkhoff D, Marks AR. beta-adrenergic receptor blockers restore cardiac calcium release channel (ryanodine receptor) structure and function in heart failure. Circulation 104: 2843–2848, 2001. [DOI] [PubMed] [Google Scholar]
- 55.Romero-Alvira D, Roche E, Placer L. Cardiomyopathies and oxidative stress. Med Hypotheses 47: 137–144, 1996. [DOI] [PubMed] [Google Scholar]
- 56.Sidney S, Sorel M, Quesenberry CP, Jr., DeLuise C, Lanes S, Eisner MD. COPD and incident cardiovascular disease hospitalizations and mortality: Kaiser Permanente Medical Care Program. Chest 128: 2068–2075, 2005. [DOI] [PubMed] [Google Scholar]
- 57.Singal PK, Bello-Klein A, Farahmand F, Sandhawalia V. Oxidative stress and functional deficit in diabetic cardiomyopathy. Adv Exp Med Biol 498: 213–220, 2001. [DOI] [PubMed] [Google Scholar]
- 58.Sun J, Li SH, Liu SM, Wu J, Weisel RD, Zhuo YF, Yau TM, Li RK, Fazel SS. Improvement in cardiac function after bone marrow cell thearpy is associated with an increase in myocardial inflammation. Am J Physiol Heart Circ Physiol 296: H43–H50, 2009. [DOI] [PubMed] [Google Scholar]
- 59.Teiger E, Than VD, Richard L, Wisnewsky C, Tea BS, Gaboury L, Tremblay J, Schwartz K, Hamet P. Apoptosis in pressure overload-induced heart hypertrophy in the rat. J Clin Invest 97: 2891–2897, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Turki J, Lorenz JN, Green SA, Donnelly ET, Jacinto M, Liggett SB. Myocardial signaling defects and impaired cardiac function of a human beta 2-adrenergic receptor polymorphism expressed in transgenic mice. Proc Natl Acad Sci USA 93: 10483–10488, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol 170: 818–829, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van den Borne SW, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol 7: 30–37, 2010. [DOI] [PubMed] [Google Scholar]
- 63.Wold LE, Ceylan-Isik AF, Ren J. Oxidative stress and stress signaling: menace of diabetic cardiomyopathy. Acta Pharmacol Sin 26: 908–917, 2005. [DOI] [PubMed] [Google Scholar]
- 64.Yan L, Vatner DE, O'Connor JP, Ivessa A, Ge H, Chen W, Hirotani S, Ishikawa Y, Sadoshima J, Vatner SF. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell 130: 247–258, 2007. [DOI] [PubMed] [Google Scholar]
- 65.Zhao Z, Babu GJ, Fefelova G, Ishikawa Y, Iwatsubo K, Lai L, Yan L, Vatner DE, Vatner SF, Xie LH. Overexpression of adenylyl cyclase type 5 (AC5) in the heart predisposes to cardiac arrhythmias. Circulation 122: A17445 (Abstract), 2010. [Google Scholar]