

Summary
Two vitamins, biotin and lipoic acid, are essential in all three domains of life. Both coenzymes function only when covalently attached to key metabolic enzymes. There they act as “swinging arms” that shuttle intermediates between two active sites (= covalent substrate channeling) of key metabolic enzymes. Although biotin was discovered over 100 years ago and lipoic acid 60 years ago, it was not known how either coenzyme is made until recently. In Escherichia coli the synthetic pathways for both coenzymes have now been worked out for the first time.
The late steps of biotin synthesis, those involved in assembling the fused rings, were well-described biochemically years ago, although recent progress has been made on the BioB reaction, the last step of the pathway in which the biotin sulfur moiety is inserted. In contrast, the early steps of biotin synthesis, assembly of the fatty acid-like “arm” of biotin were unknown. It has now been demonstrated that the arm is made by using disguised substrates to gain entry into the fatty acid synthesis pathway followed by removal of the disguise when the proper chain length is attained. The BioC methyltransferase is responsible for introducing the disguise and the BioH esterase for its removal.
In contrast to biotin, which is attached to its cognate proteins as a finished molecule, lipoic acid is assembled on its cognate proteins. An octanoyl moiety is transferred from the octanoyl-ACP of fatty acid synthesis to a specific lysine residue of a cognate protein by the LipB octanoyl transferase followed by sulfur insertion at carbons C6 and C8 by the LipA lipoyl synthetase. Assembly on the cognate proteins regulates the amount of lipoic acid synthesized and thus there is no transcriptional control of the synthetic genes. In contrast transcriptional control of the biotin synthetic genes is wielded by a remarkably sophisticated, yet simple, system, exerted through BirA a dual function protein that both represses biotin operon transcription and ligates biotin to its cognate protein.
1. INTRODUCTION
Biotin (vitamin H, viamin B7 or 5-[(3aS,4S,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoic acid) and lipoic acid (5-[(3R)-dithiolan-3-yl]pentanoic acid, also called 6,8-thioctic acid) share many similarities (Fig. 1). Both vitamins are essential for aerobic growth of E. coli and S. enterica whereas biotin is also required for growth of these bacteria under anaerobic conditions. Both biotin and lipoic acid must be covalently attached to their cognate proteins to perform their roles in cellular enzymology; the free vitamins are not physiologically useful (although free biotin plays an indirect regulatory role). The protein domains to which biotin and lipoic acid are attached have very similar 3-dimensional structures and the enzymes that perform the attachment of the two cofactors are members of the same protein family based on their structures. Thus, the speculation made many years ago (1) that biotin and lipoic arose together “late” in evolution is germane. Moreover, although the two molecules look to have little similarity when drawn as in Fig. 1, both are chiral. Biotin has a chair shape due to the C-N bonds whereas the ring of lipoic acid is skewed by the C-S bonds. Proteins recognize these structures in somewhat similar manners since the biotin binding protein, avidin, also shows significant (albeit much weaker) binding of lipoic acid and antibodies raised against one of the molecules as a hapten often bind to proteins modified with the other cofactor (2).
Figure 1.

Structures of biotin, lipoic acid, n-octanoic acid, and the reduced form of lipoic acid, dihydrolipoic acid. (A) All biotin carbon atoms are numbered as are the relevant carbons of the other molecules. (B) Stereochemistry of biotin and lipoic acid showing that both molecules have non-planar structures. The lipoic acid dithiolane ring would emerge from and protrude below the plane of the page whereas biotin has a chair structure (the viewer is looking at the back of the chair). Note that lipoic acid structure is rotated relative to that in panel A to conform with the Cahn-Ingold-Prelog rules and since biotin has three chiral centers the hydrogen atoms attached to carbon atoms 7 and 10 can be depicted as either above or below the plane of the page depending on the chiral center chosen as primary (the ring centers were chosen in this depiction). For simplicity the stereochemistry will not be given (except as relevant) in the subsequent figures of this review.
Biotin and lipoic acid also share the property that they are attached to very few protein species. E. coli has only a single biotinylated protein, the AccB subunit of the essential enzyme, acetyl-CoA carboxylase whereas S. enterica has a second inducible biotinylated protein, the α subunit of oxalacetate carboxylase (3–5). E. coli has three lipoylated proteins, these are subunits of pyruvate dehydrogenase (PDH) and 2-oxoglutarate dehydrogenase (2-OGDH), enzymes essential for aerobic growth, plus a third lipoylated protein induced by the presence of glycine that is a subunit of the glycine cleavage system of single carbon metabolism (6–8). In each of these proteins the cofactor is attached to a lysine residue ε-amino group of a domain of highly conserved structure. This domain is the N-terminal part of a lipoylated protein and the C-terminal part of a biotinylated protein and is connected to the remainder of the protein by a long proline plus alanine-rich linker region (9). The modified subunits form noncovalent interactions with other members of a protein complex of the three or four protein species that constitute the active enzyme. The cofactor-modified domains then shuttle intermediates between the multiple active sites of the enzyme complex (9). The mobility of the domains is due to the proline-alanine linkers and the domains constitute the distal ends (the “hands”) of the swinging arms long ago postulated for these enzyme complexes. These arrangements can be considered as providing substrate channeling via covalent attachment (9). Finally both biotin and lipoic acid are needed in only trace quantities. In E. coli only a few hundred molecules of biotin per cell are sufficient for growth (10) and the requirement for lipoic acid is similar. Therefore, the enzymes of these pathways are expressed at very low levels (< 350 molecules/cell [11]) and the enzymes have generally low turnover numbers.
Synthesis of Biotin
The early steps of biotin biosynthesis are not well understood in any organism, but clearly differ between E. coli and Bacillus subtilis. In both cases a seven carbon dicarboxylic acid, pimelic acid is assembled with one of its carboxyl groups in thioester linkage. Pimeloyl-CoA has long been thought to be the thioester-activated form of pimelic acid, but recent evidence indicates a role for the acyl carrier protein (ACP) of fatty acid synthesis as the thiol moiety (12, 13). In contrast the steps that follow formation of the pimeloyl-thioester are well conserved throughout biology even in organisms (e.g., Saccharomyces cerevisiae) that lack the ability to perform any early biosynthetic steps. In E. coli the atoms of biotin are derived from rather disparate sources, acetate, alanine, CO2, S-adenosylmethionine (SAM) and sulfide. Two groups have traced the origins of the biotin and dethiobiotin carbon atoms by 13C labeling followed by analysis by 13C NMR (14, 15). Using the numbering system of Figure 1, the C-3, C-5, and C-7 carbon atoms of biotin are derived from C-1 of acetate whereas the C-2 of acetate contributes the biotin C-2, C4, and C-6 carbon atoms. Acetate labeled in both carbon atoms is incorporated intact as shown by 13C coupling. Biotin carbon atoms C-9 and C10 are contributed by l-alanine. The C-1 and ureido (C-2′) carbon atoms are derived from CO2 (14). The nitrogen atom adjacent to C7 is from SAM whereas the other nitrogen atom is from alanine. The labeling pattern is consistent with formation of a pimelic acid moiety by head to tail incorporation of three intact acetate units as is the case in fatty acid (or polyketide) synthesis (14, 15) and the labeling pattern eliminates other plausible pathways from tryptophan, lysine, diaminopimelic acid or elongation of 2-oxoglutarate (15). Moreover, the 13C labeling results eliminate free pimelic acid as an intermediate in biotin biosynthesis. Pimelic acid is a symmetrical dicarboxylic acid whose carboxyl groups cannot be stereochemically distinguished and if free pimelic acid is an intermediate, then biotin carbon atoms C-1 and C-7 would have the same labeling pattern. This is not the case (14, 15) and thus, the pimelate moiety must be assembled with one of the carboxyl groups covalently linked to another moiety. A thioester seems the most likely linkage (14, 15). It should be noted that biotin is required for synthesis of malonyl-CoA, the postulated source of all of the carbon atoms of the pimelate moiety. Hence, we are presented with an evolutionary conundrum, biotin is required for biotin synthesis.
The Genes of Biotin Synthesis
Biotin requiring mutants of E. coli were first isolated many years ago. All of the mutants isolated as biotin auxotrophs were clustered at min 17 of the genetic map and defined five genes, called bioABCDF, based on mapping, cross-feeding and complementation studies (16–18). However, during deletion analysis of the maltose utilization genes, a strain that required biotin was isolated and called bioH (19). More recently, strains having a nonfunctional pfs (now called mtn), the gene encoding 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase were shown to require biotin due to accumulation of an inhibitory metabolite (see below) (20, 21). The fact that biotin auxotrophs due to mutations in the bioH or mtn genes were not isolated by classical means is probably due to the fact that E. coli biotin auxotrophs require only miniscule amounts of this cofactor (supplementation with biotin at concentrations of a few nanomolar) and that conventional undefined growth media are often very rich in biotin. Therefore, often several platings on biotin-free media are required to detect the biotin requirement of auxotrophs.
The Pathway and Proteins of Biotin Synthesis
The late steps of biotin biosynthesis (Fig. 2) were worked out many years ago whereas the early steps have only recently been elucidated. The proteins (BioA, BioB, BioD and BioF) of the late steps are well-studied enzymes of known crystal structures whereas the proteins of the early steps, BioC and BioH, were much more poorly understood. The sequence of steps in the late pathway was readily deduced since E. coli takes up each of the late intermediates. Growth of mutants with lesions in bioC and bioH proceeds when the medium is supplemented with 7-keto-8-amino pelargonic acid (KAPA) or any of the later intermediates in the pathway. No cross-feeding is observed between bioC and bioH mutant strains suggesting that the early intermediates may not pass through cell membranes, perhaps because they are protein-bound. In recent years the functions of BioC and BioH has become clear more then 50 years after the genes were discovered. The question was how to assemble a seven-carbon dicarboxylic acid in E. coli. BioC was annotated as an S-adenosyl-l-methionine (SAM)-dependent methyltransferase whereas BioH had been shown to have esterase activity on a series of short and medium chain acyl p-nitrophenyl esters(13, 22, 23) and on the methyl ester of dimethylbutyryl-S-methyl mercaptopropionate (24). The BioC annotation was especially puzzling because biotin contains no methyl groups and (as discussed above) all of the pimeloyl moiety carbon atoms are derived from acetate, alanine and CO2. Thus, it seemed that assembly of the pimeloyl moiety must require enzymes of another biosynthetic pathway that are somehow assisted in this task by BioC and BioH. Many years ago Lynen and coworkers (25) suggested that pimeloyl-CoA could be formed by the enzymes of fatty acid synthesis. They proposed that three molecules of malonyl-CoA would be condensed with the primer malonyl moiety retaining the carboxyl group introduced by acetyl-CoA carboxylase fixation of CO2. The other two malonyl-CoA molecules would lose their free carboxyl groups in the course of the two decarboxylative Claisen reactions required to give the C7 dicarboxylate. This scheme is consistent with the 13C labeling studies and is chemically reasonable because type III polyketide synthases are known that use such a malonyl-primed mechanism to make dicarboxylic acids of odd carbon lengths in which one the two carboxyl groups is in thioester linkage (26, 27). However, in fatty acid synthesis the growing chains are attached to ACP rather than CoA and unlike polyketides, where the keto groups are retained or consumed in subsequent rearrangements of the carbon chain (e.g., cyclization), pimelate synthesis requires that the keto groups be converted to methylenes. Although the condensation, reduction and dehydration enzymes of fatty acid synthesis could perform the net reduction of the keto groups to methylenes required for assembly of the pimeloyl moiety, it seemed most unlikely that the fatty acid synthetic enzymes would be active on substrates having a carboxyl group in place of the usual terminal methyl group because the fatty acid synthetic enzymes sequester the growing fatty acyl chains in tunnels or clefts that are strongly hydrophobic (28). Recently it has been shown that this conundrum is avoided by “disguising” the terminal carboxyl group such that it can be recognized by the fatty acid synthesis enzymes (Fig. 2). Introduction of the disguise is the role of BioC which converts the free carboxyl group to its methyl ester by transfer of a methyl group from SAM. Methylation cancels the carboxyl group charge and provides a methyl carbon that mimics the methyl of the normal acyl chains. This methylated species has properties (chain length, hydrophobicity) approximating those of the substrates normally accepted by the enzymes of fatty acid synthesis. Following completion of the pimelic acid moiety the methyl ester would then be cleaved by BioH to give pimeloyl-ACP. This in turn would react with l-alanine in the BioF reaction to give 7-keto-8-aminopelargonic acid (KAPA), the first intermediate in assembly of the biotin ring structures (Fig. 2). BioH thus acts to free the carboxyl group that will eventually be used to attach biotin to the metabolic enzymes where it performs its key metabolic roles (29).
Figure 2.

The current pathway of biotin synthesis in E. coli.
BioC
Prior to the recent work nothing was known of the function of BioC, a protein of 28.3 kDa. It is highly conserved among the proteobacteria and is often annotated as a SAM-dependent methyl transferase. It had been proposed that BioC acts as a carrier protein that carries an intermediate transferred by BioH (30), but recent work disproves this notion. The BioC protein had not been studied biochemically probably because it invariably forms inclusion bodies upon overexpression (31). This recalcitrant property of BioC has precluded its direct analysis, although some activity was obtained upon denaturing and refolding the protein (13). The BioCs of close relatives of E. coli were as intractable as E. coli BioC and thus the BioCs of more diverse bacteria were tested. Expression of the BioC of Bacillus cereus in E. coli restored biotin synthesis to an E. coli ΔbioC strain and this monomeric protein could be expressed in soluble form in E. coli and purified to homogeneity (32). In disagreement with prior scenarios that favored malonyl-CoA as the methyl acceptor, malonyl-ACP was a far better acceptor of methyl groups from S-adenosyl-l-methionine than was malonyl-CoA. BioC was specific for the malonyl moiety and was inhibited by S-adenosyl-l-homocysteine and sinefungin. Indeed, although the BioC kcat values of ~200 s−1 are modest, they not nearly as low as those of the enzymes late in the pathway. For example BioD and BioB, the last two enzymes of the pathway, are notably poor catalysts having reported kcat values of 0.06 and 0.002 s−1, respectively. Hence, when compared to BioD and BioB, BioC is an effective catalyst. A rationale for the disparity between the first and concluding enzymes of the biotin synthetic pathway is that BioC must have reasonable activity in order to effectively compete with the 3-ketoacyl-ACP synthases for malonyl-ACP. However, if BioC is overly active, it would convert too much malonyl-ACP to the methylated species and thereby block fatty acid synthesis. Indeed, BioC overproduction provides a very effective means to block E. coli fatty acid synthesis (32).
BioH
In contrast to E. coli BioC, E. coli BioH is a well-behaved monomeric 28.5-kDa protein which allowed determination of its crystal structure at 1.7 Å (23). BioH is a monomeric two-domain protein (23, 31). A putative catalytic triad (Ser-82, His-235, and Asp-207) similar to that of the catalytic triad of hydrolases was identified. Moreover, in the BioH crystals the serine residue was found to have been modified by a protease inhibitor. Consistent with these indications of hydrolase activity, BioH had weak esterase activity on several model substrates (23), although this activity was not shown to depend on the Ser, His, Asp triad. Others had noted two Gly-Xaa-Ser-Xaa-Gly motifs in BioH that are characteristic of acyltransferase and thioesterase proteins (30). However, the crystal structure gave no clues as to the identities of the substrates of BioH. BioH has been reported to bind CoA in vitro (31), but the significance of this finding remains unclear. BioH has recently been shown to act prior to BioF in an in vitro system and thus acts as the gatekeeper that prevents methyl pimeloyl-ACP from being elongated to azelayl-ACP methyl ester, a physiologically useless product (33). This was buttressed by 2.05 Å resolution co-crystal structure of a complex of a catalytically inactive BioH with Me-pimeloyl-ACP. The BioH-ACP interface contacts identified in the structure (four salt bridges between BioH arginine sidechains and ACP acidic residues) were demonstrated to be required for binding of its substrate by BioH (33). The BioH proteins that lacked these contacts were inactive in vitro and in vivo indicating that Me-pimeloyl-ACP is the physiological substrate of BioH, and that BioH is the gatekeeper (33). As will be further discussed below, it should be noted that in the E. coli genome the bioH gene is well removed from the other genes of the pathway and is not regulated by the BirA repressor (see below) whereas in other proteobacteria (e.g., the pseudomonads) bioH is found in a apparent biotin synthetic gene operon.
BioF
BioF is 7-keto-8-amino pelargonic acid (KAPA) synthase, a pyridoxal phosphate-dependent homodimer of 41.6 kDa of known crystal structure (34, 35). The enzyme condenses alanine with pimeloyl-CoA to give 7-keto-8-amino pelargonic acid (formal name, 8-amino-7-oxononanoic acid) plus CoA and CO2 (resulting from decarboxylation of alanine). BioF is a two-domain protein with the pyridoxal phosphate bound in a crevice between the two domains formed by residues of both domains. The mechanism of the enzyme has been studied in some detail (36). Historically the enzyme has been assayed using pimeloyl-CoA although pimeloyl-ACP could be the physiological substrate in E. coli (ACP-requiring enzymes will often use the analogous CoA compound as a model substrate). Consistent with this notion the E. coli 7-keto-8-amino pelargonic acid synthase has a much higher Michaelis constant for pimeloyl-CoA than the analogous enzyme from Bacillus sphaericus (37), an organism in which pimeloyl-CoA is thought to be the physiological substrate due to the presence of pimeloyl-CoA synthetase.
BioA
BioA is 7,8-diaminopelargonic acid (DAPA) aminotransferase (the formal name of DAPA is 7,8-diaminononanoate) that has many similarities to BioF, the preceding enzyme in the pathway. Although the BioA subunit (47.3 kDa) is slightly larger than that of BioF, it is also a homodimeric pyridoxal phosphate-dependent enzyme. Indeed, the overall structure of BioA is very similar to that of BioF (38) and this is reflected in a weak sequence homology. BioA is a transaminase that converts KAPA to DAPA and as such is not a particularly interesting enzyme (39, 40). However, the amino donor is not a standard amino acid, but rather the highly activated amino acid SAM (39, 41) which requires three ATP equivalents for its synthesis. The deaminated product derived from SAM, S-adenosyl-2-oxo-4-thiomethylbutryate, spontaneously degrades in vitro (39), and thus it seems likely that three ATP equivalents are consumed in what is an otherwise simple transamination reaction. The expense of this perplexing choice of amino donor may provide a rationale for the known tight regulation of biotin synthesis. However, it could be argued that use of a more pedestrian amino donor (B. subtilis uses lysine (42)) could alleviate the need for tight regulation.
BioD
In contrast to the preceding enzymes BioD (dethiobiotin synthase or DTBS) catalyzes an unusually interesting step, the formation of the ureido moiety of biotin (43, 44). The BioD reaction is the ATP-dependent formation of dethiobiotin from DAPA and CO2. The enzyme is a homodimeric protein (subunit of 24.1 kDa) that is structured into a single well folded domain (45–48). X-ray crystallographic studies have shown that the reaction proceeds by carbamoylation of N-7 of DAPA (45, 46) (Fig. 3). Independent NMR evidence for carbamate formation has also been obtained (49). The second partial reaction is also unusual. In this reaction the carbamate is activated by transfer of the γ-phosphoryl moiety of ATP to a carbamate oxygen to form a mixed anhydride (Fig. 3). This mixed anhydride species has been demonstrated by time-resolved crystallography (50). The final step of the dethiobiotin synthase reaction is a nucleophilic attack by the N-8 nitrogen of DAPA on a carbamoyl oxygen of the mixed anhydride (Fig. 3). This results in release of the phosphate group and formation of the ureido ring of dethiobiotin.
Figure 3.
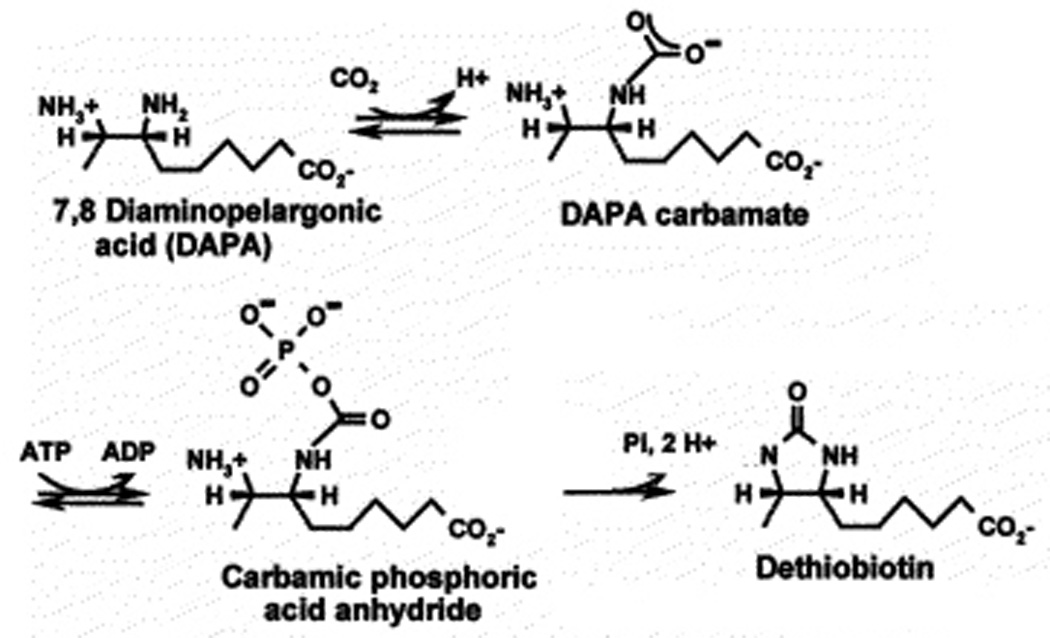
The BioD reaction.
BioB
The bête noire of biotin synthesis has long been the last step, insertion of the sulfur atom into DTB to form the thiophane ring of biotin. For many years this activity was ascribed to BioB by genetic means (biotin auxotrophs unable to grow on DTB) and could be assayed only by the ability of intact cells to convert DTB to biotin. Extensive attempts to obtain sulfur insertion in vitro all failed until Ifuku and coworkers (51) succeeded in showing biotin synthesis from DTB in a cell extract. The reaction required DTB, SAM, NADPH, BioB, and an unknown protein or proteins later shown to be flavodoxin (FldA) and flavodoxin reductase (Fpr) (51–53). This breakthrough was soon followed by demonstration of activity in a defined system containing NADPH, flavodoxin and flavodoxin reductase as the electron transfer system plus DTB, SAM, and a BioB preparation plus a reducing environment (54). BioB (a homodimer of a 38.6 kDa subunit) was found to be a very labile protein that is best purified and assayed under anaerobic conditions. The discovery that SAM was absolutely required for biotin synthesis and was not the sulfur donor (55, 56) strongly suggested that BioB was a member of the (then) small family of “radical SAM” enzymes. It has recently become apparent that this is a large family of proteins that catalyze a range of reactions that invariably involve difficult reactions often accessible only by radical chemistry. The radical is generated by reductive cleavage of SAM to give a deoxyadenosyl radical (DOA·) plus methionine. The DOA radical then cleaves a C—H bond to generate a carbon radical that allows the chemistry to proceed. The electron donor in the single electron reduction of SAM is a [4Fe-4S] cluster liganded to the cysteine residues of a perfectly conserved CXXXCXXC motif. Consistent with this picture, the BioB reaction is chemically difficult since it requires cleavage and sulfur insertion into two unreactive C—H bonds. The mechanism currently accepted by most workers in the field (57–59) is given in Fig. 4.
Figure 4.

The current model of the BioB reaction. For simplicity only DTB carbon atoms 6, 7, 9, and 10 (Fig. 1) are shown of which only carbons 6 and 9 are labeled. The reaction is shown as proceeding with the initial attack on C-9 because a derivative of DTB carrying a thiol group on C-9 has been shown to be converted to biotin both in vitro and in vivo (56, 260) and the crystal structure (24) shows C-9 in an appropriate position for the primary sulfur insertion.
The BioB species involved in the mechanism of Fig. 4 contains two [Fe-S] clusters. The number and composition of these clusters has been the subject of much disagreement in the literature. However, a variety of spectroscopic techniques plus a recent BioB crystal structure give a consistent picture. BioB contains two different clusters, the [4Fe-4S] cluster characteristic of radical SAM enzymes and a [2Fe-2S] cluster located at a different site (the [2Fe-2S] cluster was often thought to be a degradation product of a [4Fe-4S] cluster until mutagenesis experiments suggested otherwise). The crystal structure shows that BioB to be a α/β8 (TIM) barrel protein with the two [Fe-S] clusters located at either end of the barrel (58). The [4Fe-4S] cluster is located at the open end of the barrel whereas the [2Fe-2S] cluster (which utilizes an unusual arginine ligand) is at the closed end of the barrel. The crystal structures contain both SAM and DTB. The SAM is positioned such that reductive cleavage by the [4Fe-4S] cluster could readily occur while the DTB is positioned such that the C-9 carbon can accept a sulfur atom from the [2Fe-2S] cluster (58). Indeed, 9-mercaptodethiobiotin is a catalytic intermediate (60–62) This latter finding fits with the belief of most workers in the field that the [2Fe-2S] cluster is the immediate source of the biotin sulfur atom. This belief is supported by experiments in which each of the sulfur-containing small molecules of the defined in vitro reaction mixture was labeled with 35S and incorporation of the isotope into biotin was measured (see ref. (63) and references therein). No radioactive biotin was obtained. Isotopically labeled biotin was obtained only when BioB was labeled with 35S in vivo (63) or with 34S by reconstitution of the [Fe-S] clusters in vitro (64). More recent reports have shown that BioB reconstituted with Se in place of S gave selenobiotin derived from the (2Fe-2Se) cluster (65). Spectroscopic studies indicate that the [2Fe-2S] cluster disappears concomitantly with sulfur insertion (66, 67) and more recently evidence for that reduction of the [2Fe-2S] cluster accompanies formation of 9-mercaptodethiobiotin (62) consistent with a mechanism in which the [2Fe-2S] cluster simultaneously provides and oxidizes sulfide during carbon-sulfur bond formation.
For many years one of the few points of agreement in the literature was the finding that BioB itself is the sulfur source impinges, that the BioB reaction is not catalytic in vitro (57, 59, 68). Numerous and diverse justifications were put forth for the observed lack of catalysis (69–71), but no general agreement emerged. The favored and most provocative explanation for the lack of catalysis was that given above, the [2Fe-2S] cluster of the protein donates the biotin sulfur atom and this donation inactivates BioB. In this view BioB would be a reactant or substrate rather than an enzyme and, in the absence of repair of the [2Fe-2S] center, the protein would be sacrificed. The scenario of protein sacrifice was not completely unreasonable because there is no need for E. coli biotin synthase to be an efficient catalyst because E. coli (like most other organisms) requires only minuscule quantities of biotin for growth. E. coli can grow with only 100–200 molecules of biotin per cell (10, 72) and thus sacrifice of a few hundred molecules of a medium sized protein would not be a major drain on cellular resources. However, it was shown that Choi-Rhee and Cronan (73) demonstrated that E. coli BioB is catalytic in vivo. Such in vivo measurements are difficult since the endogenous expression level of biotin synthase is very low and because biotin may be split between pools of free and protein bound cofactor. The first issue was overcome by overexpressing hexahistidine-tagged biotin synthase under control of an arabinose-inducible promoter. The second issue was overcome by massively overexpressing, under control of an IPTG-inducible T7 promoter, biotin ligase (BirA) and a truncated, hexahistidine-tagged form of the acetyl CoA carboxylase biotinyl domain that can accept biotin but does not form an active enzyme complex. These investigators then used a combination of anti-pentahistidine antibodies, [35S]methionine labeling, and streptavidin to quantify the levels of each protein and of total biotinylated protein separated by denaturing and nondenaturing gel electrophoresis. The use of the two gel systems allowed the turnover number of BioB is be calculated in an unusually straightforward manner. The ratio of biotinylated domain to BioB monomer gives 20–60 equivalents of biotin produced per initial biotin synthase monomer (73). Very recently Jarrett and coworkers reported that in their in vitro assay system they observed that BioB is catalytic, 11 µM BS dimer produced 35 µM biotin over 4 h indicating at least three consecutive turnovers (74). Biotin production showed burst kinetics, a burst phase of k = 0.12 min−1, followed by a steady-state phase with a turnover number of k = 0.0089 min−1. The rate of the burst phase observed in vitro is similar to that observed in vivo suggesting that in vivo activity is not limited by FeS cluster reassembly but rather by the chemistry of biotin formation. The key to obtaining catalysis in vitro was preparation of SAM free of the contaminents present in commercial preparations and addition of Mtn to cleave the inhibitory 5′-deoxyadenosine produced in the reaction.
It should be noted that the in vivo measurement of BioB catalysis was complicated by the unexpected finding that enzyme turnover renders the enzyme susceptible to proteolytic degradation (73). A 50%–90% depletion of the level of His6-BioB was observed after incubation. This depletion was not observed in the absence of DTB or in the presence of biotin (73). The observed degradation of BioB was proposed to result from collapse of the enzyme [2Fe-2S] center due to donation of a sulfur atom to DTB. The [2Fe-2S] centre of BioB is located deep within the barrel of this α/β8 (TIM) protein (58) and thus it seems probable that a substantial unfolding of the protein would be required to allow rebuilding of the [2Fe-2S] cluster. Such unfolding would allow restoration of the [2Fe-2S] center, but at the cost of exposure of the protein to proteolytic attack while unfolded. Therefore, in this scenario catalysis by a molecule of BioB would require the protein to run a gauntlet of proteolysis until restoration of normal folding (with concomitant resistance to proteolysis) by rebuilding of the [2Fe-2S] center expended in biotin synthesis (73). The turnover numbers observed may thus be viewed as the products of a stochastic process. If the [2Fe-2S] cluster of a BioB molecule is rebuilt before proteolysis occurs, that protein will perform another turnover. If not, the protein molecule perishes and must be re-synthesized de novo. More recent work done in vitro has shown that loss of iron-sulfur clusters from BioB as a result of catalysis promotes unfolding and degradation (75). Hence, some BioB molecules may catalyze only one or a few turnovers in their lifetimes whereas others may complete >100 turnovers. The steady state level following the burst phase in the optimized in vitro system (74) may reflect the loss of active BioB molecules.
The biotin requirement of mtn (pfs) mutant strains (20) is due to inhibition of BioB by the byproduct of sulfur insertion, 5′-deoxyadenosine (21). The mtn gene encodes the 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase which was shown to also cleave 5′-deoxyadenosine to adenine plus 5′-deoxyribose (21, 74, 76). Mutants lacking Mtn activity precisely mimic BioB mutants in that they grow on biotin, but not on DTB or DAPA, and excrete DTB (21).
Remaining problems in biotin synthesis
The E. coli bioH gene differs from the other genes in the pathway in that it is neither located within the bio operon nor regulated by the BirA repressor/biotin protein ligase (77, 78). This is in contrast to many other bacteria where bioH resides within the biotin operon and is generally located immediately upstream of bioC (79). Moreover, BioH is a rather promiscuous hydrolase that also cleaves the ethyl, propyl and butyl esters of pimeloyl-ACP plus adipoyl-ACP methyl ester, although it is unable to cleave the thioester bond of these substrates. Others have reported that BioH cleaves the methyl ester of dimethylbutyryl-S-methyl mercaptopropionate (24) and a series of short and medium chain acyl p-nitrophenyl esters (22, 23, 80). Thus, the E. coli bioH gene may encode a protein that is less specific than those encoded by the more “domesticated” bioH genes. Note that the BioH function seems something of a “wild card” among biotin synthetic enzymes since in some bacteria the gene has been displaced from its site upstream of bioC by other genes (79) that have been shown functionally replace BioH (81).
The BioB reaction also requires additional study. This very intricate enzyme cannot yet be considered understood. There remain several loose ends. For example, the sequence of events that follow destruction of the BioB [2Fe-2S] cluster thought to donate the biotin sulfur atom remains unclear (67). Marquet and coworkers (67) reported that cluster destruction is accompanied by biotin formation whereas others (66) report that biotin formation is 10- to 1,000-fold slower than cluster destruction and is biphasic. It therefore seems possible that there may be several steps in the formation of biotin by BioB and different steps may be rate limiting in different enzyme preparations. A more extreme case is the claim that BioB has an intrinsic pyridoxal phosphate-dependent cysteine desulfurase activity responsible for generating the sulfur atom of biotin which would enter DTB via a persulfide (71, 82). This claim is countered by the finding that no pyridoxal phosphate is visible in the BioB crystal structure (58), other laboratories have been unable to demonstrate pyridoxal phosphate binding or cysteine desulfurase activity (27, 83) and the finding that biotin synthesis from DTB proceeds normally in cultures of E. coli starved for pyridoxal (84). It should be noted that although the BioB crystal structure is a major step forward, crystallization necessarily selects for a single protein species. Thus, the crystallized form of BioB may not fully represent all of the active enzyme species. Moreover, the present structure is of only moderate resolution (3.4 Å). An unsolved difficulty with the stoichiometry given by the BioB structure is that it contains only a single SAM molecule and there is no room for a second molecule (58). Therefore, the enzyme seems equipped to form only a single C-S bond. Using deuterium labeled DTB species it was shown that both C6 and C9 of biotin become labeled and thus it seems clear that 2 mol of AdoMet are necessary to break the positions 6 and 9 C-H bonds (85). Thus, the most likely scenario is that following synthesis of the first C-S bond, the methionine and 5′-deoxyadenosine products are released in order that a second molecule of SAM can bind. Another complication is that it is not completely clear how 9-mercaptodethiobiotin is bound in the active site. In the BioB structure DTB is located in a position where 9-mercaptodethiobiotin seems an unlikely intermediate in biotin formation. In seems probable that in order to complete the reaction BioB must attain a structure that is differs markedly from that of the extant crystal structure.
The recent finding that BioB undergoes burst kinetics during catalysis also deserves attention. Are the slow turnovers follwing the burst due to extraction of a sufur atom from the [2Fe-2S] cluster? How is the BioB [2Fe-2S] cluster rebuilt in vivo and would addition of the cellular rebuilding factors prevent decay of the enzyme to the less active state? Although BioB has recently been reported to accept a [4Fe-4S] center from two E. coli Fe-S center scaffold proteins, SufA and IscA, no [2Fe-2S] center was formed (86). It should be noted that the BioB [2Fe-2S] has a novel ligand, an arginine residue rather the Cys or His residues commonly used as ligands (58). This unusual ligand implied specificity for the guanidium ligand, but recent results indicated that substitution of Cys, Ala, His or Met for the arginine residue failed to inactivate BioB (87). Moreover, prior mutagenesis experiments indicated that two of the three conserved [2Fe-2S] cluster cysteine residues must be removed before BioB activity is lost (86, 88). The plasticity of this cluster suggests that the usual sulfur insertion pathways (the Isc and Suf systems) may not apply and, thus far, this seems to be the case. Inclusion of IscS does not allow BioB to become catalytic in vitro (69). The [2Fe-2S] cluster cannot be assembled by the Suf system in vitro (86) and E. coli strains with null mutations of either the suf or isc operons are not biotin auxotrophs (J. Imlay, personal communication). Unfortunately, suf isc double mutants are inviable so the possibility that biotin is synthesized due to redundant functions of the two systems cannot be tested.
Regulation of Biotin Synthesis
Expression of the Escherichia coli biotin synthetic (bio) operon is controlled by a simple, yet remarkably sophisticated, regulatory system in which the rate of transcription of the operon responds not only to the supply of biotin, but also to the supply of proteins (called biotin acceptor proteins) that become modified by covalent attachment of biotin (Fig. 5) (29, 89–94). This regulatory system is understood in considerable detail thanks to a combination of genetic, physiological, biochemical and biophysical investigations. The biotin operon of E. coli and other enteric bacteria is a striking example of regulation in which the transcriptional regulatory protein (BirA) is also an enzyme, in this case the biotin-protein ligase, that catalyzes the covalent attachment of the biotin to certain proteins involved in key metabolic carboxylation and decarboxylation reactions. Moreover, regulation of the E. coli biotin operon is probably the best understood example of transcriptional regulation by an enzyme unrelated to nucleic acid metabolism. Superficially, the system resembles the classical TrpR regulation of the E. coli tryptophan operon where the Trp repressor protein binds to the trpEDCBA operator only when complexed with the co-repressor, tryptophan. However in bio operon regulation, the repressor is also the biotin-protein ligase and the co-repressor is not biotin, but biotinoyl-5′-AMP (bio-AMP), the product of the first half-reaction of the ligase reaction. It is these novel features that give this regulatory system its unusually subtle properties. The bio operon is actually two transcriptional units (bioA and bioBFCD) controlled by a common operator.
Figure 5.

The biotin regulatory system of E. coli. BirA is represented by green ovals, biotin by black circles, the AMP moiety by red pentagons, AccB by dark blue ovals and AccC by light blue crescents. The arrows denote transcription from the leftward and rightward bio promoters. (A to C) BirA switches from biotin ligation function to repressor function in response to the intracellular biotin requirement which is monitored by the level of unbiotinylated AccB. If the levels unbiotinylated AccB are high, the protein functions as a biotin ligase. Once the unbiotinylated AccB has been converted to the biotinylated form, the bio-AMP is no longer consumed and remains bound to BirA. This liganded form of BirA accumulates to levels sufficiently high to form dimers that fully occupy the bio operator iresulting in transcriptional repression of the biotin biosynthetic genes. (D) Overproduction of AccC ties up unbiotinylated AccB into a complex that is a poor biotinylation substrate. Therefore, high levels of the liganded form of BirA accumulate resulting in repression of bio operon transcription.
The Model
Maximal rates of bio operon transcription (derepression) occur when the biotin supply is severely limited (e.g., biotin starvation of a bio auxotroph) (Fig. 5A) or when high levels of a biotin acceptor protein are present (Fig. 5B). Under these conditions any bio-AMP synthesized is rapidly consumed in biotinylation of the acceptor protein (apo AccB) and hence no significant levels of the BirA-bio-AMP complex accumulate. Hence, BirA remains largely monomeric so the bio operator is seldom occupied and transcription is maximal. Repression of bio operon transcription occurs when the supply of biotin is in excess of that needed to biotinylate apoAccB. Under these conditions apo-BCCP is fully biotinylated, the BirA:bio-AMP complex accumulates, followed by dimerization of the protein to form the repressor species. The dimers then bind to the bio operator and represses transcription from both promoters. The two derepression conditions act by a common mechanism in that both decrease the levels of the BirA:bio-AMP complex available to bind the bio operator (Fig. 5C). Hence, the degree of repression of bio operon transcription can be most simply viewed as an antagonism between retention of bio-AMP in the BirA active site versus consumption of the bio-AMP bound to BirA by transfer of the biotinyl moiety to unmodified acceptor proteins (93). The model of Beckett and coworkers (91) in which the unmodified acceptor protein binds monomeric BirA and thereby inhibits formation of BirA dimers, the species required for effective repression, provides a structural context for this antagonism. Because the rate of bio operon transcription is sensitive not only to the intracellular concentration of biotin, but also to the supply of the protein to which the biotin must be attached, the net result of accumulation of the unmodified protein is an increase in the rate of synthesis of the small molecule needed for the post-translational modification. The evidence for this model is strong and is discussed below.
BirA protein
The evidence that the ligase and repressor are the same protein was very firmly established by data from several laboratories. The key genetic observation was that of Campbell and co-workers who showed that E. coli mutants defective in intracellular retention of biotin (called birA) were allelic to mutants defective in repression of the bio operon (called bioR; the birA designation has been retained). Since biotin is retained in E. coli only as the protein-bound species, it followed that the birA gene encoded biotin-protein ligase activity and this was demonstrated (89, 90). Furthermore, these workers also showed that a partially purified BirA protein preparation protected a specific segment of bio operon DNA (Fig. 6B) from nuclease digestion. This DNA segment contained a region of degenerate dyad symmetry previously defined as the operator of the bio operon (see below) by transcriptional (95) and mutational studies (96). As expected (see below), protection by BirA required the presence of bio-AMP. At about the same time, Eisenberg and co-workers showed that the purified repressor protein bound to bio operon DNA and catalyzed the biotin-protein ligase reaction (97). These workers also found that binding of the repressor protein to bio operon DNA in vitro required either biotin or bio-AMP but that bio-AMP was 1,000-fold more effective than biotin and biotin was active only at non-physiological concentrations (98). Bio-AMP was also shown to be 1,000-fold more efficient than biotin in repression of bio operon transcription in a coupled transcription-translation system (99). Since these pioneering studies, it has become possible to obtain large amounts of BirA (normally a very non-abundant protein) (100, 101) that has led to biophysical studies as well as crystal structures of the unliganded (apo) protein (102) and of complexes of BirA with biotinoyl-lysine (102), biotin (103), or biotinoyl-AMP, a non-hydrolyzable analogue of bio-AMP (104). Although we lack the structure of the tertiary complex of BirA, the bio operator and bio-AMP (or an analogue), these studies show that BirA is a winged helix-turn-helix protein (102, 105) of 35.2 kDa (Fig. 6). The winged helix-turn-helix is located at the extreme N-terminus of the protein and is one of the three BirA domains, the others being a large central domain where is active site is found and a small C-terminal domain. The latter two domains show high levels of structural similarity with biotin-protein ligases from throughout biology (106). More recent work has shown that BirA requires bio-AMP to dimerize at physiological concentrations (107) and only the BirA dimer can efficiently bind the operator (108–111). Bio-AMP binding activates the assembly of the BirA-operator complex by increasing the extent of dimerization by three orders of magnitude (112, 113).
Figure 6.

The structure of BirA (A) and the sequence of the bio operon operator/promoter region (B). Panel A. The BirA structure is that of the protein liganded with a bio-AMP analogue (which for simplicity was omitted) (235). Panel B. The boxed region is the operator to which a BirA dimer binds. PA and PB are the promoters of the bioA and bioBFCD transcriptional units, respectively. The −10 and −35 promotor regions are denoted by underlines.
The biotin attachment activity of BirA (Fig. 7) proceeds through the bio-AMP intermediate formed from biotin and ATP (106). Enzyme bound bio-AMP is then attacked by the ε-amino group of a specific lysine of the acceptor protein to give the biotinylated acceptor protein (106) (Fig. 7). In the absence of an appropriate acceptor protein the bio-AMP intermediate remains bound within the BirA active site where it is protected from solvent and is quite stable (100). BirA shows very high specificity for biotin. The discrimination in favor of biotin versus DTB is ca. 50,000-fold (73, 114) although BirA-catalyzed attachment of DTB can be demonstrated (114). Both DTB and the oxidized form of biotin, biotin sulfoxide, show very weak abilities to derepress transcription of the biotin operon (115). A large number of birA mutants have been isolated based on their transcriptional phenotypes (using bio-lacZYA fusions) (77) and the mutational alterations of a considerable number of these have been determined by DNA sequencing (116). These fall into three main classes, mutants defective in regulation (the classical bioR phenotype), mutants defective in binding biotin and/or bio-AMP (the classical birA phenotype, (117)), and those having temperature-sensitive growth (77). However, there is considerable overlap among these phenotypes and some mutant proteins show all three phenotypes (77). All BirA crystal structures including that with a bio-AMP analogue show the N-terminal DNA binding domain markedly protruding from the body of the protein (Fig. 6A) and thus it is surprising that deletion of this domain has a profound effect on the ligase activity of the truncated protein due to poor binding of biotin and/or bio-AMP (118). It should be noted that BirA is an essential gene (77, 119, 120) since it is required for fatty acid synthesis and hence, membrane lipid synthesis (121).
Figure 7.

The BirA reaction is shown. This is the general reaction of biotin protein ligases (38). The lipoic acid ligase LplA has the same reaction mechanism given substitution of lipoic acid for biotin.
The biotin acceptor protein
AccB protein, the sole biotin acceptor protein of E. coli, is an unusual protein, the N-terminal half appears largely unstructured (although the extreme N-terminus is known to interact with the AccC subunit (122) whereas the C-terminal half of the protein is folded into a compact and stable structure called the biotin domain (Fig. 7). This domain has a structure very similar to that of lipoyl domains (see below). The AccB biotinoyl domain is as efficient a biotin acceptor as the full-length protein (123) and is often used for in vitro work to avoid the problems with aggregation of the full-length protein (122, 123). The structure of biotinoyl domains is strongly conserved throughout biology and expression of foreign biotinoyl domains in E. coli can derepress bio operon transcription (124). Mutants of the AccB biotinoyl domain have been isolated that are defective in interaction with BirA (125) and mutations have been introduced that allow the protein to accept lipoic acid in place of biotin (126). The work on biotin and functions is intimately involved with (and is historically derived from) that on lipoyl domain structure and will be discussed in that context below.
The biotin operator
The enzymes of E. coli biotin synthesis are encoded (with the exception of bioH) by a cluster of genes located adjacent to the attachment site of phage λ called the biotin (bio) biosynthetic operon (Fig. 5 and 6). Transcription of these bio genes is from two partially overlapping face-to-face promoters controlled by a common operator site of 40 bp that binds a dimer of the BirA protein (91, 95, 96, 127) (Fig. 6B). The leftward promoter transcribes bioA whereas the rightward promoter transcribes bioBFCD. The 5’ends of the transcripts have been mapped and mutations within the operator that ameliorate repression of either rightward or leftward transcription (or both) are known (95, 96, 128, 129). The operon and operator sequence are conserved in S. enterica, and Citrobacter freundii (129). A long-standing puzzle is that bioH is not under BirA regulation (77) especially since in other proteobacteria (e.g., pseudomonads) bioH seems part of a biotin biosynthetic operon. Also, unlike many repressors, BirA does not appear to be autoregulated because it is cotranscribed with an essential gene (murB) of peptidoglycan biosynthesis.
Physiological aspects of bio operon regulation
E. coli contains only a single species of biotin acceptor protein, the AccB subunit of acetyl-CoA carboxylase (ACC), which is the first enzyme of fatty acid biosynthesis (10, 94, 130, 131) and is therefore essential for growth. The response of the E. coli biotin regulatory system to the supply of biotin acceptor proteins is readily rationalized by the fact that biotin attains biological function only when the vitamin is covalently attached to AccB; the free vitamin cannot support ACC activity (121). AccB, which is also called biotin carboxyl carrier protein (BCCP), forms an unstable complex with AccC, the subunit that catalyses the biotin carboxylase partial reaction of acetyl-CoA carboxylase. The chromosomal locations of the genes (accA and accD) that encode the other two ACC subunits are well removed from the accBC operon and each other(10, 94, 130, 131). The AccB-AccC complex was recently shown to consist of an AccC dimer plus four copies of AccB (122). This complex is thought to bind an α 2β 2 heterotetramer of the AccA and AccD subunits to give active ACC, the enzyme required for production of malonyl-CoA, the key precursor of fatty acid synthesis (121). The rates of transcription of all four genes are controlled by cellular growth rate (132) which is physiologically reasonable since lipids (hence fatty acids) constitute a constant fraction of the cell mass. The fact that bio operon transcription is derepressed by increased synthesis of AccB nicely ties biotin synthesis to growth rate. This is because increased growth rates require increased flux through the fatty acid synthetic pathway in which ACC catalyzes a rate-limiting step (133). Indeed, biotin consumed by increased protein biotinylation has been shown to be restored by increased biotin synthesis (92).
The fact that the only acc genes that are cotranscribed are accB and accC and that this gene arrangement is very widely conserved in bacteria raised the question of its relevance to the regulation of biotin synthesis (134). It seems possible that the defined stoichiometry given by cotranscription of accB and accC might function to aid efficient biotinylation of AccB. It seemed possible that an excess of AccC might tie up apo-AccB in a complex that would be a poor substrate for BirA and thereby disrupt the regulatory system (Fig. 5D). This has been shown to be the case (134). Overproduction of AccC gave almost maximal repression at biotin concentrations that normally give only slight repression and inhibited biotinylation of AccB. As expected overproduction of both AccB and AccC to restore the normal ratio of the two proteins relieved the down-regulation given by overproduction of AccC alone and this relief required that the overproduced AccB species be competent to interact with AccC (134).
What is the regulatory switch in BirA regulation?
The present model of bio operon regulation has a very solid experimental basis obtained by both in vivo and in vitro approaches. However, there are two contrasting views of the mechanism whereby accumulation of the unmodified biotin domain derepresses transcription of the operon. In one view this is simply a competition for bio-AMP between its consumption by protein biotinylation versus its presence in the BirA active site where it triggers dimerization and subsequent operator binding (93). In the second view the biotin domain forms a heterodimeric complex with a monomer of BirA. The BirA surface used to form the heterodimer is proposed to be the same surface as that used in forming the BirA homodimer. Hence, in this view competing protein-protein interactions are responsible for derepression upon accumulation of unmodified biotin domain (91). However, a major caveat to the model is that no direct detection of the postulated AccB plus BirA:bio-AMP heterodimer has been reported and only indirect evidence for its existence is available (135)(6) (135). The two models have a conceptual distinction, the lifetime of the BirA-biotin domain interaction. In the bio-AMP competition model the interaction is ephemeral, the two proteins associate, biotin is transferred and the complex rapidly dissociates as in most enzyme reaction whereas in the competing protein-protein interaction model the BirA-biotin domain interaction is long lived.
An approach that distinguishes these models utilized the small peptides that are substrates for biotinylation by BirA (136). These peptides, which were isolated by screening large peptide libraries, are quite diverse in sequence and have as few as two residues (one being the reactive lysine residue) that are found in naturally biotinylated proteins (136). Due to their small sizes (14 residues is sufficient, (136, 137)) and diverse sequences, it seems unlikely that stable peptide-BirA complexes are made. If these sequences (attached to a partner protein) are expressed in E. coli they should derepress bio operon expression, the bio-AMP competition model is supported. If they fail to derepress, but are efficiently biotinylated, then the competing protein-protein interaction model would be supported. Although the most studied of these peptides (Pep-85) is reported to be as good a biotin acceptor as the AccB biotin domain, this peptide remains enigmatic because it seems to lack structure in solution (137) and can only be biotinylated by BirA (124). Biotin ligases from six other organisms fail to use this peptide as a biotin acceptor, although these ligases readily utilize the AccB domain as a substrate (124, 138). Thus, if the most studied peptide sequence somehow mimicked structural attributes of AccB, the sequence should be biotinylated by BPLs other than BirA. However, BirA is the only ligase known to biotinylate the Pep-85 sequence.
Two fusion proteins containing synthetic biotin accepting peptide sequences (one being Pep-85) were as efficient in derepression of bio operon transcription as the when the natural acceptor, AccB-87, was the fusion partner (138). These results argue strongly against the competing protein-protein interaction model. As noted above the strict specificity of Pep-85 for BirA argues against the peptides being structural mimics of the natural acceptor domain. Moreover, even if this were somehow the case the the peptides would have to interacted with BirA:bio-AMP with the same binding strength and kinetics as that of the natural acceptor protein despite their small size and markedly diverged sequences. Indeed, the peptides lack several residues postulated to play important roles in forming the putative heterodimer and have other residues, some of which cannot participate in hydrogen bonding) in place of resides thought to play roles in heterodimer formation. It it follows that the rules governing biotinylation are markedly different for AccB and the peptide sequences (138). The possibility that the competing protein-protein interactions model is the regulatory switch seems extremely remote. The classical work on BirA mutants did not include BirA super-repressing mutants. These would be mutants that would repress transcription under all conditions including biotin limitation and apo-domain overexpression. Some of the possible classes of mutants are: (i) BirA proteins unable to bind the biotin acceptor protein, (ii) BirA proteins that bind the acceptor protein but are unable to biotinylate it, (iii) BirA proteins that form very tight homodimers (perhaps even in the absence of bio-AMP) and (iv) BirA proteins that cannot dissociate from the operator DNA. Some of the mutants might be genetically dominant. Most of these mutants could be nonviable because fatty acid synthesis would be blocked due to lack of biotinylation of AccB thereby account for the fact that such mutants were not reported in the early investigations. Hence, the isolation of super-repressor mutants was done in a strain where expression of a heterologous biotin protein ligase active on AccB allowed fatty acid synthesis to proceed (139). This allowed mutant strains having the super-repressor phenotype by a combined selection-screening approach and resolved multiple mutations to give several birA super-repressor alleles each having a single mutation all of which showed repression dominant over the wild type allele. All of these mutant strains repressed bio operon transcription in vivo at biotin concentrations that gave derepression of the wild type strain and retained sufficient ligation activity for growth when overexpressed (139). All mutant strains except G154D BirA showed derepression of bio operon transcription upon overproduction of a biotin accepting protein. The G154D BirA was a lethal mutation in single copy and the purified protein was unable to transfer biotin from enzyme bound biotinoyl-adenylate either to the natural acceptor protein or to a biotin accepting peptide sequence. Consistent with the transcriptional repression data, each of the purified mutant proteins showed increased affinity for the biotin operator DNA in electromobility shift assays. Surprisingly although most of the mutations were located in the catalytic domain all those tested excepting G154D BirA had normal ligase activity. Most of the mutations that gave super-repressor phenotypes altered residues located close to the dimerization interface of BirA. However, two mutations were located at sites well removed from the interface. The properties of the super-repressor mutants strengthen and extend other data indicating that BirA function entails extensive interactions among the three domains of the protein and shows that normal ligase activity does not ensure normal DNA binding (139).
Finally, the crystal structure of BirA complexed with the bio operator and bio-AMP (or an analogue) seems likely to very informative. This may give information on the conformational changes in BirA that accompany bio-AMP binding (140). Co-crystals of the BirA-biotinoyl domain complex would also be of great interest. The super-repressor mutant proteins may stabilze the BirA-operator contacts and thereby facilitate crystallization of the complex.
Lipoic acid synthesis
Lipoic acid (Fig. 1) is a sulfur-containing cofactor found in most prokaryotic and eukaryotic organisms. In Escherichia coli and other organisms lipoic acid is essential for function of several key enzymes involved in oxidative and single carbon metabolism including pyruvate dehydrogenase (PDH), 2-oxoglutarate dehydrogenase (2-OGDH), branched-chain 2-oxoacid dehydrogenase, acetoin dehydrogenase and the glycine cleavage system (141). In each enzyme, a specific subunit is modified by attachment of lipoic acid to specific lysine residues within conserved domains of these subunits. In each of these domains an amide linkage is formed between the carboxyl group of lipoic acid and the ε–amino group of the specific lysine residue (142). During catalysis, the protein-bound lipoamide moieties serve as carriers of reaction intermediates among the multiple active sites of these multienzyme complexes (141).
Our knowledge of the pathways of lipoic acid synthesis, attachment and function has progressed rapidly in the last 10 years largely due to complementary genetic and biochemical analyses in E. coli. I shall first discuss the enzymes that carry and require the cofactor because they are derived from diverse areas of metabolism. Next, the mechanisms of attachment of lipoic acid and its precursor, octanoic acid, to these proteins will be reviewed. Finally, the synthesis of the cofactor itself will be discussed. This organization was chosen because the unusual biosynthetic pathway of lipoic acid is mechanistically intertwined with attachment of the cofactor.
Lipoic acid-dependent enzymes
Pyruvate dehydrogenase (PDH)
The PDH reaction mechanism is probably the most thoroughly characterized lipoic acid-dependent enzyme. PDH catalyzes the oxidative decarboxylation of pyruvate to the key metabolic intermediate, acetyl-CoA. This very large enzyme complex consists of multiple copies of each of three subunits encoded by the aceE aceF lpd operon. The first subunit (AceE) is a thiamine diphosphate-dependent decarboxylase (E1p) that catalyzes both the decarboxylation of pyruvate and the reductive acetylation of the lipoyl group that is covalently attached to the second subunit, E2p (AceF). The E2p subunit is a dihydrolipoyl acetyltransferase responsible for the transfer of the acyl group from lipoyl moiety to CoA to form acetyl-CoA. The third subunit, E3 (Lpd), is a dihydrolipoyl dehydrogenase that serves to regenerate the disulfide bond of the lipoyl moiety of E2p (143) and thereby prepares the enzyme for another cycle of catalysis. The E2p subunit to which E1 and E3 are bound strongly (but noncovalently) forms the structural core of the multienzyme complex. The oxidative decarboxylation of pyruvate to form acetyl-CoA is the link between glycolysis and the citric acid cycle and therefore PDH activity is essential to cells that rely upon respiration to provide metabolic energy. In most aerobically respiring organisms the PDH complex also supplies the acetyl-CoA necessary to sustain essential biosynthetic pathways, especially those of fatty acid and amino acid synthesis (144). Synthesis of the PDH complex varies over a 7- to 10-fold range depending on the growth conditions (145–147). It is induced by exogenous pyruvate or when pyruvate is generated endogenously e.g. by thiamine starvation, and it is partially repressed by excess glucose and during growth on acetate or on citric acid cycle intermediates. Regulation by pyruvate or a derivative of pyruvate proceeds through the PdhR repressor (148, 149). PDH synthesis is repressed during anaerobic fermentative growth where the catalytic activity is also inhibited. Under these conditions the conversion of pyruvate to acetyl-CoA is mediated by the derepression and activation of pyruvate formate lyase (146, 150).
2-Oxoglutarate dehydrogenase (2-OGDH)
The mechanism of 2-OGDH is essentially the same as that of PDH as is the structure of the complex. Indeed, the 2-OGDH complex has been reported to contain low levels of PDH subunits (151). The 2-OGDH complex contains three subunits, a 2-oxoglutarate decarboxylase component (E1o), a trans-succinylase component (E2o) and a dihydrolipoyl dehydrogenase (E3). The E1o and E2o subunits are different proteins from the corresponding subunits of the PDH complex and are encoded by the sucA and sucB genes, respectively. However, the E3 subunit is the same protein, Lpd, found in the PDH complex. In aerobically grown E. coli, this complex catalyzes a key step in the citric acid cycle and also supplies succinyl-CoA for biosynthesis of two amino acids, methionine and lysine (152). Under the appropriate conditions, E. coli strains lacking functional 2-OGDH can be supplemented with succinate or methionine plus lysine to provide metabolic bypasses of loss of this enzyme complex (152). Expression of the 2-OGDH is highly induced during aerobic growth on acetate and citric acid cycle intermediates and is severely repressed during fermentative growth where succinyl-CoA is generated by succinyl-CoA synthetase (144) although 2-OGDH is synthesized by cells gown in anaerobic media containing an electron acceptor such as nitrate or fumarate (153).
Glycine cleavage system
The third lipoylated protein of E. coli is the H protein of the glycine cleavage system, an enzyme widely distributed in bacteria and in the mitochondria of plants (where it is called glycine decarboxylase), fungi and mammals (154–156). The glycine cleavage system catalyzes the reversible cleavage of glycine, yielding carbon dioxide, ammonia, 5,10-methylenetetrahydrofolate plus a reduced pyridine nucleotide. It consists of four component proteins termed the T, H, P and L proteins. The first three proteins are encoded by the gcvT gcvH gcvP operon while L protein is the same as Lpd, the E3 protein of the 2-oxo acid dehydrogenases as discussed above (157). P protein catalyzes the pyridoxal phosphate-dependent decarboxylation of glycine and transfers the remaining methylamine moiety to one of the sulfhydryl groups of the lipoyl prosthetic group of H protein. T protein catalyzes the release of ammoniate and transfer of the one-carbon unit to tetrahydrofolate from the lipoyl residue. L protein is a lipoamide dehydrogenase that catalyzes the reoxidation of the dihydrolipoyl residue of H protein and reduction of NAD+. Thus, the lipoic acid moiety of H protein interacts with the active sites of three different enzymes in a manner analogous to that found for 2-oxoacid dehydrogenase complexes.
Structures of lipoylated and biotinylated proteins
In all 2-oxoacid dehydrogenase complexes, the core of the structure is provided by the E2 subunit to which the E1 and E3 components are bound tightly but noncovalently. In the PDH and 2-OGDH complexes of Escherichia coli and other gram-negative bacteria (158, 159) plus the 2-OGDH and branched-chain 2-oxoacid dehydrogenase complexes of mammals (160, 161), the core consists of 24 copies of the E2 chain arranged with octahedral symmetry, whereas in the PDH complexes of mammals and Gram-positive bacteria (162–165), the core comprises 60 E2 chains arranged with icosahedral symmetry. In all 2-oxoacid dehydrogenase complexes, the E2 component has a multi-domain structure comprising (from the N terminus): lipoyl domain (or domains of ca. 9 kDa), a small peripheral subunit-binding domain (ca. 4 kDa) and a much larger catalytic domain (ca 28 kDa) that houses the acyltransferase activity and aggregates to form the inner core of the complexes. These domains are separated by long (25–30 residue) segments of polypeptide chain, characteristically rich in alanine, proline and charged amino acids that form flexible but extended linkers (143).
The numbers of PDH lipoyl domains per E2 subunit varies from one to three. In the PDH complexes of Gram-negative bacteria, the number is usually three (e.g., E. coli and Azotobacter vinelandii) or two (e.g. Haemophilus influenzae, Neisseria meningitidis, Alcaligenes eutrophus, and Thiobacillus ferrooxidans) (9). All of the 2-OGDH E2o subunits described to date contain a single lipoyl domain, as is also the case for the E2b chains of all BCDH complexes (9, 141, 143, 166). A generally applicable explanation for the variation in the number of lipoyl domains has not yet been worked out. Protein engineering experiments have eliminated the straightforward explanations. In E. coli PDH, selective deletion of one or two lipoyl domains has no detectable effect on the overall catalytic activity, the system of active site coupling or the ability to complement pyruvate dehydrogenase complex mutants (167). As expected the catalytic activity is abolished when all three lipoyl domains are deleted or when the lipoyl domains are rendered unlipoylatable by conversion of the lipoylated lysine residue to glutamine (167, 168). There is no mandatory order of reductive acetylation of the repeated lipoyl domains within E2p polypeptide chains because complexes containing mixtures of wild-type and mutant lipoyl domains (+/−; −/+; +/+/−) are fully active, although the complex containing the −/−/+ version of the E2p polypeptide chain showed a 50% reduction in specific activity (168). Activity is also impaired (but not abolished) by increasing the lipoyl domain content to four to nine per E2p chain, possibly due to under-lipoylation of the domains participating in catalysis and interference from unlipoylated domains (169). High-field NMR studies were carried out with variants containing zero to nine lipoyl domains per E2p subunit. These studies suggest an explanation for the presence of three lipoyl domains per E2p subunit in the wild-type PDH complex that is based on the greater inherent mobility and thus potentially more efficient active-site coupling of this arrangement (170). The superiority of the three lipoyl domain-PDH complex has since been confirmed by physiological studies from which it was concluded that decreased lipoyl domain contents adversely affect growth rate and growth yield (171). The physiological consequences of increasing the number of lipoyl domains from three to nine per E2p chain, and the effects of inserting up to seven unlipoylated (mutant) domains between a wild-type N-terminal lipoyl domain and the E3-binding domain were also investigated and indicate that three lipoyl domains per E2p chain are optimal and that only the outermost lipoyl domain needs to be lipoylated to obtain full catalytic activity (172). It was concluded that the reason for retention of three lipoyl domains is to extend the reach of the outermost lipoyl cofactor rather than to provide extra cofactors for catalysis (172). However, given this advantage why then do many lipoylated proteins contain only a single lipoyl domain?
The conserved structure of lipoyl domains (Fig. 8A) is directly related to catalytic functions of the domain in substrate channeling and active-site coupling. First of all, although free lipoic acid is a substrate for E2p and E3 in vitro, lipoylated domain is a much better substrate (Graham et al., 1989). Attachment of the lipoyl group to the conserved lysine at the tip of the protruding β–turn gives a dramatic reach to the “business end”. Moreover, the flexible and extended linker regions that connect the lipoyl domain(s) with the catalytic domain contribute increased mobility to the swinging arm since deletion of the linker region in a modified “single lipoyl domain” E2p chain caused an almost total loss of overall activity without substantially affecting the individual enzymatic activities (173). Second E1p and E1o of E. coli (85, 114) and A. vinelandii (174) can only transfer acyl groups to their cognate E2 protein thereby providing an accurate substrate channeling mechanism such that the reductive acylation only occurs on the lipoyl group covalently attached to the appropriate E2 subunit. Third, although the attached lipoate was once thought to be freely rotating (175, 176), recent structural data showed that the lipoyl-lysine β–turn of the domain became less flexible after lipoylation of the lysine residue (177). The restricted motion of the lipoyl group would facilitate the effective E1 and E2 interaction by presenting the lipoyl group in the preferred orientation to the active sites of E1 and thereby enhance catalysis. This is in agreement with the recent crystal structure of the E1 component of the BCDH complex from P. putida (178). According to this structure, the active site where thiamine diphosphate binds is at the bottom of a long funnel-shaped tunnel, which suggests that the lipoyl group attached to the lipoyl domain must be fully extended and accurately positioned in order to reach the thiamine diphosphate cofactor. Amino acid side chains responsible for this specific positioning have been mapped to two residues that flank the lipoyl-lysine (179). Finally, the prominent surface loop connecting β–strands 1 and 2 (which lie close in space to the lipoyl-lysine) is another major determinant of the interactions of the lipoyl domain with E1 (180). Deletion of this loop results in a partially folded domain and almost completely abolishes lipoylation and reductive acylation indicating that the loop is involved in maintaining the structural integrity of the domain, post-translational modification and catalytic function (177). It is believed that the loop structure is important for stabilizing the thioester bond of the acyl-lipoyl intermediate (177, 181).
Figure 8.
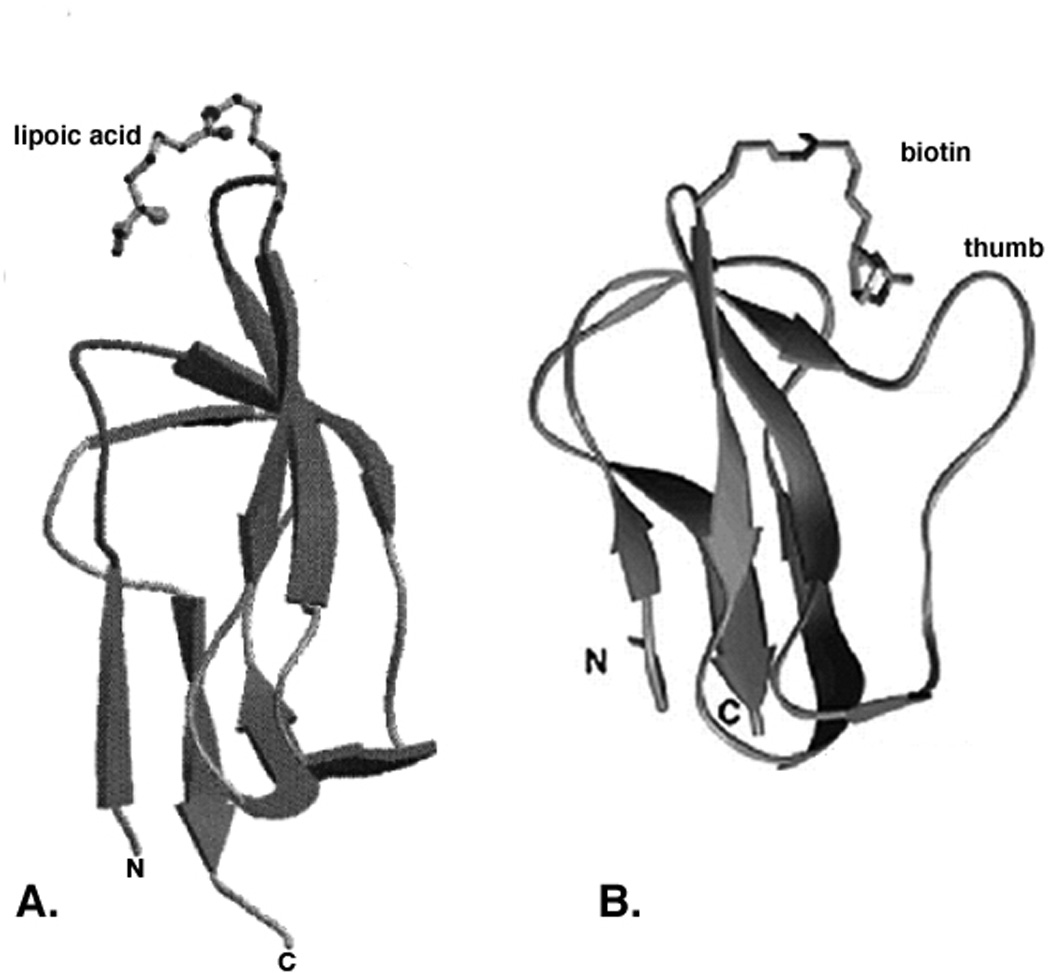
Three-dimensional structures of E. coli lipoyl and biotinyl domains. Panel A. The innermost lipoyl domain of E. coli PDH. Panel B. The BCCP biotinyl domain of E. coli acetyl-CoA carboxylase. The images are MOLSCRIPT drawings from the NMR data of Jones and coworkers (177) and the diffraction data of Athappilly and Hendrickson (193), respectively.
Subgenes that encode the lipoyl domains from a wide range of bacteria, including E. coli E2p (182) and E2o (183), Bacillus stearothermophilus E2p (184), human E2p (185), Azotobacter vinelandii E2p (166) and E2o (186), and Neisseria meningitidis E2p (187) have been overexpressed in E. coli and sufficient recombinant protein has been obtained for the domain structures to be determined by multidimensional nuclear magnetic resonance (NMR) spectroscopy. The archetypical structure, that of the single apo lipoyl domain of the E2p chain of B. stearothermophilus (188), is composed largely of two four-stranded β–sheets, with the N- and C-terminal residues of the domain close together in space in one sheet and the lysine residue earmarked for lipoylation in an exposed position in a tight type I β–turn generated by β–strand 4 and 5 in the other sheet. There is a well-defined hydrophobic core, the least well-defined regions being the exposed β–turn where the lipoyl-lysine resides and, most notably, the nearby large surface loop that connects β–strands 1 and 2 (Fig. 8A). Consistent with the high level of sequence similarity between lipoyl domains of 2-oxoacid dehydrogenase multienzyme complexes, all other lipoyl domains conform to the same structural pattern. Given the small differences in the NMR spectra of the lipoylated and unlipoylated forms of the B. stearothermophilus (175) and A. vinelandii (189) E2p domains, the structures of holo- and apo-domains have been inferred to be substantially the same.
The determination of lipoyl domain structures has allowed prediction of the structure of another lipoylated protein: the H protein of the glycine cleavage system. H proteins are about 130 resides in length (190). Although the overall sequence identity was low (<20%) (191), the conservation of key residues indicated that there was likely to be considerable structural similarity between the H protein of glycine cleavage system and the lipoyl domains of 2-oxo acid dehydrogenase complexes (192). Indeed, the X-ray crystal structure of the lipoylated pea leaf H protein agreed well with the theoretical predictions. The biotinyl domains of biotin-dependent enzymes have structures strikingly similar to those of lipoyl domains (Fig. 8B) as originally predicted by Brocklehurst and Perham (192). This is particularity true of biotin domains from enzymes other than bacterial and plant plastid acetyl-CoA carboxylases. The biotinylated subunits of the bacterial and plastid acetyl-CoA carboxylase contain a characteristic thumb structure not found in other biotinoyl domains or in lipoyl domains (10). The structure of the biotin domain of E. coli AccB has been established by X-ray crystallography (193) and NMR spectroscopy (Fig. 8B) (194–196). The structure closely resembles those of the lipoyl domain in the E2 component of 2-oxoacid dehydrogenase complexes and of the H protein in the glycine cleavage system. Like these lipoylated proteins the AccB domain is a flattened β-barrel, comprising two 4-stranded anti-parallel β–sheets, with the biotinyl-lysine residue located in the exposed β–turn between β–strands 4 and 5 (Fig. 8B). The high-resolution NMR structure of another biotinoyl domain, that of Propionibacterium shermanii transcarboxylase, has also been determined (197). This structure more closely resembles the lipoyl domain structures since it lacks the protruding thumb of the E. coli biotin domain (to which it is otherwise quite similar). Depending on the pair of domains chosen for comparision the root mean square deviation of biotinoyl and lipoyl domain backbone atoms can be as low as 1 Å and hence these proteins define a protein family (PF00364). Other work has shown that one of the proline/alanine-rich linker regions that lie between the domains of E. coli PDH can functionally replace the proline/alanine-rich linker region that lies upstream of the biotin domain of E. coli BCCP (130) underlining the interrelatedness of the biotin and lipoic acid acceptor proteins.
Protein lipoylation pathways
Post-translational modification of apoproteins with lipoic acid occurs by several mechanisms. In E. coli, two complementary systems for protein lipoylation have been characterized, by genetic and subsequent biochemical analyses. Exogenous lipoate or octanoate is transferred to unlipoylated apoproteins in an ATP-dependent process by lipoate-protein ligase (LplA) (198, 199). The second E. coli pathway requires the lipB gene product (octanoyl-ACP:protein-N-octanoyltransferase) to transfer endogenously synthesized octanoate to apoproteins which is then becomes the substrate for sulfur insertion (Figure 6) (199–203).
Lipoate-protein ligase (LplA)
Lipoate-protein ligase activity was first described by Reed and coworkers (204) in Enterococcus faecalis as well as in E. coli and these workers postulated that lipoate was attached to protein by a two-step ATP-dependent reaction with lipoyl-AMP as an activated intermediate. The reaction is exactly the same as that of BirA (Fig. 7) with the substitution of lipoic acid for biotin (hence this is not shown). Although the lipoate-protein ligases were key reagents in demonstration of the role of lipoic acid in the 2-oxoacid dehydrogenase reactions (142, 205), neither protein had been purified to homogeneity and thus the proposed mechanism could not be proved. The E. coli lplA gene was the first lipoate-protein ligase gene to be isolated and LplA was the first such ligase purified to homogeneity (199, 206). Mutants in lplA were isolated by two different approaches. In the first approach a lipA strain was mutagenized by transposon insertion and the mutagenized cells were supplemented with a mixture of succinate and acetate to bypass the lipoate requirement. The supplement was then switched to lipoate and an ampicillin enrichment was performed followed by plating onto the succinate-acetate supplemented medium. The resulting colonies were screened for strains able to grow on succinate-acetate supplemented medium, but not on lipoate supplemented medium. Three classes of such mutant strains could have resulted from this scheme, strains lacking the ligase (lplA), strains defective in lipoate transport and lpd mutants that lack the E3 subunit common to all of the lipoate-dependent enzymes of E. coli. Indeed, the selection was an unwitting repeat of the selection used for lpd mutants (207). Surprisingly, all of the mutants isolated were lplA mutants. It is unclear why no lpd mutants were isolated in the lplA selection and vice versa. The lack of lipoate transport mutants suggests that there may be no lipoate transporter in E. coli (as is believed to be the case for short chain fatty acids). Given the small size, hydrophobicity and the miniscule amount of the cofactor needed for growth no transporter may be needed. Indeed it has been reported that both enantiomers of 35S-lipoate were taken up by E. coli, although only R-lipoic acid became attached to the 2-oxoacid dehydrogenases (208). Since a protein transporter would be expected to discriminate between enantiomers, this finding argues strongly against the existence of a lipoate transporter. Mutants mapping in lplA were also isolated by a direct selection, resistance to selenolipoic acid. Selenolipoic acid is a growth-inhibitory lipoate analogue in which the sulfur atoms are replaced with selenium (209). These mutants proved to encode a ligase of somewhat compromised activity that was able to discriminate against the selenium analogue (198).
The purified LplA enzyme is a 38 kDa monomeric protein (206). Assays with a fully purified apoprotein acceptor have demonstrated that purified LplA plus lipoate and Mg-ATP are sufficient to reconstitute lipoylation in vitro (126, 199, 206, 210). Thus, it is clear that LplA catalyses both the ATP-dependent activation of lipoate to lipoyl-AMP as well as the transfer of this activated lipoyl species to apoprotein with concomitant release of AMP. This conclusion is consistent with the early findings of Reed and coworkers (204) that the E. coli enzyme could not be fractionated into separable lipoate activation and lipoyl transferase components. LplA has been shown to be capable of utilizing lipoate and several lipoate analogs as donors for the post-translational modification of E2 apoproteins in vivo (199). This rather-broad substrate specificity in vivo matches the similarly broad substrate specificity observed (211).
Very recently several crystal structures of LplA and of LplA homologues have been reported including structures of E. coli LplA (212) plus an LplA-lipoic acid complex (212). The reported structures agree well and show E. coli LplA to by two-domain protein consisting of a large N-terminal domain and a small C-terminal domain. The E. coli LplA-lipoic acid complex was difficult to interpret because lipoic acid was bound to different LplA molecules within the crystals with different modes and with poor resolution. For example in one case the lipoic acid carboxyl was hydrogen bonded to Ser-72 whereas in the other case Arg-140 was the hydrogen bond donor (212). Since enzymes rarely show such plasticity and lipoic acid is a hydrophobic molecule, it seemed possible that the observed association with a hydrophobic LplA surface in the interdomain cavity was artifactual. Moreover, in prior work Reed and coworkers had isolated LplA mutants resistant to selenolipoic acid (209). Since this is a very discrete modification of the LplA substrate, the mutant protein would be expected to have an alteration close to the pocket that binds the lipoic acid thiolane ring. However, the site of this mutation (Gly-76 to serine, (198)) was distal from the lipoate-binding site reported. This dilemma appeared resolved first by two lipoic acid-containing structures of an LplA homologue from the Archaeon, Thermoplasma acidophilum (213, 214) that can be readily superimposed on the E. coli LplA structure except that the T. acidophilum protein lacks the LplA C-terminal domain. In both T. acidophilum structures the lipoate thiolane ring was close to the glycine residue that corresponds to E. coli Gly-76, the residue giving resistance to the selenium analogue and a plausible reorganization of the molecule to prevent binding of the larger selenolipoic acid was proposed (214). Moreover, addition of lipoic acid to a complex of the T. acidophilum with ATP gave lipoyl-AMP thereby showing that the lipoic acid was bound in a physiologically meaningful manner (213). Lipoyl-AMP was bound in a U-shaped pocket and was well shielded from solvent. The T. acidophilum LplA was reported to be inactive in catalyzing the overall LplA reaction (214), although lipoyl-AMP synthesis was demonstrated (213). Since T. acidophilum LplA lacks the C-terminal domain of E. coli LplA (213, 214) this suggested that the missing domain plays a key role in transfer of the lipoyl moiety from lipoyl-AMP to the acceptor domain Indeed, a second protein has been proposed to interact with T. acidophilum LplA and allow the complete reaction (214) and this proved to be the case (215, 216). The fact that the lipoate of one of the T. acidophilum LplA structures was converted to lipoyl-AMP and that the locations of lipoate moieties of the two T. acidophilum LplA structures agreed well argues that these represent the catalytically competent lipoate binding site. It therefore follows that in the first E. coli LplA structure the lipoate molecule was bound in a catalytically inappropriate manner. Indeed, in a later report from the same group cocrystals of LplA with lipoyl-AMP and LplA with octyl-5′-AMP and the apo form of GcvH. These structures define the LplA tructural mechanism. Three large scale conformational changes occur upon completion of the lipoate adenylation reaction i) the adenylate-binding, ii) the lipoate-binding loops move to maintain the lipoyl-5′-AMP reaction intermediate and iii) the C-terminal domain rotates by about 180 degrees. These changes are prerequisites for LplA to accommodate the apoprotein for the lipoate transfer reaction. The invariant Lys133 residue plays essential roles in both lthe ipoate adenylation and transfer steps.
3.2 Octanoyl-ACP:protein N-octanoyltransferase (LipB)
During the characterization of E. coli lplA null mutant strains compelling evidence was found for a second protein lipoylation pathway that did not require the lplA gene product. When independently derived lplA null alleles were transduced into wild-type strains, the resulting mutant strains showed no growth defects on minimal glucose medium indicating that these strains possessed functional (therefore lipoylated) 2-oxoacid dehydrogenases. This was directly confirmed by bioassays that showed lplA null mutants to contain normal levels of lipoylated proteins (198). Thus, it was clear that E. coli has an lplA-independent lipoylation pathway. This was first attributed to a second ligase that had somehow been missed in the biochemical analyses perhaps due to the in vitro conditions chosen. However, no such second ligase could be found (206) and thus alternative pathways were considered. The most straightforward alternative pathway was that the fatty acid synthesis intermediate, octanoyl-ACP would be converted either directly or indirectly to lipoylated proteins. That is, lipoate synthesis would occur without a free carboxyl group. The carboxyl group would be bound in the thioester bond that links fatty acids to ACP and this bond would then be attacked by the ε-amino group of the lipoyl domain lysine residue to give the amide linkage. Several lines of evidence demonstrated that this alternative protein lipoylation pathway was dependent on the lipB gene product. The lipB gene was originally isolated as a class of lipoic acid auxotrophs (6). These mutants showed residual (leaky) growth in the absence of lipoic acid despite having putative null mutations due to transposon insertions into lipB (6, 217). This leakiness was reflected in their 2-oxo acid dehydrogenase activities and lipoylated protein contents. These strains retain about 20% of the enzyme activities and about 10% of lipoyl protein content of wild type strains (217). The leaky growth of lipB strains in the absence of lipoate was eliminated by introduction of an lplA mutation suggesting that lipB was involved in lipoyl domain modification as well as lipoate biosynthesis (198). Indeed, bioassays demonstrated that the low lipoyl protein content of the lipB null mutants was further depressed to undetectable levels in the lipB lplA double mutants (198). This finding suggested that the attenuated, but still detectable, accumulation of protein-bound lipoate by lipB null mutants was entirely due to the action of the lplA gene product. Moreover, overexpression of LplA allowed normal growth of lipB null mutant strains in the absence of lipoate thus clearly demonstrating the redundant roles of these two genes (198). Genetic and biochemical evidence demonstrated that lipB encoded the octanoyl-ACP:protein N-octanoyltransferase (which is also an lipoyl-ACP:protein N-lipoyltransferase) (Fig. 9). An enzyme activity was detected in E. coli cell extracts that catalyzed the transfer of octanoic acid (lipoic acid) from octanoyl-ACP (lipoyl-ACP) to E2 apo-domains (Fig. 9). This activity was also found in extracts of E. coli lplA null mutants and, unlike LplA, this activity was not dependent on ATP. However, transferase activity was absent in E. coli lipB mutants (218). A temperature-sensitive lipB strain was obtained and found to encode a transferase of decreased thermal stability (200) indicating that lipB encoded the transferase rather than playing a regulatory role. Finally, His-tagged LipB was purified and the purified protein had high levels of octanoyltransferase and lipoyltransferase activities (200). The untagged protein has recently been purified by conventional means (219).
Figure 9.

Overview of the LipB reaction. The thioester linkage of octanoic acid attached to the thiol of the 4′-phosphopantheteine group of ACP (the product of fatty acid synthesis) is attacked by the ε-amino group of the target lysine of a lipoyl domain resulting in the modified protein plus the free thiol form of ACP. The enzyme also uses lipoyl-ACP although this is thought to be of no physiological importance. The reaction proceeds through an octanoyl-LipB acyl enzyme intermediate (not shown).
Based on these observations, a two pathway E. coli lipoylation system was proposed (198, 201, 218) (Fig. 10). When presented with free lipoic acid in the medium E. coli incorporates extracellular lipoate (217, 220) via the LplA-dependent scavenging pathway which utilizes ATP to activate lipoic acid in the form of lipoyl-AMP. When lipoate is absent from the medium lipoyl groups must be made by de novo synthesis. An octanoyl group is first transferred from octanoyl-ACP to the apo-proteins by LipB. LipA then acts on the protein-bound octanoyl groups to catalyze the sulfur insertion step (Fig. 10). This model explains why lplA null mutants showed no growth defects unless the strain also carried a lipA or lipB mutation as well as the unidirectional redundancy between LipB and LplA functions. LplA was reported to utilize octanoyl-ACP as substrate with low but detectable efficiency (200) which was puzzling given the very different chemistries of the two reactions. However, more recent work showed that this was due to traces of ATP and octanoate present in the octanoyl-ACP preparations (F. Hermes and J. E. Cronan, unpublished data). Recently it was shown that the ability of LplA overexpression to complement ΔlipB strains is due to increased scavanging of cytosolic octanoic acid. In the absence of LplA overexpression suppression by chromosomal mutations was observed. These suppressor mutations map in LplA and result in proteins having reduced Km values for free octanoic acid which allows efficient scavanging of cytosolic octanoic acid (221).
Figure 10.
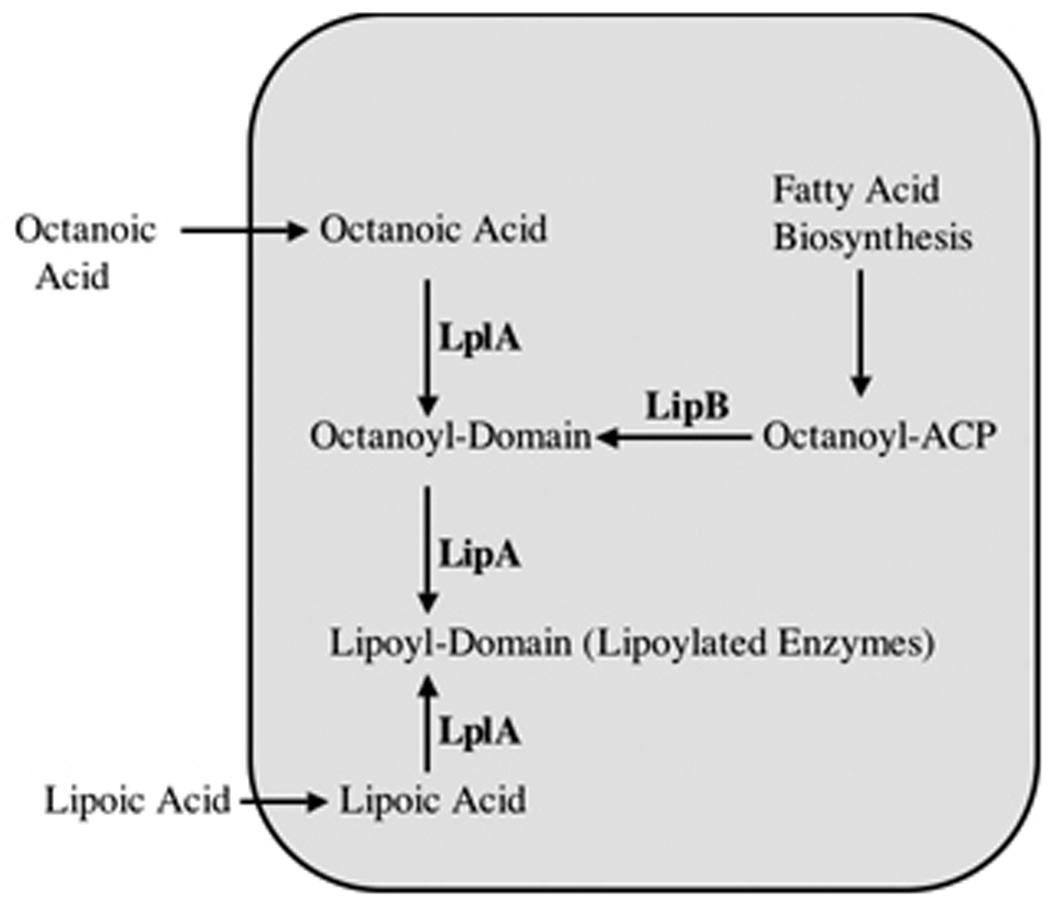
Current model for lipoic acid synthesis and utilization in E. coli. The rounded rectangle denotes an E. coli cell. Exogenous lipoic acid or octanoic acid enter by diffusion and are attached to the 2-oxo acid lipoyl domains and to H protein by LplA. The domains modified with exogenously derived octanoate can be converted to lipoyl domains by LipA, although this is probably not a reaction of physiological significance because high levels of octanoic acid are required for significant modification by this route. In contrast LplA-catalyzed attachment of lipoate is very efficient and provides a salvage or scavenging pathway for utilization of exogenous lipoic acid and seems likely to be physiologically significant. The major (and probably sole) route of lipoic acid synthesis is LipB-catalyzed transfer of octanoate from octanoyl-ACP to the 2-oxo acid lipoyl domains and H protein followed by LipA-catalyzed sulfur insertion to give lipoate.
It should be noted that strains having null mutations in both lplA and lipB contain no detectable lipoylated proteins indicating that LplA and LipB are the only E. coli enzymes capable of modifying lipoyl domains (198).
Three assays have been used to detect attachment of lipoic acid to apo forms of PDH and 2-OGDH in vitro. The first assay measured the conversion of radioactive lipoate (or octanoate) to a protein-bound form defined as being insoluble in organic solvents (199). This is a sensitive and quantitative assay, but applicable only to LplA since both the LipB substrate and product are protein bound. The second assay relies on the use of inactive unmodified apo-PDH or 2-OGDH complex purified from a lipB lplA strain completely deficient in modification of the E2 proteins. Lipoylation of the purified apo-PDH or 2-OGDH complex was detected by assay of the products of ligation reactions for either PDH or 2-OGDH activity (222). The third assay is a gel mobility shift assay (218). It follows the acylation-dependent shift in the electrophoretic mobility of a purified 87 residue apo-lipoyl domain from the E. coli PDH complex (223). This assay is much less sensitive than the other two but has the advantage that it can be used with any acyl donor because the mobility shift is due to loss of the positive charge of the lysine residue. With this assay it was found that purified LipB could convert the apo form of lipoyl domain to the holo domain with either octanoyl-ACP or lipoyl-ACP as the substrate. When LipB was tested for the ability to use ATP plus lipoic acid or octanoic acid, no modification was detected.
Another lipB phenotype is that multiple copies of the gene confer resistance to selenolipoic acid. An analogue-resistant mutant that did not map at the lplA locus (209) was shown to be a chromosomal amplification of the lipA lipB region of the chromosome (224). The increased lipB dosage resulted in greater LipB activity that resulted in increased levels of lipoylation by endogenously synthesized lipoic acid that competed with the utilization of exogenous selenolipoic acid via the LplA-dependent pathway. A very modest (two- to threefold) increase in lipB dosage was sufficient to give resistance which was explained by the known highly nonlinear relationship between the degree of protein lipoylation and the activity of the 2-oxoacid dehydrogenase complexes plus the fact that E. coli does not require full activity of the 2-oxoacid dehydrogenases for growth on minimal medium containing glucose (224). Thus, synthesis of sufficient lipoic acid to modify a few percent of the 2-oxo acid dehydrogenase complexes allowed growth in presence of selenolipoic acid.
As mentioned above the LipB reaction was recently shown to proceed via an acyl enzyme intermediate (225). The octanoyl group is transferred from the ACP thiol to the thiol of Cys-169. This protein-bound thioester is then attacked by the ε-amino group of the lipoyl domain lysine to give the modified domain.
Octanoyl-ACP + LipB ⇌ [Octanoyl-LipB] + ACP-SH
[Octanoyl-LipB] + Apo Lipoyl Domain * Octanoyl-E2 Domain
The fact that LipB could transfer either octanoate or lipoate from ACP to a lipoyl domain raised the question of the true intracellular substrate of the enzyme. This has been answered by overexpression of LipB and isolating the enzyme under conditions that retained any acyl-enzyme interemediate present. Mass spectrometry showed that only octanoate was attached to the enzyme thereby demonstrating the specificity of the enzyme (226).
The reactivity of the cysteine residue seems responsible for the both LipB crystal structure scurrently available, those of the Mycobacterium tuberculosis and T. acidophilum enzymes (227, 228). The M. tuberculosis LipB, expression which complements growth of E. coli ΔlipB mutant strains, was crystallized in a covalent complex with decanoic acid. Surprisingly, although the acyl chain was bound to the sulfur atom of a cysteine residue corresponding to Cys-169 of E. coli LipB, the bond was a thioether linkage to C3 of decanoate rather than a thioester link to the carboxyl group (227). This unexpected finding seems likely to be the result of a Michael addition of the cysteine thiol to the unsaturated bond of trans-2 decenoyl-ACP or cis-3-decenoyl-ACP, a key intermediate in E. coli unsaturated fatty acid biosynthesis. Consistent with this interpretation no such adduct was seen upon expression of the protein in Mycobacterium smegmatis, which forms unsaturated fatty acids by a pathway that does not involve decenoyl intermediates (227). However, the protein lacking the adduct failed to crystallize and thus adduct formation trapped LipB into a form amenable to crystallization. T. acidophilum LipB structure also required covalent trapping I to form crystals, an intermolecular disulfide (228). Based on the M. tuberculosis LipB crystal structure and mutagenesis studies LipB is thought to function as a novel cysteine/lysine dyad acyltranferase, in which the dyad residues function as acid/base catalysts (227).
Biosynthesis of lipoic acid
Although the functions of lipoic acid in the multienzyme complexes have been well studied over the past forty years, an understanding of lipoic acid biosynthesis pathway has only recently been achieved. Such studies have focused on E. coli. Early studies had established that octanoic (properly n-octanoic) acid (Fig. 1) is the precursor of the lipoic acid carbon chain (229). Analysis of the conversion of specifically labeled forms of octanoic acid to lipoic acid by E. coli cultures showed that sulfur atoms are introduced with loss of only two hydrogen atoms from the chain, one from C-6 and the second from C-8 (230, 231). Additional metabolic feeding studies demonstrated that E. coli lipoic acid biosynthesis does not involve either desaturation or hydroxylation of octanoic acid, but does result in inversion of stereochemistry at C-6 (231, 232). Sulfur is introduced at C-8 with racemization in agreement with the formation of an intermediate carbon radical at C-8 (230, 232–234). 8-Thiooctanoic acid and 6-thiooctanoic acid were readily converted to lipoic acid, although 6-thiooctanoic acid was converted only 10–20% as efficiently as the other positional isomer (234). Genetic studies identified a single E. coli gene responsible for the sulfur-insertion steps of lipoic acid biosynthesis, first called lip (152) and now called lipA which encodes a protein called lipoic acid synthase. E. coli strains having null mutations in lipA do not synthesize lipoic acid and the phenotypes of these mutants suggested that LipA was responsible for the formation of both C-S bonds (6] which encodes a protein called lipoic acid synthase. E. coli strains having null mutations in lipA do not synthesize lipoic acid and the phenotypes of these mutants suggested that LipA was responsible for the formation of both C-S bonds {Herbert, 1968 #114, 217, 235, 236) and encodes a protein now called lipoic acid synthase. There are strong parallels between LipA and biotin synthase (BioB), the enzyme discussed above that catalyzes the final step in the biotin biosynthesis (Fig. 4). LipA like BioB makes two C-S bonds and also removes two unactivated hydrogen atoms. The similarity in chemistry between the biosynthesis of the dithiolane ring of lipoate and the thiophane ring of biotin strongly suggests functional parallels in the mechanisms of the enzymes that produce these compounds. Indeed, the amino acid sequences of the E. coli LipA and BioB proteins show marked similarities; 40% sequence similarity and 17% sequence identity (217).
As discussed above BioB has both a [4Fe-4S] cluster and a [2Fe-2S] cluster. The canonical iron-sulfur cluster binding motif CXXXCXXC is also found in the LipA sequence leading to early predictions that it is an iron-sulfur protein (217, 236). The LipA protein has been overexpressed in E. coli and purified from both soluble lysates and insoluble aggregates that were subsequently refolded and reconstituted with ferrous iron and sulfide (217, 237, 238). The purified dimeric protein (237) has a dark reddish-brown color and displays a band at 420 nm in its light absorption spectrum, characteristic of a sulfide to iron charge transfer. Resonance Raman, electronic absorbance and Mössbauer spectroscopic results were consistent with the presence of an iron-sulfur cluster in LipA. It was suggested that LipA contains [2Fe-2S] clusters that during reduction are converted into [4Fe-4S] clusters (237, 238). The Fe-S cluster of LipA was first suggested to be a [4Fe-4S] cluster bridging the two subunits (237). However, in a different report it was suggested that the limited amount of Fe and S atoms and the presence of [2Fe-2S] clusters in the previous preparation of LipA were a direct consequence of aerobic isolation. It was reported that under strictly anaerobic conditions LipA could bind one [4Fe-4S] cluster per subunit (239). Recently it was reported that LipA contains two distinct [4Fe-4S] clusters per polypeptide (203). Thus, the types of disagreements seen in the BioB literature are also apparent for LipA showing the obvious difficulties of working with this family of proteins
Direct involvement of LipA in the sulfur insertion reaction of lipoic acid biosynthesis was difficult to establish due to the lack of an in vitro assay. Much of this difficulty was due to the assumption that free octanoic acid was the sulfur acceptor. The first indication that this was not the case was the demonstration by Jordan and Cronan (218) of the LipB transferase activity. Miller and coworkers (240) were the first to report synthesis of lipoic acid in vitro. This was based on the discovery of LplA and LipB which led to development of a defined in vitro lipoic acid synthesis system and an assay that was much more sensitive and quantitative than prior assays (240). Lipoic acid synthesis was assayed indirectly using (i) the apo form of pyruvate dehydrogenase complex (apo-PDH) as a lipoyl-accepting protein, (ii) purified LipA, and either (iii) purified LplA, ATP, octanoic acid as a substrate (for lipoic acid synthesis) or iv) LipB and octanoyl-ACP as a substrate. Activation of apo-PDH upon lipoylation was monitored spectrophotometrically via reduction of an NAD+ analogue. Within a finite range, the rate of reduced pyridine dinucleotide formation was directly dependent upon the amount of lipoylated PDH. This assay showed that LipA is responsible for both of the sulfur insertions and that octanoyl-ACP (or a derivative of octanoyl-ACP), but not octanoic acid, was a LipA substrate. Moreover, this work showed that, as suspected, the LipA reaction requires iron-sulfur clusters and SAM to perform the radical chemistry. The principal disadvantage of this assay was its indirect nature and detection of lipoylation of apo-PDH rather than of the primary lipoyl protein species per se. All attempts to isolate a free lipoyl-ACP product in the assay were unsuccessful. Thus, the exact identity of the immediate product of the LipA reaction could not be determined by this assay. Recent studies demonstrate that LipA acts on octanoylated derivatives of lipoyl-accepting proteins (201, 241, 242).
Lipoic acid synthesis proceeds by an unexpected and extraordinary pathway
The first evidence that octanoyl-domain rather than octanoyl-ACP was the substrate for sulfur insertion was the finding that lipB mutants grew well when supplemented with octanoic acid in place of lipoic acid (201). Octanoate supplementation of lipB strains required function of both the lipA and lplA genes; both lipB lipA and lipB lplA doubly mutant strains failed to grow on octanoate. Moreover, growth was specific to octanoate, fatty acids of 6, 7, 9 and 10 carbons were inactive (201). These observations argued for the existence of an LplA-dependent pathway that bypassed LipB function in the presence of octanoate. In the postulated bypass pathway (Fig. 10) LplA would attach octanoate derived from the growth medium to the unmodified E2 domains of the PDH and 2-OGDH E2 subunits. LipA would then insert two sulfur atoms into the covalently bound octanoyl moiety and thereby convert the octanoyl-E2 domains to lipoyl-E2 domains in situ. That is, lipoic acid would be assembled on its cognate proteins. The resulting active enzymes would account for the observed growth of lipB strains on octanoate (Fig. 10). This pathway was tested in vivo (201). First, an 87 residue E2 domain derived from E. coli PDH was expressed in a host strain that carried null mutations in lipA (to prevent lipoic acid synthesis), lipB (to block octanoate transfer from fatty acid synthesis) and fadE (to block β-oxidative degradation of octanoate). The use of the domain allowed detection of modification by the electrophoretic mobility shift assay and by mass spectroscopy. When this strain was cultured in a medium supplemented with octanoic acid about half of the domain became modified. In addition the LipB-dependent modification pathway was assayed in a lipA lplA null mutant strain grown in the absence of exogenous octanoate. In agreement with prior work using a lipA strain (223) octanoyl-E2 domain accumulation was detected. Therefore, the E2 domain could be octanoylated in vivo either by LplA using exogenously added octanoate or by LipB using de novo synthesized octanoate. In order to assay conversion of octanoyl-E2 domain to lipoyl-E2 domain the lipA lipB fadE strain was supplemented with deuterated octanoic acid to allow accumulation of octanoyl d15-E2 domain that was readily distinguished by mass spectroscopy from domain modified with endogenously-synthesized octanoate. Following accumulation of the d15-labeled octanoyl-E2 domain LipA function was restored by transduction with cells with phage λ particles containing a lipA cosmid that had been packaged in vivo. Using this approach two types of labeling experiments were done. In the first protocol E2 domains were labeled in vivo by growth in the presence of octanoic d15 acid. Following removal of the labeled octanoate the cells were then resuspended in growth medium and transduced with the packaged lipA-encoding cosmid. Following incubation to allow lipoate synthesis samples were taken and the E2 domain species were isolated, purified, and analyzed by electrospray mass spectroscopy (Fig. 11). In the cultures to which LipA activity was restored a readily detectable conversion of the E2 domain modified with octanoate d15 to a species of 60 additional mass units was seen. This was exactly the increase in mass (gain of two sulfur atoms of mass 32 and loss of two deuterium atoms of mass two) expected for conversion of the d15 labeled octanoyl-E2 domain to the d13-labeled lipoyl-domain. In the second protocol (a variation of the first protocol) the octanoic d15 acid was removed by washing the cells and replaced with normal (non-deuterated) octanoate. This experiment gave essentially the same result; the d15 labeled octanoyl-E2 domain was converted to d13 labeled lipoyl-E2 domain (Fig. 11). A modification of these experiments also showed that octanoyl-PDH accumulated in vivo in a lipA strain was converted to its active form upon restoration of LipA activity (201). The conversion of octanoyl-domain to lipoyl domain was also observed in vitro (201), although the extant of conversion was much less than stoichiometric with LipA. These result were recently confirmed using octanoyl-H protein as the substrate with an eight-fold increase in the yield of lipoic acid formed/LipA monomer (202).
Figure 11.
Lipoate synthesis proceeds by sulfur insertion into octanoyl-domain. The bypass pathway accounting for growth of lipB mutants on octanoate is shown in the upper right cartoon. The experimental protocol scheme and mass spectral data for testing the pathway is also shown. In the left cartoon octanoylation of the lipoyl domain by endogenously synthesized octanoyl moieties is blocked by a lipB mutation and the cells use LplA and exogenously supplied d15 octanoate to octanoylate the domain. LipA is also blocked so deuterated lipoylated domain is not made. Following accumulation of the deuterated octanoyl domain LipA function is restored (right cartoon). Following incubation to allow lipoate synthesis, the cells are harvested and the modified domains were purified then analyzed by electrospray mass spectrometry. Note the accumulation of deuterated lipoylated domain (D-Lip) in the right hand spectrum and that the mass change between deuterated octanoylated domain (D-C8) and D-Lip is 60 mass units indicating loss of two deuterons and gain of two sulfur atoms. For details see (201).
As mentioned above lipoic acid synthase is a member of the radical-SAM enzyme superfamily which utilize a reduced iron-sulfur cluster and SAM to generate 5’-deoxyadenosyl 5′-radicals (5′-dA) for further radical-based chemistry (59, 243–246). In the lipoic acid synthase reaction (Fig. 12), it is generally believed that the role of the 5′-dA is to remove one hydrogen atom from each of the C-6 and C-8 positions of octanoic acid thereby allowing for subsequent sulfur insertion (202, 240). Consistent with this prediction two molecules of SAM are required to synthesize one mole of lipoyl cofactor (202). This stoichiometry is similar to that obtained in the two studies in the BioB reaction (247, 248) and suggests that the abortive cleavage of SAM observed in these systems might result from some innate reactivity associated with this subclass of radical SAM enzymes (202).
Figure 12.
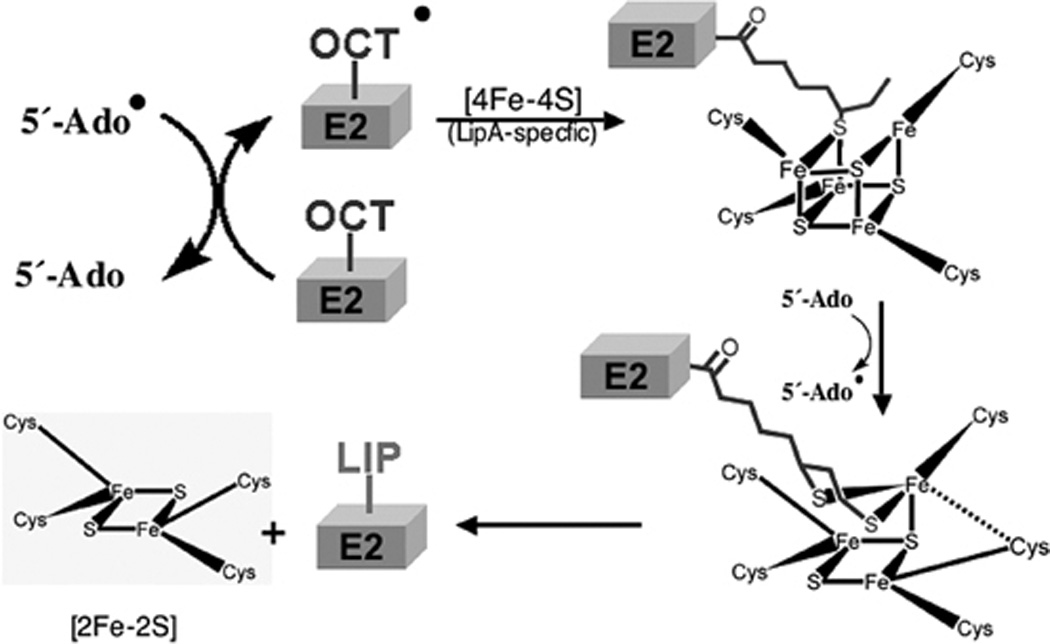
A current model of the lipoate synthase (LipA) reaction (202, 203, 240). The canonical SAM radical [4Fe-4S] cluster of LipA reduces SAM to generate the deoxyadenosine radical (5′-Ado) as seen previously in the BioB reaction (Fig. 4). The radical then removes a hydrogen atom from the C6 methylene of the octanoate moiety of an octanoyl domain (Oct-E2 on the Figure) (229) to give a carbon radical that then attacks the lipoyl synthase-specific [4Fe-4S] cluster and abstracts a reduced sulfur atom. This process is repeated at the methyl carbon (C8) to give lipoyl-domain (Lip-E2, probably as the dihydrolipoyl form due to the strongly reducing conditions under which the reaction proceeds).
As in the case of biotin the source of the sulfur atoms of lipoic acid is thought to be a [Fe-S] cluster distinct from the SAM radical [4Fe-4S] cluster. In the first successful in vitro lipoic acid synthesis assays, lipoic acid was formed in the absence of exogenous sulfur-containing compounds in the in vitro assay (202, 240). This suggested that, like biotin synthase, the protein itself has some mobilizable sulfur atoms, either from an Fe-S cluster, a persulfide or some other species. The currently favored sulfur source is an iron-sulfur cluster (240, 249). Recent work from the Booker group reported that lipoic acid synthase contains two [4Fe-4S] clusters (203). One cluster is coordinated by the radical SAM CXXXCXXC motif that functions in 5′-dA generation. The second cluster is coordinated by the CXXXXCXXXXXC motif, which, thus far, is unique to lipoic acid synthases and suggested to be the source of the sulfur atoms. This has been addressed (as in BioB (63)) by preparation of LipA from E. coli cells grown on an isotopically labeled sulfur source (34S) (250). As expected the lipoic acid formed using this enzyme in vitro was isotopically labeled with 34S. Moreover, when the reactions were performed with equimolar amounts of 32S-labeled LipA and 34S-labeled LipA the lipoic acid molecules formed contained either two 32S atoms or two 3S-atoms (250). Thus both sulfur atoms emanate from the same polypeptide thereby eliminating the possibility that the monothiolated species are released and the second sulfur atom is inserted following rebinding to LipA. If release and rebinding occurred, half of the lipoic acid formed would have one atom each of 32S and 34S. Such mechanistic experiments are facilitated if an octanoylated peptide substrate can be substituted for the octanoylated lipoyl domain as is the case for an octanoyl-tripeptide in the Sulfolobus solfataricus LipA assay (242). This system has recently been used to show that S. solfataricus LipA catalyzed lipoate biosynthesis in a stepwise manner with sulfur first being inserted at C-6 of the octanoyl chain (251). However, this intermediate remained tightly bound to LipA. This is consistent with the finding that both sulfur atoms are derived from the same LipA polypeptide and implies that the sulfur atom of the intermediate may still exist as part of the Fe/S cluster. Incorporation of the second sulfur is much slower perhaps due to rearrangement of the Fe/S cluster. A current model of the LipA reaction is given in Fig. 12.
It should be noted that the finding that lipoic acid synthesis proceeds through an octanoyl-domain intermediate explains a previously puzzling observation first made by Ail and Guest (182, 223) and subsequently by others (252). These workers found that upon overproduction of a lipoyl domain in E. coli three species of domain were obtained, the expected apo and lipoylated domains plus a third species subsequently shown to be octanoylated domain. Based on conventional biochemistry in which LipA would produce free lipoic acid, the octanoylated domain was thought to be an anomalous product resulting from the lack of sufficient free lipoic acid to modify the overexpressed domain plus a lack of specificity of the attachment enzyme (182, 223). From the present pathway it now seems clear that LipA was limiting (as was proposed), but the octanoylated domain was an accumulated intermediate rather than the aberrant byproduct of overproduction.
Finally as expected from the BioB data LipA is inhibited by the 5′-deoxyadenosine producd in the reaction. The first indication was that addition of lipoic acid and biotin to a Δmtm (Δpfs) strain gave better growth than biotin alone (21). 5′-Deoxyadenosine inhibition of LipA and BioB that is potentiated by methionine and reversed by addition of Mtn has recently been demonstrated in vitro (76).
LipA and LipB form stable complexes with the 2-oxoacid dehydrogenases of E. coli.
The experiment discussed above in which the LipB-acyl enzyme intermediate was isolated from growing cells showed an unexpected result. The intact pyruvate and 2-oxoglutarate dehydrogenase complexes specifically copurified with affinity tagged LipB (226). Similar experiment showed that LipA, but not LplA, also interacted with the 2-oxoacid dehydrogenases. Proteomic, genetic, and dehydrogenase activity data indicate that all of the 2-oxoacid dehydrogenase components are present. The interaction is specific to the dehydrogenases in that GcvH did not copurify with either LipA or LipB. Studies of LipB interaction with engineered variants of the E2 subunit of 2-oxoglutarate dehydrogenase indicate that the binding sites for LipB reside both in the lipoyl domain and catalytic core sequences (226). These results indicate that lipoic acid is not only assembled on the dehydrogenase lipoyl domains but that the enzymes that catalyze the assembly are also present “on site.” The puzzling finding that LipA and LipB do not interact with GcvH is interesting in view of bioinformatic indications that GcvH falls into a different clade than do the dehydrogenase lipoyl domains despite their similar structures (253).
Remaining questions in lipoic acid synthesis
As in the case of BioB, LipA has not been shown to be catalytic in vitro, the best preparations to date form only about 0.4 molecules of lipoic acid per LipA molecule and thus the sacrificial protein scenario of BioB might pertain to this protein. The question of whether or not LipA is catalytic in vivo remains to be tested. If results analogous to those of BioB are obtained, the question of how the novel LipA CXXXXCXXXXXC motif becomes liganded by a [4Fe-4S] cluster. In this regard coexpression of LipA with the Isc proteins results in increased LipA activity (254). However, it remains to be seen which clusters are built by this manipulation. The crystal structure of LipA with SAM and octanoylated domain would be of great benefit. This would be facilitated if an octanoylated peptide substrate can be substituted for the octanoylated lipoyl domain as is the case for Sulfolobus solfataricus LipA (242). It has been predicted that LipA is a α 6 β 6 barrel protein (a three-quarters barrel) rather than a full TIM (α6β6) barrel like BioB (245). Moreover, LipA is reported to contain distinct two [4Fe-4S] clusters whereas BioB is believed to contain one [4Fe-4S] cluster and one [2Fe-2S] cluster. This difference in the secondary FeS clusters is presumably due to the need for two sulfur atoms to make lipoate versus one to make biotin. Therefore, although many of the questions that have bedeviled the BioB literature are germane to LipA, it seems clear that there are significant differences in how the two enzymes accomplish their reactions. A LipA crystal structure would be most useful in understanding these differences. Another major question concerns the regulation of lipoic acid synthesis. Although the lipB and lipA genes lie close to one another on the E. coli chromosome and are transcribed in the same direction, the genes are separated by 1.4 kbp and this spacer region contains ybeF, an open reading frame that encodes a possible LysR-type transcription factor. Strains carrying transposon insertions into and deletions of ybeF have no phenotype indicating that lipB and lipA are not in an operon (6, 120). Is expression of these genes regulated? Lipoic acid is clearly synthesized during aerobic growth and anaerobic function of the glycine cleavage enzyme indicates that it is also made under fermentative conditions (6). Moreover, a recent report that E. coli contains high levels of 2-oxoglutarate dehydrogenase when grown anaerobically with an electron acceptor such as nitrate (153) indicates that lipoic acid synthesis must also proceed under these growth conditions. The transcription of lipA in S. enterica has been assayed by transcriptional fusions to β-galactosidase and was found to be unaffected by catabolite repression conditions or by addition of lipoic acid (255). Therefore, the lipoic acid synthesis pathway may be constitutively expressed. Given the unusually sophisticated bio operon transcriptional regulatory system, it might seem unlikely that lipoic acid synthesis is unregulated. Indeed, biotin and lipoic acid are synthesized at similar levels in E. coli. However, biotin synthesis requires six enzymes and several of the reactions of the pathway require input of metabolically expensive molecules which might justify regulation of biotin synthesis enzyme production. In contrast octanoate, the precursor of the lipoic acid carbon chain, is derived from fatty acid biosynthesis in an already activated form, octanoyl-ACP, and lipoate synthesis consumes only a tiny fraction of the total cellular fatty acid synthetic capacity. Since LipB uses a preformed activated intermediate, the only further energetic input (in the form of SAM) occurs in the LipA sulfur insertion reaction. Therefore, relative to biotin synthesis, lipoic acid synthesis has low metabolic price. Another consideration is that the lipoic acid synthesis pathway is limited by the amount of apo-lipoyl domain available and thus unlike biotin synthesis lipoate synthesis is “hard wired” and cannot “run wild” to overproduce and excrete the cofactor as is the case when the bio operon is deregulated (117). Thus, it can be reasonably argued that regulation of the lipoic acid synthetic pathway might well be more expensive than the alternative of simply allowing constitutive expression of the genes. A perplexing observation is the report that LipB acts as a negative regulator of deoxyadenosine methyltransferase (dam) gene expression in E. coli (256). These workers speculate that LipB may inactivate a repressor protein by lipoylation. However, all of the proteins that become labeled with exogenous radioactively-labeled lipoate or octanoate in vivo are known subunits of the enzymes discussed above (6, 199). Hence, the putative lipoylated repressor would have to be modified by LipB, but not by LplA. Further studies of this interesting phenomenon are needed.
How do the protein biotinylation and lipoylation reactions remain discrete?
It is gratifying that the recent crystal structures have resulted in LplA, LipB and BirA being recognized as a new protein family (PFAM 03099.13) as predicted by Reche (210) although these proteins share only a single conserved residue. Indeed the Cα carbons of E. coli LplA and BirA (minus the DNA binding domain) structures can be aligned with a root mean square deviation of 2.8Å over much of their lengths (104) whereas E. coli LplA and M. tuberculosis LipB can be aligned over the length of LipB (the smaller protein) with a Cα value of 2.5Å (227). Moreover, the cofactor ligands in these crystal structures are in register indicating similar geometries of binding. These findings together with the even more similar structures of the domains that are the substrates of these enzymes raises the question of how the cell avoids the catastrophic effects on metabolism that would result if biotinylated proteins became lipoylated and vice versa. This question of accurate modification has been addressed by Reche and Perham (126, 257) who showed that the wild type biotinoyl domain can be lipoylated by LplA in vitro but the reactions proceeds only with a molar excess of enzyme and very slowly. They also showed that mutations of the residues adjacent to the lysine residue of the biotinoyl domain (the protruding β-turn) to those found in the protruding β-turn of the lipoyl domain allow modification by lipoic acid attachment. Indeed, conversion of a single residue of the biotinoyl domain of E. coli allowed some lipoylation by LplA. Moreover, some of the hybrid domains remained substrates for biotinylation. However, these workers also found that they could leave the protruding β-turn region intact and obtain lipoylation by removal of the biotinoyl domain thumb structure which is not found in lipoyl domains (126, 257) (Fig. 8). Thus, the thumb structure seems to be the major “gate-keeper” that prevents lipoylation of AccB. This seems an appropriate choice since the thumb structure is required for function of AccB in fatty acid synthesis, but not for biotinylation of the protein (10). The determinants that prevent biotinylation of lipoyl domains have yet to be explored.
Conclusions
In the two prior editions of these volumes the biotin and lipoic acid synthetic pathways have been placed into the same chapter. It seems probable that this was because both are sulfur-containing and covalently-attached enzyme cofactors plus there was insufficient information on lipoic acid synthesis to justify a separate chapter. However, in the 16 years since the second edition of this publication the lumping of these cofactors together has come to seem prescient. We now know that there are many commonalities. Both cofactors are made as offshoots of the fatty acid synthetic pathway, sulfur insertion is done by SAM radical enzymes, the sulfur atoms inserted are derived from an enzyme iron-sulfur center, the cofactors are attached to very similar domains and the enzymes that attach these cofactors constitute a protein family despite the low sequence conservation and differing mechanisms of the enzymes. However, it should be noted that although the E. coli pathways seem to explain the synthesis of these two cofactors in most bacteria, there are well-documented exceptions. Indeed the biotin and lipoate pathways of Bacillus subtilis differ markedly from those of E. coli (253, 258, 259).
Acknowledgments
The preparation of the manuscript and the experimental work from our laboratory were supported by grant AI15650 from the National Institute of Allergy and Infectious Diseases.
REFERENCES
- 1.Visser CM, Kellogg RM. Biotin. Its place in evolution. J Mol Evol. 1978;11:171–187. doi: 10.1007/BF01733892. [DOI] [PubMed] [Google Scholar]
- 2.Green NM. Avidin. Adv Protein Chem. 1975;29:85–133. doi: 10.1016/s0065-3233(08)60411-8. [DOI] [PubMed] [Google Scholar]
- 3.Wifling K, Dimroth P. Isolation and characterization of oxaloacetate decarboxylase of I, a sodium ion pump. Arch Microbiol. 1989;152:584–588. doi: 10.1007/BF00425491. [DOI] [PubMed] [Google Scholar]
- 4.Woehlke G, Dimroth P. Anaerobic growth of Salmonella typhimurium on l(+)- and d(−)-tartrate involves an oxaloacetate decarboxylase Na+ pump. Arch Microbiol. 1994;162:233–237. doi: 10.1007/BF00301843. [DOI] [PubMed] [Google Scholar]
- 5.Woehlke G, Wifling K, Dimroth P. Sequence of the sodium ion pump oxaloacetate decarboxylase from Salmonella typhimurium. J Biol Chem. 1992;267:22798–22803. [PubMed] [Google Scholar]
- 6.Vanden Boom TJ, Reed KE, Cronan JE., Jr Lipoic acid metabolism in Escherichia coli: isolation of null mutants defective in lipoic acid biosynthesis, molecular cloning and characterization of the E. coli lip locus, and identification of the lipoylated protein of the glycine cleavage system. J Bacteriol. 1991;173:6411–6420. doi: 10.1128/jb.173.20.6411-6420.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson RL, Stauffer LT, Stauffer GV. Roles of the GcvA and PurR proteins in negative regulation of the Escherichia coli glycine cleavage enzyme system. J Bacteriol. 1993;175:5129–5134. doi: 10.1128/jb.175.16.5129-5134.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson RL, Steiert PS, Stauffer GV. Positive regulation of the Escherichia coli glycine cleavage enzyme system. J Bacteriol. 1993;175:902–904. doi: 10.1128/jb.175.3.902-904.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perham RN. Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu Rev Biochem. 2000;69:961–1004. doi: 10.1146/annurev.biochem.69.1.961. [DOI] [PubMed] [Google Scholar]
- 10.Cronan JE., Jr The biotinyl domain of Escherichia coli acetyl-CoA carboxylase. Evidence that the "thumb" structure is essential and that the domain functions as a dimer. J Biol Chem. 2001;276:37355–37364. doi: 10.1074/jbc.M106353200. [DOI] [PubMed] [Google Scholar]
- 11.Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat Biotechnol. 2007;25:117–124. doi: 10.1038/nbt1270. [DOI] [PubMed] [Google Scholar]
- 12.Stok JE, De Voss J. Expression, purification, and characterization of BioI: a carbon-carbon bond cleaving cytochrome P450 involved in biotin biosynthesis in Bacillus subtilis. Arch Biochem Biophys. 2000;384:351–360. doi: 10.1006/abbi.2000.2067. [DOI] [PubMed] [Google Scholar]
- 13.Lin S, Hanson RE, Cronan JE. Biotin synthesis begins by hijacking the fatty acid synthetic pathway. Nat Chem Biol. 2010;6:682–688. doi: 10.1038/nchembio.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ifuku O, Miyaoka H, Koga N, Kishimoto J, Haze S, Wachi Y, Kajiwara M. Origin of carbon atoms of biotin. 13C-NMR studies on biotin biosynthesis in Escherichia coli. Eur J Biochem. 1994;220:585–591. doi: 10.1111/j.1432-1033.1994.tb18659.x. [DOI] [PubMed] [Google Scholar]
- 15.Sanyal I, Lee S-L, Flint D. Biosynthesis of pimeloyl-CoA, a biotin precursor in Escherichia coli follows a modified fatty acid synthesis pathway: 13C-labeling studies. J. Am. Chem. Soc. 1994;116:2637–2638. [Google Scholar]
- 16.Cleary PP, Campbell A. Deletion and complementation analysis of biotin gene cluster of Escherichia coli. J Bacteriol. 1972;112:830–839. doi: 10.1128/jb.112.2.830-839.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Del Campillo-Campbell A, Kayajanian G, Campbell A, Adhya S. Biotin-requiring mutants of Escherichia coli K-12. J Bacteriol. 1967;94:2065–2066. doi: 10.1128/jb.94.6.2065-2066.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rolfe B, Eisenberg MA. Genetic and biochemical analysis of the biotin loci of Escherichia coli K-12. J Bacteriol. 1968;96:515–524. doi: 10.1128/jb.96.2.515-524.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwartz M. Location of the maltose A and B loci on the genetic map of Escherichia coli. J Bacteriol. 1966;92:1083–1089. doi: 10.1128/jb.92.4.1083-1089.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cadieux N, Bradbeer C, Reeger-Schneider E, Koster W, Mohanty AK, Wiener MC, Kadner RJ. Identification of the periplasmic cobalamin-binding protein BtuF of Escherichia coli. J Bacteriol. 2002;184:706–717. doi: 10.1128/JB.184.3.706-717.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi-Rhee E, Cronan JE. A nucleosidase required for in vivo function of the S-adenosyl-L-methionine radical enzyme, biotin synthase. Chem Biol. 2005;12:589–593. doi: 10.1016/j.chembiol.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 22.Kwon MA, Kim HS, Oh JY, Song BK, Song JK. Gene cloning, expression, and characterization of a new carboxylesterase from Serratia sp. SES-01: comparison with Escherichia coli BioHe enzyme. J Microbiol Biotechnol. 2009;19:147–154. doi: 10.4014/jmb.0804.287. [DOI] [PubMed] [Google Scholar]
- 23.Sanishvili R, Yakunin AF, Laskowski RA, Skarina T, Evdokimova E, Doherty-Kirby A, Lajoie GA, Thornton JM, Arrowsmith CH, Savchenko A, Joachimiak A, Edwards AM. Integrating structure, bioinformatics, and enzymology to discover function: BioH, a new carboxylesterase from I. J Biol Chem. 2003;278:26039–26045. doi: 10.1074/jbc.M303867200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie X, Wong WW, Tang Y. Improving simvastatin bioconversion in Escherichia coli by deletion of bioH. Metab Eng. 2007;9:379–386. doi: 10.1016/j.ymben.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 25.Lezius A, Ringelmann E, Lynen F. [On the biochemical function of biotin. IV. The biosynthesis of biotin.] Biochem Z. 1963;336:510–525. [PubMed] [Google Scholar]
- 26.Austin MB, Izumikawa M, Bowman ME, Udwary DW, Ferrer JL, Moore BS, Noel JP. Crystal structure of a bacterial type III polyketide synthase and enzymatic control of reactive polyketide intermediates. J Biol Chem. 2004;279:45162–45174. doi: 10.1074/jbc.M406567200. [DOI] [PubMed] [Google Scholar]
- 27.Tseng CC, McLoughlin SM, Kelleher NL, Walsh CT. Role of the active site cysteine of DpgA, a bacterial type III polyketide synthase. Biochemistry. 2004;43:970–980. doi: 10.1021/bi035714b. [DOI] [PubMed] [Google Scholar]
- 28.White SW, Zheng J, Zhang YM, Rock The structural biology of type II fatty acid biosynthesis. Annu Rev Biochem. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 29.Chapman-Smith A, Cronan JE., Jr The enzymatic biotinylation of proteins: a post-translational modification of exceptional specificity. Trends Biochem Sci. 1999;24:359–363. doi: 10.1016/s0968-0004(99)01438-3. [DOI] [PubMed] [Google Scholar]
- 30.Lemoine Y, Wach A, Jeltsch JM. To be free or not: the fate of pimelate in Bacillus sphaericus and in Escherichia coli. Mol Microbiol. 1996;19:645–647. doi: 10.1046/j.1365-2958.1996.t01-4-442924.x. [DOI] [PubMed] [Google Scholar]
- 31.Tomczyk NH, Nettleship JE, Baxter RL, Crichton HJ, Webster SP, Campopiano DJ. Purification and characterisation of the BIOH protein from the biotin biosynthetic pathway. FEBS Lett. 2002;513:299–304. doi: 10.1016/s0014-5793(02)02342-6. [DOI] [PubMed] [Google Scholar]
- 32.Lin S, Cronan JE. The BioC O-methyltransferase catalyzes methyl esterification of malonyl-acyl carrier protein, an essential step in biotin synthesis. J Biol Chem. 2012;287:37010–37020. doi: 10.1074/jbc.M112.410290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agarwal V, Lin S, Lukk T, Nair SK, Cronan JE. Structure of the enzyme-acyl carrier protein (ACP) substrate gatekeeper complex required for biotin synthesis. Proc Natl Acad Sci U S A. 2012;109:17406–17411. doi: 10.1073/pnas.1207028109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alexeev D, Alexeeva M, Baxter RL, Campopiano DJ, Webster SP, Sawyer L. The crystal structure of 8-amino-7-oxononanoate synthase: a bacterial PLP-dependent, acyl-CoA-condensing enzyme. J Mol Biol. 1998;284:401–419. doi: 10.1006/jmbi.1998.2086. [DOI] [PubMed] [Google Scholar]
- 35.Webster SP, Campopiano DJ, Alexeev D, Alexeeva M, Watt RM, Sawyer L, Baxter RL. Characterisation of 8-amino-7-oxononanoate synthase: a bacterial PLP-dependent, acyl CoA condensing enzyme. Biochem Soc Trans. 1998;26:S268. doi: 10.1042/bst026s268. [DOI] [PubMed] [Google Scholar]
- 36.Webster SP, Alexeev D, Campopiano DJ, Watt RM, Alexeeva M, Sawyer L, Baxter RL. Mechanism of 8-amino-7-oxononanoate synthase: spectroscopic, kinetic, and crystallographic studies. Biochemistry. 2000;39:516–528. doi: 10.1021/bi991620j. [DOI] [PubMed] [Google Scholar]
- 37.Ploux O, Soularue P, Marquet A, Gloeckler R, Lemoine Y. Investigation of the first step of biotin biosynthesis in I. Purification and characterization of the pimeloyl-CoA synthase, and uptake of pimelate. Biochem J. 1992;287:685–690. doi: 10.1042/bj2870685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kack H, Sandmark J, Gibson K, Schneider G, Lindqvist Y. Crystal structure of diaminopelargonic acid synthase: evolutionary relationships between pyridoxal-5'-phosphate-dependent enzymes. J Mol Biol. 1999;291:857–876. doi: 10.1006/jmbi.1999.2997. [DOI] [PubMed] [Google Scholar]
- 39.Stoner GL, Eisenberg MA. Purification and properties of 7, 8-diaminopelargonic acid aminotransferase. J Biol Chem. 1975;250:4029–4036. [PubMed] [Google Scholar]
- 40.Stoner GL, Eisenberg MA. Biosynthesis of 7, 8-diaminopelargonic acid from 7-keto-8-aminopelargonic acid and S-adenosyl-l-methionine. The kinetics of the reaction. J Biol Chem. 1975;250:4037–4043. [PubMed] [Google Scholar]
- 41.DeMoll E, White RH, Shive W. Determination of the metabolic origins of the sulfur and 3′-nitrogen atoms in biotin of Escherichia coli by mass spectrometry. Biochemistry. 1984;23:558–562. doi: 10.1021/bi00298a025. [DOI] [PubMed] [Google Scholar]
- 42.Van Arsdell SW, Perkins JB, Yocum RR, Luan L, Howitt CL, Chatterjee NP, Pero JG. Removing a bottleneck in the Bacillus subtilis biotin pathway:bioA utilizes lysine rather than S-adenosylmethionine as the amino donor in the KAPA-to-DAPA reaction. Biotechnol Bioeng. 2005;91:75–83. doi: 10.1002/bit.20488. [DOI] [PubMed] [Google Scholar]
- 43.Eisenberg MA, Krell K. Dethiobiotin synthetase. Methods Enzymol. 1979;62:348–352. doi: 10.1016/0076-6879(79)62241-3. [DOI] [PubMed] [Google Scholar]
- 44.Krell K, Eisenberg MA. The purification and properties of dethiobiotin synthetase. J Biol Chem. 1970;245:6558–6566. [PubMed] [Google Scholar]
- 45.Alexeev D, Baxter RL, Smekal O, Sawyer L. Substrate binding and carboxylation by dethiobiotin synthetase—a kinetic and X-ray study. Structure. 1995;3:1207–1215. doi: 10.1016/s0969-2126(01)00256-8. [DOI] [PubMed] [Google Scholar]
- 46.Huang W, Jia J, Gibson KJ, Taylor WS, Rendina AR, Schneider G, Lindqvist Y. Mechanism of an ATP-dependent carboxylase, dethiobiotin synthetase, based on crystallographic studies of complexes with substrates and a reaction intermediate. Biochemistry. 1995;34:10985–10995. doi: 10.1021/bi00035a004. [DOI] [PubMed] [Google Scholar]
- 47.Huang W, Lindqvist Y, Schneider G, Gibson KJ, Flint D, Lorimer G. Crystal structure of an ATP-dependent carboxylase, dethiobiotin synthetase, at 1.65 A resolution. Structure. 1994;2:407–414. doi: 10.1016/s0969-2126(00)00042-3. [DOI] [PubMed] [Google Scholar]
- 48.Sandalova T, Schneider G, Kack H, Lindqvist Y. Structure of dethiobiotin synthetase at 0.97 A resolution. Acta Crystallogr D Biol Crystallogr. 1999;55:610–624. doi: 10.1107/s090744499801381x. [DOI] [PubMed] [Google Scholar]
- 49.Gibson KJ, Lorimer GH, Rendina AR, Taylor WS, Cohen G, Gatenby AA, Payne WG, Roe DC, Lockett BA, Nudelman A, et al. Dethiobiotin synthetase: the carbonylation of 7,8-diaminonanoic acid proceeds regiospecifically via the N7-carbamate. Biochemistry. 1995;34:10976–10984. doi: 10.1021/bi00035a003. [DOI] [PubMed] [Google Scholar]
- 50.Kack H, Gibson KJ, Lindqvist Y, Schneider G. Snapshot of a phosphorylated substrate intermediate by kinetic crystallography. Proc Natl Acad Sci U S A. 1998;95:5495–5500. doi: 10.1073/pnas.95.10.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ifuku O, Kishimoto J, Haze S, Yanagi M, Fukushima S. Conversion of dethiobiotin to biotin in cell-free extracts of Escherichia coli. Biosci Biotechnol Biochem. 1992;56:1780–1785. doi: 10.1271/bbb.56.1780. [DOI] [PubMed] [Google Scholar]
- 52.Ifuku O, Koga N, Haze S, Kishimoto J, Wachi Y. Flavodoxin is required for conversion of dethiobiotin to biotin in Escherichia coli. Eur J Biochem. 1994;224:173–178. doi: 10.1111/j.1432-1033.1994.tb20009.x. [DOI] [PubMed] [Google Scholar]
- 53.Birch OM, Fuhrmann M, Shaw NM. Biotin synthase from Escherichia coli an investigation of the low molecular weight and protein components required for activity in vitro. J Biol Chem. 1995;270:19158–19165. doi: 10.1074/jbc.270.32.19158. [DOI] [PubMed] [Google Scholar]
- 54.Sanyal I, Gibson KJ, Flint DH. Escherichia coli biotin synthase: an investigation into the factors required for its activity and its sulfur donor. Arch Biochem Biophys. 1996;326:48–56. doi: 10.1006/abbi.1996.0045. [DOI] [PubMed] [Google Scholar]
- 55.DeMoll E, Shive W. The origin of sulfur in biotin. Biochem Biophys Res Commun. 1983;110:243–249. doi: 10.1016/0006-291x(83)91286-x. [DOI] [PubMed] [Google Scholar]
- 56.Florentin D, Tse Sum Bui B, Marquet A, Ohshiro T, Izumi Y. On the mechanism of biotin synthase of Bacillus sphaericus . C R Acad Sci III. 1994;317:485–488. [PubMed] [Google Scholar]
- 57.Lotierzo M, Tse Sum Bui B, Florentin D, Escalettes F, Marquet A. Biotin synthase mechanism: an overview. Biochem Soc Trans. 2005;33:820–823. doi: 10.1042/BST0330820. [DOI] [PubMed] [Google Scholar]
- 58.Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL. Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science. 2004;303:76–79. doi: 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jarrett JT. The novel structure and chemistry of iron-sulfur clusters in the adenosylmethionine-dependent radical enzyme biotin synthase. Arch Biochem Biophys. 2005;433:312–321. doi: 10.1016/j.abb.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 60.Tse Sum Bui B, Lotierzo M, Escalettes F, Florentin D, Marquet A. Further investigation on the turnover of Escherichia coli biotin synthase with dethiobiotin and 9-mercaptodethiobiotin as substrates. Biochemistry. 2004;43:16432–16441. doi: 10.1021/bi048040t. [DOI] [PubMed] [Google Scholar]
- 61.Taylor AM, Farrar CE, Jarrett JT. 9-Mercaptodethiobiotin is formed as a competent catalytic intermediate by Escherichia coli biotin synthase. Biochemistry. 2008;47:9309–9317. doi: 10.1021/bi801035b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taylor AM, Stoll S, Britt RD, Jarrett JT. Reduction of the [2Fe-2S] cluster accompanies formation of the intermediate 9-mercaptodethiobiotin in Escherichia coli biotin synthase. Biochemistry. 2011;50:7953–7963. doi: 10.1021/bi201042r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gibson KJ, Pelletier DA, Turner IM., Sr Transfer of sulfur to biotin from biotin synthase (BioB protein) Biochem Biophys Res Commun. 1999;254:632–635. doi: 10.1006/bbrc.1998.9991. [DOI] [PubMed] [Google Scholar]
- 64.Tse Sum Bui B, Florentin D, Fournier F, Ploux O, Mejean A, Marquet A. Biotin synthase mechanism: on the origin of sulphur. FEBS Lett. 1998;440:226–230. doi: 10.1016/s0014-5793(98)01464-1. [DOI] [PubMed] [Google Scholar]
- 65.Tse Sum Bui B, Mattioli TA, Florentin D, Bolbach G, Marquet A. Ii biotin synthase produces selenobiotin. Further evidence of the involvement of the [2Fe-2S]2+ cluster in the sulfur insertion step. Biochemistry. 2006;45:3824–3834. doi: 10.1021/bi052388m. [DOI] [PubMed] [Google Scholar]
- 66.Jameson GN, Cosper MM, Hernandez HL, Johnson MK, Huynh BH. Role of the [2Fe-2S] cluster in recombinant Escherichia coli biotin synthase. Biochemistry. 2004;43:2022–2031. doi: 10.1021/bi035666v. [DOI] [PubMed] [Google Scholar]
- 67.Tse Sum Bui B, Benda R, Schunemann V, Florentin D, Trautwein AX, Marquet A. Fate of the (2Fe-2S)(2+) cluster of Escherichia coli biotin synthase during reaction: a Mossbauer characterization. Biochemistry. 2003;42:8791–8798. doi: 10.1021/bi034426c. [DOI] [PubMed] [Google Scholar]
- 68.Jarrett JT. Biotin synthase: enzyme or reactant? Chem Biol. 2005;12:409–410. doi: 10.1016/j.chembiol.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 69.Kiyasu T, Asakura A, Nagahashi Y, Hoshino T. Contribution of cysteine desulfurase (NifS protein) to the biotin synthase reaction of Escherichia coli. J Bacteriol. 2000;182:2879–2885. doi: 10.1128/jb.182.10.2879-2885.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cosper MM, Jameson GN, Eidsness MK, Huynh BH, Johnson MK. Recombinant Escherichia coli biotin synthase is a [2Fe-2S](2+) protein in whole cells. FEBS Lett. 2002;529:332–336. doi: 10.1016/s0014-5793(02)03390-2. [DOI] [PubMed] [Google Scholar]
- 71.Ollagnier-de-Choudens S, Mulliez E, Fontecave M. The PLP-dependent biotin synthase from Escherichia coli: mechanistic studies. FEBS Lett. 2002;532:465–468. doi: 10.1016/s0014-5793(02)03733-x. [DOI] [PubMed] [Google Scholar]
- 72.Li SJ, Cronan JE., Jr The gene encoding the biotin carboxylase subunit of Escherichia coli acetyl-CoA carboxylase. J Biol Chem. 1992;267:855–863. [PubMed] [Google Scholar]
- 73.Choi-Rhee E, Cronan JE. Biotin synthase is catalytic in vivo, but catalysis engenders destruction of the protein. Chem Biol. 2005;12:461–468. doi: 10.1016/j.chembiol.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 74.Farrar CE, Siu KK, Howell PL, Jarrett JT. Biotin synthase exhibits burst kinetics and multiple turnovers in the absence of inhibition by products and product-related biomolecules. Biochemistry. 2010;49:9985–9996. doi: 10.1021/bi101023c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reyda MR, Dippold R, Dotson ME, Jarrett JT. Loss of iron-sulfur clusters from biotin synthase as a result of catalysis promotes unfolding and degradation. Arch Biochem Biophys. 2008;471:32–41. doi: 10.1016/j.abb.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Challand MR, Ziegert T, Douglas P, Wood RJ, Kriek M, Shaw NM, Roach PL. Product inhibition in the radical S-adenosylmethionine family. FEBS Lett. 2009;583:1358–1362. doi: 10.1016/j.febslet.2009.03.044. [DOI] [PubMed] [Google Scholar]
- 77.Barker DF, Campbell AM. Use of bio-lac fusion strains to study regulation of biotin biosynthesis in Escherichia coli. J Bacteriol. 1980;143:789–800. doi: 10.1128/jb.143.2.789-800.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koga N, Kishimoto J, Haze S, Ifuku O. Analysis of the bioH gene of Escherichia coli and its effect on biotin productivityJFerment. Bioeng. 1996;81:482–487. [Google Scholar]
- 79.Rodionov DA, Mironov AA, Gelfand MS. Conservation of the biotin regulon and the BirA regulatory signal in Eubacteria and Archaea. Genome Res. 2002;12:1507–1516. doi: 10.1101/gr.314502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sanishvili R, Yakunin AF, Laskowski RA, Skarina T, Evdokimova E, Doherty-Kirby A, Lajoie GA, Thornton JM, Arrowsmith CH, Savchenko A, Joachimiak A, Edwards AM. Integrating structure, bioinformatics, and enzymology to discover function. J Biol Chem. 2003;278:26039–26045. doi: 10.1074/jbc.M303867200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shapiro MM, Chakravartty V, Cronan JE. Remarkable diversity in the enzymes catalyzing the last step in synthesis of the pimelate moiety of biotin. PLoS One. 2012;7:e49440. doi: 10.1371/journal.pone.0049440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ollagnier-De-Choudens S, Mulliez E, Hewitson KS, Fontecave M. Biotin synthase is a pyridoxal phosphate-dependent cysteine desulfurase. Biochemistry. 2002;41:9145–9152. doi: 10.1021/bi0122011. [DOI] [PubMed] [Google Scholar]
- 83.Cosper MM, Jameson GN, Hernandez HL, Krebs C, Huynh BH, Johnson MK. Characterization of the cofactor composition of Escherichia coli biotin synthase. Biochemistry. 2004;43:2007–2021. doi: 10.1021/bi0356653. [DOI] [PubMed] [Google Scholar]
- 84.Abdel-Hamid AM, Cronan JE. In vivo resolution of conflicting in vitro results: synthesis of biotin from dethiobiotin does not require pyridoxal phosphate. Chem Biol. 2007;14:1215–1220. doi: 10.1016/j.chembiol.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 85.Escalettes F, Florentin D, Bui BTS, Lesage D, Marquet A. Biotin synthase mechanism: Evidence for hydrogen transfer from the substrate into deoxyadenosine. J Am Chem Soc. 1999;121:3571–3578. [Google Scholar]
- 86.Ollagnier-de-Choudens S, Sanakis Y, Fontecave M. SufA/IscA: reactivity studies of a class of scaffold proteins involved in [Fe-S] cluster assembly. J Biol Inorg Chem. 2004;9:828–838. doi: 10.1007/s00775-004-0581-9. [DOI] [PubMed] [Google Scholar]
- 87.Broach RB, Jarrett JT. Role of the [2Fe-2S]2+ cluster in biotin synthase: mutagenesis of the atypical metal ligand arginine 260. Biochemistry. 2006;45:14166–14174. doi: 10.1021/bi061576p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hewitson KS, Ollagnier-de Choudens S, Sanakis Y, Shaw NM, Baldwin JE, Munck E, Roach PL, Fontecave M. The iron-sulfur center of biotin synthase: site-directed mutants. J Biol Inorg Chem. 2002;7:83–93. doi: 10.1007/s007750100268. [DOI] [PubMed] [Google Scholar]
- 89.Barker DF, Campbell AM. Genetic and biochemical characterization of the birA gene and its product: evidence for a direct role of biotin holoenzyme synthetase in repression of the biotin operon in Escherichia coli. J Mol Biol. 1981;146:469–492. doi: 10.1016/0022-2836(81)90043-7. [DOI] [PubMed] [Google Scholar]
- 90.Barker DF, Campbell AM. The birA gene of Escherichia coli encodes a biotin holoenzyme synthetase. J Mol Biol. 1981;146:451–467. doi: 10.1016/0022-2836(81)90042-5. [DOI] [PubMed] [Google Scholar]
- 91.Beckett D. Multilevel regulation of protein-protein interactions in biological circuitry. Phys Biol. 2005;2:S67–73. doi: 10.1088/1478-3975/2/2/S07. [DOI] [PubMed] [Google Scholar]
- 92.Cronan JE., Jr Expression of the biotin biosynthetic operon of Escherichia coli is regulated by the rate of protein biotination. J Biol Chem. 1988;263:10332–10336. [PubMed] [Google Scholar]
- 93.Cronan JE., Jr The E. coli bio operon: transcriptional repression by an essential protein modification enzyme. Cell. 1989;58:427–429. doi: 10.1016/0092-8674(89)90421-2. [DOI] [PubMed] [Google Scholar]
- 94.Cronan JE., Jr Biotination of proteins in vivo. A post-translational modification to label, purify, and study proteins. J Biol Chem. 1990;265:10327–10333. [PubMed] [Google Scholar]
- 95.Otsuka A, Abelson J. The regulatory region of the biotin operon in Escherichia coli. Nature. 1978;276:689–694. doi: 10.1038/276689a0. [DOI] [PubMed] [Google Scholar]
- 96.Barker DF, Kuhn J, Campbell AM. Sequence and properties of operator mutations in the bio operon of Escherichia coli. Gene. 1981;13:89–102. doi: 10.1016/0378-1119(81)90046-9. [DOI] [PubMed] [Google Scholar]
- 97.Eisenberg MA. Regulation of the biotin operon in E. coli. Ann N Y Acad Sci. 1985;447:335–349. doi: 10.1111/j.1749-6632.1985.tb18449.x. [DOI] [PubMed] [Google Scholar]
- 98.Prakash O, Eisenberg MA. Biotinyl 5′-adenylate: corepressor role in the regulation of the biotin genes of Escherichia coli K-12. Proc Natl Acad Sci U S A. 1979;76:5592–5595. doi: 10.1073/pnas.76.11.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Prakash O, Eisenberg MA. In vitro synthesis and and regulation of the biotin enzymes o f Escherichia coli K-12. J Bacteriol. 1978;134:1002–1012. doi: 10.1128/jb.134.3.1002-1012.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xu Y, Beckett D. Biotinyl-5′-adenylate synthesis catalyzed by Escherichia coli repressor of biotin biosynthesis. Methods Enzymol. 1997;279:405–421. doi: 10.1016/s0076-6879(97)79045-1. [DOI] [PubMed] [Google Scholar]
- 101.Buoncristiani MR, Otsuka AJ. Overproduction and rapid purification of the biotin operon repressor from Escherichia coli. J Biol Chem. 1988;263:1013–1016. [PubMed] [Google Scholar]
- 102.Wilson KP, Shewchuk LM, Brennan RG, Otsuka AJ, Matthews BW. Escherichia coli biotin holoenzyme synthetase/bio repressor crystal structure delineates the biotin- and DNA-binding domains. Proc Natl Acad Sci U S A. 1992;89:9257–9261. doi: 10.1073/pnas.89.19.9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Weaver LH, Kwon K, Beckett D, Matthews BW. Corepressor-induced organization and assembly of the biotin repressor: a model for allosteric activation of a transcriptional regulator. Proc Natl Acad Sci U S A. 2001;98:6045–6050. doi: 10.1073/pnas.111128198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wood ZA, Weaver LH, Brown PH, Beckett D, Matthews BW. Co-repressor induced order and biotin repressor dimerization: a case for divergent followed by convergent evolution. J Mol Biol. 2006;357:509–523. doi: 10.1016/j.jmb.2005.12.066. [DOI] [PubMed] [Google Scholar]
- 105.Streaker ED, Beckett D. A map of the biotin repressor-biotin operator interface: binding of a winged helix-turn-helix protein dimer to a forty base-pair site. J Mol Biol. 1998;278:787–800. doi: 10.1006/jmbi.1998.1733. [DOI] [PubMed] [Google Scholar]
- 106.Chapman-Smith A, Cronan JE., Jr Molecular biology of biotin attachment to proteins. J Nutr. 1999;129:477S–484S. doi: 10.1093/jn/129.2.477S. [DOI] [PubMed] [Google Scholar]
- 107.Eisenstein E, Beckett D. Dimerization of the Escherichia coli biotin repressor: corepressor function in protein assembly. Biochemistry. 1999;38:13077–13084. doi: 10.1021/bi991241q. [DOI] [PubMed] [Google Scholar]
- 108.Abbott J, Beckett D. Cooperative binding of the Escherichia coli repressor of biotin biosynthesis to the biotin operator sequence. Biochemistry. 1993;32:9649–9656. doi: 10.1021/bi00088a017. [DOI] [PubMed] [Google Scholar]
- 109.Streaker ED, Beckett D. Coupling of site-specific DNA binding to protein dimerization in assembly of the biotin repressor-biotin operator complex. Biochemistry. 1998;37:3210–3219. doi: 10.1021/bi9715019. [DOI] [PubMed] [Google Scholar]
- 110.Streaker ED, Beckett D. The biotin regulatory system: kinetic control of a transcriptional switch. Biochemistry. 2006;45:6417–6425. doi: 10.1021/bi052599r. [DOI] [PubMed] [Google Scholar]
- 111.Streaker ED, Gupta A, Beckett D. The biotin repressor: thermodynamic coupling of corepressor binding, protein assembly, and sequence-specific DNA binding. Biochemistry. 2002;41:14263–14271. doi: 10.1021/bi0203839. [DOI] [PubMed] [Google Scholar]
- 112.Kwon K, Streaker ED, Beckett D. Binding specificity and the ligand dissociation process in the E. coli biotin holoenzyme synthetase. Protein Sci. 2002;11:558–570. doi: 10.1110/ps.33502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Streaker ED, Beckett D. Coupling of protein assembly and DNA binding: biotin repressor dimerization precedes biotin operator binding. J Mol Biol. 2003;325:937–948. doi: 10.1016/s0022-2836(02)01308-6. [DOI] [PubMed] [Google Scholar]
- 114.Wu SC, Wong SL. Development of an enzymatic method for site-specific incorporation of desthiobiotin to recombinant proteins in vitro. Anal Biochem. 2004;331:340–348. doi: 10.1016/j.ab.2004.03.056. [DOI] [PubMed] [Google Scholar]
- 115.Campbell A, Campillo-Campbell AD, Barker D. Repression of biotin biosynthesis in Escherichia coli during growth on biotin vitamers. J Bacteriol. 1978;135:90–98. doi: 10.1128/jb.135.1.90-98.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Buoncristiani MR, Howard PK, Otsuka AJ. DNA-binding and enzymatic domains of the bifunctional biotin operon repressor (BirA) of Escherichia coli. Gene. 1986;44:255–261. doi: 10.1016/0378-1119(86)90189-7. [DOI] [PubMed] [Google Scholar]
- 117.Campbell A, Del Campillo-Campbell A, Chang R. A mutant of Escherichia coli that requires high concentrations of biotin. Proc Natl Acad Sci USA. 1972;69:676–680. doi: 10.1073/pnas.69.3.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xu Y, Beckett D. Evidence for interdomain interaction in the Escherichia coli repressor of biotin biosynthesis from studies of an N-terminal domain deletion mutant. Biochemistry. 1996;35:1783–1792. doi: 10.1021/bi952269e. [DOI] [PubMed] [Google Scholar]
- 119.Gerdes SY, Scholle MD, Campbell JW, Balazsi G, Ravasz E, Daugherty MD, Somera AL, Kyrpides NC, Anderson I, Gelfand MS, Bhattacharya A, Kapatral V, D'Souza M, Baev MV, Grechkin Y, Mseeh F, Fonstein MY, Overbeek R, Barabasi AL, Oltvai ZN, Osterman AL. Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J Bacteriol. 2003;185:5673–5684. doi: 10.1128/JB.185.19.5673-5684.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006 0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cronan JE, Jr, Waldrop GL. Multi-subunit acetyl-CoA carboxylases. Prog Lipid Res. 2002;41:407–435. doi: 10.1016/s0163-7827(02)00007-3. [DOI] [PubMed] [Google Scholar]
- 122.Choi-Rhee E, Cronan JE. The biotin carboxylase-biotin carboxyl carrier protein complex of Escherichia coli acetyl-CoA carboxylase. J Biol Chem. 2003;278:30806–30812. doi: 10.1074/jbc.M302507200. [DOI] [PubMed] [Google Scholar]
- 123.Nenortas E, Beckett D. Purification and characterization of intact and truncated forms of the Escherichia coli biotin carboxyl carrier subunit of acetyl-CoA carboxylase. J Biol Chem. 1996;271:7559–7567. doi: 10.1074/jbc.271.13.7559. [DOI] [PubMed] [Google Scholar]
- 124.Cronan JE, Jr, Reed KE. Biotinylation of proteins in vivo: a useful posttranslational modification for protein analysis. Methods Enzymol. 2000;326:440–458. doi: 10.1016/s0076-6879(00)26069-2. [DOI] [PubMed] [Google Scholar]
- 125.Chapman-Smith A, Morris TW, Wallace JC, Cronan JE., Jr Molecular recognition in a post-translational modification of exceptional specificity. Mutants of the biotinylated domain of acetyl-CoA carboxylase defective in recognition by biotin protein ligase. J Biol Chem. 1999;274:1449–1457. doi: 10.1074/jbc.274.3.1449. [DOI] [PubMed] [Google Scholar]
- 126.Reche P, Perham RN. Structure and selectivity in post-translational modification: attaching the biotinyl-lysine and lipoyl-lysine swinging arms in multifunctional enzymes. EMBO J. 1999;18:2673–2682. doi: 10.1093/emboj/18.10.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lin KC, Shiuan D. DNaseI footprinting studies of Escherichia coli biotin repressor-operator interactions. J Biochem (Tokyo) 1993;114:670–676. doi: 10.1093/oxfordjournals.jbchem.a124235. [DOI] [PubMed] [Google Scholar]
- 128.Ketner G, Campbell A. Operator and promoter mutations affecting divergent transcription in the bio gene cluster of Escherichia coli. J. Mol. Biol. 1975;96:13–27. doi: 10.1016/0022-2836(75)90179-5. [DOI] [PubMed] [Google Scholar]
- 129.Shiuan D, Campbell A. Transcriptional regulation and gene arrangement of Escherichia coli, Citrobacter freundii and Salmonella typhimurium biotin operons. Gene. 1988;67:203–211. doi: 10.1016/0378-1119(88)90397-6. [DOI] [PubMed] [Google Scholar]
- 130.Cronan JE., Jr Interchangeable enzyme modules. Functional replacement of the essential linker of the biotinylated subunit of acetyl-CoA carboxylase with a linker from the lipoylated subunit of pyruvate dehydrogenase. J Biol Chem. 2002;277:22520–22527. doi: 10.1074/jbc.M201249200. [DOI] [PubMed] [Google Scholar]
- 131.Fall RR, Alberts AW, Vagelos PR. Analysis of bacterial biotin-proteins. Biochim Biophys Acta. 1975;379:496–503. doi: 10.1016/0005-2795(75)90156-7. [DOI] [PubMed] [Google Scholar]
- 132.Li SJ, Cronan JE., Jr Growth rate regulation of Escherichia coli acetyl coenzyme A carboxylase, which catalyzes the first committed step of lipid biosynthesis. J Bacteriol. 1993;175:332–340. doi: 10.1128/jb.175.2.332-340.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Davis MS, Solbiati J, Cronan JE., Jr Overproduction of acetyl-CoA carboxylase activity increases the rate of fatty acid biosynthesis in Escherichia coli. J Biol Chem. 2000;275:28593–28598. doi: 10.1074/jbc.M004756200. [DOI] [PubMed] [Google Scholar]
- 134.Abdel-Hamid AM, Cronan JE. Coordinate expression of the acetyl coenzyme A carboxylase genes,accB and accC is necessary for normal regulation of biotin synthesis in Escherichia coli. J Bacteriol. 2007;189:369–376. doi: 10.1128/JB.01373-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhao H, Beckett D. Kinetic partitioning between alternative protein-protein interactions controls a transcriptional switch. J Mol Biol. 2008;380:223–236. doi: 10.1016/j.jmb.2008.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schatz PJ. Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: a 13 residue consensus peptide specifies biotinylation in Escherichia coli. Biotechnology (NY) 1993;11:1138–1143. doi: 10.1038/nbt1093-1138. [DOI] [PubMed] [Google Scholar]
- 137.Beckett D, Kovaleva E, Schatz PJ. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999;8:921–929. doi: 10.1110/ps.8.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Solbiati J, Cronan JE. The switch regulating transcription of the Escherichia coli biotin operon does not require extensive protein-protein interactions. Chem Biol. 2010;17:11–17. doi: 10.1016/j.chembiol.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chakravartty V, Cronan JE. Altered regulation of Escherichia coli biotin biosynthesis in BirA superrepressor mutant strains. J Bacteriol. 2012;194:1113–1126. doi: 10.1128/JB.06549-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Brown PH, Cronan JE, Grotli M, Beckett D. The biotin repressor: modulation of allostery by corepressor analogs. J Mol Biol. 2004;337:857–869. doi: 10.1016/j.jmb.2004.01.041. [DOI] [PubMed] [Google Scholar]
- 141.Reed LJ, Hackert ML. Structure-function relationships in dihydrolipoamide acyltransferases. J Biol Chem. 1990;265:8971–8974. [PubMed] [Google Scholar]
- 142.Koike M, Reed LJ. Alpha-Keto acid dehydrogenation complexes. II. The role of protein-bound lipoic acid and flavin adenine dinucleotide. J Biol Chem. 1960;235:1931–1938. [PubMed] [Google Scholar]
- 143.Perham RN. Domains, motifs, and linkers in 2-oxo acid dehydrogenase multienzyme complexes: a paradigm in the design of a multifunctional protein. Biochemistry. 1991;30:8501–8512. doi: 10.1021/bi00099a001. [DOI] [PubMed] [Google Scholar]
- 144.Guest JR, Russell GC. Complexes and complexities of the citric acid cycle in Escherichia coli. Curr Top Cell Regul. 1992;33:231–247. doi: 10.1016/b978-0-12-152833-1.50018-6. [DOI] [PubMed] [Google Scholar]
- 145.Langley D, Guest JR. Biochemical genetics of the alpha-keto acid dehydrogenase complexes of Escherichia coli K12: genetic characterization and regulatory properties of deletion mutants. J Gen Microbiol. 1978;106:103–117. doi: 10.1099/00221287-106-1-103. [DOI] [PubMed] [Google Scholar]
- 146.Smith MW, Neidhardt FC. 2-Oxoacid dehydrogenase complexes of Escherichia coli: cellular amounts and patterns of synthesis. J Bacteriol. 1983;156:81–88. doi: 10.1128/jb.156.1.81-88.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Dietrich J, Henning U. Regulation of pyruvate dehydrogenase complex synthesis in Escherichia coli K 12. Identification of the inducing metabolite. Eur J Biochem. 1970;14:258–269. doi: 10.1111/j.1432-1033.1970.tb00285.x. [DOI] [PubMed] [Google Scholar]
- 148.Quail MA, Guest JR. Purification, characterization and mode of action of PdhR, the transcriptional repressor of the pdhR-aceEF-lpd operon of Escherichia coli. Mol Microbiol. 1995;15:519–529. doi: 10.1111/j.1365-2958.1995.tb02265.x. [DOI] [PubMed] [Google Scholar]
- 149.Quail MA, Haydon DJ, Guest JR. The pdhR-aceEF-lpd operon of Escherichia coli e xpresses the pyruvate dehydrogenase complex. Mol Microbiol. 1994;12:95–104. doi: 10.1111/j.1365-2958.1994.tb00998.x. [DOI] [PubMed] [Google Scholar]
- 150.Guest JR, Angier SJ, Russell GC. Structure, expression, and protein engineering of the pyruvate dehydrogenase complex of Escherichia coli. Ann N Y Acad Sci. 1989;573:76–99. doi: 10.1111/j.1749-6632.1989.tb14988.x. [DOI] [PubMed] [Google Scholar]
- 151.Steginsky CA, Gruys KJ, Frey PA. alpha-Ketoglutarate dehydrogenase complex of Escherichia coli A hybrid complex containing pyruvate dehydrogenase subunits from pyruvate dehydrogenase complex. J Biol Chem. 1985;260:13690–13693. [PubMed] [Google Scholar]
- 152.Herbert AA, Guest JR. Biochemical and genetic studies with lysine+methionine mutants of Escherichia coli: lipoic acid and alpha-ketoglutarate dehydrogenase-less mutants. J Gen Microbiol. 1968;53:363–381. doi: 10.1099/00221287-53-3-363. [DOI] [PubMed] [Google Scholar]
- 153.Prohl C, Wackwitz B, Vlad D, Unden G. Functional citric acid cycle in an arcA mutant of Escherichia coli during growth with nitrate under anoxic conditions. Arch Microbiol. 1998;170:1–7. doi: 10.1007/s002030050608. [DOI] [PubMed] [Google Scholar]
- 154.Douce R, Bourguignon J, Neuburger M, Rebeille F. The glycine decarboxylase system: a fascinating complex. Trends Plant Sci. 2001;6:167–176. doi: 10.1016/s1360-1385(01)01892-1. [DOI] [PubMed] [Google Scholar]
- 155.Fujiwara K, Okamura-Ikeda K, Motokawa Y. Expression of mature bovine H-protein of the glycine cleavage system in Escherichia coli and in vitro lipoylation of the apoform. J Biol Chem. 1992;267:20011–20016. [PubMed] [Google Scholar]
- 156.Okamura-Ikeda K, Ohmura Y, Fujiwara K, Motokawa Y. Cloning and nucleotide sequence of the gcv operon encoding the Escherichia coli glycine-cleavage system. Eur J Biochem. 1993;216:539–548. doi: 10.1111/j.1432-1033.1993.tb18172.x. [DOI] [PubMed] [Google Scholar]
- 157.Steiert PS, Stauffer LT, Stauffer GV. The lpd gene product functions as the L protein in the Escherichia coli glycine cleavage enzyme system. J Bacteriol. 1990;172:6142–6144. doi: 10.1128/jb.172.10.6142-6144.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Danson MJ, Hale G, Johnson P, Perham RN, Smith J, Spragg P. Molecular weight and symmetry of the pyruvate dehydrogenase multienzyme complex of Escherichia coli. J Mol Biol. 1979;129:603–617. doi: 10.1016/0022-2836(79)90471-6. [DOI] [PubMed] [Google Scholar]
- 159.Hanemaaijer R, Janssen A, de Kok A, Veeger C. The dihydrolipoyltransacetylase component of the pyruvate dehydrogenase complex from Azotobacter vinelandii. Molecular cloning and sequence analysis. Eur J Biochem. 1988;174:593–599. doi: 10.1111/j.1432-1033.1988.tb14140.x. [DOI] [PubMed] [Google Scholar]
- 160.Griffin TA, Lau KS, Chuang DT. Characterization and conservation of the inner E2 core domain structure of branched-chain alpha-keto acid dehydrogenase complex from bovine liver. Construction of a cDNA encoding the entire transacylase (E2b) precursor. J Biol Chem. 1988;263:14008–14014. [PubMed] [Google Scholar]
- 161.Hackert ML, Xu WX, Oliver RM, Wall JS, Hainfeld JF, Mullinax TR, Reed LJ. Branched-chain alpha-keto acid dehydrogenase complex from bovine kidney: radial distribution of mass determined from dark-field electron micrographs. Biochemistry. 1989;28:6816–6821. doi: 10.1021/bi00443a006. [DOI] [PubMed] [Google Scholar]
- 162.Henderson CE, Perham RN. Purificaton of the pyruvate dehydrogenase multienzyme complex of Bacillus stearothermophilus and resolution of its four component polypeptides. Biochem J. 1980;189:161–172. doi: 10.1042/bj1890161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lowe PN, Hodgson JA, Perham RN. Dual role of a single multienzyme complex in the oxidative decarboxylation of pyruvate and branched-chain 2-oxo acids in Bacillus subtilis. Biochem J. 1983;215:133–140. doi: 10.1042/bj2150133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Keha EE, Ronft H, Kresze GB. On the origin of mitochondria: a reexamination of the molecular structure and kinetic properties of pyruvate dehydrogenase complex from brewer's yeast. FEBS Lett. 1982;145:289–292. doi: 10.1016/0014-5793(82)80185-3. [DOI] [PubMed] [Google Scholar]
- 165.Allen AG, Perham RN. Two lipoyl domains in the dihydrolipoamide acetyltransferase chain of the pyruvate dehydrogenase multienzyme complex of Streptococcus faecalis. FEBS Lett. 1991;287:206–210. doi: 10.1016/0014-5793(91)80052-5. [DOI] [PubMed] [Google Scholar]
- 166.Berg A, de Kok A. 2-Oxo acid dehydrogenase multienzyme complexes. The central role of the lipoyl domain. Biol Chem. 1997;378:617–634. [PubMed] [Google Scholar]
- 167.Guest JR, Lewis HM, Graham LD, Packman LC, Perham RN. Genetic reconstruction and functional analysis of the repeating lipoyl domains in the pyruvate dehydrogenase multienzyme complex of Escherichia coli. J Mol Biol. 1985;185:743–754. doi: 10.1016/0022-2836(85)90059-2. [DOI] [PubMed] [Google Scholar]
- 168.Allen AG, Perham RN, Allison N, Miles JS, Guest JR. Reductive acetylation of tandemly repeated lipoyl domains in the pyruvate dehydrogenase multienzyme complex of Escherichia coli is random order. J Mol Biol. 1989;208:623–633. doi: 10.1016/0022-2836(89)90153-8. [DOI] [PubMed] [Google Scholar]
- 169.Machado RS, Clark DP, Guest JR. Construction and properties of pyruvate dehydrogenase complexes with up to nine lipoyl domains per lipoate acetyltransferase chain. FEMS Microbiol Lett. 1992;79:243–248. doi: 10.1111/j.1574-6968.1992.tb14047.x. [DOI] [PubMed] [Google Scholar]
- 170.Machado RS, Guest JR, Williamson MP. Mobility in pyruvate dehydrogenase complexes with multiple lipoyl domains. FEBS Lett. 1993;323:243–246. doi: 10.1016/0014-5793(93)81349-5. [DOI] [PubMed] [Google Scholar]
- 171.Dave E, Guest JR, Attwood MM. Metabolic engineering in Escherichia coli: lowering the lipoyl domain content of the pyruvate dehydrogenase complex adversely affects the growth rate and yield. Microbiology. 1995;141:1839–1849. doi: 10.1099/13500872-141-8-1839. [DOI] [PubMed] [Google Scholar]
- 172.Guest JR, Attwood MM, Machado RS, Matqi KY, Shaw JE, Turner SL. Enzymological and physiological consequences of restructuring the lipoyl domain content of the pyruvate dehydrogenase complex of Escherichia coli. Microbiology. 1997;143:457–466. doi: 10.1099/00221287-143-2-457. [DOI] [PubMed] [Google Scholar]
- 173.Miles JS, Guest JR, Radford SE, Perham RN. Investigation of the mechanism of active site coupling in the pyruvate dehydrogenase multienzyme complex of Escherichia coli by protein engineering. J Mol Biol. 1988;202:97–106. doi: 10.1016/0022-2836(88)90522-0. [DOI] [PubMed] [Google Scholar]
- 174.Berg A, Westphal AH, Bosma HJ, de Kok A. Kinetics and specificity of reductive acylation of wild-type and mutated lipoyl domains of 2-oxo-acid dehydrogenase complexes from Azotobacter vinelandii. Eur J Biochem. 1998;252:45–50. doi: 10.1046/j.1432-1327.1998.2520045.x. [DOI] [PubMed] [Google Scholar]
- 175.Dardel F, Laue ED, Perham RN. Sequence-specific 1H-NMR assignments and secondary structure of the lipoyl domain of the Bacillus stearothermophilus pyruvate dehydrogenase multienzyme complex. Eur J Biochem. 1991;201:203–209. doi: 10.1111/j.1432-1033.1991.tb16275.x. [DOI] [PubMed] [Google Scholar]
- 176.Graham LD, Packman LC, Perham RN. Kinetics and specificity of reductive acylation of lipoyl domains from 2-oxo acid dehydrogenase multienzyme complexes. Biochemistry. 1989;28:1574–1581. doi: 10.1021/bi00430a023. [DOI] [PubMed] [Google Scholar]
- 177.Jones DD, Stott KM, Howard MJ, Perham RN. Restricted motion of the lipoyl-lysine swinging arm in the pyruvate dehydrogenase complex of Escherichia coli. Biochemistry. 2000;39:8448–8459. doi: 10.1021/bi992978i. [DOI] [PubMed] [Google Scholar]
- 178.Aevarsson A, Seger K, Turley S, Sokatch JR, Hol WG. Crystal structure of 2-oxoisovalerate and dehydrogenase and the architecture of 2-oxo acid dehydrogenase multienzyme complexes. Nat Struct Biol. 1999;6:785–792. doi: 10.1038/11563. [DOI] [PubMed] [Google Scholar]
- 179.Wallis NG, Perham RN. Structural dependence of post-translational modification and reductive acetylation of the lipoyl domain of the pyruvate dehydrogenase multienzyme complex. J Mol Biol. 1994;236:209–216. doi: 10.1006/jmbi.1994.1130. [DOI] [PubMed] [Google Scholar]
- 180.Wallis NG, Allen MD, Broadhurst RW, Lessard IA, Perham RN. Recognition of a surface loop of the lipoyl domain underlies substrate channelling in the pyruvate dehydrogenase multienzyme complex. J Mol Biol. 1996;263:463–474. doi: 10.1006/jmbi.1996.0589. [DOI] [PubMed] [Google Scholar]
- 181.Jones DD, Horne HJ, Reche PA, Perham RN. Structural determinants of post-translational modification and catalytic specificity for the lipoyl domains of the pyruvate dehydrogenase multienzyme complex of Escherichia coli. J Mol Biol. 2000;295:289–306. doi: 10.1006/jmbi.1999.3335. [DOI] [PubMed] [Google Scholar]
- 182.Ali ST, Guest JR. Isolation and characterization of lipoylated and unlipoylated domains of the E2p subunit of the pyruvate dehydrogenase complex of Escherichia coli. Biochem J. 1990;271:139–145. doi: 10.1042/bj2710139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ricaud PM, Howard MJ, Roberts EL, Broadhurst RW, Perham RN. Three-dimensional structure of the lipoyl domain from the dihydrolipoyl succinyltransferase component of the 2-oxoglutarate dehydrogenase multienzyme complex of Escherichia coli. J Mol Biol. 1996;264:179–190. doi: 10.1006/jmbi.1996.0632. [DOI] [PubMed] [Google Scholar]
- 184.Dardel F, Packman LC, Perham RN. Expression in Escherichia coli of a sub-gene encoding the lipoyl domain of the pyruvate dehydrogenase complex of Bacillus stearothermophilus. FEBS Lett. 1990;264:206–210. doi: 10.1016/0014-5793(90)80249-i. [DOI] [PubMed] [Google Scholar]
- 185.Quinn J, Diamond AG, Masters AK, Brookfield DE, Wallis NG, Yeaman SJ. Expression and lipoylation in Escherichia coli of the inner lipoyl domain of the E2 component of the human pyruvate dehydrogenase complex. Biochem J. 1993;289:81–85. doi: 10.1042/bj2890081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Berg A, Vervoort J, de Kok A. Solution structure of the lipoyl domain of the 2-oxoglutarate dehydrogenase complex from Azotobacter vinelandii. J Mol Biol. 1996;261:432–442. doi: 10.1006/jmbi.1996.0474. [DOI] [PubMed] [Google Scholar]
- 187.Tozawa K, Broadhurst RW, Raine AR, Fuller C, Alvarez A, Guillen G, Padron G, Perham RN. Solution structure of the lipoyl domain of the chimeric dihydrolipoyl dehydrogenase P64K from Neisseria meningitidis. Eur J Biochem. 2001;268:4908–4917. doi: 10.1046/j.0014-2956.2001.02422.x. [DOI] [PubMed] [Google Scholar]
- 188.Dardel F, Davis AL, Laue ED, Perham RN. Three-dimensional structure of the lipoyl domain from Bacillus stearothermophilus pyruvate dehydrogenase multienzyme complex. J Mol Biol. 1993;229:1037–1048. doi: 10.1006/jmbi.1993.1103. [DOI] [PubMed] [Google Scholar]
- 189.Berg A, de Kok A, Vervoort J. Sequential 1H and 15N nuclear magnetic resonance assignments and secondary structure of the N-terminal lipoyl domain of the dihydrolipoyl transacetylase component of the pyruvate dehydrogenase complex from Azotobacter vinelandii. Eur J Biochem. 1994;221:87–100. doi: 10.1111/j.1432-1033.1994.tb18717.x. [DOI] [PubMed] [Google Scholar]
- 190.Fujiwara K, Okamura-Ikeda K, Motokawa Y. Chicken liver H-protein, a component of the glycine cleavage system. Amino acid sequence and identification of the N epsilon-lipoyllysine residue. J Biol Chem. 1986;261:8836–8841. [PubMed] [Google Scholar]
- 191.Fujiwara K, Okamura-Ikeda K, Motokawa Y. Lipoylation of H-protein of the glycine cleavage system. The effect of site-directed mutagenesis of amino acid residues around the lipoyllysine residue on the lipoate attachment. FEBS Lett. 1991;293:115–118. doi: 10.1016/0014-5793(91)81164-4. [DOI] [PubMed] [Google Scholar]
- 192.Brocklehurst SM, Perham RN. Prediction of the three-dimensional structures of the biotinylated domain from yeast pyruvate carboxylase and of the lipoylated H-protein from the pea leaf glycine cleavage system: a new automated method for the prediction of protein tertiary structure. Protein Sci. 1993;2:626–639. doi: 10.1002/pro.5560020413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Athappilly FK, Hendrickson WA. Structure of the biotinyl domain of acetyl-coenzyme A carboxylase determined by MAD phasing. Structure. 1995;3:1407–1419. doi: 10.1016/s0969-2126(01)00277-5. [DOI] [PubMed] [Google Scholar]
- 194.Roberts EL, Shu N, Howard MJ, Broadhurst RW, Chapman-Smith A, Wallace JC, Morris T, Cronan JE, Jr, Perham RN. Solution structures of apo and holo biotinyl domains from acetyl coenzyme A carboxylase of Escherichia coli determined by triple-resonance nuclear magnetic resonance spectroscopy. Biochemistry. 1999;38:5045–5053. doi: 10.1021/bi982466o. [DOI] [PubMed] [Google Scholar]
- 195.Yao X, Soden C, Jr, Summers MF, Beckett D. Comparison of the backbone dynamics of the apo- and holo-carboxy-terminal domain of the biotin carboxyl carrier subunit of Escherichia coli acetyl-CoA carboxylase. Protein Sci. 1999;8:307–317. doi: 10.1110/ps.8.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Yao X, Wei D, Soden C, Jr, Summers MF, Beckett D. Structure of the carboxy-terminal fragment of the apo-biotin carboxyl carrier subunit of Escherichia coli acetyl-CoA carboxylase. Biochemistry. 1997;36:15089–15100. doi: 10.1021/bi971485f. [DOI] [PubMed] [Google Scholar]
- 197.Reddy DV, Shenoy BC, Carey PR, Sonnichsen FD. High resolution solution structure of the 1.3S subunit of transcarboxylase from Propionibacterium shermanii. Biochemistry. 2000;39:2509–2516. doi: 10.1021/bi9925367. [DOI] [PubMed] [Google Scholar]
- 198.Morris TW, Reed KE, Cronan JE., Jr Lipoic acid metabolism in Escherichia coli: the lplA and lipB genes define redundant pathways for ligation of lipoyl groups to apoprotein. J Bacteriol. 1995;177:1–10. doi: 10.1128/jb.177.1.1-10.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Morris TW, Reed KE, Cronan JE., Jr Identification of the gene encoding lipoate-protein ligase A of Escherichia coli Molecular cloning and characterization of the lplA gene and gene product. J Biol Chem. 1994;269:16091–16100. [PubMed] [Google Scholar]
- 200.Jordan SW, Cronan JE., Jr The Escherichia coli lipB gene encodes lipoyl (octanoyl)-acyl carrier protein:protein transferase. J Bacteriol. 2003;185:1582–1589. doi: 10.1128/JB.185.5.1582-1589.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhao X, Miller JR, Jiang Y, Marletta MA, Cronan JE. Assembly of the covalent linkage between lipoic acid and its cognate enzymes. Chem Biol. 2003;10:1293–1302. doi: 10.1016/j.chembiol.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 202.Cicchillo RM, Iwig DF, Jones AD, Nesbitt NM, Baleanu-Gogonea C, Souder MG, Tu L, Booker SJ. Lipoyl synthase requires two equivalents of S-adenosyl-L-methionine to synthesize one equivalent of lipoic acid. Biochemistry. 2004;43:6378–6386. doi: 10.1021/bi049528x. [DOI] [PubMed] [Google Scholar]
- 203.Cicchillo RM, Lee KH, Baleanu-Gogonea C, Nesbitt NM, Krebs C, Booker SJ. Escherichia coli lipoyl synthase binds two distinct [4Fe-4S] clusters per polypeptide. Biochemistry. 2004;43:11770–11781. doi: 10.1021/bi0488505. [DOI] [PubMed] [Google Scholar]
- 204.Reed LJ, Leach FR, Koike M. Studies on a lipoic acid-activating system. J Biol Chem. 1958;232:123–142. [PubMed] [Google Scholar]
- 205.Reed LJ, Koike M, Levitch ME, Leach FR. Studies on the nature and reactions of protein-bound lipoic acid. J Biol Chem. 1958;232:143–158. [PubMed] [Google Scholar]
- 206.Green DE, Morris TW, Green J, Cronan JE, Jr, Guest JR. Purification and properties of the lipoate protein ligase of Escherichia coli. Biochem J. 1995;309:853–862. doi: 10.1042/bj3090853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Guest JR, Creaghan IT. Gene-protein relationships of the alpha-keto acid dehydrogenase complexes of Escherichia coli K12: isolation and characterization of lipoamide dehydrogenase mutants. J Gen Microbiol. 1973;75:197–210. doi: 10.1099/00221287-75-1-197. [DOI] [PubMed] [Google Scholar]
- 208.Oehring R, Bisswanger H. Incorporation of the enantiomers of lipoic acid into the pyruvate dehydrogenase complex from Escherichia coli in vivo. Biol Chem Hoppe Seyler. 1992;373:333–335. doi: 10.1515/bchm3.1992.373.1.333. [DOI] [PubMed] [Google Scholar]
- 209.Reed KE, Morris TW, Cronan JE., Jr Mutants of Escherichia coli K-12 that are resistant to a selenium analog of lipoic acid identify unknown genes in lipoate metabolism. Proc Natl Acad Sci USA. 1994;91:3720–3724. doi: 10.1073/pnas.91.9.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Reche PA. Lipoylating and biotinylating enzymes contain a homologous catalytic module. Protein Sci. 2000;9:1922–1929. doi: 10.1110/ps.9.10.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Brookfield DE, Green J, Ali ST, Machado RS, Guest JR. Evidence for two protein-lipoylation activities in Escherichia coli. FEBS Lett. 1991;295:13–16. doi: 10.1016/0014-5793(91)81373-g. [DOI] [PubMed] [Google Scholar]
- 212.Fujiwara K, Toma S, Okamura-Ikeda K, Motokawa Y, Nakagawa A, Taniguchi H. Crystal structure of lipoate-protein ligase A from Escherichia coli Determination of the lipoic acid-binding site. J Biol Chem. 2005;280:33645–33651. doi: 10.1074/jbc.M505010200. [DOI] [PubMed] [Google Scholar]
- 213.Kim DJ, Kim KH, Lee HH, Lee SJ, Ha JY, Yoon HJ, Suh SW. Crystal structure of lipoate-protein ligase A bound with the activated intermediate: insights into interaction with lipoyl domains. J Biol Chem. 2005;280:38081–38089. doi: 10.1074/jbc.M507284200. [DOI] [PubMed] [Google Scholar]
- 214.McManus E, Luisi BF, Perham RN. Structure of a putative lipoate protein ligase from Thermoplasma acidophilum and the mechanism of target selection for post-translational modification. J Mol Biol. 2006;356:625–637. doi: 10.1016/j.jmb.2005.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Christensen QH, Cronan JE. The Thermoplasma acidophilum LplA-LplB complex defines a new class of bipartite lipoate-protein ligases. J Biol Chem. 2009;284:21317–21326. doi: 10.1074/jbc.M109.015016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Posner MG, Upadhyay A, Bagby S, Hough DW, Danson MJ. A unique lipoylation system in the Archaea. Lipoylation in Thermoplasma acidophilum requires two proteins. FEBS J. 2009;276:4012–4022. doi: 10.1111/j.1742-4658.2009.07110.x. [DOI] [PubMed] [Google Scholar]
- 217.Reed KE, Cronan JE., Jr Lipoic acid metabolism in Escherichia coli: sequencing and functional characterization of the lipA and lipB genes. J Bacteriol. 1993;175:1325–1336. doi: 10.1128/jb.175.5.1325-1336.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Jordan SW, Cronan JE., Jr A new metabolic link. The acyl carrier protein of lipid synthesis donates lipoic acid to the pyruvate dehydrogenase complex in Escherichia coli and mitochondria. J Biol Chem. 1997;272:17903–17906. doi: 10.1074/jbc.272.29.17903. [DOI] [PubMed] [Google Scholar]
- 219.Nesbitt NM, Baleanu-Gogonea C, Cicchillo RM, Goodson K, Iwig DF, Broadwater JA, Haas JA, Fox BG, Booker SJ. Expression, purification, and physical characterization of Escherichia coli lipoyl(octanoyl)transferase. Protein Expr Purif. 2005;39:269–282. doi: 10.1016/j.pep.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 220.Herbert AA, Guest JR. Lipoic acid content of Escherichia coli and other microorganisms. Arch Microbiol. 1975;106:259–266. doi: 10.1007/BF00446532. [DOI] [PubMed] [Google Scholar]
- 221.Hermes FA, Cronan JE. Scavenging of cytosolic octanoic acid by mutant LplA lipoate ligases allows growth of Escherichia coli strains lacking the LipB octanoyltransferase of lipoic acid synthesis. J Bacteriol. 2009;191:6796–6803. doi: 10.1128/JB.00798-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Jordan SW, Cronan JE., Jr Biosynthesis of lipoic acid and posttranslational modification with lipoic acid in Escherichia coli. Methods Enzymol. 1997;279:176–183. doi: 10.1016/s0076-6879(97)79021-9. [DOI] [PubMed] [Google Scholar]
- 223.Ali ST, Moir AJ, Ashton PR, Engel PC, Guest JR. Octanoylation of the lipoyl domains of the pyruvate dehydrogenase complex in a lipoyl-deficient strain of Escherichia coli. Mol Microbiol. 1990;4:943–950. doi: 10.1111/j.1365-2958.1990.tb00667.x. [DOI] [PubMed] [Google Scholar]
- 224.Jordan SW, Cronan JE., Jr Chromosomal amplification of the Escherichia coli lipB region confers high-level resistance to selenolipoic acid. J Bacteriol. 2002;184:5495–5501. doi: 10.1128/JB.184.19.5495-5501.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Zhao X, Miller JR, Cronan JE. The reaction of LipB, the octanoyl-[acyl carrier protein]:protein N-octanoyltransferase of lipoic acid synthesis, proceeds through an acyl-enzyme intermediate. Biochemistry. 2005;44:16737–16746. doi: 10.1021/bi051865y. [DOI] [PubMed] [Google Scholar]
- 226.Hassan BH, Cronan JE. Protein-protein interactions in assembly of lipoic acid on the 2-oxoacid dehydrogenases of aerobic metabolism. J Biol Chem. 2011;286:8263–8276. doi: 10.1074/jbc.M110.194191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ma Q, Zhao X, Nasser Eddine A, Geerlof A, Li X, Cronan JE, Kaufmann SH, Wilmanns M. The Mycobacterium tuberculosis LipB enzyme functions as a cysteine/lysine dyad acyltransferase. Proc Natl Acad Sci USA. 2006;103:8662–8667. doi: 10.1073/pnas.0510436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kim do J, Lee SJ, Kim HS, Kim KH, Lee HH, Yoon HJ, Suh SW. Structural basis of octanoic acid recognition by lipoate-protein ligase B. Proteins. 2008;70:1620–1625. doi: 10.1002/prot.21843. [DOI] [PubMed] [Google Scholar]
- 229.Parry RJ. Biosynthesis of some sulfur-containing natural products. Investigations of the mechanism of carbon-sulfur bond formation. Tetrahedron. 1983;39:1215–1238. [Google Scholar]
- 230.White RH. Stable isotope studies on the biosynthesis of lipoic acid in Escherichia coli. Biochemistry. 1980;19:15–19. doi: 10.1021/bi00542a003. [DOI] [PubMed] [Google Scholar]
- 231.Parry RJ. Biosynthesis of lipoic acid. 1. Incorporation of specifically tritiated octanoic acid into lipoic acid. J. Am. Chem. Soc. 1977;99:6464–6466. [Google Scholar]
- 232.Parry RJ. Biosynthesis of lipoic acid. 2. Stereochemistry of sulfur introduction as C-6 of octanoic acid. J. Am. Chem. Soc. 1978;100:5243–5244. [Google Scholar]
- 233.White RH. Stoichiometry and stereochemistry of deuterium incorporated into fatty acids by cells of Escherichia coli grown on [methyl-2H3]acetate. Biochemistry. 1980;19:9–15. doi: 10.1021/bi00542a002. [DOI] [PubMed] [Google Scholar]
- 234.White RH. Biosynthesis of lipoic acid: Extent of incorporation of deuterated hydroxy- and thiooctanoic acids into lipoic acid. J. Am. Chem. Soc. 1980;102:6605–6607. [Google Scholar]
- 235.Hayden MA, Huang IY, Iliopoulos G, Orozco M, Ashley GW. Biosynthesis of lipoic acid: characterization of the lipoic acid auxotrophs Escherichia coli W1485-lip2 and JRG33-lip9. Biochemistry. 1993;32:3778–3782. doi: 10.1021/bi00065a033. [DOI] [PubMed] [Google Scholar]
- 236.Hayden MA, Huang I, Bussiere DE, Ashley GW. The biosynthesis of lipoic acid. Cloning of lip a lipoate biosynthetic locus of Escherichia coli. J Biol Chem. 1992;267:9512–9515. [PubMed] [Google Scholar]
- 237.Busby RW, Schelvis JPM, Yu DS, Babcock GT, Marletta MA. Lipoic acid biosynthesis: LipA is an iron sulfur protein. J. Am. Chem. Soc. 1999;121:4706–4707. [Google Scholar]
- 238.Ollagnier-de Choudens S, Fontecave M. The lipoate synthase from Escherichia coli is an iron-sulfur protein. FEBS Lett. 1999;453:25–28. doi: 10.1016/s0014-5793(99)00694-8. [DOI] [PubMed] [Google Scholar]
- 239.Ollagnier-De Choudens S, Sanakis Y, Hewitson KS, Roach P, Baldwin JE, Munck E, Fontecave M. Iron-sulfur center of biotin synthase and lipoate synthase. Biochemistry. 2000;39:4165–4173. doi: 10.1021/bi992090u. [DOI] [PubMed] [Google Scholar]
- 240.Miller JR, Busby RW, Jordan SW, Cheek J, Henshaw TF, Ashley GW, Broderick JB, Cronan JE, Jr, Marletta MA. Escherichia coli LipA is a lipoyl synthase: in vitro biosynthesis of lipoylated pyruvate dehydrogenase complex from octanoyl-acyl carrier protein. Biochemistry. 2000;39:15166–15178. doi: 10.1021/bi002060n. [DOI] [PubMed] [Google Scholar]
- 241.Booker SJ. Unraveling the pathway of lipoic acid biosynthesis. Chem Biol. 2004;11:10–12. doi: 10.1016/j.chembiol.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 242.Bryant P, Kriek M, Wood RJ, Roach PL. The activity of a thermostable lipoyl synthase from Sulfolobus solfataricus with a synthetic octanoyl substrate. Anal Biochem. 2006;351:44–49. doi: 10.1016/j.ab.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 243.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Cheek J, Broderick JB. Adenosylmethionine-dependent iron-sulfur enzymes: versatile clusters in a radical new role. J Biol Inorg Chem. 2001;6:209–226. doi: 10.1007/s007750100210. [DOI] [PubMed] [Google Scholar]
- 245.Nicolet Y, Drennan CL. AdoMet radical proteins--from structure to evolution--alignment of divergent protein sequences reveals strong secondary structure element conservation. Nucleic Acids Res. 2004;32:4015–4025. doi: 10.1093/nar/gkh728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Frey PA. Radical mechanisms of enzymatic catalysis. Annu Rev Biochem. 2001;70:121–148. doi: 10.1146/annurev.biochem.70.1.121. [DOI] [PubMed] [Google Scholar]
- 247.Shaw NM, Birch OM, Tinschert A, Venetz V, Dietrich R, Savoy LA. Biotin synthase from Escherichia coli: isolation of an enzyme-generated intermediate and stoichiometry of S-adenosylmethionine use. Biochem J. 1998;330:1079–1085. doi: 10.1042/bj3301079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Guianvarc'h D, Florentin D, Tse Sum Bui B, Nunzi F, Marquet A. Biotin synthase, a new member of the family of enzymes which uses S-adenosylmethionine as a source of deoxyadenosyl radical. Biochem Biophys Res Commun. 1997;236:402–406. doi: 10.1006/bbrc.1997.6952. [DOI] [PubMed] [Google Scholar]
- 249.Marquet A. Enzymology of carbon-sulfur bond formation. Curr Opin Chem Biol. 2001;5:541–549. doi: 10.1016/s1367-5931(00)00249-0. [DOI] [PubMed] [Google Scholar]
- 250.Cicchillo RM, Booker SJ. Mechanistic investigations of lipoic acid biosynthesis in Escherichia coli: both sulfur atoms in lipoic acid are contributed by the same lipoyl synthase polypeptide. J Am Chem Soc. 2005;127:2860–2861. doi: 10.1021/ja042428u. [DOI] [PubMed] [Google Scholar]
- 251.Douglas P, Kriek M, Bryant P, Roach PL. Lipoyl synthase inserts sulfur atoms into an octanoyl substrate in a stepwise manner. Angew Chem Int Ed Engl. 2006;45:5197–5199. doi: 10.1002/anie.200601910. [DOI] [PubMed] [Google Scholar]
- 252.Hipps DS, Perham RN. Expression in Escherichia coli of a sub-gene encoding the lipoyl and peripheral subunit-binding domains of the dihydrolipoamide acetyltransferase component of the pyruvate dehydrogenase complex of Bacillus stearothermophilus. Biochem J. 1992;283:665–671. doi: 10.1042/bj2830665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Christensen QH, Martin N, Mansilla MC, de Mendoza D, Cronan JE. A novel amidotransferase required for lipoic acid cofactor assembly in Bacillus subtilis. Mol Microbiol. 2011;80:350–363. doi: 10.1111/j.1365-2958.2011.07598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kriek M, Peters L, Takahashi Y, Roach PL. Effect of iron-sulfur cluster assembly proteins on the expression of Escherichia coli lipoic acid synthase. Protein Expr Purif. 2003;28:241–245. doi: 10.1016/s1046-5928(02)00680-0. [DOI] [PubMed] [Google Scholar]
- 255.Smith RL, Pelley JW, Jeter RM. Characterization of lip expression in Salmonella typhimurium: analysis of lip::lac operon fusions. J Gen Microbiol. 1991;137(Pt 10):2307–2312. doi: 10.1099/00221287-137-10-2307. [DOI] [PubMed] [Google Scholar]
- 256.Vaisvila R, Rasmussen LJ, Lobner-Olesen A, von Freiesleben U, Marinus MG. The LipB protein is a negative regulator of dam gene expression in Escherichia coli. Biochim Biophys Acta. 2000;1494:43–53. doi: 10.1016/s0167-4781(00)00209-8. [DOI] [PubMed] [Google Scholar]
- 257.Reche P, Li YL, Fuller C, Eichhorn K, Perham RN. Selectivity of post-translational modification in biotinylated proteins: the carboxy carrier protein of the acetyl-CoA carboxylase of Escherichia coli. Biochem J. 1998;329:589–596. doi: 10.1042/bj3290589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Lin S, Cronan JE. Closing in on complete pathways of biotin biosynthesis. Mol Biosyst. 2011;7:1811–1821. doi: 10.1039/c1mb05022b. [DOI] [PubMed] [Google Scholar]
- 259.Martin N, Christensen QH, Mansilla MC, Cronan JE, de Mendoza D. A novel two-gene requirement for the octanoyltransfer reaction of Bacillus subtilis lipoic acid biosynthesis. Mol Microbiol. 2011;80:335–349. doi: 10.1111/j.1365-2958.2011.07597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Marquet A, Frappier F, Guillerm G, Azoulay M, Florentin D, Tabet J-C. Biotin biosynthesis: Synthesis and biological evaluation of the putative intermediate thiols. J. Am. Chem. Soc. 1993;115:2139–2145. [Google Scholar]