

Abstract
In the search for novel Gram-negative agents, we performed a comprehensive search of the AstraZeneca collection and identified a tetrahydropyran-based matrix metalloprotease (MMP) inhibitor that demonstrated nanomolar inhibition of UDP-3-O-(acyl)-N-acetylglucosamine deacetylase (LpxC). Crystallographic studies in Aquifex aeolicus LpxC indicated the tetrahydropyran engaged in the same hydrogen bonds and van der Waals interactions as other known inhibitors. Systematic optimization of three locales on the scaffold provided compounds with improved Gram-negative activity. However, the optimization of LpxC activity was not accompanied by reduced inhibition of MMPs. Comparison of the crystal structure of the native product, UDP-3-O-(acyl)-glucosamine, in Aquifex aeolicus to the structure of a tetrahydropyran-based inhibitor indicates pathways for future optimization.
Keywords: Antibacterial, LpxC, Gram-negative bacteria, MMP, hydrophobe, Pseudomonas aeruginosa, Aquifex aeolicus
The need for new antibacterials active against Gram-negative bacteria is becoming critical. Current therapies are becoming ineffective due to increasing resistance, leaving healthcare practicioners with limited treatment options for serious bacterial infections.1−3 To compound the issue, the presence of an additional cell membrane, incorporating lipopolysaccharide (LPS) in the outer leaflet, confers an intrinsic degree of resistance to Gram-negative pathogens.4−6 The presence of the LPS layer, in addition to promiscuous efflux pumps, prevents agents effective at killing Gram-positive bacteria from penetrating the Gram-negative cellular envelope.
UDP-3-O-(acyl)-N-acetylglucosamine deacetylase (LpxC) has become the focus of a number of programs aimed at the development of novel Gram-negative agents.7 LpxC is a cytosolic zinc metallo-enzyme that catalyzes the deacetylation of UDP-3-O-(acyl)-N-acetylglucosamine, the first nonreversible step in the biosynthesis of lipid A, the main component and the substrate to which LPS is attached in the outer membrane of most Gram-negative bacteria.8,9 Because the biosynthesis of lipid A is essential for maintenance of the outer membrane, LpxC is an attractive target for the development of new agents.
A number of inhibitors of LpxC have been reported in the literature.10−15 One of the most well documented inhibitors is CHIR-090 (1, Figure 1).16−18 Key structural features of CHIR-090, which are consistent in other reported inhibitors, are a zinc-binding group (hydroxamate) and a hydrophobic tail (hydrophobe), which sits in a narrow hydrophobic tunnel, usually occupied by the fatty acid tail of the natural substrate. The major difference between the various classes of inhibitors is the core that links the hydroxamate to the hydrophobe. CHIR-090 contains a threonine-based core, which has been shown to engage in a key hydrogen bond with Lys238 in Pseudomonas aeruginosa (P. aeruginosa) LpxC. Pfizer has reported on a series of methylsulfone-based inhibitors, which also take advantage of this hydrogen bonding interaction.12,13
Figure 1.

CHIR-090 (1) and tetrahydropyran-based MMP inhibitor (2).
In our efforts to identify novel inhibitors of LpxC, we performed a screen of all hydroxamates present in the AstraZeneca compound collection. Through this effort we identified compound 2, a tetrahydropyran-based hydroxamate, which is a derivative of a known inhibitor (RS-130830) of matrix metalloprotease (MMP)-2, -8, -9, and -13.19 Tetrahydropyran-based compound 2 demonstrated potent inhibition of P. aeruginosa LpxC (7.4 nM, Table 1), moderate cellular activity against P. aeruginosa PAO1 (MIC = 50 μM), but poor activity against wild-type Escherichia coli (E. coli ARC523 MIC >200 μM). We were attracted to the tetrahydropyran since this structural motif had not been employed in other LpxC inhibitors, yet it has similarity when compared to the natural substrate, UDP-3-O-(acyl)-N-acetylglucosamine. The oxygen of the tetrahydropyran has the potential to take advantage of the key Lys238 hydrogen bond in P. aeruginosa. In addition, the MMP literature has shown a cyclic core inhibits metabolism of the hydroxamate.19−21 With this initial hit, we looked to maintain the tetrahydropyran core and optimize the hydrophobe. Our initial focus was improvement of Gram-negative activity and minimization of any potential off-target activity, particularly, against MMPs.
Table 1. Antibacterial Activity of Tetrahydropyran-Based LpxC Inhibitors.
| MIC (μM) |
|||||
|---|---|---|---|---|---|
| P. aeruginosa LpxC IC50 (nM)a | P. aeruginosa ARC546 (ΔMexABCDXY-PAO1) | P. aeruginosa ARC545 (PAO1) | E. coli ARC524 (ΔTolC-W3110) | E. coli ARC523 (W3110) | |
| 1 | 0.31 ± 0.15 (n = 4) | <0.2 | 1.56 | <0.2 | <0.2 |
| 2 | 7.4 ± 2.6 | 6.25 | 50 | 50 | >200 |
| 9 | 4.4 ± 1.0 | 1.56 | 100 | 1.56 | 12.5 |
| 11a | 8.7 ± 2.8 | 6.25 | 100 | >200 | >200 |
| 11b | 14 ± 2.1 | 6.25 | 200 | 200 | >200 |
| 11c | 92 ± 43 | 25 | >200 | >200 | >200 |
| 11d | 33 ± 6.5 | >200 | >200 | 50 | 200 |
| 11e | 139 ± 21 | 25 | >200 | 6.25 | 200 |
| 18a | 3.0 (n = 1) | 0.78 | 50 | 0.39 | 25 |
| 18b | 7.5 ± 2.4 | 3.13 | 100 | 12.5 | 100 |
| 18c | 2.7 ± 1.1 | 0.78 | 50 | 1.56 | 25 |
| 18d | 2.6 ± 0.9 | 3.13 | >200 | 0.39 | 12.5 |
| 18e | 5.8 ± 1.2 | 3.13 | 100 | 6.25 | 200 |
| 18f | 10 ± 2.8 | 6.25 | 100 | 12.5 | 50 |
| 18g | 6.1 ± 3.2 | 1.56 | 100 | 3.13 | 50 |
| 18h | 2.0 ± 0.7 | 0.78 | 50 | 0.78 | 6.25 |
| 18i | 1.7 (n = 2) | 12.5 | >200 | 25 | >200 |
| 23 | 0.20 ± 0.1 | 0.39 | 25 | <0.2 | 3.13 |
| 25 | 3.1 ± 2.9 (n = 4) | <0.2 | 25 | <0.2 | 1.56 |
| 27a | 82.5 (n = 1) | 25 | >200 | 25 | >200 |
| 27b | 8.6 ± 2.8 | 3.13 | >200 | 6.25 | 100 |
All errors are reported as ±1 standard deviation. n = 3 unless otherwise noted.
On the basis of the wealth of structural information and SAR available for LpxC, we proposed that a more linear hydrophobe would optimize substrate binding in the pocket.22,23 The phenylacetylenephenyl hydrophobe, similar to CHIR-090, was introduced to the tetrahydropyran core to see if we could improve the activity against LpxC as compared to the initial hit 2. For the initial studies, an ether linkage of the phenylacetylenephenyl to the tetrahydropyran core was chosen as the starting point, but we planned on investigating alternative linkers as well.
The synthesis of our first target is highlighted in Scheme 1. The tetrahydropyran core building block (4) was derived from alkylation of the lithium enolate of 3 with diiodomethane. We synthesized phenol 7 in four steps, employing Sonogashira coupling and reductive amination. Alkylation of 7 with alkyl iodide 4 followed by hydroxamate formation afforded the target inhibitor 9.
Scheme 1. Synthesis of Tetrahydropyran-Based LpxC Inhibitor 9.

Reagents and conditions: (a) LDA, CH2I2, THF, −40 °C to rt (80%); (b) 3,4-dihydro-2H-pyran, PTSA, K2CO3, Et2O, 0 °C (80%); (c) 4-ethynylbenzaldehyde, 10 mol % Pd(PPh3)2Cl2, 5 mol % CuI, Et3N, MeCN, 60 °C (79%); (d) morpholine, NaBH(OAc)3, AcOH, DCE, 0 °C to rt; (e) HCl, MeOH; (f) 4, K2CO3, DMF, 120 °C (51% over three steps); (g) LiOH, MeOH/THF/H2O (1:1:1), 60 °C (95%); (h) NH2OTHP, diethylcyanophosphonate, Et3N, DCM; (i) HCl, MeOH (88% over two steps).
We were pleased to find that compound 9 demonstrated excellent biochemical potency against P. aeruginosa LpxC (4.4 nM, Table 1). In addition, compound 9 exhibited similar cellular activity against P. aeruginosa as compound 2 and improved activity against E. coli. On the basis of their excellent activity, we obtained crystal structures of compounds 2 and 9 in Aquifex aeolicus (A. aeolicus) LpxC in order to confirm binding modes and to generate a tool to aid in further design and optimization.
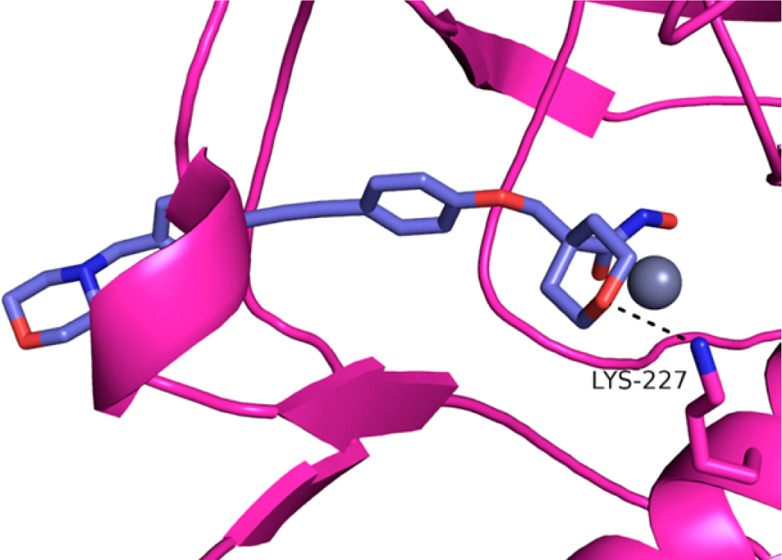
As expected, the hydroxamates of each compound superimposed and interacted with the zinc atom of the LpxC enzyme (Figure 2A,B). Examination of the core section of the molecules indicated a hydrogen bond interaction between the tetrahydropyran oxygen and Lys227 in A. aeolicus (equivalent to Lys238 in P. aeruginosa), similar to the interaction observed with other LpxC inhibitors.22−25 The carbon backbone of the tetrahydropyran engaged in van der Waals interactions with the hydrophobic phenylalanine present in the UDP pocket. Examination of the hydrophobe portion of the molecules indicated some differences in binding. Whereas compound 2 had a kink in its hydrophobe, compound 9 was linear and extended further into the hydrophobic tunnel. This difference explains the contrast in E. coli activity between 2 and 9. It has been shown that compounds that bind in this area of the pocket, like compound 9, are more potent inhibitors of E. coli.26
Figure 2.
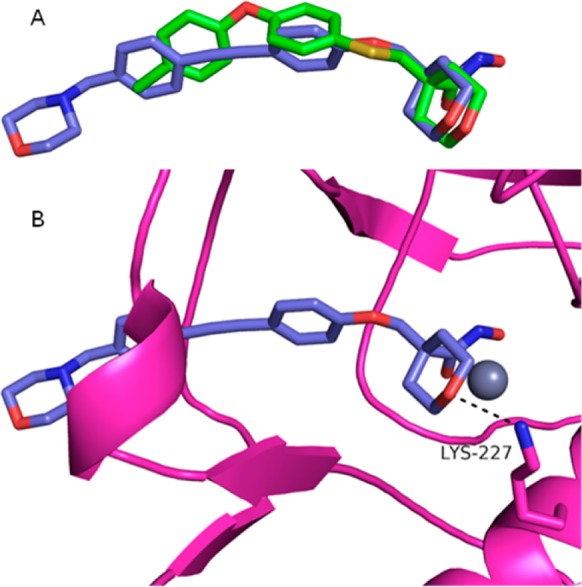
Superimposed cocrystal structures of A. aeolicus LpxC. Compound 2 in green (PDB 4U3B); compound 9 in purple (PDB 4U3D); zinc ion is the gray sphere; compound 9A. aeolicus LpxC protein in pink. (A) Protein α-carbon superposition of compound 2 versus compound 9 cocrystal structures; protein model removed for clarity. (B) Compound 9 cocrystal structure with A. aeolicus Lys227 interaction (3.0 Å).
With the binding mode of the tetrahydropyran core confirmed, we examined whether the diphenylacetylene-based hydrophobe was optimal. The synthesis of the inhibitors with modified hydrophobes can be found in Scheme 2. The target compounds (11a–11e) were synthesized by alkylation of the desired hydrophobe-phenol with alkyl iodide 4, followed by three steps to access the hydroxamates. The phenols 10a, 10b, and 10c were accessed from commercial sources. Phenol 10d was synthesized by acylation of 4-hydroxybenzaldehyde (12) followed by olefination to provide dibromo-olefin 13. Subsequent Sonogashira reaction and deprotection of the phenol provided 10d. Diacetylene substituted phenol 10e was synthesized via Ni-catalyzed coupling with 2-methylbut-3-yn-2-ol.
Scheme 2. Synthesis of Hydrophobe Analogues.

Reagents and conditions: (a) 4, K2CO3, DMF, 120 °C; (b) LiOH, MeOH/THF/H2O (1:1:1), 60 °C; (c) NH2OTHP, diethylcyanophosphonate, Et3N, DCM; (d) HCl, MeOH; (e) AcCl, Et3N, DCM 0 °C (74%); (f) CBr4, PPh3, DCM, 0 °C to rt (28%); (g) C6H5CCH, 4 mol % Pd2(dba)3, (4-MeOPh)3P, Et3N, DMF, 85 °C (60%); (h) LiOH, MeOH/THF/H2O (80%); (i) 3,4-dihydro-2H-pyran, PTSA, K2CO3, Et2O (80%); (j) Me3SiCCH, 5 mol % Pd(PPh3)2Cl2, 5 mol % CuI, Et3N, MeCN (67%); (k) K2CO3, MeOH (73%); (l) 2-methylbut-3-yn-2-ol, 5 mol % NiCl2·6H2O, 5 mol % CuI, TMEDA, THF (28%); (m) PPTS, EtOH, reflux (quant.).
The results from the hydrophobe screen can be found in Table 1. Of the nonacetylene-based hydrophobes, biphenyl 11a and ether 11b were the most active with LpxC IC50s <20 nM and good MICs against a P. aeruginosa MexAB efflux pump knockout mutant. Diacetylene compounds 11d and 11e, which presumably fill the hydrophobic tunnel, each had activity against an efflux-deficient strain of E. coli in which TolC, E. coli’s main efflux pump, has been knocked out.27 However, both compounds had poor activity against P. aeruginosa with efflux mutant MICs >25 μM. On the basis of this data, we decided to continue our studies with the phenyl acetylene phenyl-based hydrophobe utilized in compound 9.
We then looked to optimize the activity of compound 9 through modifications of the hydrophobe terminus. In order to optimize the Gram-negative activity we planned to introduce more basic substitutents since the outer membrane of P. aeruginosa has been shown to be penetrated by basic compounds.28,29 To minimize off-target MMP activity, we planned to introduce disruptive interactions in the terminus of the hydrophobe region since the MMPs and LpxC vary in their electrostatics in this region.30−32 The chemistry to access these modified inhibitors 18a–18i can be found in Scheme 3.
Scheme 3. Synthesis of Modified Phenylacetylenephenyl Hydrophobes.

Reagents and conditions: (a) 4, K2CO3, DMF, 120 °C; (b) amine, Na(OAc)3BH, AcOH, DCE, 0 °C to rt; (c) LiOH, MeOH/THF/H2O (1:1:1), 60 °C; (d) NH2OTHP, 2-chloro-1-methyl-pyridinium iodide, DIPEA, DMAP, DCM, 0 °C; (e) 4 N HCl, MeOH.
All of the new inhibitors exhibited potent P. aeruginosa IC50s, ranging from 1.7 to 10 nM (Table 1). The compounds exhibited a range of cellular activity with substituted piperazines 18a, 18c, and 18h showing the lowest P. aeruginosa and E. coli MICs. Compound 18h demonstrated the best overall profile.
We sought to improve the Gram-negative activity further by modifying the ether linkage. On the basis of the promising activity of the original MMP inhibitor hit 2, we decided to focus on thio-ethers. Additionally, we reasoned that thio-ethers would provide the opportunity to improve overall properties by varying the oxidation state of the sulfur linkage.25,26,28,29
Targets 23 and 25 were accessed starting from 4-iodobenzenesulfonyl chloride (19, Scheme 4) in a similar manner to the other targets. Racemic sulfoxide 25 was accessed via peroxide oxidation of methyl ester 22.
Scheme 4. Synthesis of Modified Linkers.

Reagents and conditions: (a) (i) Zn, Me2SiCl2, DCE, DMA, 75 °C; (ii) AcCl, 50 °C (65%); (b) 4-ethynylbenzaldehyde, 10 mol % Pd(PPh3)2Cl2, 5 mol % CuI, Et3N, MeCN, 60 °C (74%); (c) morpholine, NaB(OAc)3H, AcOH, DCE, 0 °C to rt (quant.); (d) 4, K2CO3, DMF, 120 °C (57%); (e) LiOH, MeOH/THF/H2O (1:1:1), 60 °C; (f) NH2OTHP, diethylcyanophosphonate, Et3N, DCM; (g) HCl, MeOH; (h) H2O2, AcOH, 0 °C (53%).
As can be seen in Table 1, the S-linked compounds exhibited excellent biochemical potency and improved P. aeruginosa and E. coli cellular activity compared to the ethers, with racemic sulfoxide 25 (Table 1) having the best cellular activity of any analogue to date. Modifying the linkage resulted in improved penetration of the cell membranes as evidenced by an equivalent P. aeruginosa ARC546 MIC to CHIR-090 (1). However, efflux was still an issue.
With the identification of lead structures for LpxC inhibition, we turned our attention to off-target activity since this would be critical for the program to move forward. Because of the fact that MMPs and LpxC are both zinc-dependent metalloenzymes and our initial hit was derived from a series designed to inhibit MMPs, we examined the potential for off-target activity of our LpxC inhibitors against representative MMPs. As can be seen in Table 2, the tetrahydropyran-based LpxC inhibitors exhibited significant inhibition of MMP-2 and MMP-13, as opposed to CHIR-090 (1), which showed low inhibition. Modifications of the hydrophobe and linker did nothing to help reduce the MMP inhibition as compared to the initial hit 2. This data indicates the selectivity against MMPs is due to the identity of the core. The lack of MMP selectivity by our compounds with an elongated hydrophobe suggests that the MMP binding pocket retains inherent plasticity to accommodate ligands of variable size and rigidity.33
Table 2. MMP Activity of Tetrahydropyran-Based Inhibitors.
| % inhibition (10 μM) |
||
|---|---|---|
| compd | MMP-2 | MMP-13 |
| 1 | 12a | 30a |
| 2 | NT | 99 ± 0.4b |
| 9 | 94 | 82 |
| 18d | 92 | 75 |
| 18h | 93 | 80 |
| 25 | 97 | 96 |
% inhibition at 30 μM.
Inhibition at 15 μM; n = 3; error is reported as ±1 standard deviation.
To understand these results we analyzed the catalytic domain of human collagenase-1 and -3 (MMP-1 and -13, respectively) cocrystal structures where each MMP was in complex with a tetrahydropyran biphenylether compound.33 We also obtained a crystal structure of the native product, UDP-3-O-(acyl)-glucosamine, complexed with A. aeolicus LpxC and compared it to the structures of compounds 2 and 9 and the MMP complexes (Figure 3A).34 Having established that the selectivity of 1 did not stem from the hydrophobe, we turned our attention to the remaining parts of the molecule in order to increase selectivity over MMP. Since modifications to the hydroxamate were expected to be detrimental to potency, we focused on modification of the tetrahydropyran ring. Analysis of the product-bound cocrystal structure of A. aeolicus LpxC indicated that the product pyrophosphate group covalently linked to the glucosamine directly interacted with Lys227 and a protein loop between β6′ and α2′ of Domain II (Figure 3A). We hypothesized that similar interactions could be targeted by vectors extending target engagement while conferring selectivity against MMPs.
Figure 3.
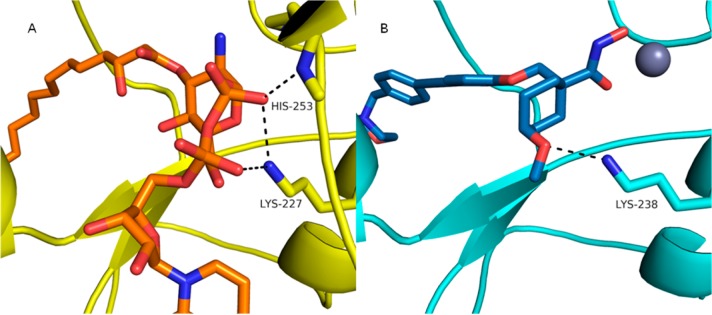
Cocrystal structure of A. aeolicus LpxC and P. aeruginosa docking model. UDP-3-O-(acyl)-glucosamine in orange; product bound form of A. aeolicus LpxC protein in yellow; zinc ion is the gray sphere; P. aeruginosa docking model in cyan; modeled compound 27b in P. aeruginosa in blue. (A) UDP-3-O-(acyl)-glucosamine bound cocrystal structure in A. aeolicus LpxC (4OZE). (B) Compound 27b modeled with P. aeruginosa Lys238 interaction.
With this information in hand we examined modifications to the tetrahydropyran core (Scheme 5). Each modification maintained the acceptor functionality of the tetrahydropyran oxygen in order to keep the interaction with Lys238 in P. aeruginosa as suggested in the docking model (Figure 3B).
Scheme 5. Synthesis of Modified Tetrahydropyran Cores.

Reagents and conditions: (a) LDA, CH2I2, THF, −40 °C to rt (86%); (b) 7, K2CO3, DMF, 120 °C (56%); (c) LiOH, MeOH/THF/H2O (1:1:1), 60 °C (>98%); (d) NH2OTHP, 2-chloro-1-methyl-pyridinium iodide, DIPEA, DMAP, DCM, 0 °C; (e) 4 N HCl, MeOH (14% over two steps).
As can be seen in Table 1, compounds 27a and 27b were less potent than the optimal tetrahydropyran-based compounds (9, 18a, 18c, 18h, and 25). Alcohol 27a exhibited a significant loss in enzyme potency as well as cellular activity. Methyl ether 27b exhibited excellent biochemical potency (7 nM IC50) but poor antibacterial activity against wild-type P. aeruginosa and E. coli, presumably due to high efflux activity. On the basis of their poor LpxC inhibition, we did not follow-up with MMP testing.
In summary, we identified a new class of tetrahydropyran-based LpxC inhibitors. We were able to improve upon the activity of the original tetrahydropyran-based hydroxamate 2 through optimization of the hydrophobe and linker. We identified compounds 23 and 25, which exhibited nanomolar potencies against P. aeruginosa LpxC enzyme, 25 μM MIC against wild-type P. aeruginosa (PAO1), and excellent activity against E. coli (MIC < 3.13 μM). Unfortunately we were not able to dial out the potent MMP inhibition. Attempts to modify the tetrahydropyran core provided compounds (27a and 27b, Scheme 5) with diminished potency. In general these compounds demonstrate good penetration of the P. aeruginosa cell membranes, as evidenced by their MICs against the efflux knockout. However, they are still not as potent as other analogues like CHIR-090 (1), and penetration needs improvement. Future designs will focus on alternative substitutions off the tetrahydropyran core. It has been observed with other series that building into the UDP pocket can result in higher efflux ratios; however, this may be a result of poor influx.11 We plan on building into the UDP pocket, trying to imitate the natural substrate, with the goals of increasing LpxC affinity, improving cellular activity, and reducing activity against MMPs.
Acknowledgments
We are thankful for Kumar Srinivas and colleagues at Syngene International, Ltd., Bangalore, India for compound synthesis, Nikunj Agrawal, Barbara Arsenault, Jesse Brock, Michele Johnstone, and Linda Otterson for MICs, Eurofins Panlabs Taiwan, Ltd., for MMP data, Camil Joubran and Sharon Tentarelli for generating characterization data.
Glossary
Abbreviations
- MMP
matrix metalloprotease
- UDP
uridine diphosphate
- MIC
minimum inhibitory concentration
- THP
tetrahydropyran
- LDA
lithium diisopropylamide
- THF
tetrahydrofuran
- MeCN
acetonitrile
- DCE
1,2-dichloroethane
- DMF
N,N-dimethylformamide
- DCM
dichloromethane
- PTSA
para-toluenesulfonic acid
- TMEDA
N,N,N′,N′-tetramethylethylenediamine
- DMAP
4-(dimethylamino)pyridine
- DIPEA
N,N-diisopropylethylamine
- DMA
N,N-dimethylacetamide
- NT
not tested
Supporting Information Available
Biochemical P. aeruginosa LpxC assay; cloning, expression, and purification of LpxC enzyme; crystallization, X-ray data collection, and structure determination; and compound synthesis. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Protein data bank codes are as follows; compound 2, 4U3B; compound 9, 4U3D; UDP-3-O-(acyl)-glucosamine bound cocrystal structure in A. aeolicus LpxC, 4OZE.
Author Present Address
§ Moderna Therapeutics, Inc., 200 Technology Square, Cambridge, MA 02139, United States.
Author Present Address
∞ Semichem, Inc., Shawnee, Kansas 66216, United States.
Author Contributions
All authors have given approval to this manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Giamarellou H.; Poulakou G. Multidrug-resistant Gram-negative infections: what are the treatment options?. Drugs 2009, 69, 1879–1901. [DOI] [PubMed] [Google Scholar]
- Alekshun M. N.; Levy S. B. Molecular mechanisms of antibacterial multidrug resistance. Cell 2007, 128, 1037–1050. [DOI] [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 2007, 6, 29–40. [DOI] [PubMed] [Google Scholar]
- Silver L. L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhavy T. J.; Kahne D.; Walker S. The bacterial cell envelope. Cold Spring Harb Perspect. Biol. 2010, 2, a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver L. L. Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discovery 2007, 6, 41–55. [DOI] [PubMed] [Google Scholar]
- Onishi H. R.; Pelak B. A.; Gerckens L. S.; Silver L. L.; Kahan F. M.; Chen M. H.; Patchett A. A.; Galloway S. M.; Hyland S. A.; Anderson M. S.; Raetz C. R. Antibacterial agents that inhibit lipid A biosynthesis. Science 1996, 274, 980–982. [DOI] [PubMed] [Google Scholar]
- Barb A. W.; Zhou P. Mechanism and inhibition of LpxC: an essential zinc-dependent deacetylase of bacterial lipid A synthesis. Curr. Pharm. Biotechnol. 2008, 9, 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raetz C. R.; Guan Z.; Ingram B. O.; Six D. A.; Song F.; Wang X.; Zhao J. Discovery of new biosynthetic pathways: the lipid A story. J. Lipid Res. 2009, 50SupplS103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szermerski M.; Melesina J.; Wichapong K.; Loppenberg M.; Jose J.; Sippl W.; Holl R. Synthesis, biological evaluation and molecular docking studies of benzyloxyacetohydroxamic acids as LpxC inhibitors. Bioorg. Med. Chem. 2014, 22, 1016–1028. [DOI] [PubMed] [Google Scholar]
- Hale M. R.; Hill P.; Lahiri S.; Miller M. D.; Ross P.; Alm R.; Gao N.; Kutschke A.; Johnstone M.; Prince B.; Thresher J.; Yang W. Exploring the UDP pocket of LpxC through amino acid analogs. Bioorg. Med. Chem. Lett. 2013, 23, 2362–2367. [DOI] [PubMed] [Google Scholar]
- Montgomery J. I.; Brown M. F.; Reilly U.; Price L. M.; Abramite J. A.; Arcari J.; Barham R.; Che Y.; Chen J. M.; Chung S. W.; Collantes E. M.; Desbonnet C.; Doroski M.; Doty J.; Engtrakul J. J.; Harris T. M.; Huband M.; Knafels J. D.; Leach K. L.; Liu S.; Marfat A.; McAllister L.; McElroy E.; Menard C. A.; Mitton-Fry M.; Mullins L.; Noe M. C.; O’Donnell J.; Oliver R.; Penzien J.; Plummer M.; Shanmugasundaram V.; Thoma C.; Tomaras A. P.; Uccello D. P.; Vaz A.; Wishka D. G. Pyridone methylsulfone hydroxamate LpxC inhibitors for the treatment of serious gram-negative infections. J. Med. Chem. 2012, 55, 1662–1670. [DOI] [PubMed] [Google Scholar]
- Brown M. F.; Reilly U.; Abramite J. A.; Arcari J. T.; Oliver R.; Barham R. A.; Che Y.; Chen J. M.; Collantes E. M.; Chung S. W.; Desbonnet C.; Doty J.; Doroski M.; Engtrakul J. J.; Harris T. M.; Huband M.; Knafels J. D.; Leach K. L.; Liu S.; Marfat A.; Marra A.; McElroy E.; Melnick M.; Menard C. A.; Montgomery J. I.; Mullins L.; Noe M. C.; O’Donnell J.; Penzien J.; Plummer M. S.; Price L. M.; Shanmugasundaram V.; Thoma C.; Uccello D. P.; Warmus J. S.; Wishka D. G. Potent inhibitors of LpxC for the treatment of Gram-negative infections. J. Med. Chem. 2012, 55, 914–923. [DOI] [PubMed] [Google Scholar]
- Mansoor U. F.; Vitharana D.; Reddy P. A.; Daubaras D. L.; McNicholas P.; Orth P.; Black T.; Siddiqui M. A. Design and synthesis of potent Gram-negative specific LpxC inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 1155–1161. [DOI] [PubMed] [Google Scholar]
- Liang X.; Lee C. J.; Zhao J.; Toone E. J.; Zhou P. Synthesis, structure, and antibiotic activity of aryl-substituted LpxC inhibitors. J. Med. Chem. 2013, 56, 6954–6966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClerren A. L.; Endsley S.; Bowman J. L.; Andersen N. H.; Guan Z.; Rudolph J.; Raetz C. R. A slow, tight-binding inhibitor of the zinc-dependent deacetylase LpxC of lipid A biosynthesis with antibiotic activity comparable to ciprofloxacin. Biochemistry 2005, 44, 16574–16583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barb A. W.; McClerren A. L.; Snehelatha K.; Reynolds C. M.; Zhou P.; Raetz C. R. Inhibition of lipid A biosynthesis as the primary mechanism of CHIR-090 antibiotic activity in Escherichia coli. Biochemistry 2007, 46, 3793–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barb A. W.; Jiang L.; Raetz C. R.; Zhou P. Structure of the deacetylase LpxC bound to the antibiotic CHIR-090: Time-dependent inhibition and specificity in ligand binding. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 18433–18438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher J. F.; Mobashery S. Recent advances in MMP inhibitor design. Cancer Metastasis Rev. 2006, 25, 115–136. [DOI] [PubMed] [Google Scholar]
- Reiter L. A.; Robinson R. P.; McClure K. F.; Jones C. S.; Reese M. R.; Mitchell P. G.; Otterness I. G.; Bliven M. L.; Liras J.; Cortina S. R.; Donahue K. M.; Eskra J. D.; Griffiths R. J.; Lame M. E.; Lopez-Anaya A.; Martinelli G. J.; McGahee S. M.; Yocum S. A.; Lopresti-Morrow L. L.; Tobiassen L. M.; Vaughn-Bowser M. L. Pyran-containing sulfonamide hydroxamic acids: potent MMP inhibitors that spare MMP-1. Bioorg. Med. Chem. Lett. 2004, 14, 3389–3395. [DOI] [PubMed] [Google Scholar]
- Weisburger J. H.; Weisburger E. K. Biochemical formation and pharmacological, toxicological, and pathological properties of hydroxylamines and hydroxamic acids. Pharmacol. Rev. 1973, 25, 1–66. [PubMed] [Google Scholar]
- Gennadios H. A.; Whittington D. A.; Li X.; Fierke C. A.; Christianson D. W. Mechanistic inferences from the binding of ligands to LpxC, a metal-dependent deacetylase. Biochemistry 2006, 45, 7940–7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernick M.; Gennadios H. A.; Whittington D. A.; Rusche K. M.; Christianson D. W.; Fierke C. A. UDP-3-O-((R)-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase functions through a general acid-base catalyst pair mechanism. J. Biol. Chem. 2005, 280, 16969–16978. [DOI] [PubMed] [Google Scholar]
- Liang X.; Lee C. J.; Chen X.; Chung H. S.; Zeng D.; Raetz C. R.; Li Y.; Zhou P.; Toone E. J. Syntheses, structures and antibiotic activities of LpxC inhibitors based on the diacetylene scaffold. Bioorg. Med. Chem. 2011, 19, 852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. J.; Liang X.; Chen X.; Zeng D.; Joo S. H.; Chung H. S.; Barb A. W.; Swanson S. M.; Nicholas R. A.; Li Y.; Toone E. J.; Raetz C. R.; Zhou P. Species-specific and inhibitor-dependent conformations of LpxC: implications for antibiotic design. Chem. Biol. 2011, 18, 38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. J.; Liang X.; Gopalaswamy R.; Najeeb J.; Ark E. D.; Toone E. J.; Zhou P. Structural basis of the promiscuous inhibitor susceptibility of Escherichia coli LpxC. ACS Chem. Biol. 2014, 9, 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair J. M.; Piddock L. J. Structure, function and inhibition of RND efflux pumps in Gram-negative bacteria: an update. Curr. Opin. Microbiol. 2009, 12, 512–519. [DOI] [PubMed] [Google Scholar]
- O’Shea R.; Moser H. E. Physicochemical properties of antibacterial compounds: implications for drug discovery. J. Med. Chem. 2008, 51, 2871–2878. [DOI] [PubMed] [Google Scholar]
- Nikaido H.; Vaara M. Molecular basis of bacterial outer membrane permeability. Microbiol. Rev. 1985, 49, 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuccinardi T.; Nuti E.; Ortore G.; Rossello A.; Avramova S. I.; Martinelli A. Development of a receptor-based 3D-QSAR study for the analysis of MMP2, MMP3, and MMP9 inhibitors. Bioorg. Med. Chem. 2008, 16, 7749–7758. [DOI] [PubMed] [Google Scholar]
- Pirard B.; Matter H. Matrix metalloproteinase target family landscape: a chemometrical approach to ligand selectivity based on protein binding site analysis. J. Med. Chem. 2006, 49, 51–69. [DOI] [PubMed] [Google Scholar]
- Lukacova V.; Zhang Y.; Kroll D. M.; Raha S.; Comez D.; Balaz S. A comparison of the binding sites of matrix metalloproteinases and tumor necrosis factor-alpha converting enzyme: implications for selectivity. J. Med. Chem. 2005, 48, 2361–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovejoy B.; Welch A. R.; Carr S.; Luong C.; Broka C.; Hendricks R. T.; Campbell J. A.; Walker K. A.; Martin R.; Van Wart H.; Browner M. F. Crystal structures of MMP-1 and -13 reveal the structural basis for selectivity of collagenase inhibitors. Nat. Struct. Biol. 1999, 6, 217–221. [DOI] [PubMed] [Google Scholar]
- Olivier N.; Miller M. D. PDB 4OZE. www.rcsb.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.