

Abstract
Antagonism of αvβ6 is emerging as a potential treatment of idiopathic pulmonary fibrosis based on strong target validation. Starting from an αvβ3 antagonist lead and through simple variation in the nature and position of the aryl substituent, the discovery of compounds with improved αvβ6 activity is described. The compounds also have physicochemical properties commensurate with oral bioavailability and are high quality starting points for a drug discovery program. Compounds 33S and 43E1 are pan αv antagonists having ca. 100 nM potency against αvβ3, αvβ5, αvβ6, and αvβ8 in cell adhesion assays. Detailed structure activity relationships with these integrins are described which also reveal substituents providing partial selectivity (defined as at least a 0.7 log difference in pIC50 values between the integrins in question) for αvβ3 and αvβ5.
Keywords: Integrins, antagonist, pulmonary, fibrosis, αvβ6, αvβ3, β-amino acids
Once diagnosed, life expectancy for patients with idiopathic pulmonary fibrosis (IPF) is usually only a few years and the mortality rate exceeds that of many cancer indications.1,2 A recent study in the U.K. also suggests that the incidence of IPF is rising, with >5,000 new cases diagnosed each year.3 Although pirfenidone is now marketed for the treatment of IPF in some countries,4 there remains an urgent need for effective new medicines. Recent research into IPF together with estimated peak sales for an effective treatment at around $2 billion per annum have driven significant commercial activity around potential biological and small molecule fibrosis assets in the pharmaceutical sector.5
There is now reasonable evidence that the RGD (arginine–glycine–aspartic acid) integrin receptor αvβ6 may play an important role in the initiation and progression of IPF inter alia.6,7 This receptor is predominantly expressed in injured lung tissue, and inhibition of the receptor with αvβ6 antibodies or its absence in knockout mice leads to substantial protection from the development of fibrosis in both bleomycin8 and radiation9 induced animal models. Selective αvβ6 antibodies are in development for fibrotic diseases.5
The αvβ6 receptor is a cell surface heterodimeric receptor composed of a noncovalently bound complex between the αv and β6 proteins. Three closely related integrins where only the β subunit is varied are αvβ3, αvβ5, and αvβ8. Despite substantial research and drug discovery over the past decade on antagonists of the αvβ3 receptor for osteoporosis and other indications,10−13 there is remarkably little literature on potent and selective small molecule antagonists of αvβ6. The selectivity profiles of the αvβ3 antagonists in the literature rarely include cross-screening data against αvβ6 or other αv integrins, with the exception of a Pfizer series of compounds for which a broad range of integrin data is reported (although the potencies are quite weak as αvβ6 antagonists).14 Other exceptions are the Merck KGa,15 Merck AG,16 Merck,17 and Monsanto18 compounds (1–4, Figure 1) and JNJ-2607671319 (where the Arg guanidine and Asp acid mimetics are apparent at either end of a chain) and very recently αv integrin data in patents from Ruminski and Griggs at the University of St. Louis.20
Figure 1.

Exemplar αv integrin antagonists from the literature together with selected reported activities and activities in the assays described herein.
We describe here detailed structure activity relationship (SAR) studies of novel analogues of 4 describing for the first time their profiles against αvβ3, αvβ5, αvβ6, and αvβ8 in cell adhesion assays. By varying the substituent pattern on the aryl ring, we have discovered pan αv antagonists such as 33S and 43E1 (“pan” defined here as having antagonism against all the αv integrins tested, namely αvβ3, αvβ5, αvβ6, and αvβ8), antagonists with partial selectivity (defined as at least a 0.7 log difference in pIC50 values between the integrins in question) for αvβ3 (such as 27), and partial selectivity for αvβ5 (such as 35). The profiles of antagonists with partial dual selectivity for αvβ3 and αvβ5 (such as the known compounds 4(17) and 42(17)) are also described. Seemingly fairly subtle changes on just the aryl ring of the structures exert a profound effect on the overall selectivity profile, and in this series, it appears considerably more marked than in the Merck KGa series.15 Measured lipophilicity values for compounds (chrom. logD)21 are also provided. (When the sum of chrom. logD plus aromatic ring count is <6, there is an increased likelihood of a compound being more developable as an oral drug.21)
The combination of the αvβ6 activity of these compounds with their physicochemical properties, which are commensurate with potential oral bioavailability, identifies them as high quality starting points for a drug discovery program and among the best currently known in the literature.
Derivatives of ethyl or methyl 3-amino-3-phenylpropanoate 7 were prepared in two steps from the appropriate aldehydes 5 by the Rodionov reaction22 followed by esterification to 7, or were purchased from commercial sources (Scheme 1). Ethyl 3-amino-3-(3-pyridyl)propanoate 10 was made in the same way from 3-formylpyridine. Indane-5-carboxaldehyde was prepared in 38% yield by treatment of indane with hexamethylenetetramine/TFA, as described by Arora et al.23
Scheme 1. Synthesis of 3-Aryl-3-aminopropanoic Ester Intermediates.

Reagents and conditions. (a) malonic acid, ammonium acetate, MeOH or EtOH or iPrOH, reflux; (b) SOCl2, EtOH, −15 °C then reflux; (c) H2 (4 bar), 5% Rh/Al2O3, EtOH, 80 °C; (d) N-(benzyloxycarbonyloxy)succinimide, EtNPri2, CH2Cl2, room temp., 2 h; (e) N,N′-carbonyldiimidazole, THF, room temp., then EtOH; (f) H2 (1 bar), 10% Pd/C, EtOH, room temp., 24 h. All compounds are racemic unless indicated in the text.
Hydrogenation of amino ester hydrochloride 7 (R = H) gave the cyclohexyl derivative 8. The 4-cyano analogue 7 (R = 4-CN) was obtained in low yield after the esterification, owing to a competing Pinner reaction. To circumvent this problem in the case of the 3-cyano analogue, the amino group was protected as the benzyl carbamate (CBZ), followed by esterification under mildly basic conditions and CBZ-group deprotection to give 9.
Amide coupling of (1,8-naphthyridin-2-yl)pentanoic acid 11(17) to amino esters 7 (R = 2-OMe, 3-Me, 3-OCF3, and 3,4-(OMe)2, Scheme 2) gave the corresponding esters 12 in 31–67% yields. Selective hydrogenation of the naphthyridine ring gave the corresponding compounds 13 (R = 2-OMe, 3-Me, 3-OCF3, and 3,4-(OMe)2) (44–96%). For all the other compounds, the tetrahydronaphthyridine pentanoic acid 14(17) could be coupled directly to the amino esters 7–9 to give compounds 13. Hydrolysis of the esters 13 under basic conditions then afforded the target acids (see Table 1 for numbering).
Scheme 2. Synthesis of Integrin Antagonists.

Reagents and conditions. (a) N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide, N-methylmorpholine, 1-hydroxybenzotriazole hydrate, 20 °C; (b) H2 (1 bar), 10% Pd/C, EtOH, 20 °C; (c) 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxide hexafluorophosphate, Et3N, CH2Cl2, room temp.; (d) NaOH(aq), EtOH, 20 °C; (e) LiOH, THF/H2O, 20 °C. All compounds are racemic unless indicated in the text.
Table 1. Activity of Aryl Substituted Analogues in αv Integrin Cell Adhesion Assaysa.
| Compd | R | αvβ6 cell pIC50 | αvβ3 cell pIC50 | αvβ5 cell pIC50 | αvβ8 cell pIC50 | Chrom. log Db | Partial selectivityc for ... |
|---|---|---|---|---|---|---|---|
| 15 | H | 5.7 | 7.0 | 7.1 | 6.1 | 1.56 | αvβ3 and αvβ5 |
| 16 | 2-F | 6.0 | 6.5 | 7.0 | 5.4 | 1.88 | |
| 17 | 2-Cl | <5.0 | 5.7 | 5.8 | <5.0 | 1.92 | |
| 18 | 2-Me | 6.4 | 7.3 | 7.7 | 5.9 | 1.85 | αvβ3 and αvβ5 |
| 19 | 2-OMe | <5.0 | 6.3 | 6.3 | <5.0 | 1.83 | αvβ3 and αvβ5 |
| 4 | 3-F | 6.1 | 7.5 | 7.8 | 5.8 | 1.92 | αvβ3 and αvβ5d |
| 20 | 3-Cl | 6.6 | 7.1 | 7.6 | 6.3 | 2.30 | |
| 21 | 3-CH3 | 6.4 | 6.8 | 7.3 | 6.2 | 1.95 | |
| 22 | 3-OMe | 6.5 | 7.4 | 7.2 | 6.3 | 1.66 | αvβ3 and αvβ5 |
| 23 | 4-F | 6.3 | 7.0 | 7.2 | 6.3 | 1.75 | |
| 24 | 4-Cl | 6.4 | 6.9 | 6.9 | 6.2 | 2.14 | |
| 25 | 4-Me | 6.1 | 7.4 | 7.0 | 6.0 | 1.97 | αvβ3 and αvβ5 |
| 26 | 4-OMe | 6.3 | 7.6 | 7.3 | 6.3 | 1.64 | αvβ3 and αvβ5 |
| 27 | 4-CN | 6.4 | 7.1 | 6.4 | 6.1 | 1.47 | αvβ3 |
| 28 | 4-CF3 | 6.2 | 6.6 | 6.5 | 6.1 | 2.72 | pan |
| 29 | 4-OCF3 | 6.2 | 6.8 | 6.3 | 6.1 | 2.85 | |
| 30 | 4-SO2Me | 6.2 | 6.9 | 6.4 | 6.1 | 1.08 | |
| 31 | 4-Ph | 6.4 | 7 | 6.2 | 6.5 | 3.10 | |
| 32 | 3-CN | 6.6 | 6.7 | 7.4 | 6.0 | 1.67 | αvβ5 |
| 33 | 3-CF3 | 7.0 | 6.7 | 7.4 | 6.8 | 2.73 | |
| 33S | (S)-3-CF3 | 7.1 | 7.0 | 7.2 | 6.9 | 2.72 | pan, αvβ6 lead |
| 33R | (R)-3-CF3 | 5.2 | <5d | 5.7d | <5d | 2.61 | |
| 34 | 3-OCF3 | 6.7 | 6.4 | 7.1 | 6.6 | 3.11 | |
| 35 | 2,3-Cl2 | 5.1 | 5.4 | 6.3 | 5 | 2.73 | αvβ5 |
| 36 | 3,4-Cl2 | 6.7 | 7.3 | 7.0 | 6.7 | 2.62 | pan |
| 37 | 3,5-Cl2 | 6.6 | 6.4 | 6.7 | 6.6 | 2.93 | |
| 37E1 | 3,5-Cl2 (Ent. 1) | 5.2d | <5d | 6.0d | <5 | 2.97 | |
| 37E2 | 3,5-Cl2 (Ent. 2) | 6.8 | 6.9 | 6.6 | 7.0 | 2.80 | pan |
| 38 | 3,4-Me2 | 6.7 | 7.2 | 7.2 | 6.3 | 2.29 | |
| 39 | 3,4-CH2CH2CH2 | 6.8 | 6.9 | 7.1 | 6.4 | 2.52 | |
| 40 | 3,5-Me2 | 6.3 | 6.4 | 6.4 | 6.2 | 2.43 | |
| 41 | 3,4-(OMe)2 | 6.5 | 7 | 7.2 | 6.4 | 1.37 | |
| 42 | 3,4-OCH2O- | 6.6 | 7.8 | 7.9 | 6.1 | 1.47 | αvβ3 and αvβ5d |
| 43 | 3-CF3-4-Cl | 7.0 | 6.5 | 6.9 | 6.6 | 3.1 | pan |
| 43E1 | 3-CF3-4-Cl (Ent. 1) | 7.2 | 6.8 | 7.2 | 6.9 | 3.28 | pan, αvβ6 lead |
| 43E2 | 3-CF3-4-Cl (Ent. 2) | <5 | 5.8e | 6.2e | <5e | 3.25 |
All compounds racemic unless shown. All biological data are means from at least n = 2 and within ±0.42 of the mean. pIC50 values are the negative log of the IC50. The lower limit of the assays is around pIC50 5.
Chromatographic log D; see ref21.
Defined as at least a 0.7 log difference in pIC50 values between the integrins in question;
See ref (17);
Values are n = 1.
For the preparation of the single enantiomers of 33, commercially available assigned (R) and (S) enantiomers of the 3-CF3 amino ester 7 were used. For the enantiomers of 37 and 43, the precursor ethyl esters 13 (R = 3,5-Cl2 and 3-CF3,4-Cl, respectively) were separated by preparative chiral HPLC and then hydrolyzed to afford the target compounds.
The compounds were tested in αvβ6, αvβ3, αvβ5, and αvβ8 cell adhesion assays as previously described.24 Lipophilicity was determined using an HPLC chromatographic logD protocol.21
Preliminary unpublished work led to the selection of template 4 as suitable for exploring αvβ6 activity and in particular investigating what the impact of different aryl substituents might be. Although structure activity relationships (SAR) have been described by Merck for this template,17 these predominantly relate to αvβ3 with little, if any, αvβ6 data published. Data from the present study are presented in Table 1. In order to establish preliminary αv SAR rapidly, fluoro, chloro, methyl, and methoxy analogues were prepared in the ortho, meta, and para positions.
The parent phenyl compound 15 is a micromolar αvβ6 antagonist and substantially more potent against αvβ3 and αvβ5.25,26 In every case, the fluoro, chloro, methyl, and methoxy compounds are similarly more potent against αvβ3 and αvβ5 than αvβ6, with activity against αvβ8 being similar to or less than the αvβ6 values. The αvβ3 and αvβ5 values are generally similar to each other. The superior antagonism against αvβ3 is perhaps unsurprising given the series emanates from one designed as αvβ3 antagonists.
The SARs are idiosyncratic, although there are compounds with approximately 10-fold selectivity for αvβ3 and αvβ5 over αvβ6 and αvβ8, such as R = H (15), m-F (4), m-OMe (22), and p-OMe (26). More intriguingly, perhaps, given the relative lack of such compounds in the literature, some show suggestions of selectivity for αvβ5 with the o-F (16), m-Cl (20), and m-Me (21) having 0.5 log selectivity over αvβ3 and about a log selectivity over αvβ6 and αvβ8. Recent studies suggest there may be a role for αvβ5 antagonists in the treatment of sepsis.27 Based on these data, the next iteration of analogues focused on further monosubstituents in the meta and para positions, as these showed more consistent αvβ6 activity and are more synthetically accessible compared to the ortho analogues.
Data from further mono-para substituents were explored (Table 1, 27–31). Activities against αvβ6 are similar (pIC50 6.1–6.4) despite varying size and electronic properties. In contrast, there is a 10-fold range in activity against αvβ3 (pIC50 6.6–7.6) with a p-OMe 26 showing the most potent activity and a p–CF328 the least, suggesting the electronic properties of the substituent may play a role. A similar pattern is seen with αvβ5 whereas antagonism of αvβ8 is flat and essentially similar to αvβ6. The p-CN analogue 27 suggests some selectivity for αvβ3. However, these data do not indicate monosubstitution in the para position is useful for increasing αvβ6 activity, and so no further analogues were prepared.
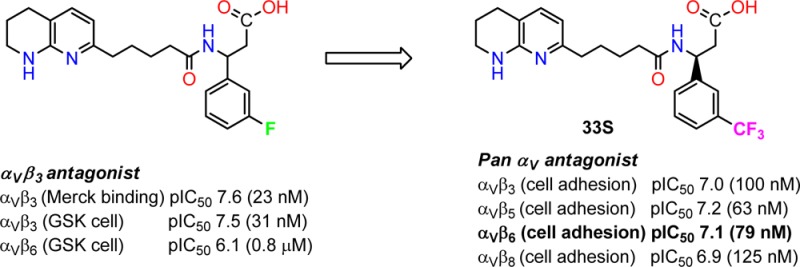
In contrast, monosubstitution in the meta-position (Table 1, 32–34) proved to be more influential on αvβ6 antagonism, with around a 10-fold range of activity (pIC50 6.1–7.1). The size of the substituent is more influential (compare m-F 4 (pIC50 6.1) with m-CF333 (pIC50 7.0)) than electronic properties (compare m-MeO 22 and m-Me 21 with m-CN 32 and m-Cl 20 (pIC50 values of 6.5 and 6.4 vs 6.6 and 6.6, respectively)). The enantiomers of the most potent m-CF3 analogue 33 (racemate pIC50 7.0) were prepared from commercially available pure (S) and (R) enantiomers of 7, thus allowing 33S and 33R to be assigned as the (S) and (R) trifluoromethyl enantiomers, respectively, and with corresponding αvβ6 pIC50 values of 7.1 and 5.2. Given the high sequence homology between αvβ3 and αvβ6, this is consistent with the more potent m-fluoro analogue 4 against αvβ3 also having (S) stereochemistry, as described elswhere.28
An approximately 10-fold range in activity for m-substituents is also seen for αvβ3 (pIC50 6.4–7.5) and αvβ5 (pIC50 7.1–7.8), with antagonist activities generally being greater than those observed for αvβ6. The SAR is idiosyncratic, suggesting that multiple low energy binding conformations may be possible. As seen with the para-substituents, the αvβ8 SAR essentially tracks the αvβ6 SAR.
The m-CN analogue 32 shows an approximate 0.7–1.4 log selectivity for αvβ5 (pIC50 7.4) over the other αv integrins. The m-CF3 analogue 33S is the most potent antagonist against αvβ6 (pIC50 7.1) and equipotent against αvβ3, αvβ5, and αvβ8 (pIC50 7.0, 7.2, and 6.9). Meta-substitution is clearly highly influential on the selectivity profile, and further work is ongoing.
Given the impact of monoaryl substitution on potency and selectivity, we decided to explore also the impact of disubstitution because if there are multiple binding pockets in this region of the active site, then synergistic effects might be observed. As the meta position had proved most sensitive, we preserved substitution at this position while varying the position of the second substituent, selecting analogues (35–43) which could be prepared from readily available starting materials.
A greater range in potency was seen with disubstituted analogues compared with monosubstituted analogues. Preparation of substitution patterns of dichloro analogues gives an interesting range of selectivity profiles. The 2,3-dichloro 35 shows micromolar potency for αvβ5 and 10-fold selectivity over the other αv integrins. The 3,4-dichloro 36 and the 3,5-dichloro 37 restore αvβ6 activity (pIC50 6.7 and 6.6, respectively), with the 3,5 analogue 37 being a pan αv antagonist. Separation of the 3,5-Cl2 enantiomers29 gave αvβ6 pIC50 activities of 6.8 (37E2) and 5.2 (37E1), with 37E2 remaining predominantly a pan antagonist.
As expected, the known 3,4-methylenedioxy analogue 42(17) has approximately nanomolar potency and greater than 10-fold selectivity for αvβ3 and αvβ5 over αvβ6 and αvβ8; it also has increased potency by 0.6–0.7 log units over the 3,4-dimethoxy derivative 41, which is less selective for αvβ3 and αvβ5. The presence of the oxygens is important, as both the corresponding indane 39 and 3,4-dimethyl derivative 38 is almost a log unit less potent against αvβ3 and αvβ5 and are also less selective. The same applies to the 3,4-dichloro analogue 36. The 3-trifluoromethyl-4-chloro analogue 43 is also a pan αv antagonist, with the more active enantiomer 43E1(29) having a similar profile.
Presented here are SAR studies of a series of integrin antagonists against αvβ3, αvβ5, αvβ6, and αvβ8. Although 4 and 42 have previously been described as αvβ3 antagonists,17 the studies described here show a more detailed picture of their profile with both compounds potent against αvβ3 but also being equipotent against αvβ5. The SARs presented here clearly show that, by simple variation of the position and nature of the aryl substituent, the cell adhesion potency against αvβ6 can be increased and, comparatively, potency against αvβ3 and αvβ5 reduced. Comparison of the lead compounds described here (e.g., 33S and 43E1) with the standards 1 and 3 from the literature (cf. Figure 1) shows they have similar αvβ6 activity but with structural features perhaps more commensurate with oral bioavailability properties. Their lipophilicities are reasonable (chrom. logD values of 2.72 for 33S and 3.28 for 43E1), and they possess good permeability and solubility (data not shown). Indeed, analogues of these compounds prepared by Merck have been shown to have good oral bioavailability in dog.17,28
Although a crystal structure of αvβ6 is currently not available, a homology model is available,30 and the αvβ3 crystal structure has been described.31 From the latter, it is known that the RGD motif (or ligand mimetic) binds at the interface between the αv and the β3 (or β6) subunits, with the arginine (or mimetic) binding to the αv unit and the aspartic acid (or mimetic) binding to the β3 (or β6) subunit. There is considerable sequence similarity between the β3 and β6 subunits, so the gross topology is also likely to be similar. Based on this, an extended conformation for these ligands in αvβ6 is likely to be the conformation for antagonism and is also consistent with why small differences in aryl group substitution might have a profound impact on the selectivity between the different αv integrins observed.
Further studies to improve αvβ6 potency are underway. Clearly sufficient potency is required to drive an antifibrotic response at a realistic clinical dose, but a compromise may be required between the ideal potency and ideal selectivity. Although our focus here is IPF, a pan αv antagonist has recently been reported32 as being efficacious in a number of models of fibrotic diseases (see Supporting Information).
Described here are SAR relationships against αvβ3, αvβ5, αvβ6, and αvβ8 with the aim of ultimately identifying an orally bioavailable selective and potent αvβ6 for treating IPF. The lead compounds 33S and 43E1 have been derived from αvβ3 antagonists to pan αv antagonists with antagonisms around pIC50 7 (100 nM). This has been achieved while maintaining physicochemical properties commensurate with oral bioavailability. Compounds which are partially selective for αvβ5 have also been identified. Further studies will be reported in due course.
Acknowledgments
The authors wish to thank Dr. Simon Peace for helpful discussions and the following project students whose compounds are not described here: Christine Butcher, Adam Eason, Nick Herbert, Dominic Howgego, William Restorick, and Jack Sorrell.
Supporting Information Available
Experimental details and spectroscopic data for the compounds described in this paper. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
All authors have given approval to the final version of the manuscript.
M.J.F. holds a University of Nottingham Teaching Fellowship funded by GSK, who also provided funds for consumables associated with this work.
The authors declare no competing financial interest.
Notes
The results in this paper are from fourth year undergraduate research projects at the University of Nottingham in collaboration with GlaxoSmithKline with the aim of giving students experience of industrial medicinal chemistry. The selection of the targets and the syntheses were carried out predominantly by the students under the guidance of the last three authors.
Supplementary Material
References
- Vancheri C.; Failla M.; Crimi N.; Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur. Respir. J. 2010, 35, 496–504. [DOI] [PubMed] [Google Scholar]
- Du Bois R. M. Strategies for treating idiopathic pulmonary fibrosis. Nat. Rev. Drug Discovery 2010, 9, 129–140. [DOI] [PubMed] [Google Scholar]
- Navaratnam V.; Fleming K. M.; West J.; Smith C. J. P.; Jenkins R. G.; A Fogarty A.; Hubbard R. B. The rising incidence of idiopathic pulmonary fibrosis in the UK. Thorax 2011, 66, 462–467. [DOI] [PubMed] [Google Scholar]
- Jenkins G. Pirfenidone should be prescribed for patients with idiopathic pulmonary fibrosis. Thorax 2013, 68, 603–605. [DOI] [PubMed] [Google Scholar]
- Allison M. Stromedix acquisition signals growing interest in fibrosis. Nat. Biotechnol. 2012, 30, 375–376. [DOI] [PubMed] [Google Scholar]
- Henderson N. C.; Sheppard D. Integrin mediated regulation of TGFβ in fibrosis. Biochim. Biophys. Acta 2013, 1832, 891–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger J. S.; Huang X.; Kawakatsu H.; Griffiths M. J.; Dalton S. L.; Wu J.; Pitter J. F.; Kaminski N.; Garat C.; Matthay M. A.; Rifkin D. B.; Sheppard D. The integrin αvβ6 binds and activates latent TGFβ: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [DOI] [PubMed] [Google Scholar]
- Horan G. S.; Wood S.; Ona V.; Li D. J.; Lukashev M. E.; Weinreb P. H.; Simon K. J.; Hahm K.; Allaire N. E.; Rinaldi N. J. Partial inhibition of integrin αvβ6 prevents pulmonary fibrosis without exacerbating inflammation. Am. J. Respir. Crit. Care Med. 2008, 177, 56–65. [DOI] [PubMed] [Google Scholar]
- Puthawala K.; Hadjiangelis N.; Jacoby S. C.; Bayongan E.; Zhao Z.; Yang Z.; Devitt M. L.; Horan G. S.; Weinreb P. H.; Lukashev M. E. Inhibition of integrin αvβ6, an activator of latent transforming growth factor-β, prevents radiation-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdih A.; Dolenc M. S. Small molecule antagonists of integrin receptors. Curr. Med. Chem. 2010, 17, 2371–2392. [DOI] [PubMed] [Google Scholar]
- Sheldrake H. M.; Patterson L. H. Strategies to inhibit tumour associated integrin receptors: rationale for dual and multi antagonists. J. Med. Chem. 2014, 57, 6301–6315. [DOI] [PubMed] [Google Scholar]
- Goswami S. Importance of integrin receptors in the field of pharmaceutical and medical science. Adv. Biol. Chem. 2013, 3, 224–252. [Google Scholar]
- Goodman S. L.; Picard M. Integrins as therapeutic targets. Trends Pharmacol. Sci. 2012, 33, 405–412. [DOI] [PubMed] [Google Scholar]
- Nagarajan S. R.; Lu H.-F.; Gasiecki A. F.; Khanna I. K.; Parikh M. D.; Desai B. N.; Rogers T. E.; Clare M.; Chen B. B.; Russell M. A.; Keene J. L.; Duffin T.; Engleman V. W.; Finn M. B.; Freeman S. K.; Klover J. A.; Nickols G. A.; Nickols M. A.; Shannon K. E.; Steininger C. A.; Westlin W. F.; Westlin M. M.; Williams M. L. Discovery of (+)-(2-{4-[2-(5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl)ethoxy]phenyl}-cyclopropyl)acetic acid as potent and selective αvβ3 inhibitor: Design, synthesis, and optimization. Bioorg. Med. Chem. 2007, 15, 3390–3412and references therein. [DOI] [PubMed] [Google Scholar]
- Goodman S. L.; Holzemann G.; Sulyok G. A. G.; Kessler H. Nanomolar small molecule inhibitors for αvβ6, αvβ5 and αvβ3 integrins. J. Med. Chem. 2002, 45, 1045–1051. [DOI] [PubMed] [Google Scholar]
- Popov Y.; Patsenker E.; Stickel F.; Zaks J.; Bhaskar K. R.; Niedobitek G.; Kolb A.; Friess H.; Schuppan D. Integrin αvβ6 is a marker of the progression of biliary and portal liver fibrosis and a novel marker for antifibrotic therapies. J. Hepatol. 2008, 48, 453–464. [DOI] [PubMed] [Google Scholar]
- Coleman P. J.; Askew B. C.; Hutchinson J. H.; Whitman D. B.; Perkins J. J.; Hartman G. D.; Rodan G. A.; Leu C.-T.; Prueksaritanont T.; Fernandez-Metzler C.; Merkle K. M.; Lynch R.; Lynch J. L.; Rodan S. R.; Duggan M. E. Non-peptide αvβ3 antagonists. Part 4: potent and orally bioavailable chain-shortened RGD mimetics. Bioorg. Med. Chem. Lett. 2002, 12, 2463–2465. [DOI] [PubMed] [Google Scholar]
- Carron C. P.; Meyer D. M.; Pegg J. A.; Engleman V. W.; Nickols M. A.; Settle S. L.; Westlin W. F.; Ruminski P. G.; Nickols G. A. A peptidomimetic antagonist of the integrin αvβ3 inhibits Leydig cell tumor growth and the development of hypercalcemia of malignancy. Cancer Res. 1998, 58, 1930–1935. [PubMed] [Google Scholar]
- Santulli R. J.; Kinney W. A.; Ghosh S.; DeCorte B. L.; Li L.; Tuman R. W. A.; Zhou Z.; Huebert N.; Bursell S. E.; Clermont A. C.; Grant M. B.; Shaw L. C.; Mousa S. A.; Galemmo R. A. Jr; Johnson D. L.; Maryanoff B. E.; Damiano B. P. Studies with an orally bioavailable αV integrin antagonist in animal models of ocular vasculopathy: retinal neovascularization in mice and retinal vascular permeability in diabetic rats. J. Pharmacol. Exp. Ther. 2008, 324, 894–901. [DOI] [PubMed] [Google Scholar]
- Ruminski P. G.; Griggs D. W.. Preparation of 3,5-phenyl substituted beta amino acid derivatives as integrin receptor antagonists. US patent 2014, US 20140051715.; Ruminski P. G.; Griggs D. W.. Preparation of N-glycyl-beta amino acid derivatives as integrin antagonists. PCT Int. Appl. 2014, WO 2014015054.
- Young R. J.; Green D. V.; Luscombe C. N.; Hill A. P. Getting physical in drug discovery II: the impact of chromatographic hydrophobicity measurements and aromaticity. Drug Discovery Today 2011, 16, 822–830. [DOI] [PubMed] [Google Scholar]
- Rodionov V. M.; Bezinger N. N. New synthesis of alkyl esters of β-amino acids. Izv. Akad. Nauk SSSR 1952, 696–702. [Google Scholar]
- Arora S. K.; Banerjee R.; Kamboj R. K.; Loriya R.; Mathai S.; Joshi M.; Suthar R.; Cheerlavancha R.; Gote G.; Bagul R.; Wetal R.; Patel S.; Dixit R.; Waghchowe A.; Goel R.; Seedhara S. K. H.. Preparation of substituted benzylidenethiazolamines as novel protein tyrosine phosphatase-1B inhibitors. PCT Int. Appl. 2009, WO2009109999.
- Ludbrook S. B.; Barry S. T.; Chris J. Delves C. J.; Horgan C. M. T. The integrin αvβ3 is a receptor for the latency-associated peptides of transforming growth factors β1 and β3. Biochem. J. 2003, 369, 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Based on an analysis of the data from the assays used here, about a 0.5 log unit difference in activity should be regarded as a significant difference.
- Analogues where the phenyl ring is replaced with a 3-pyridyl derived from 10 or a cyclohexyl ring derived from 8 have little or no activity against αvβ6.
- Su G.; Atakilit A.; Li J. T.; Wu N.; Luong J.; Chen R.; Bhattacharya M.; Sheppard D. Effective treatment of mouse sepsis with an inhibitory antibody targeting integrin αvβ5. Crit. Care Med. 2013, 41, 546–553. [DOI] [PubMed] [Google Scholar]
- Coleman P. J.; Brashear K. M.; Askew B. C.; Hutchinson J. H.; McVean C. A.; Duong L. T.; Feuston B. P.; Fernandez-Metzler C.; Gentile M. A.; Hartman G. D.; Kimmel D. B.; Leu C.-T.; Lipfert L.; Merkle K.; Pennypacker B.; Prueksaritanont T.; Rodan G. A.; Wesolowski G. A.; Rodan S. B.; Duggan M. E. Nonpeptide αvβ3 Antagonists. Part 11: discovery and preclinical evaluation of potent αvβ3 antagonists for the prevention and treatment of osteoporosis. J. Med. Chem. 2004, 47, 4829–4837. [DOI] [PubMed] [Google Scholar]
- These racemates were separated as the corresponding esters and then hydrolyzed (see Supporting Information). By analogy with 33S and 33R, it is likely that the more active enantiomers have (S) stereochemistry.
- Bochen A.; Marelli U. K.; Otto E.; Pallarola D.; Mas-Moruno C.; Di Leva F. S.; Boehm H.; Spatz J. P.; Novellino E.; Kessler H.; Marinelli L. Biselectivty of isoDGR peptides for fibronectin binding integrin subtypes α5β1 and αvβ6: conformational control through flanking amino acids. J. Med. Chem. 2013, 56, 1509–1519. [DOI] [PubMed] [Google Scholar]; Detailed molecular modeling studies to rationalize the observed results have not yet been carried out. The residues in the β6 and β3 subunits where the aryl ring binds differ, and the homology model in the above reference is used to rationalize the affinities of different cyclic peptides. Thus, understanding tenfold differences in the β6 and β3 affinities of aryl substituents may not be straightforward. However, brief notes for modeling representative compounds in the known αvβ3 crystal structure are given in the Supporting Information.
- Springer T. A.; Dustin M. L. Integrin inside-out signaling and the immunological synapse. Curr. Opin. Cell Biol. 2012, 24, 107–115and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson N. C.; Arnold T. D.; Katamura Y.; Giacomini M. M.; Rodriguez J. D.; McCarty J. H.; Pellicoro A.; Raschperger E.; Betsholtz C.; Ruminski P. G.; Griggs D. W.; Prinsen M. J.; Maher J. J.; Iredale J. P.; Lucy-Hulbert A.; Adams R. H.; Sheppard D. Targeting of αv integrin indentifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.