

Abstract
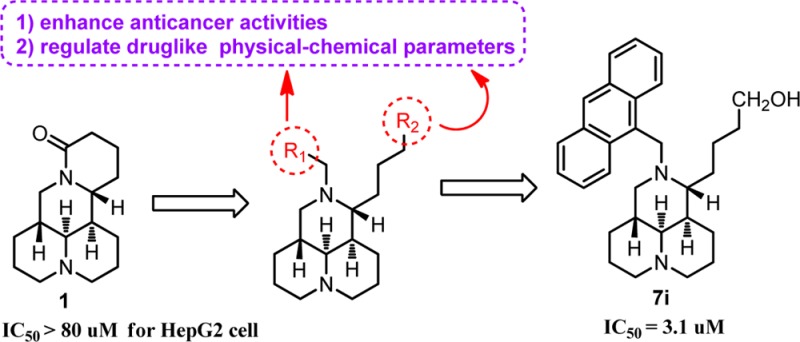
New N-substituted sophoridinic acid/ester and sophoridinol derivatives were synthesized and evaluated for their cytotoxic activity in human HepG2 hepatoma cells from the lead sophoridine (1). Among the newly synthesized compounds, sophoridinol 7i displayed a potential antiproliferative activity with an IC50 of 3.1 μM. Importantly, it exerted an almost equipotent effect against both wild MCF-7 and adriamycin (AMD)-resistant MCF-7 (MCF-7/AMD) breast carcinoma cell lines. Its mode of action was to arrest the cell cycle at the G0/G1 phase, consistent with that of the parent 1. In addition, compound 7i also showed a reasonable ClogP value and favorable pharmacokinetic property with an area under the concentration–time curve (AUC) of 10.3 μM·h in rats, indicating an ideal druggable characteristic. We consider sophoridinol derivatives to be a novel family of promising antitumor agents with an advantage of inhibiting drug-resistant cancer cells.
Keywords: Sophoridine, sophoridinol, structure−activity relationship, antiproliferative, drug resistance
The Chinese traditional medicine Fufang Kushen injection, which was approved by Chinese FDA (CFDA) in 1995 as an anticancer drug, has been widely used to treat nonsmall cell lung carcinoma, live cancer, and gastric cancer in combination with other anticancer drugs, such as vinorelbine, cisplatin, and taxol et al.1−6 The main chemical ingredients of Fufang Kushen injection are oxymatrine, matrine, and sophoridine (1, Figure 1),7−9 and all of them are quinolizidine natural products extracted from Sophora flavescens. Compound 1 alone was approved by CFDA in 2005 to cure the cancer patients with malignant trophoblastic tumors. The mechanism of action of 1 is to inhibit the DNA topoisomerase I (topo I) activity, induce cell cycle arrest at the G0/G1 phase, and cause apoptotic cell death.10−13 What’s more, it has many of druggable advantages such as special chemical scaffold, flexibility structure, high solubility, and good safety profiles, suggesting that it is an ideal lead compound for further modifications and optimizations.14−16
Figure 1.
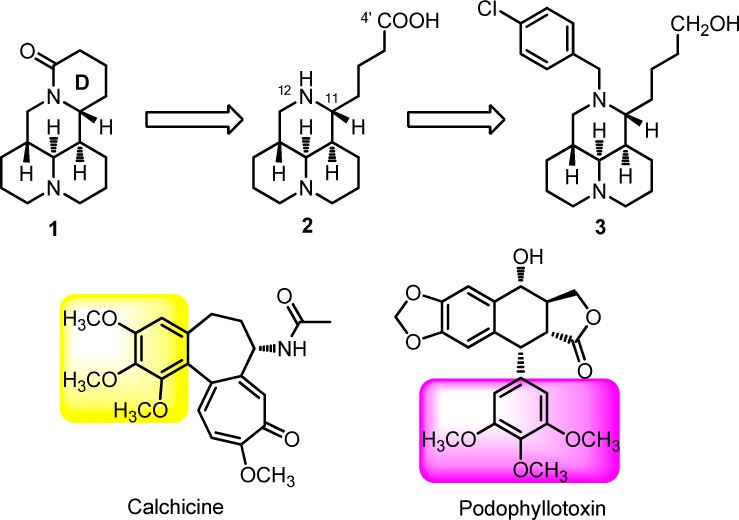
Chemical structures of sophoridine (1), sophoridinic acid (2), 12-N-p-chlorobenzyl sophoridinol (3), colchicine, and podophyllotoxin.
The structure–activity relationship (SAR) study of 1 as anticancer agent was carried out with the modifications of D ring opening in our laboratory,17,18 and SAR results revealed that sophoridinic acid (2, Figure 1) analogues with a 3-ring core scaffold was more favorable than 1 with a 4-ring scaffold. The representative compound 3 (Figure 1), 12-N-p-chlorobenzyl sophoridinol, had a greatly improved antiproliferative activity in HepG2 hepatoma cells with an IC50 of 9.3 μM compared to the parent 1 (IC50 > 80 μM).17 The mode of action of 3 was to inhibit DNA topo I activity and arrested the cell cycle at the G0/G1 phase, consistent with that of 1.17 The unique chemical scaffold and promising anticancer activity of 3 provoked our strong interest to continue SAR analysis in an effort to develop a novel class of promising anticancer candidates with high druggability.
In the present study, a variety of substituents were introduced on the 12-nitrogen atom and 4′-carboxyl regions of compound 2, respectively, from which 27 new sophoridinic acid/ester and sophoridinol derivatives were generated and examined for their anticancer activities. Herein, we describe the synthesis, in vitro antitumor assay, SAR analysis, primary mechanism of action, and in vivo pharmacokinetic (PK) evaluation of the new compounds.
Twenty-seven target compounds were prepared using commercially available 1 as the starting material as described in Scheme 1. The sophoridinic ester 5 was obtained through a four-step procedure including hydrolysis, esterization, condensation, and reduction reactions as reported previously.17 The sophoridinic acid 6 was acquired by the hydrolysis of 5 in 3 N HCl in a good yield, while the sophoridinol 7 was obtained via reduction of 5 with LiAlH4 in THF with a yield of over 80%. The sophoridinic ketone 8 was synthesized via addition-hydrolysis reaction of 5 with an equivalent of p-Cl-PhMgBr in dry THF at room temperature in a 77% yield. Similarly, the products in series 9 were also gained through the addition-hydrolysis reaction of 5 with 4–5 equiv of Grignard reagents in THF at refluxing temperature with yields of 57%–69%. The product 10 was acquired by the dehydration of 9 in a 63% yield. All crude products were purified with flash column chromatography on silica gel using CH2Cl2 and MeOH/NH3·H2O as gradient eluents.
Scheme 1.
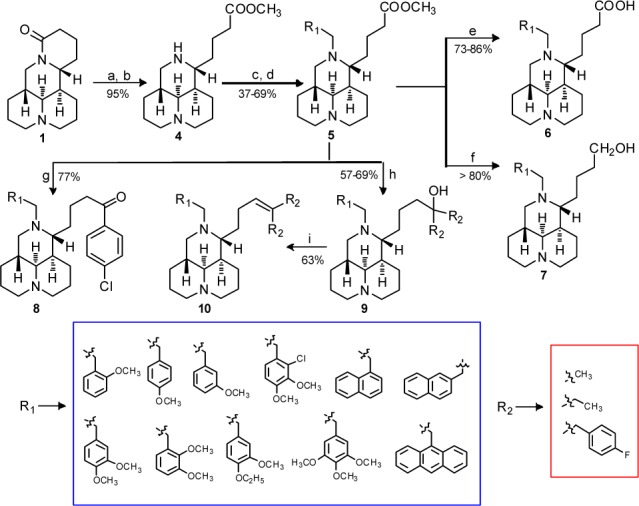
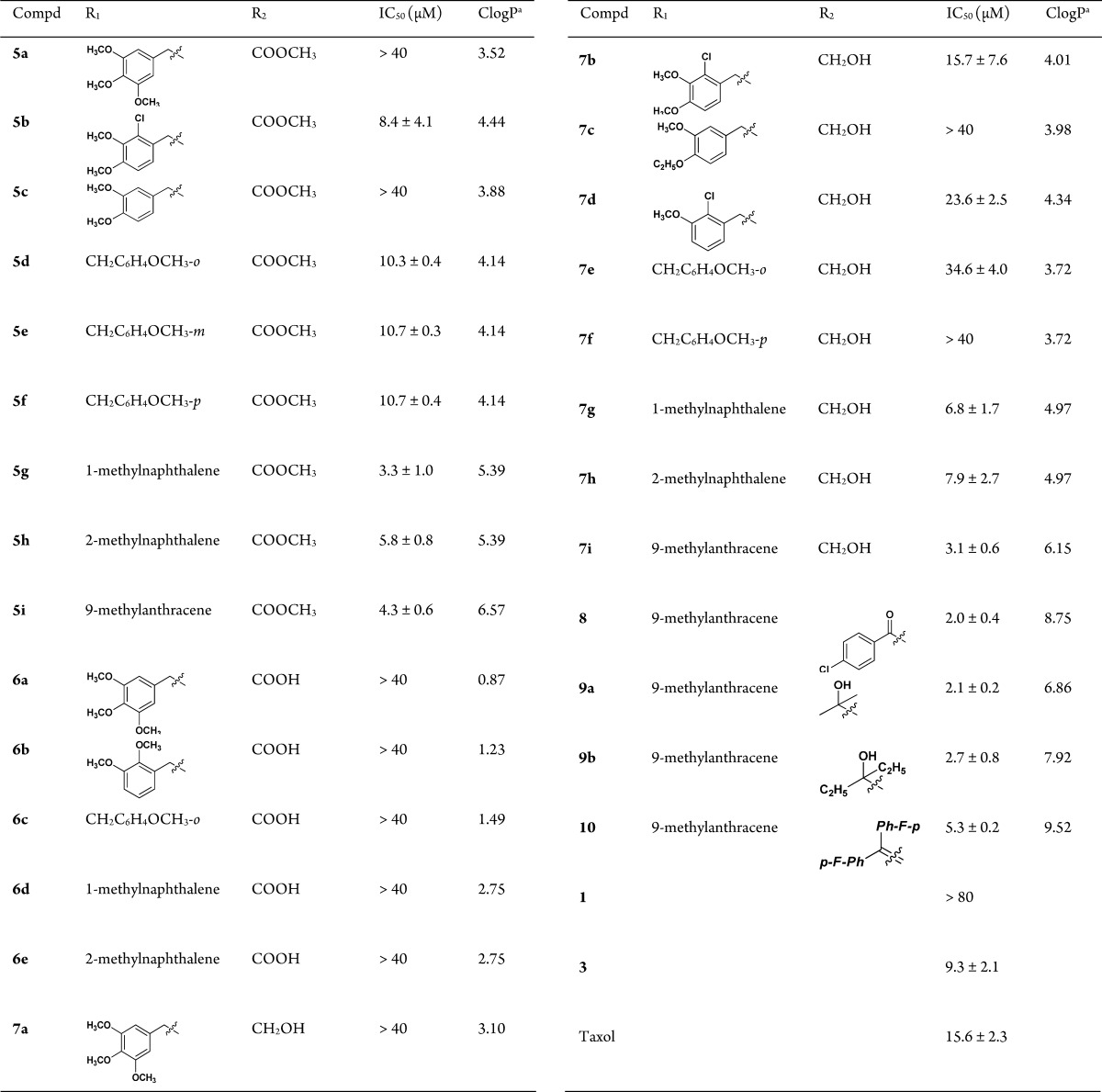
All the desired compounds were evaluated for their cytotoxic activities in human HepG2 hepatoma cell lines using MTT assay with Taxol as the positive control.19 Structures of the 27 sophoridinic acid/ester and sophoridinol derivatives and their antiproliferative activities were depicted in Table 1.
Table 1. Structure–Activity Relationships of the Newly Synthesized Compounds for Their Antiproliferative Activities in HepG2 Cells.
ClogP value was generated by ChemBioOffice software (version 12.0).
SAR analysis was first focused on the substituents at the 12-nitrogen atom. Since trimethoxyphenyl group (Figure 1) is crucial for the anticancer activity in many natural medicines against cancer such as colchicine and podophyllotoxin, this group was introduced into the nitrogen atom at position 12 to create N-trimethoxybenzyl sophoridinic ester/acid (5a and 6a) and sophoridinol (7a). However, as described in Table 1, none of them was active, indicating that the trimethoxyphenyl herein was not beneficial for the anticancer activity. Next, mono/dimethoxyphenyl groups were employed as substituents at 12-N position to generate corresponding new derivatives (5b–f, 6b–c, and 7b–f). The results showed that most of the sophoridinic acids/alcohols lost their cytotoxic activities partially or completely, while the sophoridinic esters 5b and 5d–f afforded moderate antiproliferative activities with IC50 values ranging from 8.4 to 10.7 μM. It seemed that the improved anticancer activities of compounds 5b and 5d–f were consistent with their relatively higher CLogP values (>4) calculated by ChemBioOffice software (version 12.0).
Then, naphthalenyl and anthracenyl with high lipophilicity were added on the 12-nitrogen atom aiming at enhancing the ClogP values, thereby enhancing the activity against tumor, with which many new corresponding compounds (5g–i, 6d–e, and 7g–i) were made and examined. As anticipated, the sophoridinic esters (5g–i) and sophoridinols (7g–i) displayed higher potency with IC50 values ranging from 3.1 to 7.9 μM, much better than that of Taxol (IC50 = 15.6 μM). Especially, compound 7i with a 9-anthracene at the 12-N position exhibited a potent activity against HepG2 cancer with an IC50 of 3.1 μM. Subsequently, the 9-anthracenyl on the 12-nitrogen atom was retained, and a couple of sophoridinic ketone (8), sophoridinols (9a–b), and sophoridinic alkene (10) were constructed and measured. The results showed that all of them had favorable antiproliferative activities with IC50 between 2.0 and 5.3 μM owing to the improvement of ClogP values ranging from 6.8 and 9.5. Among the active compounds, sophoridinic esters 5g–i had metabolic instabilities in vivo owing to the metabolically labile ester group. Therefore, sophoridinols 7g and 7i bearing potent anticancer effects as well as reasonable ClogP values were selected out as the representative compounds for further investigation.
Evaluation of compounds 7g and 7i against drug-resistant cancer cell lines were carried out. In this experiment, we tested their activity against human wild MCF-7 and adriamycin (AMD)-resistant MCF-7 (MCF-7/AMD) breast carcinoma cell lines,20 with AMD as a reference control. As described in Table 2, AMD exhibited a promising activity against wild MCF-7 with an IC50 of 0.88 μM, compared with that of 94 μM in the MCF-7/AMD cells. Notably, compounds 7g and 7i afforded an almost equipotent antiproliferative effect against both MCF-7 (IC50 of 9.0 and 5.6 μM) and drug-resistance MCF-7/AMD cells (IC50 of 9.5 and 5.9 μM), suggesting a different anticancer mechanism of action from AMD.
Table 2. IC50 (μM) Values of 7g and 7i in MCF-7 and MCF-7/AMD Breast Carcinoma Cell Lines.
| MCF-7 | MCF-7/AMD | |
|---|---|---|
| 7g | 9.07 ± 1.22 | 9.51 ± 1.85 |
| 7i | 5.63 ± 0.33 | 5.94 ± 1.07 |
| AMD | 0.88 ± 0.15 | 94.1 ± 7.79 |
To verify the possible change of mechanism of action after the structure modifications, flow cytometric analysis in the HepG2 cells was carried out to learn whether or not compound 7i is able to cause G0/G1 arrest. The HepG2 cells were treated for 24 h without or with 7i at the different concentrations of 1.25, 2.5, and 5.0 μg/mL, respectively. As shown in Figure 2, compound 7i arrested the HepG2 cells at the G0/G1 phase as anticipated, indicating a similar mechanism of action with its parent compound 1.
Figure 2.

Cell cycle analysis of 7i. HepG2 cells were incubated without (control) or with compounds at different concentrations (1.25, 2.5, and 5.0 μg/mL) for 24 h. Cells were then analyzed for their cell cycle distribution using flow cytometry. (*) P < 0.05; (**) P < 0.01 as compared to that of control group.
Single dose PK investigation for compound 7i was performed in adult male Sprague–Dawley (SD) rats. Compound 7i was administered via oral routes (25 mg/kg), and nine blood samples (0.3 mL) were collected over a 24 h period. As described in Figure 3 and Table 3, the compound 7i was rapidly absorbed with a Tmax of 4.6 h, a favorable half-life of 12 h, and a mean residence time (MRT) of 5.4 h. The area under the concentration–time curve (AUC) of 7i was 10.3 μM·h, which is greater than the IC50 value (3.1 μM) of antiproliferative activity in vitro. The PK profiles of 7i were not as good as those of compound 3,17 and this might be due to the higher ClogP value (ClogP = 6.15) of this compound.
Figure 3.

Mean plasma concenreation-time curve of compound 7i after single oral administration of 25 mg/kg in SD rats (n = 3). Pharmacokinetic parameters were calculated by noncompartmental analysis using WinNonlin, version 5.3.
Table 3. Pharmacokinetic Profilea of 7i in Rats after a Single Oral Administration (n =3).
| dosage (mg/kg) | Tmax (h) | Cmax (μM) | AUC0–t (μM·h) | AUC0–∞ (μM·h) | MRT (h) | t1/2 (h) |
|---|---|---|---|---|---|---|
| 25 | 4.6 ± 1.2 | 3.4 ± 0.5 | 10.3 ± 1.4 | 10.5 ± 1.2 | 5.4 ± 0.1 | 12.7 ± 0.3 |
PK parameters were calculated by noncompartmental analysis using WinNonlin, version 5.3.
Taken together, 27 new sophoridinic acid/ester and sophoridinol derivatives were constructed and evaluated for their antitumor activities in vitro. SAR analysis revealed that (i) two suitable substituents on the 12-nitrogen atom and carboxyl region were helpful for keeping good antitumor activity. (ii) The two moieties could be introduced decorating various substituents in order to regulate the druglike physical–chemical properties of the compounds. The SAR results provide useful information on further strategic optimization. Among them, sophoridinol 7i showed an equipotent effect against wild MCF-7 and MCF-7/AMD breast carcinoma cell lines. Its mechanism of action was to arrest the cell cycle at the G0/G1 phase consistent with that of 1. In addition, it also showed a good PK profile in vivo, indicating a good druggable characteristic. Thus, we consider sophoridinol analogues to be a new family of promising antitumor agents with an advantage of inhibiting drug-resistant cancer cells.
Glossary
Abbreviations
- SAR
structure–activity relationship
- MTT
tetrazolium bromide
- AMD
adriamycin
- THF
tetrahydrofuran
- TLC
thin layer chromatography
Supporting Information Available
Synthetic procedure, analytical data, cytotoxicity assay, cell cycle analysis, and PK evaluation. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ (C.B. and C.Z.) These authors contributed equally to this work.
This work was supported by the National Mega-Project for Innovation Drugs (2012ZX09301002-001-017) and Student Fund of Innovation Project of Peking Union Medical College.
The authors declare no competing financial interest.
Supplementary Material
References
- Sun M.; Cao H.; Sun L.; Dong S.; Bian Y.; Han J.; Zhang L.; Ren S.; Hu Y.; Liu C.; Xu L.; Liu P. Antitumor activities of kushen: literature review. J. Evidence-Based Complementary Altern. Med. 2012, 2012, 373219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei R.; Yang D. Y.; Jiang W. Z.; Dai Y. Y.; Wan L. Y.; Yang Z. Efficacy of Yanshu injection (a compound Chinese traditional medicine) combined with concurrent radiochemotherapy in patients with stage III nasopharyngeal carcinoma. Zhonghua Zhongliu Zazhi 2011, 33, 391–394. [PubMed] [Google Scholar]
- Wang C. Y.; Bai X. Y.; Wang C. H. Traditional Chinese medicine: a treasured natural resource of anticancer drug research and development. Am. J. Chin. Med. 2014, 42, 543–559. [DOI] [PubMed] [Google Scholar]
- Liu J.; Liu Y. Influence of erbanxiao solution on inhibiting angiogenesis in stasis toxin stagnation of non-small cell lung cancer. J. Tradit. Chin. Med. 2013, 33, 303–306. [DOI] [PubMed] [Google Scholar]
- Guan C. N.; Cai L. Z.; Yue L. Q.; Zhang Y. Clinicel study on treatment of advanced primary liver cancer by Yanshu injection combining with chemotherapy. Zhongguo Zhongyao Zazhi 2006, 31, 510–512. [PubMed] [Google Scholar]
- Sun Q.; Ma W.; Gao Y.; Zheng W.; Zhang B.; Peng Y. Meta-analysis: therapeutic effect of transcatheter arterial chemoembolization combined with compound kushen injection in hepatocellular carcinoma. Afr. J. Tradit. Complementary Altern. Med. 2012, 9, 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Xue M.; Huang X.; Wang S.; Jiang Z.; Zhang L. Pharmacokinetic of four alkaloids of Yanshu injection in Beagel dogs. Zhongguo Zhongyao Zazhi 2012, 37, 1845–1849. [PubMed] [Google Scholar]
- Qi L.; Zhang J.; Zhang Z. Determination of four alkaloids in compound Kushen Injection by high performance liquid chromatography with ionic liquid as mobile phase additive. Sepu 2013, 31, 249–253. [DOI] [PubMed] [Google Scholar]
- Tian J.; Wang W. H.; Gao H. M.; Wang Z. M. Determination of matrine, sophoridine and oxymatrine in Compound Kushen Injection by HPLC. Zhongguo Zhongyao Zazhi 2007, 32, 222–224. [PubMed] [Google Scholar]
- Zheng K.; Li C.; Shan X.; Liu H.; Fan W.; Wang Z. A study on isolation of chemical constituents from Sophora flavescens Ait. and their anti-glioma effects. Afr. J. Tradit. Complementary Altern. Med. 2014, 11, 156–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H.; Luo H.; Huang F.; Li X.; Gao Q. Inhibition of proliferation and influence of proto-oncogenes expression by matrine in C6 cell. Zhongyaocai 2004, 27, 416–419. [PubMed] [Google Scholar]
- Guo L.; Xue T. Y.; Xu W.; Gao J. Z. Matrine promotes G0/G1 arrest and down-regulates cyclin D1 expression in human rhabdomyosarcoma cells. Panminerva Med. 2013, 55, 291–296. [PubMed] [Google Scholar]
- Li X. M.; Wu Y. G.; Pan D. X.; Wu L. K.; Yu Y. H. Sophoridine is a new antitumor medicine with new molecular structure. Zhongguo Xinyao Zazhi 2006, 15, 654–657. [Google Scholar]
- Ye G.; Zhu H. Y.; Li Z. X.; Ma C. H.; Fan M. S.; Sun Z. L.; Huang C. G. LC-MS characterization of efficacy substances in serum of experimental animals treated with Sophora flavescens extracts. Biomed. Chromatogr. 2007, 21, 655–660. [DOI] [PubMed] [Google Scholar]
- Li Y. M.; Min G.; Xue Q.; Chen L.; Liu W.; Chen H. High-performance liquid chromatographic method for simultaneous determination of sophoridine and matrine in rat plasma. Biomed. Chromatogr. 2004, 18, 619–624. [DOI] [PubMed] [Google Scholar]
- Hu P. Y.; Zheng Q.; Chen H.; Wu Z. F.; Yue P. F.; Yang M. Pharmacokinetics and distribution of sophoridine nanoliposomes in rats. Zhongguo Xinyao Zazhi 2012, 21, 2662–2666. [Google Scholar]
- Bi C. W.; Zhang C. X.; Li Y. H.; Tang S.; Deng H. B.; Zhao W. L.; Wang Z.; Shao R. G.; Song D. Q. Novel N-substituted sophoridinol derivatives as anticancer agents. Eur. J. Med. Chem. 2014, 81, 95–105. [DOI] [PubMed] [Google Scholar]
- Li X.; Zhao W. L.; Jiang J. D.; Ren K. H.; Du N. N.; Li Y. B.; Wang Y. X.; Bi C. W.; Shao R. G.; Song D. Q. Synthesis, structure-activity relationship and biological evaluation of anticancer activity for novel N-substituted sophoridinic acid derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 5251–5254. [DOI] [PubMed] [Google Scholar]
- Hu G. Q.; Yang Y.; Yi L.; Wang G. Q.; Duan N. N.; Wen X. Y.; Cao T. Y.; Xie S. Q.; Huang W. L. Design, synthesis and antitumor activity of C3/C3 bis-fluoroquonolones cross-linked with [1,2,4]triazolo[3,4-b] [1,3,4]thiadiazole. Acta Pharm. Sin. B 2011, 1, 172–177. [Google Scholar]
- Emirdağ-Öztürk S.; Karayıldırım T.; Capcı-Karagöz A.; Alankuş-Çalışkan O.; Ozmen A.; Poyrazoğlu-Çoban E. Synthesis, antimicrobial and cytotoxic activities, and structure–activity relationships of gypsogenin derivatives against human cancer cells. Eur. J. Med. Chem. 2014, 82, 510–512. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.