

Abstract
In the present study, we have analysed the ability of Streptococcus pyogenes [Group A streptococcus (GAS)] to activate the NACHT-domain-, leucine-rich repeat- and PYD-containing protein 3 (NALP3) inflammasome complex in human monocyte-derived macrophages and the molecules and signalling pathways involved in GAS-induced inflammatory responses. We focused upon analysing the impact of dynamin-dependent endocytosis and the role of major streptococcal virulence factors streptolysin O (SLO) and streptolysin S (SLS) in the immune responses induced by GAS. These virulence factors are involved in immune evasion by forming pores in host cell membranes, and aid the bacteria to escape from the endosome–lysosome pathway. We analysed cytokine gene expression in human primary macrophages after stimulation with live or inactivated wild-type GAS as well as with live SLO and SLS defective bacteria. Interleukin (IL)-1β, IL-10, tumour necrosis factor (TNF)-α and chemokine (C-X-C motif) ligand (CXCL)-10 cytokines were produced after bacterial stimulation in a dose-dependent manner and no differences in cytokine levels were seen between live, inactivated or mutant bacteria. These data suggest that streptolysins or other secreted bacterial products are not required for the inflammatory responses induced by GAS. Our data indicate that inhibition of dynamin-dependent endocytosis in macrophages attenuates the induction of IL-1β, TNF-α, interferon (IFN)-β and CXCL-10 mRNAs. We also observed that pro-IL-1β protein was expressed and efficiently cleaved into mature-IL-1β via inflammasome activation after bacterial stimulation. Furthermore, we demonstrate that multiple signalling pathways are involved in GAS-stimulated inflammatory responses in human macrophages.
Keywords: cytokine, gene regulation, inflammasome, innate immunity, signal transduction
Introduction
The Gram-positive bacterium Streptococcus pyogenes (GAS) is an important human pathogen causing a wide range of infections, from mild superficial infections (e.g. pharyngitis, tonsillitis) to severe invasive infections (e.g. necrotizing fasciitis, sepsis, toxic shock syndrome), which are all characterized by a robust inflammatory response. The disease burden of GAS infections is huge, and it is estimated that approximately 700 million mild cases occur globally each year; nearly 650 000 of these infections progress to severe invasive infections. More than 500 000 people die annually from severe GAS infections, of which ca. 160 000 deaths are due to invasive infections [1,2]. Mild infections can be treated easily with antibiotics. However, if left untreated these infections can lead to severe post-streptococcal diseases such as rheumatic fever and glomerulonephritis. Especially in the developing countries, GAS is a major cause of mortality. The GAS family is highly variable, with multiple clades and genetic variants. The antigenically variable M surface protein is used for serological typing of GAS strains. The M protein is encoded by the emm gene, of which more than 200 different gene types have been identified. The M1 serotype is one of the most frequently isolated serotypes in severe invasive GAS infections [3].
GAS encodes a wide range of virulence factors that are involved in the activation of the immune system, as well as allowing the bacterium to escape from the immune surveillance of the host [4]. These factors can, for example, interfere with the recognition of the bacterium, prevent complement activation and disturb the recruitment of neutrophils to the site of infection. One of the fundamental mechanisms of immune evasion is the ability of streptococcal virulence factors to inhibit phagocytosis or phagosome function [3]. It has been reported previously that GAS can survive inside human macrophages through M protein-dependent disturbance of phagosomal maturation, resulting in impaired fusion with lysosomes [5]. Streptolysins are responsible for the ability of GAS to lyse red blood cells [6]. Streptolysin O (SLO) is encoded by the slo gene, and resembles other bacterial cytolysins (e.g. listeriolysin, pneumolysin) in its sequence, sensitivity to oxygen and cholesterol binding [7,8]. SLO is involved in the escape of GAS from the endosome–lysosome pathway. Streptolysin S (SLS), encoded by the sagA gene, is unrelated to SLO and other cholesterol-binding cytolysins and its functions are associated with intracellular invasion of GAS. Both SLO and SLS can form pores in host cell membranes and induce apoptosis. The expression of SLO is higher in GAS strains that have been isolated from severe invasive cases compared with serotype-matched bacteria isolated from non-invasive cases [3].
Macrophages play a central role in innate and adaptive immune responses. Macrophages function mainly locally in different tissues by recognizing, taking up and destroying microbial pathogens [9]. They are also important in maintaining inflammatory responses in inflamed tissues. During activation, macrophages produce cytokines that regulate host immune responses. Recognition of microbes by cell surface and intracellular pattern recognition receptors (PRRs) is an important step in host defence against bacteria, viruses and other infectious agents. PRRs are expressed in different cellular compartments, such as on the cell surface, in endosomes or in the cytoplasm. Upon activation by microbes, their genetic material or structural components PRRs activate specific signalling pathways leading to expression of multiple genes involved in innate immune responses. PRRs include Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), C-type lectin receptors (CLRs), RIG-I receptors (RLRs) and other receptors that contribute to activation of the immune system in response to diverse stimuli [10]. Ligand binding to a TLR leads to activation of multiple signalling cascades and to the production of proinflammatory cytokines, anti-microbial peptides and co-stimulatory and adhesion molecules. At present, no specific receptor for GAS has been identified. Interestingly, in a mouse model TLR-2, TLR-4 and TLR-9, generally considered to be important in microbe recognition, were not required for GAS-induced signalling [11], suggesting that alternative molecules are involved in GAS recognition.
NLRs mediate cytoplasmic recognition of bacterial products. The NLR family member, NALP3 inflammasome, is a protein complex composed of NLR protein, adaptor protein ASC (apoptotic speck-containing protein with a CARD) and pro-caspase-1 [12]. The NALP3 inflammasome mediates caspase-1 activation in response to microbial and endogenous stimuli, which drives proteolytic processing of inflammatory cytokines interleukin (IL)-1β and IL-18 [13]. IL-1β and IL-18 play important roles in inflammatory conditions [13,14]. These cytokines are synthesized as inactive precursors and their processing into bioactive form depends on cleavage by proteases [13,15]. Synthesis and release of IL-1β differ between human monocytes and macrophages. Monocytes have constitutively activated caspase-1, leading to the release of active IL-1β after a single stimulation event with bacterial ligands such as lipopolysaccharide (LPS). In contrast, macrophages need two distinct stimuli; one stimulus induces transcription and translation of pro-IL-1β, a second stimulus is needed for caspase-1 activation with subsequent IL-1β processing and secretion of bioactive mature IL-1β [16].
The purpose of this study was to investigate host cell–bacteria interactions in the human macrophage model system during the early phase of GAS infection. We cultured human monocyte-derived macrophages from healthy adults and stimulated these cells in-vitro with live, inactivated and live mutant bacteria. The cells were stimulated with wild-type GAS and SLO or SLS virulence factor defective mutants to compare and analyse the importance of these virulence factors in GAS infections. In addition, we analysed the mechanisms and signalling pathways involved in GAS-induced cytokine gene expression and inflammasome activation. These data provide us with a better understanding of the mechanisms of GAS–human macrophage interactions, which may be helpful in controlling excessive GAS-induced inflammatory responses or designing novel strategies to enhance anti-bacterial immunity.
Materials and methods
Monocyte purification and differentiation into macrophages
Human peripheral blood mononuclear cells (PBMCs) from leukocyte-rich buffy coats obtained from healthy anonymous blood donors (Finnish Red Cross Blood Transfusion Service, Helsinki, Finland) were isolated by density gradient centrifugation over a Ficoll-Paque gradient (GE-Healthcare BioSciences, Uppsala, Sweden). Monocytes were further purified from PBMCs by adherence on plastic plates (Falcon, Becton Dickinson, Franklin Lakes, NJ, USA) at a concentration of 2·5 × 106 cells/24 wells or 5 × 106 cells/six wells for 1 h at +37°C. To differentiate monocytes into macrophages, cells were grown in macrophage serum-free substitution medium (Gibco Invitrogen, Grand Island, NY, USA) supplemented with 0·6 μg/ml penicillin, 60 μg/ml streptomycin and 10 ng/ml recombinant human granulocyte-macrophage colony-stimulating factor (GM-CSF) (Biosource, Camarillo, CA, USA), as described previously [17]. The identity and expression of macrophage markers in these differentiated cells has been previously characterized in detail [18]. Fresh medium was changed every 2 days, and experiments were started after 7 days of cultivation.
Bacterial strains and viruses
Streptococcus pyogenes serotype M1 (ATCC 700294) is a clinical strain originally isolated from an infected wound. GAS mutants lacking slo (EC997) [11] or sagA (EC695), the genes encoding cytolysins, streptolysin O and streptolysin S, respectively, were provided by Emmanuelle Charpentier. Bacteria were stored in skimmed milk at −70°C, and grown to the end of logarithmic growth phase before they were used in experiments. Bacterial cultivations were performed on sheep blood agar and Nefrit broth, as described previously [19]. Bacteria were passaged three times before they were used in stimulation experiments. The number of bacteria was determined by counting in a Petroff–Hausser chamber. The bacterial preparations were confirmed to be endotoxin-free by a Limulus test (detection limit 0·125 IU/ml).
Sendai virus, which was used as control, was grown in embryonated chicken eggs and stored in −70°C. The haemagglutination titre of the virus stock was 4096 and the infectivity in macrophages and dendritic cells (DCs) was 6 × 109 plaque-forming units (pfu)/ml. Multiplicity of infection (MOI) values of 5 were used in cell stimulation experiments.
Reagents
Pharmacological kinase-specific inhibitors were used to block different signalling pathways. Macrophages were treated with inhibitors for 30 min prior to bacterial stimulation. Signalling inhibitors PD98059 [mitogen-activated protein kinase (MEK1) inhibitor, used at 10 μmol/l] and LY294002 (PI3K inhibitor, 50 μmol/l) were obtained from Calbiochem (San Diego, CA, USA) and SB202190 (p38 inhibitor, 10 μmol/l), SP600125 [stress-activated protein kinase/c-Jun N-terminal kinase (JNK) inhibitor, 10 μmol/l] and pyrrolidine dithiocarbamate [PDTC, specific nuclear factor (NF)-κB inhibitor, 100 μmol/l] were purchased from Alexis Biochemicals (Lausen, Switzerland). The functional concentrations of inhibitors used in the present study have been determined previously and used in a number of studies [19–23]. Similarly, macrophages were treated with the dynamin inhibitor dynasore (D7693; Sigma-Aldrich, St Louis, MO, USA) for 30 min prior to bacterial stimulation. The optimal concentration of 80 μM was determined previously [21,24,25]. Kinase-specific inhibitors or dynasore had no effect on cell viability (data not shown). This was confirmed by fluorescence activated cell sorter (FACS) analysis with a dead cell discriminator (Caltag Laboratories, Burlingame, CA, USA) containing propidium iodide (PI), which labels dead cells by integrating with the DNA in cells with disrupted cell membranes.
Stimulation experiments
All stimulation experiments were carried out with cells obtained from three or four blood donors and conducted in RPMI-1640 medium. For stimulation experiments bacteria were collected and suspended into RPMI-1640 medium (Sigma, St Louis, MO, USA). Heat inactivation of bacteria was performed in 95°C dry heat block for 5 min and ultraviolet (UV) inactivation by exposing bacteria to UV light at an energy level of 2 × 0·12 J. Complete inactivation of bacteria was verified by the sheep blood agar plate-counting method. Streptolysin-defective mutant strains SLO− (EC997) and SLS− (EC695) were used as live bacteria in all experiments. Macrophages were stimulated with different bacteria : host cell ratios for 3, 9 or 24 h, as indicated in the figure legends.
Enzyme-linked immunosorbent assay (ELISA) and Western blot analysis
The levels of secreted cytokines from cell culture supernatants were determined by a sandwich ELISA method, as described previously [26]. Tumour necrosis factor (TNF)-α, IL-10 and chemokine (C-X-C motif) ligand (CXCL)-10 cytokine levels were determined by using antibody pairs and standards obtained from BD PharMingen (San Diego, CA, USA). IL-1β cytokine levels were determined using antibody pairs and standards from R&D Systems (Abingdon, UK). Whole cell lysates were prepared in Laemmli sample buffer for Western blot analysis. The proteins were separated on 15% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), blotted and membranes were stained with specific antibodies against IL-1β [27] and β-actin (SC-10731; Santa Cruz Biotechnology, Santa Cruz, CA, USA).
RNA isolation and quantitative reverse transcription–polymerase chain reaction (qRT–PCR)
For RNA analysis, cells were collected and lysed with TRI reagent (Sigma-Aldrich) and total cellular RNA was isolated with the RNeasy Mini kit (Qiagen, Crawley, UK). In mRNA analysis, pooled samples from four blood donors were used unless indicated otherwise. Synthesis of cDNA was performed as described [28] using Multiscribe RT (Applied Biosystems, Foster City, CA, USA) with oligo(dT) primers (Applied Biosystems). To analyse mRNA levels, cDNA amplification was performed using Master Mix Buffer with Assays-on-Demand gene expression assay primers and probes for IL-1β (Hs00174097_m1), TNF-α (Hs00174128_m1), interferon (IFN)-β (Hs00277188_s1), CXCL-10 (Hs00171042_m1), NALP3 (Hs00918082_m1) and β-actin (Hs99999903_m1; all from Applied Biosystems). Each cDNA sample was amplified in duplicate with Stratagene MxPro 3005P. The threshold cycle values were normalized against β-actin and relative amounts of mRNAs were calculated with the ΔΔ comparative threshold (Ct) method as instructed by the manufacturer using dimethylsulphoxide (DMSO)-treated cells as a calibrator.
Statistical analysis
Statistical analysis was performed with Student's t-test and P-values < 0·05 were considered statistically significant.
Results
Pro- and anti-inflammatory cytokines are secreted from human macrophages in response to stimulation with GAS
The ability of pathogenic GAS to induce cytokine production was analysed from macrophages stimulated with live, heat-inactivated or UV-inactivated wild-type bacteria as well as with live SLO- or SLS-defective bacteria. The ability of GAS to activate the NALP3 inflammasome and to induce the maturation of IL-1β has not been characterized previously in human macrophages, while extensive studies have been conducted with mouse models and mouse macrophages [29,30]. Macrophage responses to live GAS were compared to those induced by inactivated bacteria or live virulence factor defective mutant bacteria in order to analyse the effects of bacterial viability and the contribution of virulence factors SLO and SLS to the immune responses induced by GAS. Macrophages were stimulated with different doses of bacteria to determine the optimal dose for further stimulation experiments (Fig. 1a). MOI ranging from 1 to 100 bacteria/macrophage was used for stimulations. Proinflammatory IL-1β and TNF-α, anti-inflammatory IL-10 and CXCL-10 production was measured by ELISA after 24 h stimulation to obtain a view of the overall cytokine responses induced by GAS stimulation. IL-10 was included as a representative of an anti-inflammatory cytokine and CXCL-10 as an IFN-inducible gene generally expressed after inflammatory stimuli. All bacteria induced cytokine production similarly, and no differences in secreted IL-1β, TNF-α, IL-10 or CXCL-10 levels were seen after 24 h stimulation between live, inactivated or mutant bacteria. Cytokine production was clearly dependent upon the bacterial dose and MOI 10 was chosen for further experiments, as this dose was sufficient to induce submaximal cytokine response without toxic effects on cells.
Fig. 1.

Group A streptococcus (GAS) induces cytokine production in human macrophages in a dose-dependent manner. (a) Macrophages were stimulated with live, ultraviolet (UV)-inactivated, heat-inactivated, live streptolysin O (SLO)-mutant or live streptolysin S (SLS)-mutant GAS with different multiplicity of infection (MOI) values (1–100). Cell culture supernatants were collected 24 h after bacterial stimulation and cytokine levels were determined by enzyme-linked immunosorbent assay (ELISA). The experiment was carried out with cells obtained from four different blood donors, the columns represent the means and error bars indicate standard deviations. The data are from a representative experiment of three. (b) Samples from individual blood donors were analysed separately for MOI 10 to demonstrate the individual variation between donors. The means are marked in the figure with horizontal lines. n.d. = not detectable.
Individual blood donors responded to bacterial stimulation with different cytokine levels (Fig. 1b). Four blood donors were analysed individually for their IL-1β, TNF-α, IL-10 and CXCL-10 cytokine production levels induced after bacterial stimulation with MOI 10 at 24 h. Some donors were high responders, while others induced lower or undetectable cytokine production levels. However, the cytokine production trends were quite similar between individual donors.
Inhibition of dynamin-dependent endocytosis attenuates cytokine mRNA expression
The role of dynamin-dependent endocytosis and endosomal signalling in the induction of immune responses against GAS was analysed using dynasore, a dynamin inhibitor. Because our interest was in the early events of GAS infection, we analysed cytokine mRNA expression in GAS-stimulated cells with or without dynasore at an early time-point of 3 h after bacterial stimulation (Figs 2 and 3 and Supporting information, F2ig. S2). We were interested in proinflammatory cytokine responses, and therefore IL-10 mRNA expression was not analysed. In addition, unlike proinflammatory and IFN mRNAs, which are induced rapidly, IL-10 mRNA was induced at later time-points, making it an unsuitable marker for an early cytokine gene. To confirm that there were no kinetic differences in cytokine mRNA expression levels induced by live, inactivated or live mutant bacteria in the early stages of infection, TNF-α mRNA expression was analysed at 3 and 9 h after stimulation (Supporting information, Fig. S1). TNF-α levels are presented as it is one of the early genes activated in response to microbial stimulation. No kinetic differences in TNF-α mRNA expression levels were seen between different stimulants. The concentration of dynasore (80 μM) used in this study has been determined previously as optimal for human primary cells and cell lines [21,24]. Dynasore was reconstituted in DMSO, therefore DMSO-treated cells were used as a control. Cytokine mRNA expression rather than protein production was measured, as dynasore could also interfere with protein secretion. In addition, the early time-point of 3 h is not optimal for detecting secreted cytokines. Sendai virus was included as a negative control for dynamin-dependent endocytosis (Supporting information, Fig. S2), as it is known to be able to fuse directly with the plasma membrane to enter the host cells in a dynamin-independent manner [31].
Fig. 2.
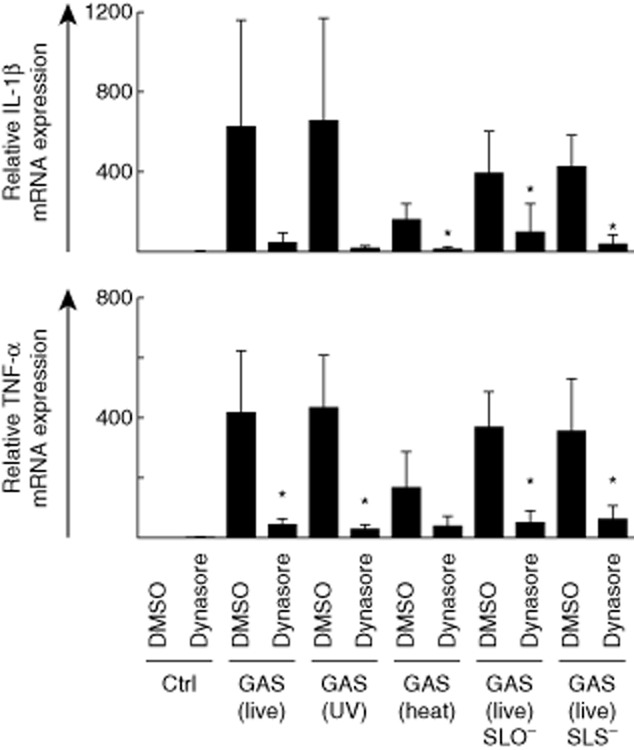
Proinflammatory cytokine mRNA expression is induced in group A streptococcus (GAS)-stimulated macrophages and inhibited by the dynamin inhibitor dynasore. Macrophages were left untreated or treated with dynasore (80 μM) for 30 min prior to stimulation with live, ultraviolet (UV)-inactivated, heat-inactivated, streptolysin O (SLO)-mutant or streptolysin S (SLS)-mutant GAS [multiplicity of infection (MOI) 10] for 3 h. Interleukin (IL)-1β and tumour necrosis factor (TNF)-α mRNA levels were analysed by quantitative reverse transcription–polymerase chain reaction (qRT–PCR). Relative mRNA levels were normalized against β-actin and the relative level of mRNA was calculated with the ΔΔ comparative threshold (Ct) method using dimethylsulphoxide (DMSO)-treated cells as a calibrator. Results are the means (± standard deviation) of four independent experiments performed with cells of four blood donors in each experiment (altogether n = 16). *P < 0·05 between DMSO-treated and dynasore-treated GAS-infected cells.
Fig. 3.
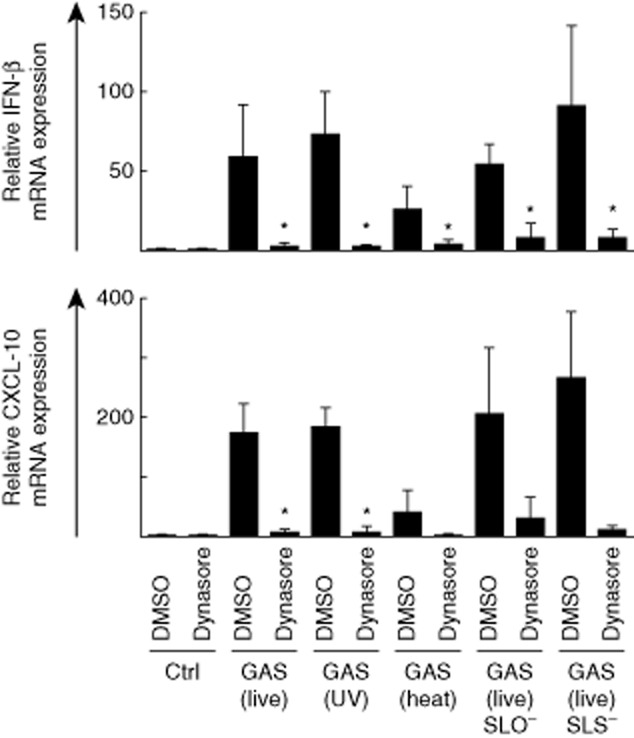
Interferon (IFN)-β and IFN-inducible chemokine (C-X-C motif) ligand (CXCL)-10 mRNA expression is induced in group A streptococcus (GAS)-stimulated macrophages and inhibited by dynasore. Macrophages were left untreated or treated with dynasore (80 μM) for 30 min prior to stimulation with live, ultraviolet (UV)-inactivated, heat-inactivated, live streptolysin O (SLO)-mutant or live streptolysin S (SLS)-mutant GAS [multiplicity of infection (MOI)10] for 3 h. IFN-β and CXCL-10 mRNA levels were analysed by quantitative reverse transcription–polymerase chain reaction (qRT–PCR). Relative mRNA levels were normalized against β-actin and the relative level of mRNA was calculated with the ΔΔ comparative threshold (Ct) method using dimethylsulphoxide (DMSO)-treated cells as a calibrator. Results are the means (± standard deviation) of four independent experiments performed with cells of four blood donors in each experiment (altogether n = 16). *P < 0·05 between DMSO-treated and dynasore-treated GAS-infected cells.
Proinflammatory IL-1β and TNF-α gene expression was inhibited efficiently in dynasore-treated GAS-stimulated macrophages compared with DMSO-treated control cells (Fig. 2). The effects of dynasore on IL-1β gene expression were statistically significant in macrophages stimulated with heat-inactivated GAS, as well as in cells stimulated with SLO- or SLS-defective bacteria as analysed by Student's t-test (P < 0·05). TNF-α gene expression was also significantly (P < 0·05) inhibited in dynasore-treated macrophages stimulated with all bacteria except for heat-inactivated GAS. No significant differences between live, inactivated or mutant bacteria were observed in cytokine mRNA expression in dynasore-treated or untreated cells. Heat-inactivated bacteria induced slightly lower cytokine mRNA expression levels than other bacteria, which could be due to possible destruction of bacterial surface components or aggregation of bacteria during the inactivation process. The effects of dynasore were also analysed with three individual blood donors (Supporting information, Fig. S2a). Individual donors induced differential levels of IL-1β and TNF-α mRNAs, which were inhibited efficiently by dynasore treatment. Sendai virus, the negative control for dynamin-dependent endocytosis, did not induce IL-1β or TNF-α cytokine mRNAs at 3 h (Supporting information, Fig. S2a). This is due probably to the lack of viral replication, which is not taking place efficiently at the 3-h time-point in human macrophages.
IFN responses were also analysed in GAS-stimulated cells in the presence or absence of dynasore at 3 h after bacterial stimulation (Fig. 3). No significant differences between live, UV-inactivated or mutant bacteria were observed in IFN-β or CXCL-10 mRNA expression levels. IFN-β expression was reduced significantly in dynasore-treated cells stimulated with all bacteria (P < 0·05). IFN-inducible CXCL-10 gene expression was also affected significantly in dynasore-treated cells stimulated with live or UV-inactivated GAS (Fig. 3). In cells stimulated with heat-inactivated and mutant GAS, CXCL-10 mRNA levels were also clearly reduced after dynasore treatment. However, this did not reach statistical significance, due to a marked variation between the donors. In accordance with IL-1β and TNF-α gene expression (Fig. 2), heat-inactivated bacteria induced lower, although not statistically significant, IFN-β and CXCL-10 mRNA expression levels compared to other stimuli.
Three donors were also analysed for their individual IFN-β and CXCL-10 mRNA expression levels (Supporting information, Fig. S2b). IFN-β mRNA was strongly induced in bacteria-stimulated cells in all donors and the levels were attenuated by dynasore treatment. Sendai virus induced strong IFN-β mRNA expression which, to a lesser extent, was attenuated by dynasore (Supporting information, Fig. S2b), demonstrating that Sendai virus enters the cells primarily via the dynamin-independent endocytic pathway by fusing directly with the plasma membrane, as shown previously [31]. In addition, Sendai virus-induced CXCL-10 mRNA expression was attenuated by dynasore-treatment similarly to GAS-induced responses. Because CXCL-10 is an IFN-inducible gene with a promoter region consisting of various transcription factor binding sites it is likely that dynasore interferes with some of the signalling pathways needed for CXCL-10 mRNA expression. In addition, dynasore has been associated previously with the internalization of cytokine receptors, including interferon receptor chains [32]. This probably also affects Sendai virus-induced CXCL-10 mRNA expression, as it is considered to be an IFN-inducible gene. Thus, dynasore may interfere with cytokine networks and their positive and negative feedback loops.
The NALP3 inflammasome-complex is activated in GAS-stimulated macrophages
Inflammasome activation in human macrophages in response to bacterial stimulation was analysed by Western blotting. Activation of the NALP3 inflammasome leads to self-cleavage and activation of caspase-1 and cleavage of pro-IL-1β into its mature biologically active form. The kinetics of inflammasome activation was analysed by detecting the maturation of IL-1β at different time-points after stimulation with live wild-type GAS (Fig. 4). Cleavage of pro-IL-1β into various intermediate and mature forms was seen at 6 h after bacterial stimulation and the levels continued to increase up to 24 h after stimulation. It is noteworthy that the expression of pro-IL-1β continued to take place with a similar kinetics.
Fig. 4.
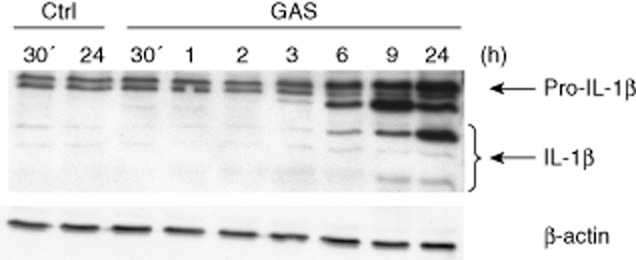
Kinetics of interleukin (IL)-1β production in group A streptococcus (GAS)-stimulated macrophages. Macrophages were stimulated with live GAS [multiplicity of infection (MOI) 10] and the cells were collected at times indicated in the figure. Cell lysates were prepared, proteins were separated on 15% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting was performed with rabbit anti-human IL-1β or anti-β-actin antibodies. Analysis of β-actin expression functions as a loading control.
The role of dynamin-dependent endocytosis in inflammasome activation after bacterial stimulation was analysed by stimulating the cells with live, heat-inactivated, UV-inactivated, SLO-mutant and SLS-mutant bacteria for 24 h with MOI 10 in the absence or presence of dynasore. In dynasore-treated cells the accumulation of pro- and mature-IL-1β levels were clearly reduced (Fig. 5). The induction of pro-IL-1β protein expression or the cleavage of it into the mature form were similarly decreased in cells stimulated with live, inactivated or virulence factor-defective live bacteria (Fig. 5).
Fig. 5.
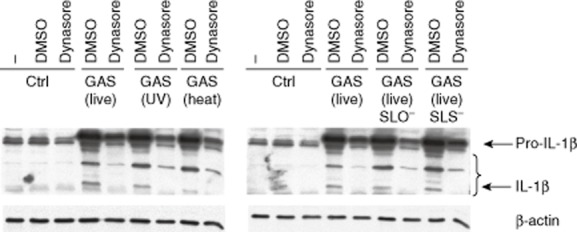
Maturation of interleukin (IL)-1β is inhibited in dynasore-treated group A streptococcus (GAS)-stimulated human macrophages. Macrophages were left untreated or treated with dynasore (80 μM) for 30 min prior to stimulation with live, ultraviolet (UV)-inactivated, heat-inactivated, live streptolysin O (SLO)-mutant or live streptolysin S (SLS)-mutant GAS [multiplicity of infection (MOI) 10]. Cell lysates were prepared after 24 h stimulation, proteins were separated on 15% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting was performed with rabbit anti-human IL-1β antibody. Analysis of β-actin expression functions as a loading control.
NALP3 mRNA expression was expressed constitutively in macrophages and a clear, up to fourfold increase in NALP3 gene expression was seen at the early time-point of 3 h after bacterial stimulation (Fig. 6). Interestingly, dynasore treatment almost completely inhibited bacteria-induced NALP3 mRNA expression, indicating that some early steps in macrophage–GAS interactions can trigger NALP3 gene expression. However, this phenomenon did not reach statistical significance due to individual variation between donors.
Fig. 6.
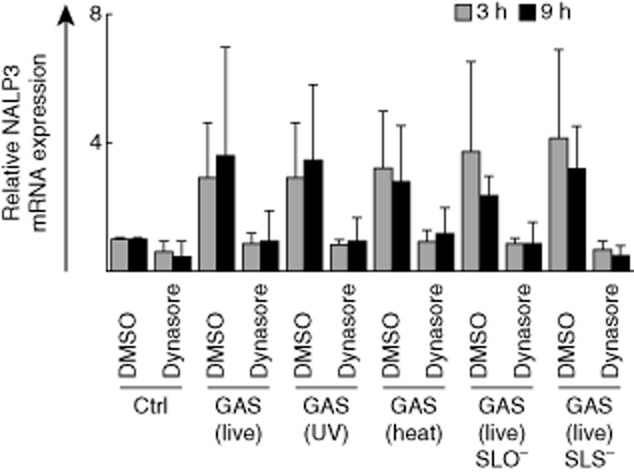
NACHT-domain-, leucine-rich repeat- and PYD-containing protein 3 (NALP3) is constitutively expressed in human macrophages. Macrophages were left untreated or treated with dynasore (80 μM) for 30 min prior to stimulation with live, ultraviolet (UV)-inactivated, heat-inactivated, live streptolysin O (SLO)-mutant, or live streptolysin S (SLS)-mutant group A streptococcus (GAS) [multiplicity of infection (MOI) 10]. Macrophages were collected at 3 or 9 h after GAS-stimulation and cells from different blood donors were pooled. Total cellular RNA was isolated and NALP3 mRNA levels were analysed by quantitative reverse transcription–polymerase chain reaction (qRT–PCR). Relative mRNA levels were normalized against β-actin and the relative level of mRNA was calculated with the ΔΔ comparative threshold (Ct) method using dimethylsulphoxide (DMSO) pretreated cells as a calibrator. Results are the means (± standard deviation) of three independent experiments each performed with cells of four blood donors (n = 12).
Multiple signalling pathways are involved in GAS-stimulated innate immune responses in human macrophages
Next, we analysed the contribution of different signalling pathways in immune responses induced by GAS by stimulating the cells with live wild-type bacteria in the presence or absence of different pharmacological kinase-specific signalling inhibitors (Fig. 7). Macrophages were left untreated or pretreated with different signalling inhibitors followed by stimulation with live GAS. Cells were collected at 3 h after stimulation, total cellular RNA was isolated and proinflammatory IL-1β and TNF-α mRNAs (Fig. 7a), as well as IFN-β and CXCL-10 mRNAs (Fig. 7b), were measured. Mitogen-activated protein kinase (MAPK) p38 inhibitor SB202190 decreased GAS-induced proinflammatory IL-1β and TNF-α as well as IFN-β and CXCL-10 mRNA expression levels. Only the effects on IL-1β mRNA levels were statistically significant (P < 0·05). JNK inhibitor SP600125 was also able to decrease IL-1β, TNF-α and CXCL-10 mRNA expression levels, but this inhibitor had no effect on IFN-β mRNA levels in GAS-stimulated cells. The SP600125 inhibitor had significant (P < 0·05) effects only on IL-1β mRNA levels. PI3K inhibitor LY294002 had minor effects on TNF-α and IFN-β mRNA expression levels. MEK1 inhibitor PD98059 and NF-κB inhibitor PDTC had no inhibitory effects on IL-1β, TNF-α, IFN-β and CXCL-10 mRNA levels; rather, some stimulatory effects were seen. These data suggest that compensatory pathways are activated after treatment with these inhibitors. NALP3 mRNA expression was measured in GAS-stimulated cells after treatment with the same inhibitors (Fig. 7c). NALP3 mRNA expression was clearly induced in GAS-stimulated cells. However, in contrast to cytokine and IFN mRNAs, whose expression was reduced to some extent by multiple inhibitors, only the JNK inhibitor SP600125 weakly reduced NALP3 expression levels. This suggests that the expression of NALP3 mRNA is regulated by signalling pathways apart from those that are regulating cytokine mRNA expression.
Fig. 7.

Multiple signalling pathways are involved in group A streptococcus (GAS)-induced cytokine gene expression. Macrophages were left untreated or treated with pharmacological signalling inhibitors for 30 min prior to stimulation with live GAS [multiplicity of infection (MOI) 10]. Cells were collected at 3 h after stimulation, total cellular RNA was isolated and (a) interleukin (IL)-1β, tumour necrosis factor (TNF)-α, (b) interferon (IFN)-β, chemokine (C-X-C motif) ligand (CXCL)-10 and (c) NACHT-domain-, leucine-rich repeat- and PYD-containing protein 3 (NALP3) mRNA levels were determined by quantitative reverse transcription–polymerase chain reaction (qRT–PCR). Relative mRNA levels were normalized against β-actin and the relative level of mRNA was calculated with the ΔΔ comparative threshold (Ct) method using dimethylsulphoxide (DMSO)-treated cells as a calibrator. The used inhibitors were: PD98059 [mitogen-activated protein kinase (MEK)1 inhibitor, 10 μmol/l], SB202190 (p38 inhibitor, 10 μmol/l), SP600125 [stress-activated protein kinase/c-Jun N-terminal kinase (JNK) inhibitor, 10 μmol/l], LY294002 (PI3K inhibitor, 50 μmol/l), and pyrrolidine dithiocarbamate (PDTC) [nuclear factor (NF)-κB inhibitor, 100 μmol/l]. Results are the means (± standard deviation) of three independent experiments performed with cells of four blood donors (n = 12). *P < 0·05 between DMSO-treated and inhibitor-treated GAS-infected cells. Note the differences in scales.
Because production of the mature form of IL-1β requires other cellular processing steps, we analysed whether the signalling inhibitors would also have an effect on the accumulation of mature IL-1β. IL-1β protein levels in inhibitor-treated, GAS-stimulated macrophages were analysed by Western blotting (Fig. 8a). All inhibitors efficiently inhibited the production of mature form of IL-1β, but not that of pro-IL-1β, suggesting that the second signal of inflammasome activation was disturbed by these inhibitors. IL-1β cytokine secretion in GAS-stimulated cells after treatment with inhibitors was analysed from cell culture supernatants at 9 h after stimulation (Fig. 8b). All inhibitors decreased IL-1β cytokine secretion levels significantly (P < 0·05), MAP kinase inhibitors SP600125 and SB202190 and PI3K inhibitor LY294002 having the strongest effects.
Fig. 8.
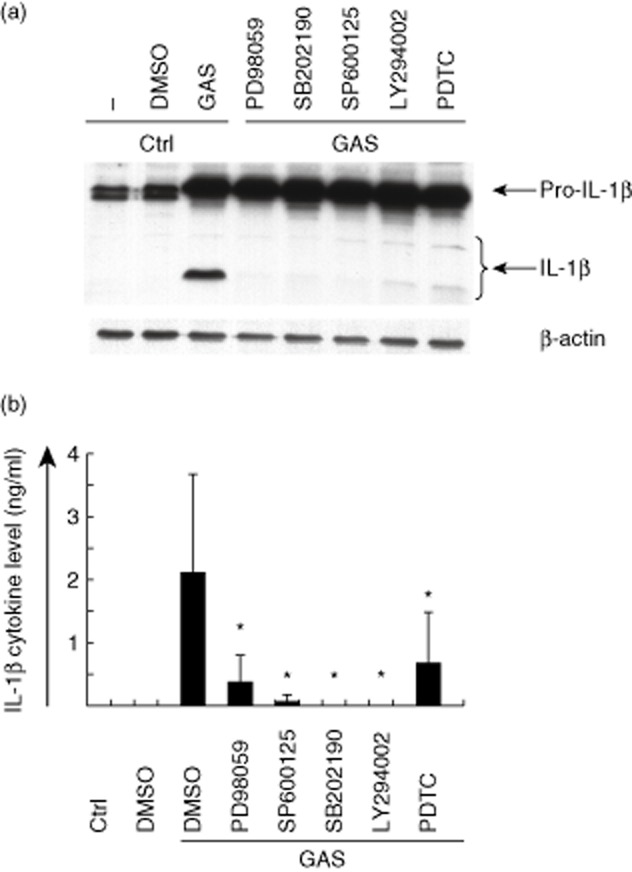
Pharmacological signalling inhibitors interfere with the processing and secretion of interleukin (IL)-1β. (a) Macrophages from three blood donors were left untreated or treated with different signalling inhibitors 30 min prior to stimulation with live group A streptococcus (GAS) [multiplicity of infection (MOI) 10] for 9 h. Cell lysates were prepared, proteins were separated on 15% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting was performed with rabbit anti-human-IL-1β antibody. Staining of β-actin functions as a loading control. (b) Cell culture supernatants were collected at 9 h after GAS-stimulation and secreted IL-1β levels were determined by enzyme-linked immunosorbent assay (ELISA). The used inhibitors were: PD98059 [specific mitogen-activated protein kinase (MEK)1 inhibitor, 10 μmol/l], SB202190 (p38 inhibitor, 10 μmol/l), SP600125 [stress-activated protein kinase/janus kinase (JNK0 inhibitor, 10 μmol/l], LY294002 (PI3K inhibitor, 50 μmol/l), and pyrrolidine dithiocarbamate (PDTC) [nuclear factor (NF)-κB inhibitor, 100 μmol/l]. The experiment was carried out with cells obtained from three different blood donors, the columns represent the means and error bars indicate standard deviations of the means. *P < 0·05 between dimethylsulphoxide (DMSO)-treated and inhibitor-treated GAS-infected cells.
Discussion
S. pyogenes is a human pathogen capable of causing a wide range of different diseases. GAS has multiple virulence factors that contribute to bacterial carriage, invasion, pathogenesis of the infection and induction and inhibition of host inflammatory and anti-microbial responses [3,5,33]. Although the bacterium itself and its virulence determinants have been studied extensively, GAS–host cell interactions, especially in the human system, have remained less well characterized. In the present study we have analysed the mechanisms of GAS-activated host cell signalling pathways and the expression of cytokine genes in human monocyte-derived macrophages at the early phase of GAS infection. We show that in the human macrophage model system live and both heat- or UV-inactivated, as well as SLO- and SLS-deletion mutant GAS bacteria, readily activate host cytokine responses. Dynamin inhibitor dynasore efficiently attenuated GAS-induced cytokine gene expression at the early steps of infection. PI3K-, NF-κB- and multiple MAPK-inhibitors had weak inhibitory effects on GAS-induced cytokine mRNA expression. Inflammasome-dependent pro-IL-1β processing and secretion into the cell culture supernatant was clearly attenuated by different kinase-specific inhibitors. This indicates that GAS–macrophage interaction triggers a cascade of multiple host cell signalling and activation processes that contribute to the activation of host innate immune system.
Phagocytic cells have an essential role in the host defence against microbial infections and the important role of macrophages in the early control of GAS infections has been reported previously in mice [34]. An intracellular reservoir of GAS, residing predominantly in macrophages, was found in patients suffering from invasive tissue infections [35]. Intracellular streptococci have also been demonstrated in epithelial cells [36,37] and neutrophils [38]. In the present study, we observed that stimulation of human macrophages with GAS resulted in the production of proinflammatory cytokines such as IL-1β, TNF-α, anti-inflammatory IL-10, as well as in the gene expression of IFN-β and IFN-inducible chemokine CXCL-10. Interestingly, unlike in human DCs [39], live and inactivated Gram-positive bacteria induced cytokine production equally well, suggesting that macrophages take up and respond to GAS regardless of secreted bacterial factors or active bacterial internalization mechanisms. Our results are in line with a previous study demonstrating that live and inactivated (paraformaldehyde-fixed) bacteria were taken up by human macrophages in a similar fashion [5]. It has been reported that in mouse macrophages live, but not killed, GAS induced caspase-1 activation and IL-1β secretion [30]. However, in our human macrophage model system both UV- and heat-inactivated bacteria induced cytokine responses, including IL-1β secretion, practically as efficiently as live GAS at 24 h after infection. This apparent discrepancy between our results and those of Harder and co-workers [30] may be related to differences between mouse and human cells. We have reported previously, using the same human macrophage system, that other inactivated Gram-positive bacteria such as probiotic lactobacilli are also able to induce pro-IL-1β production and IL-1β maturation equally, or even more efficiently, than live bacteria [40]. The differences in these studies may be related to the host cell species (human versus mouse) as well as to the different bacteria used in these two studies.
GAS is generally considered to be an extracellular pathogen, but there are previous findings suggesting intracellular survival of the bacterium [35,38,41,42]. Various intracellular survival strategies have been proposed for GAS, including streptolysin O-mediated evasion of lysosomal degradation in epithelial cells [42]. The role of virulence factors SLO and SLS in GAS pathogenesis is unclear. We show that virulence factor-defective SLO and SLS mutants did not differ in their ability to induce IL-1β, TNF-α, CXCL-10 or IFN-β cytokine gene expression compared to wild-type GAS, suggesting that these virulence factors are probably not regulating inflammatory responses in human macrophages. Maturation of IL-1β is indicative of NALP3-dependent inflammasome activation. In addition to TLR stimulation, which triggers pro-IL-1β mRNA expression and protein production, the activation of NALP3 inflammasome in macrophages requires a second signal that leads to caspase-1 activation and cleavage of pro-IL-1β into its mature active form. Certain pore-forming toxins have been shown to be responsible for inducing the second signal of inflammasome activation [43,44]. In GAS infections, pore-forming SLO has been shown to mediate the delivery of microbial molecules to the host cytosol and cytosolic escape of GAS [41], which could trigger NALP3-dependent caspase-1 activation. In mouse cells, GAS-induced caspase-1 activation and IL-1β secretion was dependent upon NF-κB activation and the pore-forming toxin SLO [30]. SLO has also been reported to be a critical modulator of GAS internalization, intracellular trafficking and bacterial killing by human oropharyngeal keratinocytes. Streptolysin-defective bacteria were internalized into lysosomes and were killed by pharyngeal epithelial cells, while wild-type bacteria were not targeted into lysosomes for intracellular killing [42]. Our results, showing that there is no difference in the maturation of IL-1β after stimulation with virulence factor SLO- or SLS-defective bacteria, suggest that other factors apart from streptolysins are responsible for the inflammasome activation in human macrophages. Previous studies in macrophages have also identified that serine proteases (e.g. proteinase-3, elastase and cathepsin-G) and some bacterial proteases can process the inactive precursor of IL-1β into its active forms [14,45]. Recently, non-canonical inflammasome activation by caspase-11 was also identified in mice [46].
Gram-positive bacteria are generally thought to be recognized by TLR-2. The streptococcal surface protein M1 can interact with TLR-2 in human blood monocytes, resulting in the expression of IL-6, IL-1β and TNF-α [47]. Transcriptional systems activated downstream of TLR signalling include NF-κB, IFN regulatory factor (IRFs) and MAPK-activated factors [10]. Using different signalling inhibitors, we observed that the p38 and JNK MAPK pathways are particularly involved in IL-1β cytokine responses induced by GAS (Fig. 7). Different inhibitors had also some, but not statistically significant, inhibitory effects on TNF-α, IFN-β and CXCL-10 gene expression. These results support the idea that multiple signalling pathways are involved in immune responses induced by GAS.
Bacteria can induce IFN responses via TLR-9 [48] and the initially cell surface localized TLR-2 and TLR-4 can turn on IFN-β gene expression after they are internalized in a ligand-specific manner [49,50]. Some bacteria, such as Listeria monocytogenes, can efficiently induce IFN-β gene expression in mice [51]. Previously, we observed that GAS can induce some IFN-β gene expression in human macrophages [17]. Activation of IFN-β gene requires the recruitment of activated IRF, NF-κB and AP-1 factors to the promoter [52]. In mice infected with intracellular L. monocytogenes, IFN production was independent of TLRs but required IRF3 activation [53]. Listeriolysin (LLO)-defective L. monocytogenes, which is incapable of escaping from phagosomes into the cytosol, was unable to induce IFN-β gene expression in mice [53]. In mouse macrophages streptococcal DNA was the bacterial factor involved in IFN-β production, and this phenomenon appeared to be dependent upon myeloid differentiation primary response gene 88 (MyD88), stimulator of IFN genes (STING), TANK-binding-kinase 1 (TBK1) and IRF3 signalling components [29]. Our results, showing that in GAS-stimulated human macrophages neither SLO nor SLS was required for IFN mRNA expression, are in line with a previous study performed in murine BMDMs [11]. Interestingly, in other Gram-positive bacterial infections, either cytolysin itself or cytolysin-mediated cytoplasmic escape of bacteria was implicated in triggering IFN production [53–56]. In mice, MyD88-dependent IFN production occurred without involvement of TLR-2, TLR-4 or TLR-9 [29], suggesting the involvement of an as-yet unknown receptor in the immune responses induced by GAS.
To study the role of endocytosis and endosomal signalling in immune responses induced by GAS, we inhibited the dynamin-dependent endocytosis with dynasore, a specific inhibitor. Dynamin participates in the severing of membrane-bound vesicles such as clathrin-coated pits and caveolae from cell membrane [57], and is also involved in actin comet formation and transport of macropinosomes [25]. Dynamin inhibits the endocytic functions of dynamin and vesicle formation from other organelles [24]. Although many internalization mechanisms require dynamin, there are also dynamin-independent pathways. Lipid raft-dependent endocytosis is a dynamin-independent pathway required for internalization by some viruses, bacteria and bacterial toxins [58]. In the present study we noted that inhibition of dynamin-dependent endocytosis with dynasore led to decreased expression of IL-1β, TNF-α, IFN-β, CXCL-10 and NALP3 mRNAs in GAS-infected cells at an early time-point of 3 h. In addition, the proteolytic cleavage of pro-IL-1β was decreased in dynasore-treated macrophages, suggesting the disturbance of the second signal of inflammasome activation by dynasore. It has been shown previously that dynamin-dependent endocytosis is also involved in type III IFN, CXCL-10 and IL-6 expression in Salmonella-infected human monocyte-derived DCs. It was also shown that dynasore prevented bacteria-induced phosphorylation of IRF-3 and the internalization of Salmonella [21]. However, we were unable to detect the phosphorylation of IRF3 in our macrophage system after GAS stimulation. These differences might result from different cells and bacteria used in these studies. Unlike GAS, Salmonella is rich in LPS and it has an active internalization mechanism, which might activate different signalling pathways in host cells.
In the present study, carried out in human primary macrophages, we show that GAS is able to induce strong inflammatory responses and efficient inflammasome activation. As this activation occurred in a similar fashion by live and dead bacteria and independently of major streptococcal virulence factors SLO or SLS, it seems that no secreted bacterial components or the ability to escape from the endosome–lysosome pathway are required. We also show that dynamin-dependent endocytosis is required for the induction of inflammatory responses after GAS stimulation, but the exact mechanism remains unknown. The data provided by this study may be essential in developing novel ways to control excessive inflammatory reactions during bacterial infections such as those induced by S. pyogenes.
Acknowledgments
The technical assistance of Hanna Valtonen and Teija Aalto is greatly acknowledged. The work was supported by a project grant no. 252252 and a special EraNet pathogenomics programme grant no. 130098 of the Medical Research Council of the Academy of Finland, and by the Sigrid Juselius and Oskar Öfflund Foundations.
Disclosure
The authors declare no conflicts of interest.
Author contributions
S. L. designed the study, performed laboratory experiments, analysed the results and wrote the paper. S. M. M. performed laboratory experiments and took part in writing of the manuscript. M. M. took part in designing the study and writing of the manuscript. E. C. took part in designing the study and writing of the manuscript. I. J. designed the study, analysed the results, and wrote the paper.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web-site:
Fig. S1. Kinetics of tumour necrosis factor (TNF)-α mRNA expression in bacteria-stimulated macrophages. Macrophages were stimulated with live, ultraviolet (UV)-inactivated, heat-inactivated, live streptolysin O (SLO)-mutant or live streptolysin S (SLS)-mutant group A streptococcus (GAS) (multiplicity of infection 10). Macrophages were collected at 3 or 9 h after stimulation and cells from different blood donors were pooled. Total cellular RNA was isolated and TNF-α mRNA levels were analysed by quantitative reverse transcription–polymerase chain reaction (qRT–PCR). Relative mRNA levels were normalized against β-actin and the relative level of mRNA was calculated with the ΔΔ comparative threshold (Ct) method using untreated cells as a calibrator. Data are from a representative experiment of three.
Fig. S2. Proinflammatory cytokine mRNA expression and interferon (IFN)-β and IFN-inducible chemokine (C-X-C motif) ligand (CXCL)-10 mRNA expression are induced in group A streptococcus (GAS)-stimulated macrophages and inhibited by the dynamin inhibitor dynasore. Macrophages were left untreated or treated with dynasore (80 μM) for 30 min prior to stimulation with live, Group A streptococ cus (GAS)-inactivated, heat-inactivated, live streptolysin O (SLO)-mutant, live streptolysin S (SLS)-mutant GAS [multiplicity of infection (MOI) 10] or Sendai virus (MOI 5) for 3 h. (a) Interleukin (IL)-1β and tumour necrosis factor (TNF)-α mRNA levels and (b) IFN-β and CXCL-10 levels were analysed by quantitative reverse transcription–polymerase chain reaction (qRT–PCR). Relative mRNA levels were normalized against β-actin and the relative level of mRNA was calculated with the ΔΔ comparative threshold (Ct) method using dimethylsulphoxide (DMSO)-treated cells as a calibrator. Results are from three independent blood donors and the means are indicated in the figure by horizontal lines. Data are from a representative experiment. Note the differences in scales.
References
- 1.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 2.Ralph AP, Carapetis JR. Group A streptococcal diseases and their global burden. Curr Top Microbiol Immunol. 2013;368:1–27. doi: 10.1007/82_2012_280. [DOI] [PubMed] [Google Scholar]
- 3.Cole JN, Barnett TC, Nizet V, Walker MJ. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol. 2011;9:724–736. doi: 10.1038/nrmicro2648. [DOI] [PubMed] [Google Scholar]
- 4.Kwinn LA, Nizet V. How group A Streptococcus circumvents host phagocyte defenses. Future Microbiol. 2007;2:75–84. doi: 10.2217/17460913.2.1.75. [DOI] [PubMed] [Google Scholar]
- 5.Hertzen E, Johansson L, Wallin R, et al. M1 protein-dependent intracellular trafficking promotes persistence and replication of Streptococcus pyogenes in macrophages. J Innate Immun. 2010;2:534–545. doi: 10.1159/000317635. [DOI] [PubMed] [Google Scholar]
- 6.Maloy KJ. The interleukin-23/interleukin-17 axis in intestinal inflammation. J Intern Med. 2008;263:584–590. doi: 10.1111/j.1365-2796.2008.01950.x. [DOI] [PubMed] [Google Scholar]
- 7.Alouf JE. Streptococcal toxins (streptolysin O, streptolysin S, erythrogenic toxin) Pharmacol Ther. 1980;11:661–717. doi: 10.1016/0163-7258(80)90045-5. [DOI] [PubMed] [Google Scholar]
- 8.Nizet V. Streptococcal beta-hemolysins: genetics and role in disease pathogenesis. Trends Microbiol. 2002;10:575–580. doi: 10.1016/s0966-842x(02)02473-3. [DOI] [PubMed] [Google Scholar]
- 9.Stout RD, Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76:509–513. doi: 10.1189/jlb.0504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 11.Gratz N, Siller M, Schaljo B, et al. Group A streptococcus activates type I interferon production and MyD88-dependent signaling without involvement of TLR2, TLR4, and TLR9. J Biol Chem. 2008;283:19879–19887. doi: 10.1074/jbc.M802848200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 13.Netea MG, Simon A, van de Veerdonk F, Kullberg BJ, Van der Meer JW, Joosten LA. IL-1beta processing in host defense: beyond the inflammasomes. PLOS Pathog. 2010;6:e1000661. doi: 10.1371/journal.ppat.1000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dinarello CA. Interleukin-1 beta, interleukin-18, and the interleukin-1 beta converting enzyme. Ann NY Acad Sci. 1998;856:1–11. doi: 10.1111/j.1749-6632.1998.tb08307.x. [DOI] [PubMed] [Google Scholar]
- 15.Schonbeck U, Mach F, Libby P. Generation of biologically active IL-1 beta by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1 beta processing. J Immunol. 1998;161:3340–3346. [PubMed] [Google Scholar]
- 16.Netea MG, Nold-Petry CA, Nold MF, et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood. 2009;113:2324–2335. doi: 10.1182/blood-2008-03-146720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miettinen M, Lehtonen A, Julkunen I, Matikainen S. Lactobacilli and Streptococci activate NF-kappa B and STAT signaling pathways in human macrophages. J Immunol. 2000;164:3733–3740. doi: 10.4049/jimmunol.164.7.3733. [DOI] [PubMed] [Google Scholar]
- 18.Lehtonen A, Ahlfors H, Veckman V, Miettinen M, Lahesmaa R, Julkunen I. Gene expression profiling during differentiation of human monocytes to macrophages or dendritic cells. J Leukoc Biol. 2007;82:710–720. doi: 10.1189/jlb.0307194. [DOI] [PubMed] [Google Scholar]
- 19.Latvala S, Pietila TE, Veckman V, et al. Potentially probiotic bacteria induce efficient maturation but differential cytokine production in human monocyte-derived dendritic cells. World J Gastroenterol. 2008;14:5570–5583. doi: 10.3748/wjg.14.5570. discussion 5581–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Latvala S, Miettinen M, Kekkonen RA, Korpela R, Julkunen I. Lactobacillus rhamnosus GG and Streptococcus thermophilus induce suppressor of cytokine signalling 3 (SOCS3) gene expression directly and indirectly via interleukin-10 in human primary macrophages. Clin Exp Immunol. 2011;165:94–103. doi: 10.1111/j.1365-2249.2011.04408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pietila TE, Latvala S, Osterlund P, Julkunen I. Inhibition of dynamin-dependent endocytosis interferes with type III IFN expression in bacteria-infected human monocyte-derived DCs. J Leukoc Biol. 2010;88:665–674. doi: 10.1189/jlb.1009651. [DOI] [PubMed] [Google Scholar]
- 22.Makela SM, Osterlund P, Julkunen I. TLR ligands induce synergistic interferon-beta and interferon-lambda1 gene expression in human monocyte-derived dendritic cells. Mol Immunol. 2011;48:505–515. doi: 10.1016/j.molimm.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 23.Makela SM, Strengell M, Pietila TE, Osterlund P, Julkunen I. Multiple signaling pathways contribute to synergistic TLR ligand-dependent cytokine gene expression in human monocyte-derived macrophages and dendritic cells. J Leukoc Biol. 2009;85:664–672. doi: 10.1189/jlb.0808503. [DOI] [PubMed] [Google Scholar]
- 24.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 25.Kirchhausen T, Macia E, Pelish HE. Use of dynasore, the small molecule inhibitor of dynamin, in the regulation of endocytosis. Methods Enzymol. 2008;438:77–93. doi: 10.1016/S0076-6879(07)38006-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miettinen M, Vuopio-Varkila J, Varkila K. Production of human tumor necrosis factor alpha, interleukin-6, and interleukin-10 is induced by lactic acid bacteria. Infect Immun. 1996;64:5403–5405. doi: 10.1128/iai.64.12.5403-5405.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pirhonen J, Sareneva T, Julkunen I, Matikainen S. Virus infection induces proteolytic processing of IL-18 in human macrophages via caspase-1 and caspase-3 activation. Eur J Immunol. 2001;31:726–733. doi: 10.1002/1521-4141(200103)31:3<726::aid-immu726>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 28.Sillanpaa M, Kaukinen P, Melen K, Julkunen I. Hepatitis C virus proteins interfere with the activation of chemokine gene promoters and downregulate chemokine gene expression. J Gen Virol. 2008;89:432–443. doi: 10.1099/vir.0.83316-0. [DOI] [PubMed] [Google Scholar]
- 29.Gratz N, Hartweger H, Matt U, et al. Type I interferon production induced by Streptococcus pyogenes-derived nucleic acids is required for host protection. PLOS Pathog. 2011;7:e1001345. doi: 10.1371/journal.ppat.1001345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harder J, Franchi L, Munoz-Planillo R, Park JH, Reimer T, Nunez G. Activation of the Nlrp3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-kappa B activation but proceeds independently of TLR signaling and P2X7 receptor. J Immunol. 2009;183:5823–5829. doi: 10.4049/jimmunol.0900444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marsh M, Helenius A. Virus entry: open sesame. Cell. 2006;124:729–740. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Claudinon J, Monier MN, Lamaze C. Interfering with interferon receptor sorting and trafficking: impact on signaling. Biochimie. 2007;89:735–743. doi: 10.1016/j.biochi.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 33.Molloy EM, Cotter PD, Hill C, Mitchell DA, Ross RP. Streptolysin S-like virulence factors: the continuing sagA. Nat Rev Microbiol. 2011;9:670–681. doi: 10.1038/nrmicro2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldmann O, Rohde M, Chhatwal GS, Medina E. Role of macrophages in host resistance to group A streptococci. Infect Immun. 2004;72:2956–2963. doi: 10.1128/IAI.72.5.2956-2963.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thulin P, Johansson L, Low DE, et al. Viable group A streptococci in macrophages during acute soft tissue infection. PLOS Med. 2006;3:e53. doi: 10.1371/journal.pmed.0030053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.LaPenta D, Rubens C, Chi E, Cleary PP. Group A streptococci efficiently invade human respiratory epithelial cells. Proc Natl Acad Sci USA. 1994;91:12115–12119. doi: 10.1073/pnas.91.25.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rohde M, Muller E, Chhatwal GS, Talay SR. Host cell caveolae act as an entry-port for group A streptococci. Cell Microbiol. 2003;5:323–342. doi: 10.1046/j.1462-5822.2003.00279.x. [DOI] [PubMed] [Google Scholar]
- 38.Staali L, Morgelin M, Bjorck L, Tapper H. Streptococcus pyogenes expressing M and M-like surface proteins are phagocytosed but survive inside human neutrophils. Cell Microbiol. 2003;5:253–265. doi: 10.1046/j.1462-5822.2003.00272.x. [DOI] [PubMed] [Google Scholar]
- 39.Veckman V, Miettinen M, Pirhonen J, Siren J, Matikainen S, Julkunen I. Streptococcus pyogenes and Lactobacillus rhamnosus differentially induce maturation and production of Th1-type cytokines and chemokines in human monocyte-derived dendritic cells. J Leukoc Biol. 2004;75:764–771. doi: 10.1189/jlb.1003461. [DOI] [PubMed] [Google Scholar]
- 40.Miettinen M, Pietila TE, Kekkonen RA, et al. Nonpathogenic Lactobacillus rhamnosus activates the inflammasome and antiviral responses in human macrophages. Gut Microbes. 2012;3:510–522. doi: 10.4161/gmic.21736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakagawa I, Amano A, Mizushima N, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 42.Hakansson A, Bentley CC, Shakhnovic EA, Wessels MR. Cytolysin-dependent evasion of lysosomal killing. Proc Natl Acad Sci USA. 2005;102:5192–5197. doi: 10.1073/pnas.0408721102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Freche B, Reig N, van der Goot FG. The role of the inflammasome in cellular responses to toxins and bacterial effectors. Semin Immunopathol. 2007;29:249–260. doi: 10.1007/s00281-007-0085-0. [DOI] [PubMed] [Google Scholar]
- 44.Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 45.Coeshott C, Ohnemus C, Pilyavskaya A, et al. Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc Natl Acad Sci USA. 1999;96:6261–6266. doi: 10.1073/pnas.96.11.6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kayagaki N, Wong MT, Stowe IB, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 47.Pahlman LI, Morgelin M, Eckert J, et al. Streptococcal M protein: a multipotent and powerful inducer of inflammation. J Immunol. 2006;177:1221–1228. doi: 10.4049/jimmunol.177.2.1221. [DOI] [PubMed] [Google Scholar]
- 48.Parker D, Prince A. Staphylococcus aureus induces type I IFN signaling in dendritic cells via TLR9. J Immunol. 2012;189:4040–4046. doi: 10.4049/jimmunol.1201055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barbalat R, Lau L, Locksley RM, Barton GM. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat Immunol. 2009;10:1200–1207. doi: 10.1038/ni.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stockinger S, Materna T, Stoiber D, et al. Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes. J Immunol. 2002;169:6522–6529. doi: 10.4049/jimmunol.169.11.6522. [DOI] [PubMed] [Google Scholar]
- 52.Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 53.Stockinger S, Reutterer B, Schaljo B, et al. IFN regulatory factor 3-dependent induction of type I IFNs by intracellular bacteria is mediated by a TLR- and Nod2-independent mechanism. J Immunol. 2004;173:7416–7425. doi: 10.4049/jimmunol.173.12.7416. [DOI] [PubMed] [Google Scholar]
- 54.O'Riordan M, Yi CH, Gonzales R, Lee KD, Portnoy DA. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc Natl Acad Sci USA. 2002;99:13861–13866. doi: 10.1073/pnas.202476699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 56.Park JM, Ng VH, Maeda S, Rest RF, Karin M. Anthrolysin O and other gram-positive cytolysins are toll-like receptor 4 agonists. J Exp Med. 2004;200:1647–1655. doi: 10.1084/jem.20041215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol. 2007;8:603–612. doi: 10.1038/nrm2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonazzi M, Cossart P. Bacterial entry into cells: a role for the endocytic machinery. FEBS Lett. 2006;580:2962–2967. doi: 10.1016/j.febslet.2006.04.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Kinetics of tumour necrosis factor (TNF)-α mRNA expression in bacteria-stimulated macrophages. Macrophages were stimulated with live, ultraviolet (UV)-inactivated, heat-inactivated, live streptolysin O (SLO)-mutant or live streptolysin S (SLS)-mutant group A streptococcus (GAS) (multiplicity of infection 10). Macrophages were collected at 3 or 9 h after stimulation and cells from different blood donors were pooled. Total cellular RNA was isolated and TNF-α mRNA levels were analysed by quantitative reverse transcription–polymerase chain reaction (qRT–PCR). Relative mRNA levels were normalized against β-actin and the relative level of mRNA was calculated with the ΔΔ comparative threshold (Ct) method using untreated cells as a calibrator. Data are from a representative experiment of three.
Fig. S2. Proinflammatory cytokine mRNA expression and interferon (IFN)-β and IFN-inducible chemokine (C-X-C motif) ligand (CXCL)-10 mRNA expression are induced in group A streptococcus (GAS)-stimulated macrophages and inhibited by the dynamin inhibitor dynasore. Macrophages were left untreated or treated with dynasore (80 μM) for 30 min prior to stimulation with live, Group A streptococ cus (GAS)-inactivated, heat-inactivated, live streptolysin O (SLO)-mutant, live streptolysin S (SLS)-mutant GAS [multiplicity of infection (MOI) 10] or Sendai virus (MOI 5) for 3 h. (a) Interleukin (IL)-1β and tumour necrosis factor (TNF)-α mRNA levels and (b) IFN-β and CXCL-10 levels were analysed by quantitative reverse transcription–polymerase chain reaction (qRT–PCR). Relative mRNA levels were normalized against β-actin and the relative level of mRNA was calculated with the ΔΔ comparative threshold (Ct) method using dimethylsulphoxide (DMSO)-treated cells as a calibrator. Results are from three independent blood donors and the means are indicated in the figure by horizontal lines. Data are from a representative experiment. Note the differences in scales.