

Abstract
Long-term epilepsy associated tumors (LEAT) represent a well known cause of focal epilepsies. Glioneuronal tumors are the most frequent histological type consisting of a mixture of glial and neuronal elements and most commonly arising in the temporal lobe. Cortical dysplasia or other neuronal migration abnormalities often coexist. Epilepsy associated with LEAT is generally poorly controlled by antiepileptic drugs while, on the other hand, it is high responsive to surgical treatment. However the best management strategy of tumor-related focal epilepsies remains controversial representing a contemporary issues in epilepsy surgery. Temporo-mesial LEAT have a widespread epileptic network with complex epileptogenic mechanisms. By using an epilepsy surgery oriented strategy LEAT may have an excellent seizure outcome therefore surgical treatment should be offered early, irrespective of pharmacoresistance, avoiding both the consequences of uncontrolled seizures as well as the side effects of prolonged pharmacological therapy and the rare risk of malignant transformation.
Keywords: Epilepsy, Low grade tumors, Long-term epilepsy associated tumors, Glioneuronal tumors, Ganglioglioma, Dysembryoplastic neuroepithelial tumor, Lesionectomy, Epilepsy surgery
Core tip: Long-term epilepsy associated tumors (LEAT) represent a frequent cause of focal epilepsies, particularly in children and young adults. Epilepsy associated with LEAT is generally poorly controlled by antiepileptic drugs while it is extremely responsive to surgical treatment. Temporo-mesial LEAT have a widespread epileptic network and complex epileptogenic mechanisms. The best management strategy of tumor-related focal epilepsies remains controversial representing a contemporary issues in epilepsy surgery.
INTRODUCTION
Brain tumors, mostly low grade tumors, are associated with epilepsy in more than a half of cases and approximately 30% of tumor-associated epilepsy are pharmacoresistant.
Recent advances in neuroimaging and neurophysiology have allowed the recognition of subtle epilepsy-associated focal structural lesions, and have improved our understanding of the complex functional relevance of these lesions for seizure generation. Among this group of lesions the concept of long-term epilepsy associated tumors (LEAT) describes the wide group of low grade tumors in patients associated with chronic focal epilepsy[1-5]. Indeed, developmental brain lesions, in particular glioneuronal tumors (GNT), often associated with malformations of cortical development, in particular focal cortical dysplasia (FCD)[1,3,4], are among the most common causes of pharmacologically intractable epilepsy. In the setting of epilepsy surgery a brain tumor is the second most common cause of focal epilepsy[6] and it could be encountered in approximately 30% of patients operated on for refractory focal epilepsy[5,7,8].
Epilepsy associated-tumor is a debilitating condition, causing distress and adversely affecting the quality of life[3,5,9-17].
Epileptic seizure incidence varies according to tumour location and hystotype. Furthermore low-grade tumors often are more epileptogenic than high-grade tumours[8,10,12,13,16,18].
Epilepsy associated with brain tumours can be divided into two groups: tumors without other symptoms (usually low-grade tumors affecting children or young patients) or tumors together with neurological deficits (more frequently high-grade tumours in middle-aged and older patients)[10].
In the group of epilepsy associated with low grade tumors it is useful to further distinguish between the preeminence of oncological and epileptic logical aspects. In the group of the “diffuse low grade glioma” (LGG) the oncological aspects should prevail according to the progressive course of the neoplastic brain disease[10-12,15,16,19-22]. On the contrary in the group of LEAT, mainly represented by glioneuronal tumors (GNTs), epilepsy control should be the main goal[1,3-5,8,23-28].
The group of LEAT, is currently enlarging not only for the recognition of new, often rare, histotypes but also for the identification of tumors having hybrid and/or mixed features[3,29-32]. The biologic behaviour of LEAT is generally benign even if some tumors could present recurrence or malignant transformation[5,33]. New tumoral entities have been recently introduced and there is ongoing debate on improving consensus for diagnosis of LEAT between specialized centers[3,30]. While LEAT rarely coexist with hippocampal sclerosis (2%-25% of cases ) they may often be associated with FCD (40%-80% of cases)[2,23,24,29,34-36].
LONG TERM EPILEPSY ASSOCIATED TUMORS
The histological characteristics of these tumors influence their propensity to generate seizures.
Gangliogliomas
Gangliogliomas (GG) are the most common neoplasm causing chronic focal epileptic disorders (about 40%)[5,37-39]. GG can occur in any part of the central nervous system, although the temporal lobe is the most common location[5,33,40] followed by the frontal lobe, the optic pathway, the spinal cord, the brainstem, the cerebellum and the pineal gland.
Dysembryoplastic neuroepithelial tumors
Dysembryoplastic neuroepithelial tumors (DNT) are grade I WHO tumors with cystic components, described by Daumas-Duport et al[41] in 1988 as a typically cortical tumor affecting children and young adults with long-standing, drug-resistant epilepsy. Most frequently DNT are sited in the cortex of temporal lobe above all at the temporo-mesial site[42-46]. Rarely DNTs have been described in ectopic locations (septum pellucidum and the caudate nucleus)[47] in the pons, thalamus, basal ganglia, cerebellum, third ventricle, and brainstem. Familial occurrence of these neoplasms have been described.
Pleomorphic Xanthoastrocytoma
Recently also pleomorphic Xanthoastrocytoma (PXA) has been considered part of this group of tumors. In fact, in addition to the astrocytic nature, there is growing evidence that PXA exhibits some histological, immunophenotypic and ultrastructural neuronal features[48,49]. Furthermore occasionally, FCD can be associated with PXA.
Papillary glioneuronal tumor
Firstly described by Kim et al[50] 1997, Papillary glioneuronal tumor (PGNT) most frequently arises in a supratentorial locationwith rare case showing multilobar involvement[3]. At MRI they appear as a cystic enhancing lesion with solid areas and often a mural nodule. PGNTs affect young adults.
Pilocytic astrocytoma
Supratentorial pilocytic astrocytoma (PA) is frequently presents with chronic epilepsy. PA are included within the common histological entities encountered in series of tumor-associated epilepsy cases[3,5,30,51].
Diffuse astrocytoma
Diffuse astrocytomas mostly arise in the cerebral hemispheres of young adults (frontal and temporal cerebral lobes) and seizures represent one of the most common symptoms. It has been described in the group of LEAT[52]. Some author stated that initial presentation with seizures could influence long-term survival[30,53].
Oligodendroglioma
Oligodendrogliomas usually arise in the cerebral hemispheres of young adults. They belong to LEAT group. Seizures represent a common presenting symptom[3,5,30].
Angiocentric gliomas
low grade cerebral tumor mostly affecting children and young adults and it is more and more frequently identified in the setting of chronic epilepsy[3]. Angiocentric gliomas (AG) have a cerebro-cortical location, often with involvement of the fronto-parietal and temporal lobe.
Extraventricular neurocytoma
This rare entity, may be considered in the spectrum of GNT associated with focal epilepsy[29].
LEAT with mixed tumor features
“Hybrid” tumors constituted by mixed forms of ganglioglioma and DNT but also PXA and ganglioglioma, PXA with DNT and, PXA with an oligodendroglioma have long been recognised, representing an increasing group of tumors in epilepsy surgical series. Cases where a PA developed within a DNT, as well as PA grew in combination with a low-grade oligodendroglioma, have been noted[54,55].
TUMOR SITE
In the setting of low grade tumors associated with epilepsy, diffuse LGG (WHO grade II gliomas) are mainly found in the insular, fronto-insular, temporo-insular regions (namely paralimbic structures) representing the iso-mesocortical transition zone[56].
Instead, LEAT mainly arise in the temporo-mesial structures (namely limbic lobe) in the site of allo-isocortical transition, harboring more frequently a neuronal differentiation maybe due to their proximity to the hippocampal granular layer where neurogenesis during adult life takes place[56-62].
In our findings[8,26,28,36] according with others[62-65] in the mesial temporal lobe both the lesion and hippocampus seem to be epileptogenic even if there are no other MRI abnormality (hippocampal sclerosis) and pathological examination shows normal findings.
PATHOGENESIS OF TUMOR-ASSOCIATED SEIZURES
Epileptogenesis of brain tumors depends on the histotype and location, even if the complexity of structural and molecular changes implies a multifactoriality of the pathogenesis[10,28,66,67] and may differ according to histologies (GNT vs diffuse low grade gliomas)[4,10,15,16,68,69]. The comprehension of the epileptogenesis in GNT is crucial to treat effectively pharmacologically intractable epilepsy (as discussed above) represents the initial, and often the only, clinical manifestation of the tumor and critically affects the patient’s daily life.
LEAT are often large tumors, with a high propensity to develop seizures when located in temporal or frontal lobes[10,70]. GNT are composed of peculiar cellular components with hyperexcitable neurons, functionally integrated into excitatory circuitries, and neurochemical characteristics that can be relevant for epileptogenesis[34]. Data provided by intralesional EEG recording have demonstrated intrinsic epileptogenicity of GG and DNT[71,72]. In addition, immunocytochemical studies showing high expression of specific glutamate receptors (GluR) subtypes suggest a hyperexcitability in the neuronal constituent of GNT[73,74]. An additional mechanism that can sustain epileptogenesis is related to an unbalance between excitation and inhibition due to to a prominent expression of mGluR5 and downregulation of several gammaaminobutyric acid (GABA-A) a receptor (GABA-AR) subunits that suggest an impairment of GABAergic inhibition[75,76]. Furthermore, a disturbed ion homeostasis and transport could represent an additional potential mechanism leading to increased excitability in GNT[9,77].
Another potential epileptogenic mechanism is related to a possible role of inflammation in the pathophysiology of human epilepsy[78]. Proinflammatory molecules have been shown in experimental models to decrease the seizure threshold[78,79] and may be involved in the generation of seizures in brain tumors, particularly in GNT[80]. Different mechanisms can cause an increment of neuronal excitability, for instance by enhancing the extracellular glutamate concentrations, as well as modifying the function of both glutamate and GABA receptors. Furthermore in GNT, particularly in GG, inflammatory changes have been showed to be associated with evidence of alterations in blood-brain barrier (BBB), with albumin extravasation and uptake in tumor astrocytes[66,81]. Interestingly, some data have shown also a prominent upregulation of the mTOR pathway, known to be a key regulator of cellular changes involved in epileptogenesis in GNT, particularly in GG[82,83].
Several other additional mechanisms have been hypothesized to account for enhanced excitability in GG, such as for example, hypoxia and acidosis, ionic changes, and deposition of hemosiderin in the peritumoral region[10]. Enzymatic changes may also occur in peritumoral tissue, impairing neurotransmitter synthesis and storage, and contributing to tumor-associated epilepsy. Finally, association with cortical dysplasia (as discussed below) also has to be considered in the evaluation of the epileptogenicity of GG. Indeed the identification of a coexistent pathology may be clinically relevant since it has been extensively reported that the tumor itself may be electrically silent and the origin of seizures is from a pathological tissue adjacent to the tumor[42,77,84]. The implication is that excising the tumor and leaving in place the nearby abnormal epileptogenic tissue, may give unsatisfactory results on the seizure outcome. Young age and long duration of illness are associated with an increased risk of secondary epileptogenesis. GNT can be intrinsically epileptogenic, even when associated with FCD[71].
CLINICAL AND EEG FEATURES OF FOCAL EPILEPSY ASSOCIATED WITH LEATs
Clinical features
Focal epilepsy is the most common and often the only symptom of LEAT. Neurological deficits are relatively uncommon, varying from 0% to 15% according to different series: the neurological sparing might depend on the indolent and slow course of LEAT that might allow compensation of possible brain impairment by slowly developing plastic processes, particularly in the young age. Epilepsy can appear at any age: however, the majority of cases present with an epilepsy onset in adolescence and young adulthood. In DNET, seizures appear almost always and more than half of the patients have focal seizures with alterated consciousness, with or without secondary generalization[85]. Regarding GGs, 80%-90% of patients seizures represent the only clinical symptom (mainly secondarily generalized tonic-clonic seizures)[86,87]. As already reported, the most common location of GG is the temporal lobe where they are frequently positive to CD34 glycoprotein staining. On the contrary it is not reported the association with CD34 for GG located in other sites of the brain[37,86-88]. It could be argued that this protein might represent a marker of dysplastic differentiation and that it could contribute to epileptogenesis. Seizure semiology is related to the site of tumor. In general, complex partial seizures with aura are more common in LEAT located in the temporal lobe, whereas secondary generalization is more common in epilepsies associated with extratemporal LEAT[39]. However, the extension of the tumor-related epileptogenic area may vary according to the anatomical location of the neoplasm: in fact, several data suggest that epileptogenic zone may be more widespread and complex in focal epilepsies associated to LEAT in the mesial temporal lobe in comparison to neocortical temporal lateral locations[36,40,61,65]. Occurrence of status epilepticus has been reported to be rare. Clinical parameters that differentiated patients operated on in childhood from patients operated on in adulthood were: (1) aura that was reported more often in the adult group, but it should be noted that this finding might at least partially depend on the fact that, in general, children are less able to refer their auras; and (2) mean age at seizure: probably due to the fact that developing brain has a low seizure threshold which leads to early and frequent seizures. Moreover, in pediatric age, malformations of cortical development are most often the basis of lesional epilepsy that is characterized by a high seizure frequency that can facilitate an early diagnosis and that can lead to early evaluation for a surgical approach. No differences between the clinical features of epilepsy associated with DNET and with GG have been reported[42,89]. A favorable seizure outcome has been observed in cases with a short duration of epilepsy, only partial seizure and the lack of secondary generalization . Response of GNT-associated epilepsy to antiepileptic treatment is variable, but drug-resistance is quite common[5,39,42,90].
Task Force of the ILAE Commission on Therapeutic Strategies defined drug resistant epilepsy as the failure of adequate trials of two tolerated, appropriately chosen and used antiepileptic drug (AEDs) schedules (whether as monotherapies or in combination) to achieve sustained seizure freedom[91].
Several explanations could be at the basis for drug resistance. AEDs could be affected by the biochemical milieu of the peri-tumoral space.
Furthermore significant interactions between AEDs and other drugs (i.e., chemoterapeutics) may decrease antiepileptic effectiveness, while increasing side effects interfering with the hepatic cytochrome P450 system. Furthermore AEDs resistance might result from over expression of multi-drug resistance-related proteins (MRPs) in tumors (particularly in capillary endothelial cells and astrocytes), which restrict the penetration of lipophilic substances into the pathologic tissue[10].
EEG features
Usually interictal EEG shows spikes and/or, sharp waves, sometimes intermixed with slow activities; in some instances normal EEG have been reported. These abnormalities, in preoperative EEG are commonly lateralized to the tumor side, less often to the correct lobe. However, Morris et al[39] (1998) reported that the occurrence of interictal EEG abnormalities and ictal EEG onset in correspondence of the site of the tumor may not be predictive of seizure outcome; indeed, in some cases a post-operative poor seizure outcome has been reported in patients with EEG interictal and ictal findings perfectly concordant with tumor location. On the other hand, also patients with EEG slow or epileptiform abnormalities distant from the tumor site or with ictal EEG onset non-localized or widespread to a whole hemisphere improved regarding seizure outcome after tumor resection[39]. In temporal lobe GNT, long-term video-EEG monitoring may allow recording of seizures and identification of the epileptogenic zone; indeed, several data suggest that in mesial temporal lobe GNT a tailored resection that include, besides the tumor, the epileptogenic area as defined by the anatomic and electroclinical correlations performed on the ictal video-EEG data, provides better post-operative seizure outcome as compared to simple lesionectomy[36,45,61,92,93]. In cases of undetermined lateralization of seizure focus, invasive EEG investigations may provide useful information, although in GNT-associated focal epilepsy the main goal of intracerebral recordings is usually to map eloquent cortex in proximity of the neoplasm. Several reports focused on prediction of poor postoperative outcome by identifying ECoG spike discharge patterns in FCD and the persistence of seizure patterns or continuous epileptiform discharges in post-resection ECoG recordings . There are little evidences about the ECoG discharge patterns in patients with GNTs because of the small numbers of patients investigated[89,93,94]. Different disorders (LEAT and FCD) may have similar electrocorticographic abnormalities probably due to the common developmental origin. In these cases have been observed continuous spiking (more often in FCD), bursts, and recruiting discharges. When continuous spiking is found in GNT, it is likely to be due to associated dysplastic regions with a high neuronal density[45,93]. A recent study employed MEG to investigate possible differences in whole brain topology of epileptic glioma patients, comparing them to patients with non-glial lesions and healthy controls. LGG patients showed decreased network synchronizability compared to healthy controls in the theta frequency range (4-8 Hz), similar to patients with non glial lesions. Network characteristics are associated with clinical presentation (seizure frequency in LGG), and with poorer cognitive performance (both low grade and high grade glioma) suggesting that histology could partly determine differences in epileptogenesis and epileptic probably due to differences in cortical plasticity. Interestingly, it would seem that low grade glioma and non tumoral lesions have a decreased synchronizability that could predispose to a high occurrence of seizures and cognitive decline.
IMAGING
Gangliogliomas and gangliocytoma
The differentation between GG and gangliocytoma (GC) is mainly based on histology. They may have variable tumour size (2-3 cm) and a typical location at the periphery of cerebral hemispheres. There is usually little associated mass effect and peripheral vasogenic edema and superficial lesions may expand cortex and remodel bone.
The MR signal of GG is variable and inhomogeneous due to the presence of a combination of solid, cystic and calcified components[46,95] (Figures 1-4).
Figure 1.
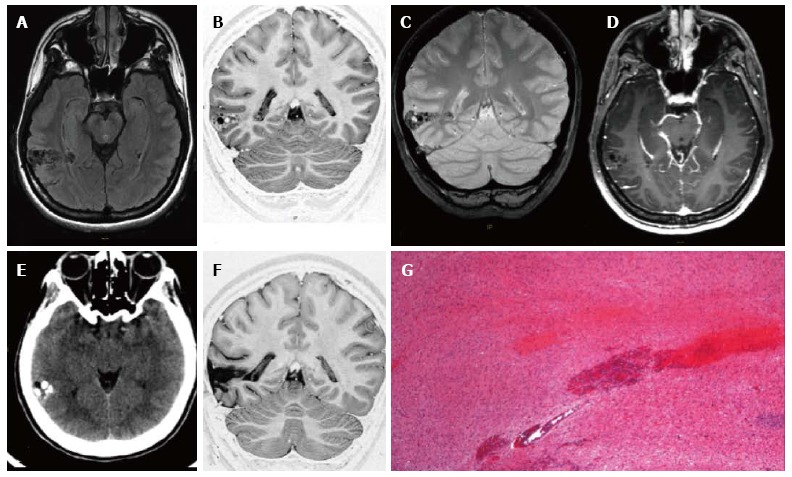
Ganglioglioma World Health Organization grade I of the right posterior middle temporal gyrus. Axial FLAIR T2-w (A) and coronal IR T1-w (B) images show inhomogeneous cortical-subcortical mass extending within the deep white matter and reaching the ependymal layer. The tumor presents a combination of solid, cystic and calcified components. The latter is better identified on coronal T2*-w sequence (C). Post-contrast axial T1-w image (D) shows no pathological enhancement and axial CT scan (E) confirms the calcified component. Coronal IR T1-w image (F) demonstrates lesion resec-tion; G: Histological examination evidences a biphasic neoplastic population, with neu-ronal and glial elements.
Figure 4.
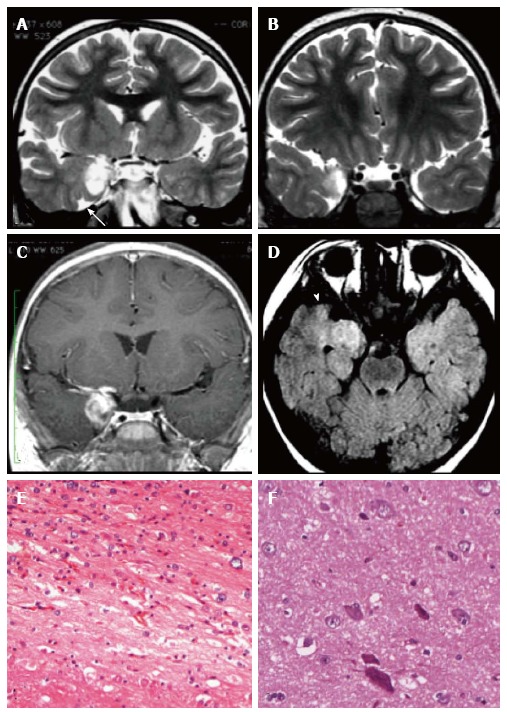
Ganglioglioma and associated focal cortical dysplasia IIa. Coronal FSE T2-w (A, B) demonstrate a temporo-mesial heterogeneously hyperintense lesion. Post-contrast coronal T1-w (C) shows enhancement of the tumor and adjacent leptomeninges. A signal abnormality extending from the surface of the ventricle to the pole (arrowheads in D) and adjacent anomalous sulci (arrow in A) were suspicious for FCD, subsequently histologically confirmed. Microscopy evidenced a tumor composed of ganglion cells intimately intermixed with astrocytic elements (E) and focal cortical dysplasia with dysmorphic neurons (FCD Type IIa) (F).
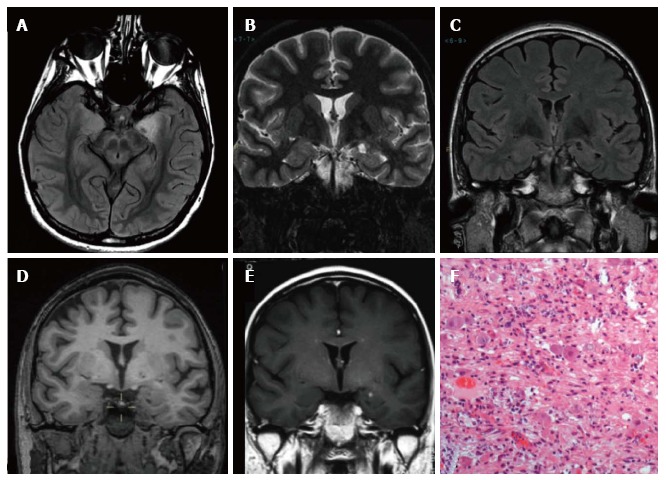
Figure 2.
Gangliocytoma and Mesial Temporal Sclerosis MTS (dual pathology). Coronal Flair T2 (A) and T1-w images (B) demonstrate a right hippocampal atrophy with signal hyperintensity on FLAIR. The ipsilateral temporal horn is dilated. Axial T1-w pre- (C) and post-contrast injection (D) show a non-enhancing multicystic lesion with calcification near the optic tract. The right mammillary body is atrophic (arrowhead); E: Neoplastic ganglion cells exhibit disorganized clusters and show abnormal cytologic features; F: Hippocampal specimen displays ILAE hippocampal sclerosis type 1, with severe pyramidal cell loss in both CA1, CA3 and CA4 sectors.
Calcifcations can be more conspicuous as areas of hypointensity on T2* gradient echo weighted images, or even more evident at unenhanced CT (Figure 1E).
Medium contrast enhancement could be variable: nodular, intense and homogenous (Figure 3E); “ringlike” appearance (Figure 4C) but also nonenhancing (Figure 1D). Although extremely rare GG may show focal leptomeningeal involvement (Figure 4C).
Figure 3.
Ganglioglioma World Health Organization grade I of the left temporo-mesial cortex. The tumor shows heterogeneous cortical-subcortical high signal on axial proton density weighted image (A). It appears partially cystic on coronal IR T1 (B) FLAIR T2 (C), T1 (D) weighted sequences. Post-contast T1-w image displays nodular, intense and homogenous enhancement (E). Low-magnification view shows a vaguely lobulated, hypocellular vascularized neoplasia, with scattered lymphocytic infiltrates (F).
Dysembryoplastic neuroepithelial tumor
DNET are well-demarcated, wedge shaped, multinodular, “bubbly” intracortical tumors often similar to other LGG.
DNET may show a multicystic morphology more frequently than GG. Absent or very slow increase in size over time is typical of DNET, and recurrence is also extremely rare. Contrast enhancement could be found in about 30% of cases.
On CT scan the tumor appears as a cortical-subcortical hypoattenuating mass with sporadic calcifications. Scalloping of the adjacent inner table of the skull may also be present. At MR imaging, DNET most commonly manifest as pseudocystic, multinodular cortical masses that are hypointense on T1-weighted images and hyperintense on T2-weighted images with minimal or without mass effect and surrounding vasogenic edema (Figures 5 and 6).
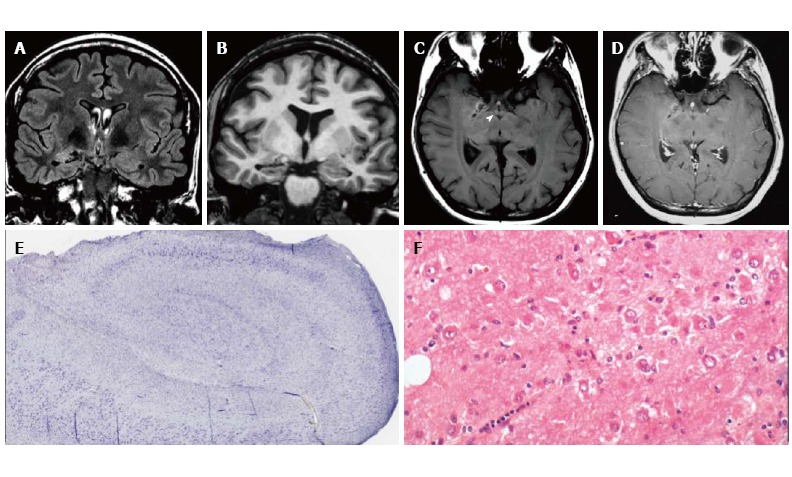
Figure 5.
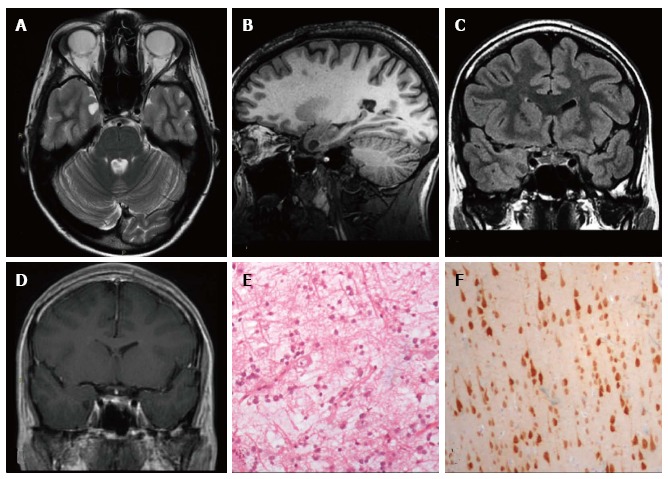
DNET of the right uncus. Axial T2-w (A) and sagittal 3D T1-w (B) reveal a cystic cortical mass well-demarcated, without perilesional oedema or mass effect. On coronal FLAIR T2-w image (C) the tumor is variably hypo- and isointense. Post-contrast axial T1-w sequence (D) shows no enhancement. Histological examination shows a tu-mor characterized by the “specific glioneuronal element”, typical of DNET, (E), while the cortex adjacent to the tumor displays cortical lamination abnormalities compatible with FCD type IIIb (F); the latter was not depicted at MR study.
Figure 6.
Extra temporal DNET. Sagittal 3D T1 (A) coronal IR T1 (B) and FLAIR T2-w (C) demonstrate a cystic wedge-shaped lesion in the right fronto-orbital gyrus. On FLAIR T2-w the tumor is slightly hypointense with a faint hyperintense rim. On post contrast coronal T1-w images there is no enhancement uptake (D); E: Post-surgical scan on cor-onal IR T1-w; F: Microscopic study evidences the presence of floating neurons, a feature of DNET, in microcystic areas lined with oligo-like cells.
Some lesions may expand involving cortical gyri and, producing a soap bubble appearance at the cortical margin. (Figures 5 and 6).
Pleomorphic Xanthoastrocytoma
PXA classically, although not specifically, appear as cystic supratentorial mass containing a mural nodule and involvong cortex and adjacent leptomeninges[96]. PXA is usually a circumscribed and slow growing lesion, that rarely recurs; size and morphology are variable.
At unenhanced CT the tumor appears as a hypo or isoattenuating mass. Calcifications are rare. On MRI T1- WI PXA are usually hypo to isointense displaying inhomogenous, mainly iso-hyperintense, signal intensity on T2-weighted sequences. Peritumoral edema is relatively uncommon. They usually enhance after gadolinium injection (Figure 7). Leptomeninges contrast enhcement is highly characteristic[97].
Figure 7.
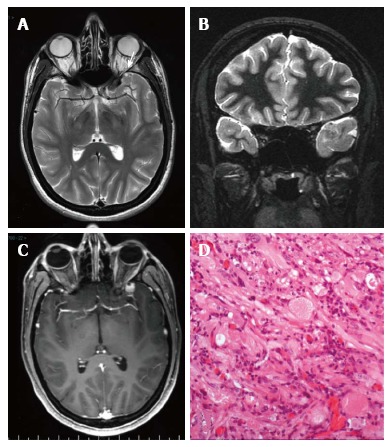
Pleomorphic xanthoastrocytoma World Health Organization grade II. Axial T2 w (A) and coronal IR T1-w (B) images show temporo-polar mixed signal intensity cortical mass with a small cystic component anteriorly (arrow in A). Post-contrast coronal SE T1w (C) shows a well-delineated, peripherally located enhancing nodule. (D) Microscopically the tumor is characterized by huge cytologic atypia, a vaguely fascicular arrangement and scattered eosinophilic granular bodies.
Extraventricular neurocytoma
Extraventricular neurocytoma may be difficult to differentiate from other types of low-grade tumor, such as GGs or DNET. It could be a well circumscribed, heterogeneous and variably enhancing mass. CT and MRI aspects depend on the cellularity and degree of calcification. They may have peritumoral oedema and intralesional cyst but rarely intralesional bleeding[29,98,99].
Pilocytic astrocytoma
Pilocytic astrocytomas are the most common form of glioma in childhood and most frequently manifest in the first two decades of life[100]. They may arise anywhere within the neuraxis, but among the pediatric population they are more frequently found infratentorially. Optic nerves, optic chiasm and hypothalamus, basal ganglia, and thalamus represent other common localtions. Cerebral hemispheres involvement has been less frequently described[100].
Pilocytic Astrocytomas are commonly characterized by fluid accumulation, with subsequent cyst formation and by mural nodule or a rim of tissue surrounding the cyst that enhances on post-contrast imaging. Predominantly solid mass lesions with minimal or no cyst have been described[101] (Figure 8).
Figure 8.
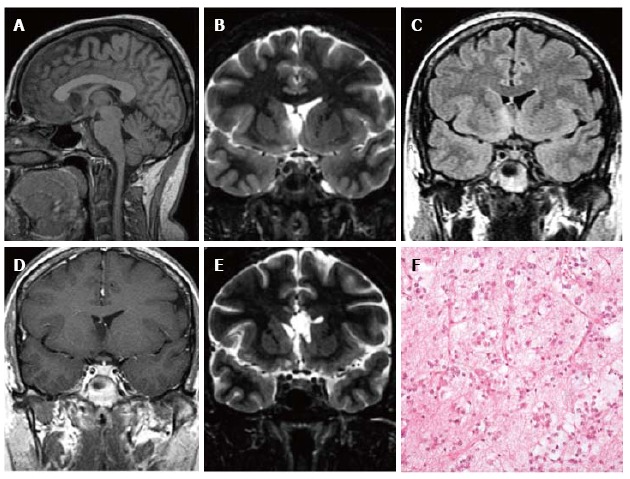
Pilocytic astrocytoma World Health Organization grade I of the right frontal lobe. Axial FLAIR T2-w (A) and coronal lR T1-w (B) images show a cortical-subcortical lesion, with cystic component, with minimal mass effect. The tumor appears well de-marcated (C) on 3D sagittal sequence and displays nodular and homogeneous en-hancement on post-contrast axial T1-w images (D). Perfusion study doesn’t show any rCBV increase within the lesion (E). MR Spectroscopy (MRS) study (F) reveals elevation in choline and reduction in NAA. (G) Histological examination shows a tumor com-posed of areas rich in myxoid material, elongated glial elements with uniform nuclei and numerous eosinophilic granular bodies.
Calcification may be seen in up to 25% of cases and haemorrhage has been reported. On MRI the solid portion of the neoplasm is typically isointense to hypointense on T1 weighted images and hyperintense on T2 weighted sequences to grey matter and it enhances after gadolinium chelates injection (Figure 8D). The signal intensity of the cystic portion is often not suppressed on FLAIR T2 weighted images due to its protein content.
MR spectroscopy reveals elevation in choline and reduction in NAA, with minimal elevation in lipid peak (Figure 8F). Lactate peak could be elevated, representing alteration in mitochondrial metabolism or variability in glucose uptake[102].
Diffuse astrocytoma
Diffuse astrocytomas are diffusely infiltrating primary brain neoplasms of astrocytic origin that are classified as WHO grade II.
Diffuse astrocytomas are usually at unenhanced CT iso to hypoattenuating lesions. They do not enhance on post-contrast imaging. Calcifications may be present in approximately 20%; cyst formation is rare.
MRI demonstrates astrocytomas as relatively homogenous mass involving and expanding more typically cerebral hemispheres cortex and adjacent white matter. The lesions are high in water content, thus appearing as hyperintense on T2 weighted images and hypo isointense on T1 weighted sequences. They usually lack peritumoral oedema (Figure 9).
Figure 9.
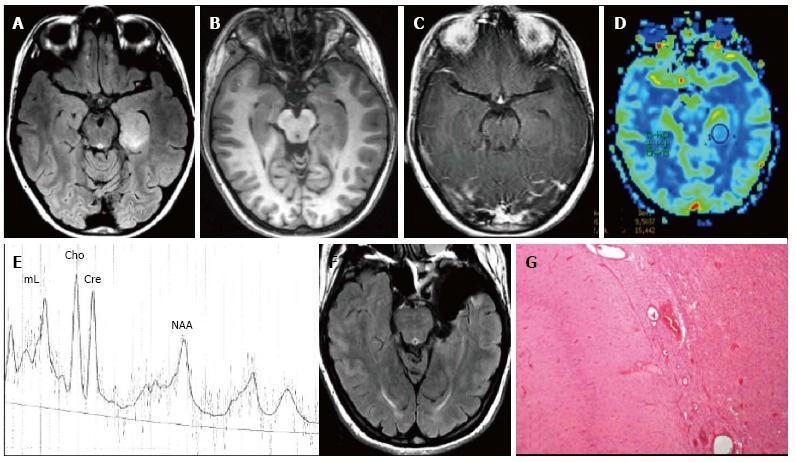
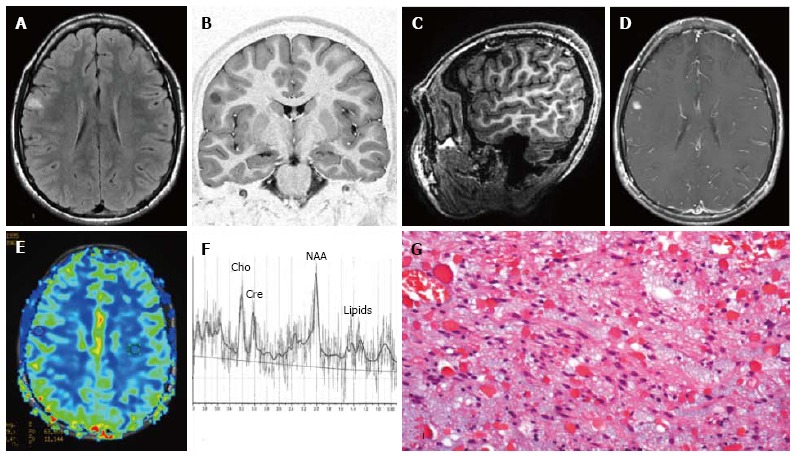
Temporo-mesial astrocytoma World Health Organization grade II. Axial FLAIR T2-w (A) shows a left temporal hyperintense mass, involving mainly the hippocampus. The lesion is slightly hypointense on T1-w image (B) and does not demonstrate enhancement after gadolinium injection (C). Perfusion study reveals no significant rCBV increase (D). MRS study shows a faint NAA reduction, a slight Cho elevation and high mI, expression of low grade glioma (E). Postsurgical axial FLAIR T2-w (F). (G) This histological picture exhibits in the left side a portion of hippocampus and in the right side an infiltrating astrocytoma, composed of fibrillary elements with varying degree of hypercellularity.
Well differentiated Astrocytomas has a variable appearance after contrast agent administration, but usually shows no significant contrast enhancement (Figure 9C). In general, contrast enhancement is not recognized as a reliable indicator of the grade of infiltrative astrocytomas. MR perfusion imaging however seems to be more informative for distinguish low- and high-grade astrocytoma and for identifying low-grade lesions that could more likely behave aggressively[103].
On dynamic contrast enhanced T2* weighted MR perfusion imaging study rCBV is typically less than 1.75 (Figure 9D)[104].
MR Spectroscopy should display Cho elevation and NAA reduction. A High myo-inositol to Creatine ratio (Myo/Cre) in also present (Figure 9E)[105].
FOCAL CORTICAL DYSPLASIA AND LEAT
LEAT and focal cortical dysplasia FCD are common findings in drug-resistant focal epilepsies, and frequently coexist[1,6,8,24,34,36,104,106-109].
Rarely MRI features of LEAT could be misinterpreted as FCD. Generally most important neuroradiological findings in FCD are increased cortical thickness, blurring of the cortical-white matter junction, increased signal on T2-W, a radially oriented linear or conical transmantle stripe of T2 hyperintensity, cortical thinning, and localized brain atrophy[34,110-112] (Figure 10).
Figure 10.
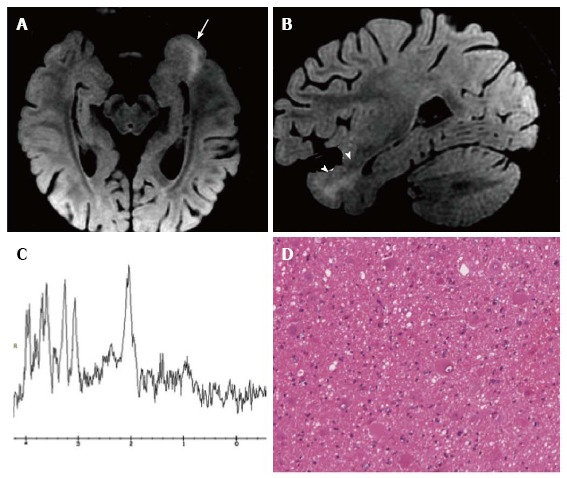
Focal cortical dysplasia with balloon cells (Taylor). Axial (A) and sagittal (B) reformatted fat-saturated 3D FLAIR images show a left temporo-mesial cortical thicken-ing (arrow) and white matter tapering to the temporal horn of the lateral ventricle (arrowheads). MR spectroscopy shows normal metabolite concentrations (C). (D) Histol-ogy demonstrates the presence of typical balloon cells, showing large and opalescent glassy eosinophilic cytoplasm.
Some limitations are encountered in the correct identification of different FCD types or subtypes[111-114]. A high number of false negatives is detected with FCD type I and slightly fewer with FCD type IIa (about 50% sensitivity). FCD IIb is much more easily identified (about 90% sensitivity)[112,113] (Figure 10).
Association between LEAT and FCD poses further issues into its correct identification. In a limited serie of patients with LEAT, who underwent surgery at our Institution associated FCD has been correctly identified in the majority of cases[113] (Figure 4). In a few cases peritumoral edema and neoplastic infiltration both caused subcortical white matter signal alterations, determining false positives (FP) and false negatives (FN) FCD results. Indeed, the signal abnormality is able to mimic a blurring, but it could hide a FCD contiguous to the tumour (Figure 11).
Figure 11.
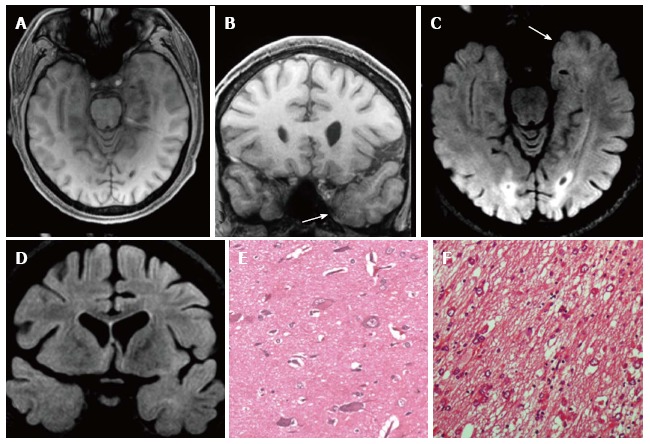
Gangliogliomas World Health Organization grade I of the left temporo-mesial region and focal cortical dysplasia IIa subtype associated. Axial and coronal T1-w images (A-B) show thickening of amygdala and uncus (arrow in B). Axial and coronal FLAIR T2-w images (C-D) present blurring and adjacent subcortical high signal abnormality compatible with a focal cortical dysplasia (FCD). Microscopy evidenced a glioneuronal tumor, with scattered binucleated ganglion cells, compatible with a gan-glioglioma (E) and dysmorphic neurons in the adjacent cortex (FCD Type IIa) (F).
These limitations were more evident when tumour size is larger (Figure 12). MRI sensibility can be reduced by an incomplete protocol too.
Figure 12.
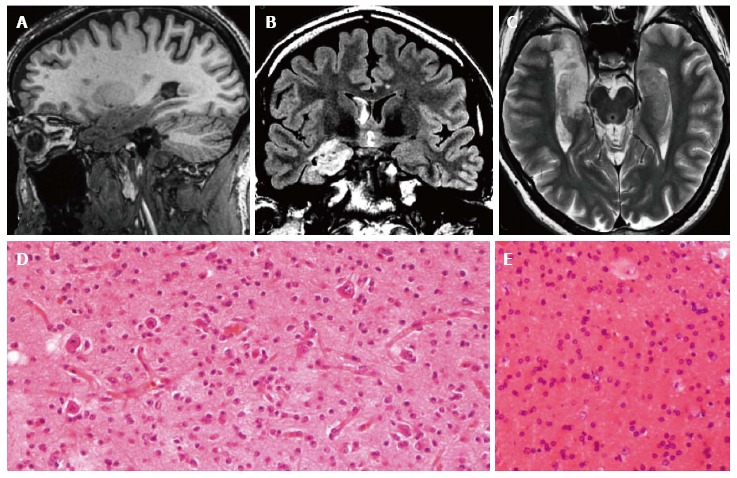
Gangliogliomas and focal cortical dysplasia IIa. Sagittal 3D T1-w (A), coronal FLAIR T2-w (B) and axial T2-w (C) reveal an inhomogeneous mass, involving the right hippocampus and the temporal pole. Due to the size of the tumor, the associated dysplasia is not clearly visible. Histological examination demonstrates the presence of a glioneuronal tumor with small ganglion cells in a desmoplastic stroma (D) and of dysmorphic neurons in the adjacent cortex (focal cortical dysplasia Type IIa) (E).
MOLECULAR ASPECT OF LEAT
The following molecular markers may facilitate differential diagnosis of LEAT: (1) IDH1 and IDH2 mutations: common in low grade diffuse gliomas (70%-80%), while they are generally not present in PA and GNT[115]; (2) LOH 1p/19q: constitutes the keystone in diagnosis of oligodendrogliomas (> 70% of tumors), while it has not been detected in DNT, a useful difference in those cases in which histological aspects do not permit a conclusive diagnosis[116]; and (3) BRAF V600E mutations: frequently found in PXA, GG and PA, whereas diffuse grade II gliomas harbor only rarely these mutations[117].
As recently observed these BRAF-mutant grade II diffuse gliomas seem to present with refractory seizures and frequently are located within the temporal lobe.It as been proposed that BRAF mutations could be strictly linked to epileptogenesis[118].
Interestingly we found that BRAF mutations could be present in the FCD associated with LEAT, suggesting a pathogenetic role of BRAF mutations in cyto-architectural dysplasia and in the tumorigenesis of LEAT[109].
SURGICAL STRATEGIES FOR LEAT
Epilepsies associated to LEAT are usually unsatisfactorily controlled by antiepileptic drugs, whereas excellent results can be achieved by surgery[4,5,26,28,42,65,92,119]. Various surgical approaches have been adopted for the radical resection of these tumors. The choice of surgical approach is also related to the goal of surgical strategy.
The surgical strategy may be directed only to oncological issues and/or to resolve epilepsy. In this last condition we must have an epilepsy surgery oriented approach.
A non-invasive presurgical study and neuropsycological assessment, may define the extension of the epileptogenic zone and may address the choice of the better surgical strategy to optimise seizure control (lesionectomy or tailored resection)[24,26,28,36,120].
Rarely in the setting of epilepsy associated tumor may be necessary an invasive presurgical study (using subdural grid, depth electrodes, or stereo-EEG)[24,64,121-123]. LEAT are certainly the prototype of cases where epilepsy is the main problem. However, in recent years, even in cases where the main problem is oncological, it is becoming equally important trying to preserve brain functions and to best cure even epilepsy (especially when tumors involve mesiotemporal structures, the insular lobe, or the central area) in order to improve the quality of life[15,63,64,124].
Several authors analyzed epileptological outcome according to surgical treatment in tumor-related chronic epilepsy. While some argue that lesionectomy alone is enough for good seizure control others say that the best management should include additional resection of epileptogenic zones adjacent to the tumor[26,28,36,39,40,44,62,119,120,125,126].
In Epilepsy-associated tumors it reaches a special meaning for the epileptogenicity and surgical strategies, the site of the lesion, i.e., temporal, mesio-temporal (or limbic), temporo-lateral, paralimbic, extratemporal, eloquent areas[8,26,28,36,40,44,45,56,63,119,125,126].
Regarding limbic and paralimbic system, the role of hippocampus in the epilepsy network is pivotal since that could play a pivotal role in epileptogenesis even without obvious neuroradiological and pathological changes (i.e., hippocampal sclerosis)[26,28,36,61,64,127].
The limbic system consists of the following elements, which are all directly or indirectly interconnected: the temporo-mesial structures (hippocampus, the parahippocampus,giryus) and the cingulate gyrus[58,60,61,128]. The paralimbic system is composed of 3 independent anatomical parts: the orbitofrontal cortex, the temporopolar cortex, and the insula[58,60,128]. The limbic system is connected via the entorhinal cortex and the uncinate fasciculus to the paralimbic system. In the setting of low-grade tumors associated with epilepsy WHO Grade II gliomas are mainly found in the paralimbic system while glioneuronal tumors are found in the limbic system (temporomesial structures)[8,57].
LEAT are frequently associated with type I or type IIa cortical dysplasia which, in contrast to focal cortical dysplasia type IIb, are more difficult to identify with MRI[1,8,24,36,109,111,113]. It means that the anatomical structural lesion can be larger than what have been detected by MRI[110,113].
For this reason the target of the surgical resection should be the epileptogenic zone defined according to neuroradiological, clinical, neurophysiological and neuropsycological findings[40,129].
In case of LEAT located near or in eloquent area, the surgical approach is usually directed only to the anatomical structural lesion. Regarding the more frequent site of these tumors, i.e., the temporal lobe, several approach have been used. The surgical strategy used in temporal lobe tumors includes lesionectomy, extended lesionectomy, tailored resection, anterior temporal lobectomy.
The majority of authors agree that lesionectomy alone provides the best seizure outcome results in LEAT located in the extratemporal and temporo-lateral site[40,42,44] while its results for temporomesial lesions are questionable[5,8,26,28,40]. Some authors suggested that the involvement of temporo-mesial regions may extend and make more complex the epileptogenic zone.
For this reason the amount of tissue removed in temporomesial surgeries is considered crucial to gain good postoperative results[5,8,60,62,63,65,125,130,131].
One important and persistent problem are the conflicting needs of the necessary extent of resection and the avoidance of neuropsychological deficits[126,131,132].
SEIZURE OUTCOME
Focal epilepsy associated with LEAT and particularly GG and DNT shows the best seizure outcome after surgery[5,18,28,36,92,95,119,132-134]. Some authors observed improvement of seizure outcome in young patients whereas others found no correlation with age at the time of surgery[4,28,38,132].
A recent literature meta-analysis about epileptogenic gangliogliomas in adult showed that an early surgical intervention of less than 3 years from the onset of seizure is significantly associated with improved seizure control[90].
Several studies reported that lesionectomy plus temporal tailored resection seems to offer the best results for seizure outcome[8,24,26,28,36,40]. Several authors insist that the the temporal pole has a pivotal role in epileptogenesis in temporomesial epilepsy[130,135,136].
The higher effectiveness of an extended resection beyond the LEAT might depend on the frequent association of this tumor type with other epileptogenic pathologies, such as the spectrum of cortical dysplasias that might represent the origin of a widespread epileptic network[8,24,34,36,68].
The new class FCD Type IIIb, which includes cases with abnormal cortical layering associated with a glial or glioneuronal tumor, has been introduced by the ILAE classification[34].
However in light of the frequent association of FCD Type II with LEAT and the immunohistochemical evidence of a common pathogenesis linking LEAT and FCD Type II[8,24,36,105], the possibility of creating a unifying class also for this kind of FCD should be considered.
With respect to oncological behavior, LEAT are usually indolent WHO Grade I lesions, although several reports have demonstrated that gangliogliomas may potentially have an evolving course and may demonstrate malignant transformation[33,40,70]. Pleomorphic xanthoastrocytoma can carry a higher risk of early recurrence when it is characterized by numerous mitoses and/or necrosis[137-139].
CONCLUSION
We believe that the adjective “long-term” included in the acronym LEAT could be prospectively confusing or misleading. Nowadays patients with LEAT are operated 5 years earlier compared to mid 1990s (mean of 7.4 years vs 12.9 years, respectively)[1,2].
The further increase in knowledge and a better recognition of these lesions among the scientific community (neurologists, epileptologists, neuropediatrics, neuroradiologists, epilepsy surgeons, neuropathologists) will lead to modify the present concept of “pharmacoresistance” vs a “tailored concept” of pharmacoresistance related to the underliyng pathology submitting many more patients to an early surgical treatment[107,132,140].
As a pathology-based approach to epilepsy surgery will be increasingly adopted[2,28,36,107,132,140,141] an early surgical treatment will become unavoidable.
In the near future this prototype of “surgically remediable cause of epilepsy” will be properly operated early, irrespective of the concept of pharmacoresistance, making the adjective “long-term” obsolete and not appropriated.
An early surgical strategy can achieve various aims, namely to obtain a definite diagnosis, to contrast epilepsy progression (including psychosocial consequences and/or adverse effects of pharmacological treatment) and even to prevent the risk, present although uncommon, of tumor growth and malignant transformation. In addition, early surgery may reduce the risk of sudden unexplained death (SUDEP) or seizure-related injuries.
Such predictable future approach will modify the clinical history of these patients, and features of epilepsy as “chronic” or “long term”, nowadays adopted in the definition of epilepsy-associated tumors, will loose sense. Neurophysiological aspects together with a proper histological and molecular characterization will become increasingly necessary for an accurate diagnosis of these epileptomas[25,34,85,142].
Therefore, what characterizes and makes up special for this group of tumors it is not the “chronic” or “long term” epilepsy history, but their pathological-biological features (i.e., sharing of immunopositivity for CD34 and of BRAF (V600E) mutation[109,143].
Footnotes
P- Reviewer: Kakisaka Y, Spalice A, Sotelo J S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
References
- 1.Blumcke I, Aronica E, Urbach H, Alexopoulos A, Gonzalez-Martinez JA. A neuropathology-based approach to epilepsy surgery in brain tumors and proposal for a new terminology use for long-term epilepsy-associated brain tumors. Acta Neuropathol. 2014;128:39–54. doi: 10.1007/s00401-014-1288-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blumcke I, Russo GL, Najm I, Palmini A. Pathology-based approach to epilepsy surgery. Acta Neuropathol. 2014;128:1–3. doi: 10.1007/s00401-014-1301-3. [DOI] [PubMed] [Google Scholar]
- 3.Thom M, Blümcke I, Aronica E. Long-term epilepsy-associated tumors. Brain Pathol. 2012;22:350–379. doi: 10.1111/j.1750-3639.2012.00582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aronica E, Leenstra S, van Veelen CW, van Rijen PC, Hulsebos TJ, Tersmette AC, Yankaya B, Troost D. Glioneuronal tumors and medically intractable epilepsy: a clinical study with long-term follow-up of seizure outcome after surgery. Epilepsy Res. 2001;43:179–191. doi: 10.1016/s0920-1211(00)00208-4. [DOI] [PubMed] [Google Scholar]
- 5.Luyken C, Blümcke I, Fimmers R, Urbach H, Elger CE, Wiestler OD, Schramm J. The spectrum of long-term epilepsy-associated tumors: long-term seizure and tumor outcome and neurosurgical aspects. Epilepsia. 2003;44:822–830. doi: 10.1046/j.1528-1157.2003.56102.x. [DOI] [PubMed] [Google Scholar]
- 6.Englot DJ, Chang EF. Rates and predictors of seizure freedom in resective epilepsy surgery: an update. Neurosurg Rev. 2014;37:389–404; discussion 404-405. doi: 10.1007/s10143-014-0527-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tassi L, Meroni A, Deleo F, Villani F, Mai R, Russo GL, Colombo N, Avanzini G, Falcone C, Bramerio M, et al. Temporal lobe epilepsy: neuropathological and clinical correlations in 243 surgically treated patients. Epileptic Disord. 2009;11:281–292. doi: 10.1684/epd.2009.0279. [DOI] [PubMed] [Google Scholar]
- 8.Giulioni M, Rubboli G, Marucci G, Martinoni M, Marliani AF, Bartiromo F, Calbucci F. Focal epilepsies associated with glioneuronal tumors: review article. Panminerva Med. 2013;55:225–238. [PubMed] [Google Scholar]
- 9.Beaumont A, Whittle IR. The pathogenesis of tumour associated epilepsy. Acta Neurochir (Wien) 2000;142:1–15. doi: 10.1007/s007010050001. [DOI] [PubMed] [Google Scholar]
- 10.van Breemen MS, Wilms EB, Vecht CJ. Epilepsy in patients with brain tumours: epidemiology, mechanisms, and management. Lancet Neurol. 2007;6:421–430. doi: 10.1016/S1474-4422(07)70103-5. [DOI] [PubMed] [Google Scholar]
- 11.Chang EF, Potts MB, Keles GE, Lamborn KR, Chang SM, Barbaro NM, Berger MS. Seizure characteristics and control following resection in 332 patients with low-grade gliomas. J Neurosurg. 2008;108:227–235. doi: 10.3171/JNS/2008/108/2/0227. [DOI] [PubMed] [Google Scholar]
- 12.Sherman JH, Moldovan K, Yeoh HK, Starke RM, Pouratian N, Shaffrey ME, Schiff D. Impact of temozolomide chemotherapy on seizure frequency in patients with low-grade gliomas. J Neurosurg. 2011;114:1617–1621. doi: 10.3171/2010.12.JNS101602. [DOI] [PubMed] [Google Scholar]
- 13.Michelucci R, Pasini E, Meletti S, Fallica E, Rizzi R, Florindo I, Chiari A, Monetti C, Cremonini AM, Forlivesi S, et al. Epilepsy in primary cerebral tumors: the characteristics of epilepsy at the onset (results from the PERNO study--Project of Emilia Romagna Region on Neuro-Oncology) Epilepsia. 2013;54 Suppl 7:86–91. doi: 10.1111/epi.12314. [DOI] [PubMed] [Google Scholar]
- 14.Maschio M. Brain tumor-related epilepsy. Curr Neuropharmacol. 2012;10:124–133. doi: 10.2174/157015912800604470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pallud J, Audureau E, Blonski M, Sanai N, Bauchet L, Fontaine D, Mandonnet E, Dezamis E, Psimaras D, Guyotat J, et al. Epileptic seizures in diffuse low-grade gliomas in adults. Brain. 2014;137:449–462. doi: 10.1093/brain/awt345. [DOI] [PubMed] [Google Scholar]
- 16.Rudà R, Bello L, Duffau H, Soffietti R. Seizures in low-grade gliomas: natural history, pathogenesis, and outcome after treatments. Neuro Oncol. 2012;14 Suppl 4:iv55–iv64. doi: 10.1093/neuonc/nos199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajneesh KF, Binder DK. Tumor-associated epilepsy. Neurosurg Focus. 2009;27:E4. doi: 10.3171/2009.5.FOCUS09101. [DOI] [PubMed] [Google Scholar]
- 18.Chang EF, Christie C, Sullivan JE, Garcia PA, Tihan T, Gupta N, Berger MS, Barbaro NM. Seizure control outcomes after resection of dysembryoplastic neuroepithelial tumor in 50 patients. J Neurosurg Pediatr. 2010;5:123–130. doi: 10.3171/2009.8.PEDS09368. [DOI] [PubMed] [Google Scholar]
- 19.Rudà R, Trevisan E, Soffietti R. Epilepsy and brain tumors. Curr Opin Oncol. 2010;22:611–620. doi: 10.1097/CCO.0b013e32833de99d. [DOI] [PubMed] [Google Scholar]
- 20.Sanai N, Chang S, Berger MS. Low-grade gliomas in adults. J Neurosurg. 2011;115:948–965. doi: 10.3171/2011.7.JNS101238. [DOI] [PubMed] [Google Scholar]
- 21.Duffau H. Awake surgery for incidental WHO grade II gliomas involving eloquent areas. Acta Neurochir (Wien) 2012;154:575–584; discussion 584. doi: 10.1007/s00701-011-1216-x. [DOI] [PubMed] [Google Scholar]
- 22.Duffau H, Peggy Gatignol ST, Mandonnet E, Capelle L, Taillandier L. Intraoperative subcortical stimulation mapping of language pathways in a consecutive series of 115 patients with Grade II glioma in the left dominant hemisphere. J Neurosurg. 2008;109:461–471. doi: 10.3171/JNS/2008/109/9/0461. [DOI] [PubMed] [Google Scholar]
- 23.Aronica E, Crino PB. Epilepsy related to developmental tumors and malformations of cortical development. Neurotherapeutics. 2014;11:251–268. doi: 10.1007/s13311-013-0251-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cossu M, Fuschillo D, Bramerio M, Galli C, Gozzo F, Pelliccia V, Casaceli G, Tassi L, Lo Russo G. Epilepsy surgery of focal cortical dysplasia-associated tumors. Epilepsia. 2013;54 Suppl 9:115–122. doi: 10.1111/epi.12455. [DOI] [PubMed] [Google Scholar]
- 25.Japp A, Gielen GH, Becker AJ. Recent aspects of classification and epidemiology of epilepsy-associated tumors. Epilepsia. 2013;54 Suppl 9:5–11. doi: 10.1111/epi.12436. [DOI] [PubMed] [Google Scholar]
- 26.Giulioni M, Rubboli G, Marucci G, Martinoni M, Volpi L, Michelucci R, Marliani AF, Bisulli F, Tinuper P, Castana L, et al. Seizure outcome of epilepsy surgery in focal epilepsies associated with temporomesial glioneuronal tumors: lesionectomy compared with tailored resection. J Neurosurg. 2009;111:1275–1282. doi: 10.3171/2009.3.JNS081350. [DOI] [PubMed] [Google Scholar]
- 27.Giulioni M. Epilepsy. J Neurosurg. 2013;118:915–917. doi: 10.3171/2012.11.JNS12574. [DOI] [PubMed] [Google Scholar]
- 28.Babini M, Giulioni M, Galassi E, Marucci G, Martinoni M, Rubboli G, Volpi L, Zucchelli M, Nicolini F, Marliani AF, et al. Seizure outcome of surgical treatment of focal epilepsy associated with low-grade tumors in children. J Neurosurg Pediatr. 2013;11:214–223. doi: 10.3171/2012.11.PEDS12137. [DOI] [PubMed] [Google Scholar]
- 29.Giulioni M, Martinoni M, Rubboli G, Marucci G, Marliani AF, Battaglia S, Badaloni F, Pozzati E, Calbucci F. Temporo-mesial extraventricular neurocytoma and cortical dysplasia in focal temporal lobe epilepsy. J Clin Neurosci. 2011;18:147–148. doi: 10.1016/j.jocn.2010.03.058. [DOI] [PubMed] [Google Scholar]
- 30.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prayson RA. Brain tumors in adults with medically intractable epilepsy. Am J Clin Pathol. 2011;136:557–563. doi: 10.1309/AJCP0RBUQAQPZOUE. [DOI] [PubMed] [Google Scholar]
- 32.Prayson RA. Tumours arising in the setting of paediatric chronic epilepsy. Pathology. 2010;42:426–431. doi: 10.3109/00313025.2010.493870. [DOI] [PubMed] [Google Scholar]
- 33.Luyken C, Blümcke I, Fimmers R, Urbach H, Wiestler OD, Schramm J. Supratentorial gangliogliomas: histopathologic grading and tumor recurrence in 184 patients with a median follow-up of 8 years. Cancer. 2004;101:146–155. doi: 10.1002/cncr.20332. [DOI] [PubMed] [Google Scholar]
- 34.Blümcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A, Jacques TS, Avanzini G, Barkovich AJ, Battaglia G, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia. 2011;52:158–174. doi: 10.1111/j.1528-1167.2010.02777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bauer R, Dobesberger J, Unterhofer C, Unterberger I, Walser G, Bauer G, Trinka E, Ortler M. Outcome of adult patients with temporal lobe tumours and medically refractory focal epilepsy. Acta Neurochir (Wien) 2007;149:1211–126; discussion 1211-1226;. doi: 10.1007/s00701-007-1366-z. [DOI] [PubMed] [Google Scholar]
- 36.Giulioni M, Marucci G, Martinoni M, Volpi L, Riguzzi P, Marliani AF, Bisulli F, Tinuper P, Tassinari CA, Michelucci R, et al. Seizure outcome in surgically treated drug-resistant mesial temporal lobe epilepsy based on the recent histopathological classifications. J Neurosurg. 2013;119:37–47. doi: 10.3171/2013.3.JNS122132. [DOI] [PubMed] [Google Scholar]
- 37.Blümcke I, Wiestler OD. Gangliogliomas: an intriguing tumor entity associated with focal epilepsies. J Neuropathol Exp Neurol. 2002;61:575–584. doi: 10.1093/jnen/61.7.575. [DOI] [PubMed] [Google Scholar]
- 38.Im SH, Chung CK, Cho BK, Lee SK. Supratentorial ganglioglioma and epilepsy: postoperative seizure outcome. J Neurooncol. 2002;57:59–66. doi: 10.1023/a:1015761507357. [DOI] [PubMed] [Google Scholar]
- 39.Morris HH, Matkovic Z, Estes ML, Prayson RA, Comair YG, Turnbull J, Najm I, Kotagal P, Wyllie E. Ganglioglioma and intractable epilepsy: clinical and neurophysiologic features and predictors of outcome after surgery. Epilepsia. 1998;39:307–313. doi: 10.1111/j.1528-1157.1998.tb01378.x. [DOI] [PubMed] [Google Scholar]
- 40.Giulioni M, Gardella E, Rubboli G, Roncaroli F, Zucchelli M, Bernardi B, Tassinari CA, Calbucci F. Lesionectomy in epileptogenic gangliogliomas: seizure outcome and surgical results. J Clin Neurosci. 2006;13:529–535. doi: 10.1016/j.jocn.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 41.Daumas-Duport C, Scheithauer BW, Chodkiewicz JP, Laws ER, Vedrenne C. Dysembryoplastic neuroepithelial tumor: a surgically curable tumor of young patients with intractable partial seizures. Report of thirty-nine cases. Neurosurgery. 1988;23:545–556. doi: 10.1227/00006123-198811000-00002. [DOI] [PubMed] [Google Scholar]
- 42.Cataltepe O, Turanli G, Yalnizoglu D, Topçu M, Akalan N. Surgical management of temporal lobe tumor-related epilepsy in children. J Neurosurg. 2005;102:280–287. doi: 10.3171/ped.2005.102.3.0280. [DOI] [PubMed] [Google Scholar]
- 43.Minkin K, Klein O, Mancini J, Lena G. Surgical strategies and seizure control in pediatric patients with dysembryoplastic neuroepithelial tumors: a single-institution experience. J Neurosurg Pediatr. 2008;1:206–210. doi: 10.3171/PED/2008/1/3/206. [DOI] [PubMed] [Google Scholar]
- 44.Giulioni M, Galassi E, Zucchelli M, Volpi L. Seizure outcome of lesionectomy in glioneuronal tumors associated with epilepsy in children. J Neurosurg. 2005;102:288–293. doi: 10.3171/ped.2005.102.3.0288. [DOI] [PubMed] [Google Scholar]
- 45.Schramm J, Aliashkevich AF. Surgery for temporal mediobasal tumors: experience based on a series of 235 patients. Neurosurgery. 2007;60:285–294; discussion 294-295. doi: 10.1227/01.NEU.0000249281.69384.D7. [DOI] [PubMed] [Google Scholar]
- 46.Adachi Y, Yagishita A. Gangliogliomas: Characteristic imaging findings and role in the temporal lobe epilepsy. Neuroradiology. 2008;50:829–834. doi: 10.1007/s00234-008-0410-x. [DOI] [PubMed] [Google Scholar]
- 47.Giulioni M, Rubboli G, Marucci G, Martinoni M, Marliani AF, Riguzzi P, Calbucci F. Focal epilepsy associated with dysembryoplastic neuroepithelial tumor in the area of the caudate nucleus. Clin Neurol Neurosurg. 2012;114:1119–1122. doi: 10.1016/j.clineuro.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 48.Perry A, Giannini C, Scheithauer BW, Rojiani AM, Yachnis AT, Seo IS, Johnson PC, Kho J, Shapiro S. Composite pleomorphic xanthoastrocytoma and ganglioglioma: report of four cases and review of the literature. Am J Surg Pathol. 1997;21:763–771. doi: 10.1097/00000478-199707000-00004. [DOI] [PubMed] [Google Scholar]
- 49.Powell SZ, Yachnis AT, Rorke LB, Rojiani AM, Eskin TA. Divergent differentiation in pleomorphic xanthoastrocytoma. Evidence for a neuronal element and possible relationship to ganglion cell tumors. Am J Surg Pathol. 1996;20:80–85. doi: 10.1097/00000478-199601000-00009. [DOI] [PubMed] [Google Scholar]
- 50.Kim DH, Suh YL. Pseudopapillary neurocytoma of temporal lobe with glial differentiation. Acta Neuropathol. 1997;94:187–191. doi: 10.1007/s004010050692. [DOI] [PubMed] [Google Scholar]
- 51.Korshunov A, Meyer J, Capper D, Christians A, Remke M, Witt H, Pfister S, von Deimling A, Hartmann C. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118:401–405. doi: 10.1007/s00401-009-0550-z. [DOI] [PubMed] [Google Scholar]
- 52.Bodi I, Selway R, Bannister P, Doey L, Mullatti N, Elwes R, Honavar M. Diffuse form of dysembryoplastic neuroepithelial tumour: the histological and immunohistochemical features of a distinct entity showing transition to dysembryoplastic neuroepithelial tumour and ganglioglioma. Neuropathol Appl Neurobiol. 2012;38:411–425. doi: 10.1111/j.1365-2990.2011.01225.x. [DOI] [PubMed] [Google Scholar]
- 53.Kim YH, Nobusawa S, Mittelbronn M, Paulus W, Brokinkel B, Keyvani K, Sure U, Wrede K, Nakazato Y, Tanaka Y, et al. Molecular classification of low-grade diffuse gliomas. Am J Pathol. 2010;177:2708–2714. doi: 10.2353/ajpath.2010.100680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiong J, Ding L, Chen H, Chen H, Wang Y. Mixed glioneuronal tumor: a dysembryoplastic neuroepithelial tumor with rosette-forming glioneuronal tumor component. Neuropathology. 2013;33:431–435. doi: 10.1111/neup.12000. [DOI] [PubMed] [Google Scholar]
- 55.Prayson RA, Napekoski KM. Composite ganglioglioma/dysembryoplastic neuroepithelial tumor: a clinicopathologic study of 8 cases. Hum Pathol. 2012;43:1113–1118. doi: 10.1016/j.humpath.2011.08.023. [DOI] [PubMed] [Google Scholar]
- 56.Mandonnet E, Capelle L, Duffau H. Extension of paralimbic low grade gliomas: toward an anatomical classification based on white matter invasion patterns. J Neurooncol. 2006;78:179–185. doi: 10.1007/s11060-005-9084-y. [DOI] [PubMed] [Google Scholar]
- 57.Capizzano AA, Kirby P, Moritani T. Limbic Tumors of the Temporal Lobe: Radiologic-Pathologic Correlation. Clin Neuroradiol. 2014 doi: 10.1007/s00062-014-0287-5. [DOI] [PubMed] [Google Scholar]
- 58.Yaşargil MG, von Ammon K, Cavazos E, Doczi T, Reeves JD, Roth P. Tumours of the limbic and paralimbic systems. Acta Neurochir (Wien) 1992;118:40–52. doi: 10.1007/BF01400725. [DOI] [PubMed] [Google Scholar]
- 59.Lövblad KO, Schaller K. Surgical anatomy and functional connectivity of the limbic system. Neurosurg Focus. 2009;27:E3. doi: 10.3171/2009.5.FOCUS09103. [DOI] [PubMed] [Google Scholar]
- 60.Wen HT, Rhoton AL, de Oliveira E, Cardoso AC, Tedeschi H, Baccanelli M, Marino R. Microsurgical anatomy of the temporal lobe: part 1: mesial temporal lobe anatomy and its vascular relationships as applied to amygdalohippocampectomy. Neurosurgery. 1999;45:549–591; discussion 591-592. doi: 10.1097/00006123-199909000-00028. [DOI] [PubMed] [Google Scholar]
- 61.Schramm J, Aliashkevich AF. Temporal mediobasal tumors: a proposal for classification according to surgical anatomy. Acta Neurochir (Wien) 2008;150:857–864; discussion 864. doi: 10.1007/s00701-008-0013-7. [DOI] [PubMed] [Google Scholar]
- 62.Schramm J. Temporal lobe epilepsy surgery and the quest for optimal extent of resection: a review. Epilepsia. 2008;49:1296–1307. doi: 10.1111/j.1528-1167.2008.01604.x. [DOI] [PubMed] [Google Scholar]
- 63.Ghareeb F, Duffau H. Intractable epilepsy in paralimbic Word Health Organization Grade II gliomas: should the hippocampus be resected when not invaded by the tumor? J Neurosurg. 2012;116:1226–1234. doi: 10.3171/2012.1.JNS112120. [DOI] [PubMed] [Google Scholar]
- 64.Duffau H. Brain mapping in tumors: intraoperative or extraoperative? Epilepsia. 2013;54 Suppl 9:79–83. doi: 10.1111/epi.12449. [DOI] [PubMed] [Google Scholar]
- 65.Clusmann H, Schramm J, Kral T, Helmstaedter C, Ostertun B, Fimmers R, Haun D, Elger CE. Prognostic factors and outcome after different types of resection for temporal lobe epilepsy. J Neurosurg. 2002;97:1131–1141. doi: 10.3171/jns.2002.97.5.1131. [DOI] [PubMed] [Google Scholar]
- 66.Shamji MF, Fric-Shamji EC, Benoit BG. Brain tumors and epilepsy: pathophysiology of peritumoral changes. Neurosurg Rev. 2009;32:275–284; discussion 284-286. doi: 10.1007/s10143-009-0191-7. [DOI] [PubMed] [Google Scholar]
- 67.Williamson A, Patrylo PR, Lee S, Spencer DD. Physiology of human cortical neurons adjacent to cavernous malformations and tumors. Epilepsia. 2003;44:1413–1419. doi: 10.1046/j.1528-1157.2003.23603.x. [DOI] [PubMed] [Google Scholar]
- 68.Aronica E, Redeker S, Boer K, Spliet WG, van Rijen PC, Gorter JA, Troost D. Inhibitory networks in epilepsy-associated gangliogliomas and in the perilesional epileptic cortex. Epilepsy Res. 2007;74:33–44. doi: 10.1016/j.eplepsyres.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 69.You G, Sha Z, Jiang T. The pathogenesis of tumor-related epilepsy and its implications for clinical treatment. Seizure. 2012;21:153–159. doi: 10.1016/j.seizure.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 70.Lee MC, Kang JY, Seol MB, Kim HS, Woo JY, Lee JS, Jung S, Kim JH, Woo YJ, Kim MK, et al. Clinical features and epileptogenesis of dysembryoplastic neuroepithelial tumor. Childs Nerv Syst. 2006;22:1611–1618. doi: 10.1007/s00381-006-0162-z. [DOI] [PubMed] [Google Scholar]
- 71.Barba C, Coras R, Giordano F, Buccoliero AM, Genitori L, Blümcke I, Guerrini R. Intrinsic epileptogenicity of gangliogliomas may be independent from co-occurring focal cortical dysplasia. Epilepsy Res. 2011;97:208–213. doi: 10.1016/j.eplepsyres.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 72.Chassoux F, Landré E, Mellerio C, Laschet J, Devaux B, Daumas-Duport C. Dysembryoplastic neuroepithelial tumors: epileptogenicity related to histologic subtypes. Clin Neurophysiol. 2013;124:1068–1078. doi: 10.1016/j.clinph.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 73.Wolf HK, Birkholz T, Wellmer J, Blümcke I, Pietsch T, Wiestler OD. Neurochemical profile of glioneuronal lesions from patients with pharmacoresistant focal epilepsies. J Neuropathol Exp Neurol. 1995;54:689–697. doi: 10.1097/00005072-199509000-00011. [DOI] [PubMed] [Google Scholar]
- 74.Aronica E, Yankaya B, Jansen GH, Leenstra S, van Veelen CW, Gorter JA, Troost D. Ionotropic and metabotropic glutamate receptor protein expression in glioneuronal tumours from patients with intractable epilepsy. Neuropathol Appl Neurobiol. 2001;27:223–237. doi: 10.1046/j.0305-1846.2001.00314.x. [DOI] [PubMed] [Google Scholar]
- 75.Samadani U, Judkins AR, Akpalu A, Aronica E, Crino PB. Differential cellular gene expression in ganglioglioma. Epilepsia. 2007;48:646–653. doi: 10.1111/j.1528-1167.2007.00925.x. [DOI] [PubMed] [Google Scholar]
- 76.Fassunke J, Majores M, Tresch A, Niehusmann P, Grote A, Schoch S, Becker AJ. Array analysis of epilepsy-associated gangliogliomas reveals expression patterns related to aberrant development of neuronal precursors. Brain. 2008;131:3034–3050. doi: 10.1093/brain/awn233. [DOI] [PubMed] [Google Scholar]
- 77.Becker AJ, Blümcke I, Urbach H, Hans V, Majores M. Molecular neuropathology of epilepsy-associated glioneuronal malformations. J Neuropathol Exp Neurol. 2006;65:99–108. doi: 10.1097/01.jnen.0000199570.19344.33. [DOI] [PubMed] [Google Scholar]
- 78.Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol. 2011;7:31–40. doi: 10.1038/nrneurol.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vezzani A, Auvin S, Ravizza T, and Aronica E: Glia-neuronal interactions in ictogenesis and epileptogenesis: role of inflammatory mediators. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies [Internet]. 4th ed. Bethesda (MD): National Center for Biotechnology Information (US); 2012. Available from: http: //www.ncbi.nlm.nih.gov/books/NBK98146/ [PubMed] [Google Scholar]
- 80.de Groot M, Toering ST, Boer K, Spliet WG, Heimans JJ, Aronica E, Reijneveld JC. Expression of synaptic vesicle protein 2A in epilepsy-associated brain tumors and in the peritumoral cortex. Neuro Oncol. 2010;12:265–273. doi: 10.1093/neuonc/nop028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Prabowo AS, Iyer AM, Veersema TJ, Anink JJ, Schouten-van Meeteren AY, Spliet WG, van Rijen PC, Ferrier CH, Capper D, Thom M, et al. BRAF V600E mutation is associated with mTOR signaling activation in glioneuronal tumors. Brain Pathol. 2014;24:52–66. doi: 10.1111/bpa.12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vezzani A. Before epilepsy unfolds: finding the epileptogenesis switch. Nat Med. 2012;18:1626–1627. doi: 10.1038/nm.2982. [DOI] [PubMed] [Google Scholar]
- 83.Wong M, Crino PB. mTOR and Epileptogenesis in Developmental Brain Malformations. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, et al., editors. Jasper's Basic Mechanisms of the Epilepsies [Internet]. 4th ed. Bethesda (MD): National Center for Biotechnology Information (US); 2012. Available from: http: //www.ncbi.nlm.nih.gov/books/NBK98145/ [PubMed] [Google Scholar]
- 84.Prayson RA, Fong J, Najm I. Coexistent pathology in chronic epilepsy patients with neoplasms. Mod Pathol. 2010;23:1097–1103. doi: 10.1038/modpathol.2010.94. [DOI] [PubMed] [Google Scholar]
- 85.Thom M, Toma A, An S, Martinian L, Hadjivassiliou G, Ratilal B, Dean A, McEvoy A, Sisodiya SM, Brandner S. One hundred and one dysembryoplastic neuroepithelial tumors: an adult epilepsy series with immunohistochemical, molecular genetic, and clinical correlations and a review of the literature. J Neuropathol Exp Neurol. 2011;70:859–878. doi: 10.1097/NEN.0b013e3182302475. [DOI] [PubMed] [Google Scholar]
- 86.Compton JJ, Laack NN, Eckel LJ, Schomas DA, Giannini C, Meyer FB. Long-term outcomes for low-grade intracranial ganglioglioma: 30-year experience from the Mayo Clinic. J Neurosurg. 2012;117:825–830. doi: 10.3171/2012.7.JNS111260. [DOI] [PubMed] [Google Scholar]
- 87.Southwell DG, Garcia PA, Berger MS, Barbaro NM, Chang EF. Long-term seizure control outcomes after resection of gangliogliomas. Neurosurgery. 2012;70:1406–1413; discussion 1406-1413. doi: 10.1227/NEU.0b013e3182500a4c. [DOI] [PubMed] [Google Scholar]
- 88.Kerkhof M, Vecht CJ. Seizure characteristics and prognostic factors of gliomas. Epilepsia. 2013;54 Suppl 9:12–17. doi: 10.1111/epi.12437. [DOI] [PubMed] [Google Scholar]
- 89.Ozlen F, Gunduz A, Asan Z, Tanriverdi T, Ozkara C, Yeni N, Yalcinkaya C, Ozyurt E, Uzan M. Dysembryoplastic neuroepithelial tumors and gangliogliomas: clinical results of 52 patients. Acta Neurochir (Wien) 2010;152:1661–1671. doi: 10.1007/s00701-010-0696-4. [DOI] [PubMed] [Google Scholar]
- 90.Yang I, Chang EF, Han SJ, Barry JJ, Fang S, Tihan T, Barbaro NM, Parsa AT. Early surgical intervention in adult patients with ganglioglioma is associated with improved clinical seizure outcomes. J Clin Neurosci. 2011;18:29–33. doi: 10.1016/j.jocn.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 91.Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, Moshé SL, Perucca E, Wiebe S, French J. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51:1069–1077. doi: 10.1111/j.1528-1167.2009.02397.x. [DOI] [PubMed] [Google Scholar]
- 92.Clusmann H, Kral T, Gleissner U, Sassen R, Urbach H, Blümcke I, Bogucki J, Schramm J. Analysis of different types of resection for pediatric patients with temporal lobe epilepsy. Neurosurgery. 2004;54:847–859; discussion 859-860. doi: 10.1227/01.neu.0000114141.37640.37. [DOI] [PubMed] [Google Scholar]
- 93.Gelinas JN, Battison AW, Smith S, Connolly MB, Steinbok P. Electrocorticography and seizure outcomes in children with lesional epilepsy. Childs Nerv Syst. 2011;27:381–390. doi: 10.1007/s00381-010-1279-7. [DOI] [PubMed] [Google Scholar]
- 94.Ferrier CH, Aronica E, Leijten FS, Spliet WG, van Huffelen AC, van Rijen PC, Binnie CD. Electrocorticographic discharge patterns in glioneuronal tumors and focal cortical dysplasia. Epilepsia. 2006;47:1477–1486. doi: 10.1111/j.1528-1167.2006.00619.x. [DOI] [PubMed] [Google Scholar]
- 95.Zentner J, Wolf HK, Ostertun B, Hufnagel A, Campos MG, Solymosi L, Schramm J. Gangliogliomas: clinical, radiological, and histopathological findings in 51 patients. J Neurol Neurosurg Psychiatry. 1994;57:1497–1502. doi: 10.1136/jnnp.57.12.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lipper MH, Eberhard DA, Phillips CD, Vezina LG, Cail WS. Pleomorphic xanthoastrocytoma, a distinctive astroglial tumor: neuroradiologic and pathologic features. AJNR Am J Neuroradiol. 1993;14:1397–1404. [PMC free article] [PubMed] [Google Scholar]
- 97.Crespo-Rodríguez AM, Smirniotopoulos JG, Rushing EJ. MR and CT imaging of 24 pleomorphic xanthoastrocytomas (PXA) and a review of the literature. Neuroradiology. 2007;49:307–315. doi: 10.1007/s00234-006-0191-z. [DOI] [PubMed] [Google Scholar]
- 98.Tortori-Donati P, Fondelli MP, Rossi A, Cama A, Brisigotti M, Pellicanò G. Extraventricular neurocytoma with ganglionic differentiation associated with complex partial seizures. AJNR Am J Neuroradiol. 1999;20:724–727. [PMC free article] [PubMed] [Google Scholar]
- 99.Yang GF, Wu SY, Zhang LJ, Lu GM, Tian W, Shah K. Imaging findings of extraventricular neurocytoma: report of 3 cases and review of the literature. AJNR Am J Neuroradiol. 2009;30:581–585. doi: 10.3174/ajnr.A1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koeller KK, Rushing EJ. From the archives of the AFIP: pilocytic astrocytoma: radiologic-pathologic correlation. Radiographics. 2004;24:1693–1708. doi: 10.1148/rg.246045146. [DOI] [PubMed] [Google Scholar]
- 101.Pencalet P, Maixner W, Sainte-Rose C, Lellouch-Tubiana A, Cinalli G, Zerah M, Pierre-Kahn A, Hoppe-Hirsch E, Bourgeois M, Renier D. Benign cerebellar astrocytomas in children. J Neurosurg. 1999;90:265–273. doi: 10.3171/jns.1999.90.2.0265. [DOI] [PubMed] [Google Scholar]
- 102.Hwang JH, Egnaczyk GF, Ballard E, Dunn RS, Holland SK, Ball WS. Proton MR spectroscopic characteristics of pediatric pilocytic astrocytomas. AJNR Am J Neuroradiol. 1998;19:535–540. [PMC free article] [PubMed] [Google Scholar]
- 103.Danchaivijitr N, Waldman AD, Tozer DJ, Benton CE, Brasil Caseiras G, Tofts PS, Rees JH, Jäger HR. Low-grade gliomas: do changes in rCBV measurements at longitudinal perfusion-weighted MR imaging predict malignant transformation? Radiology. 2008;247:170–178. doi: 10.1148/radiol.2471062089. [DOI] [PubMed] [Google Scholar]
- 104.Palmini A, Najm I, Avanzini G, Babb T, Guerrini R, Foldvary-Schaefer N, Jackson G, Lüders HO, Prayson R, Spreafico R, et al. Terminology and classification of the cortical dysplasias. Neurology. 2004;62:S2–S8. doi: 10.1212/01.wnl.0000114507.30388.7e. [DOI] [PubMed] [Google Scholar]
- 105.Englot DJ, Berger MS, Barbaro NM, Chang EF. Predictors of seizure freedom after resection of supratentorial low-grade gliomas. A review. J Neurosurg. 2011;115:240–244. doi: 10.3171/2011.3.JNS1153. [DOI] [PubMed] [Google Scholar]
- 106.Giulioni M, Martinoni M, Marucci G. Tailored pharmacoresistance related to underlying structural lesions. Epilepsy Behav. 2014;36:22–23. doi: 10.1016/j.yebeh.2014.04.023. [DOI] [PubMed] [Google Scholar]
- 107.Ortiz-González XR, Venneti S, Biegel JA, Rorke-Adams LB, Porter BE. Ganglioglioma arising from dysplastic cortex. Epilepsia. 2011;52:e106–e108. doi: 10.1111/j.1528-1167.2011.03124.x. [DOI] [PubMed] [Google Scholar]
- 108.Castillo M, Smith JK, Kwock L. Correlation of myo-inositol levels and grading of cerebral astrocytomas. AJNR Am J Neuroradiol. 2000;21:1645–1649. [PMC free article] [PubMed] [Google Scholar]
- 109.Marucci G, Martinoni M, Giulioni M. Relationship between focal cortical dysplasia and epilepsy-associated low-grade tumors: an immunohistochemical study. APMIS. 2013;121:22–29. doi: 10.1111/j.1600-0463.2012.02938.x. [DOI] [PubMed] [Google Scholar]
- 110.Urbach H. MRI of long-term epilepsy-associated tumors. Semin Ultrasound CT MR. 2008;29:40–46. doi: 10.1053/j.sult.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 111.Tassi L, Colombo N, Garbelli R, Francione S, Lo Russo G, Mai R, Cardinale F, Cossu M, Ferrario A, Galli C, et al. Focal cortical dysplasia: neuropathological subtypes, EEG, neuroimaging and surgical outcome. Brain. 2002;125:1719–1732. doi: 10.1093/brain/awf175. [DOI] [PubMed] [Google Scholar]
- 112.Colombo N, Tassi L, Deleo F, Citterio A, Bramerio M, Mai R, Sartori I, Cardinale F, Lo Russo G, Spreafico R. Focal cortical dysplasia type IIa and IIb: MRI aspects in 118 cases proven by histopathology. Neuroradiology. 2012;54:1065–1077. doi: 10.1007/s00234-012-1049-1. [DOI] [PubMed] [Google Scholar]
- 113.Tarsi A, Marliani AF, Bartiromo F, Giulioni M, Marucci G, Martinoni M, Volpi L, Leonardi M. MRI findings in low grade tumours associated with focal cortical dysplasia. Neuroradiol J. 2012;25:639–648. doi: 10.1177/197140091202500601. [DOI] [PubMed] [Google Scholar]
- 114.Chamberlain WA, Cohen ML, Gyure KA, Kleinschmidt-DeMasters BK, Perry A, Powell SZ, Qian J, Staugaitis SM, Prayson RA. Interobserver and intraobserver reproducibility in focal cortical dysplasia (malformations of cortical development) Epilepsia. 2009;50:2593–2598. doi: 10.1111/j.1528-1167.2009.02344.x. [DOI] [PubMed] [Google Scholar]
- 115.Bourne TD, Schiff D. Update on molecular findings, management and outcome in low-grade gliomas. Nat Rev Neurol. 2010;6:695–701. doi: 10.1038/nrneurol.2010.159. [DOI] [PubMed] [Google Scholar]
- 116.Ray WZ, Blackburn SL, Casavilca-Zambrano S, Barrionuevo C, Orrego JE, Heinicke H, Dowling JL, Perry A. Clinicopathologic features of recurrent dysembryoplastic neuroepithelial tumor and rare malignant transformation: a report of 5 cases and review of the literature. J Neurooncol. 2009;94:283–292. doi: 10.1007/s11060-009-9849-9. [DOI] [PubMed] [Google Scholar]
- 117.Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, Schmieder K, Wesseling P, Mawrin C, Hasselblatt M, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011;121:397–405. doi: 10.1007/s00401-011-0802-6. [DOI] [PubMed] [Google Scholar]
- 118.Chi AS, Batchelor TT, Yang D, Dias-Santagata D, Borger DR, Ellisen LW, Iafrate AJ, Louis DN. BRAF V600E mutation identifies a subset of low-grade diffusely infiltrating gliomas in adults. J Clin Oncol. 2013;31:e233–e236. doi: 10.1200/JCO.2012.46.0220. [DOI] [PubMed] [Google Scholar]
- 119.Zaghloul KA, Schramm J. Surgical management of glioneuronal tumors with drug-resistant epilepsy. Acta Neurochir (Wien) 2011;153:1551–1559. doi: 10.1007/s00701-011-1050-1. [DOI] [PubMed] [Google Scholar]
- 120.Cossu M, Lo Russo G, Francione S, Mai R, Nobili L, Sartori I, Tassi L, Citterio A, Colombo N, Bramerio M, et al. Epilepsy surgery in children: results and predictors of outcome on seizures. Epilepsia. 2008;49:65–72. doi: 10.1111/j.1528-1167.2007.01207.x. [DOI] [PubMed] [Google Scholar]
- 121.Rosenow F, Menzler K. Invasive EEG studies in tumor-related epilepsy: when are they indicated and with what kind of electrodes? Epilepsia. 2013;54 Suppl 9:61–65. doi: 10.1111/epi.12446. [DOI] [PubMed] [Google Scholar]
- 122.Cardinale F, Cossu M, Castana L, Casaceli G, Schiariti MP, Miserocchi A, Fuschillo D, Moscato A, Caborni C, Arnulfo G, et al. Stereoelectroencephalography: surgical methodology, safety, and stereotactic application accuracy in 500 procedures. Neurosurgery. 2013;72:353–366; discussion 366. doi: 10.1227/NEU.0b013e31827d1161. [DOI] [PubMed] [Google Scholar]
- 123.Cossu M, Cardinale F, Castana L, Citterio A, Francione S, Tassi L, Benabid AL, Lo Russo G. Stereoelectroencephalography in the presurgical evaluation of focal epilepsy: a retrospective analysis of 215 procedures. Neurosurgery. 2005;57:706–718; discussion 706-718. [PubMed] [Google Scholar]
- 124.Smits A, Duffau H. Seizures and the natural history of World Health Organization Grade II gliomas: a review. Neurosurgery. 2011;68:1326–1333. doi: 10.1227/NEU.0b013e31820c3419. [DOI] [PubMed] [Google Scholar]
- 125.Peace PK. Five year review of Küntscher nailing for femoral shaft fractures with emphasis on complications. Acta Orthop Belg. 2002;41:346–354. [PubMed] [Google Scholar]
- 126.Schramm J, Clusmann H. The surgery of epilepsy. Neurosurgery. 2008;62 Suppl 2:463–481; discussion 481. doi: 10.1227/01.neu.0000316250.69898.23. [DOI] [PubMed] [Google Scholar]
- 127.Morioka T, Hashiguchi K, Nagata S, Miyagi Y, Yoshida F, Shono T, Mihara F, Koga H, Sasaki T. Additional hippocampectomy in the surgical management of intractable temporal lobe epilepsy associated with glioneuronal tumor. Neurol Res. 2007;29:807–815. doi: 10.1179/016164107X223566. [DOI] [PubMed] [Google Scholar]
- 128.Wen HT, Rhoton AL, de Oliveira E, Castro LH, Figueiredo EG, Teixeira MJ. Microsurgical anatomy of the temporal lobe: part 2--sylvian fissure region and its clinical application. Neurosurgery. 2009;65:1–35; discussion 36. doi: 10.1227/01.NEU.0000336314.20759.85. [DOI] [PubMed] [Google Scholar]
- 129.Lüders HO, Najm I, Nair D, Widdess-Walsh P, Bingman W. The epileptogenic zone: general principles. Epileptic Disord. 2006;8 Suppl 2:S1–S9. [PubMed] [Google Scholar]
- 130.Chabardès S, Kahane P, Minotti L, Tassi L, Grand S, Hoffmann D, Benabid AL. The temporopolar cortex plays a pivotal role in temporal lobe seizures. Brain. 2005;128:1818–1831. doi: 10.1093/brain/awh512. [DOI] [PubMed] [Google Scholar]
- 131.Helmstaedter C, Roeske S, Kaaden S, Elger CE, Schramm J. Hippocampal resection length and memory outcome in selective epilepsy surgery. J Neurol Neurosurg Psychiatry. 2011;82:1375–1381. doi: 10.1136/jnnp.2010.240176. [DOI] [PubMed] [Google Scholar]
- 132.Ramantani G, Kadish NE, Anastasopoulos C, Brandt A, Wagner K, Strobl K, Mayer H, Schubert-Bast S, Stathi A, Korinthenberg R, et al. Epilepsy surgery for glioneuronal tumors in childhood: avoid loss of time. Neurosurgery. 2014;74:648–657; discussion 657. doi: 10.1227/NEU.0000000000000327. [DOI] [PubMed] [Google Scholar]
- 133.Ogiwara H, Nordli DR, DiPatri AJ, Alden TD, Bowman RM, Tomita T. Pediatric epileptogenic gangliogliomas: seizure outcome and surgical results. J Neurosurg Pediatr. 2010;5:271–276. doi: 10.3171/2009.10.PEDS09372. [DOI] [PubMed] [Google Scholar]
- 134.Radhakrishnan A, Abraham M, Radhakrishnan VV, Sarma SP, Radhakrishnan K. Medically refractory epilepsy associated with temporal lobe ganglioglioma: characteristics and postoperative outcome. Clin Neurol Neurosurg. 2006;108:648–654. doi: 10.1016/j.clineuro.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 135.Chan CH, Bittar RG, Davis GA, Kalnins RM, Fabinyi GC. Long-term seizure outcome following surgery for dysembryoplastic neuroepithelial tumor. J Neurosurg. 2006;104:62–69. doi: 10.3171/jns.2006.104.1.62. [DOI] [PubMed] [Google Scholar]
- 136.Takahashi A, Hong SC, Seo DW, Hong SB, Lee M, Suh YL. Frequent association of cortical dysplasia in dysembryoplastic neuroepithelial tumor treated by epilepsy surgery. Surg Neurol. 2005;64:419–427. doi: 10.1016/j.surneu.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 137.Giannini C, Scheithauer BW, Burger PC, Brat DJ, Wollan PC, Lach B, O’Neill BP. Pleomorphic xanthoastrocytoma: what do we really know about it? Cancer. 1999;85:2033–2045. [PubMed] [Google Scholar]
- 138.Nakajima T, Kumabe T, Shamoto H, Watanabe M, Suzuki H, Tominaga T. Malignant transformation of pleomorphic xanthoastrocytoma. Acta Neurochir (Wien) 2006;148:67–71; discussion 71. doi: 10.1007/s00701-005-0549-8. [DOI] [PubMed] [Google Scholar]
- 139.Giannini C, Scheithauer BW, Lopes MB, Hirose T, Kros JM, VandenBerg SR. Immunophenotype of pleomorphic xanthoastrocytoma. Am J Surg Pathol. 2002;26:479–485. doi: 10.1097/00000478-200204000-00010. [DOI] [PubMed] [Google Scholar]
- 140.Wirrell EC, Wong-Kisiel LC, Nickels KC. Seizure outcome after AED failure in pediatric focal epilepsy: impact of underlying etiology. Epilepsy Behav. 2014;34:20–24. doi: 10.1016/j.yebeh.2014.02.032. [DOI] [PubMed] [Google Scholar]
- 141.Dahiya S, Haydon DH, Alvarado D, Gurnett CA, Gutmann DH, Leonard JR. BRAF(V600E) mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol. 2013;125:901–910. doi: 10.1007/s00401-013-1120-y. [DOI] [PubMed] [Google Scholar]
- 142.Duncan JS, de Tisi J. MRI in the diagnosis and management of epileptomas. Epilepsia. 2013;54 Suppl 9:40–43. doi: 10.1111/epi.12442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chappé C, Padovani L, Scavarda D, Forest F, Nanni-Metellus I, Loundou A, Mercurio S, Fina F, Lena G, Colin C, et al. Dysembryoplastic neuroepithelial tumors share with pleomorphic xanthoastrocytomas and gangliogliomas BRAF(V600E) mutation and expression. Brain Pathol. 2013;23:574–583. doi: 10.1111/bpa.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]