

Abstract
Papillomaviruses are a family of slowly evolving DNA viruses and their evolution is commonly linked to that of their host species. However, whilst bovine papillomavirus-1 (BPV-1) primarily causes warts in its natural host, the cow, it can also cause locally aggressive and invasive skin tumours in equids, known as sarcoids, and thus provides a rare contemporary example of cross-species transmission of a papillomavirus. Here, we describe the first phylogenetic analysis of BPV-1 in equine sarcoids to our knowledge, allowing us to explore the evolutionary history of BPV-1 and investigate its cross-species association with equids. A phylogenetic analysis of the BPV-1 transcriptional promoter region (the long control region or LCR) was conducted on 15 bovine and 116 equine samples from four continents. Incorporating previous estimates for evolutionary rates in papillomavirus implied that the genetic diversity in the LCR variants was ancient and predated domestication of both equids and cattle. The phylogeny demonstrated geographical segregation into an ancestral group (African, South American and Australian samples), and a more recently derived, largely European clade. Whilst our data are consistent with BPV-1 originating in cattle, we found evidence of multiple, probably relatively recent, cross-species transmission events into horses. We also demonstrated the high prevalence of one particular sequence variant (variant 20), and suggest this may indicate that this variant shows a fitness advantage in equids. Although strong host specificity remains the norm in papillomaviruses, our results demonstrate that exceptions to this rule exist and can become epidemiologically relevant.
Introduction
As slowly evolving, host-adapted dsDNA viruses, papillomaviruses are unlikely candidates for infecting novel host species (Shadan & Villarreal, 1993). Papillomaviruses do appear to evolve primarily through co-speciation with their hosts (Bernard, 2006, 2013; Chan et al., 1992; Halpern, 2000; Shadan & Villarreal, 1993); however, studies have indicated that cross-species transmission has also played an important role in their evolution (Chan et al., 1997; García-Pérez et al., 2014; Gottschling et al., 2007, 2011). Currently, the only papillomaviruses that show clear and ongoing evidence of transmission across species barriers are bovine papillomavirus 1 (BPV-1) and BPV-2, which are associated with benign warts in their natural bovine host, and with sarcoids in equids (Nasir & Campo, 2008). There are also numerous reports of BPV infection in other animals including water buffalo, captive tapir, giraffe and antelope (Kidney & Berrocal, 2008; Roperto et al., 2013; Silvestre et al., 2009; Tomita et al., 2007), and BPV has been shown experimentally to infect rabbits, hamsters and mice (Boiron et al., 1964; Robl & Olson, 1968).
Equine sarcoids are locally invasive and aggressive fibroblastic tumours, and are one of the most common skin tumours to affect equids worldwide (Knottenbelt, 2005). They represent a significant health and welfare issue, as lesions are difficult and often costly to treat and rarely resolve without intervention. Although they do not appear to metastasize, sarcoids can grow very large and can cause loss of value, reduced performance and ‘loss of use’, all of which may lead to euthanasia. This can be especially important in developing countries, where working equids are a key source of traction and transport in local communities.
When sarcoids were first described in 1936, it was hypothesized that they were caused by an infectious agent, and they were linked to cattle warts caused by BPV (Jackson, 1936), which give rise to benign papillomas and malignant lesions of the skin or mucosal epithelium in bovids (Campo, 2006). Since Jackson’s observations, many studies have demonstrated the presence of BPV-1 and -2 DNA, RNA and proteins in equine sarcoids, and the evidence demonstrating that infection by BPV-1 and -2 causes equine sarcoids is now extensive (reviewed by Nasir & Brandt, 2013; Nasir & Campo, 2008). The BPV-1/2 genomes are circular and episomally maintained (Amtmann et al., 1980; Yuan et al., 2007, 2008).
In contrast to BPV-1/-2-induced lesions in cattle, to date no BPV viral particles have been detected in equine sarcoids, and the disease has been thought to be the result of a non-productive BPV-1/-2 infection. A subset of sarcoids has been shown to contain BPV-1 DNA in a complex with the L1 capsid protein, which may represent viral particle precursors (Brandt et al., 2008; Hartl et al., 2011), raising the potential for ongoing transmission. Several studies have described the occurrence of sarcoids in groups of individuals, suggesting that BPV infection can indeed be transmitted between equine hosts (Bogaert et al., 2007; Marais & Page, 2011; Ragland et al., 1970; Reid et al., 1994), and Nasir & Campo (2008) have shown that co-stabling of sarcoid-affected and healthy donkeys can results in the transmission of BPV-1, with insects suspected as possible transmission vectors (Finlay et al., 2009).
Despite the first scientific description of equine sarcoids dating back almost a century, it is as yet unclear whether the ability of BPVs to infect equine hosts was acquired in the recent past or whether this cross-species association could have a much longer history. Here, for the first time to our knowledge, we examined the population genetic structure and evolutionary history of viruses associated with equine sarcoids worldwide, by applying phylogenetic analyses to a global sample of BPV-1 long control region (LCR) sequence data. Specifically, we sought to address the following research questions: (i) How genetically variable is the BPV-1 LCR from equine sarcoids, and when did this variability arise? (ii) Does the BPV-1 LCR from equine sarcoids exhibit any global phylogeographic structure? (iii) What does the BPV-1 LCR phylogeny reveal about the frequency of cross-species transmission between cattle and equids? (iv) Do the data provide evidence for continued transmission and adaptation of BPV-1 LCR in the equine host?
Results
Diversity and phylogeographical structure
A 695 bp sequence of the BPV-1 LCR (nt 7252–7947) was determined from 116 equine sarcoid and 15 bovine papilloma samples, from which a total of 21 different sequence variants (SVs) were examined (Table 1). Of the 21 SVs detected, one corresponded to the BPV-1 reference sample and the rest were assigned numbers from 1 to 20 (GenBank accession numbers are shown in Table 1). Six SVs were identified in 35 sarcoid lesions examined in a previous study (SVs 15, 16, 17, 18 and 20; Nasir et al. (2007), and these samples were included in this analysis and are detailed in Table 1. The remaining 15 SVs and the samples that comprise them were novel to this study. In this study, using the methods described, we were able to consistently isolate large DNA fragments from paraffin-embedded tissues (see Fig. S1 available in the online Supplementary Material).
Table 1. Numbers, origins, and accession IDs for BPV-1 LCR SVs.
Numbers of samples that were reused for this analysis from the previous study by Nasir et al. (2007) are indicated in parentheses along with their original denomination. Cattle samples all originated from Bos taurus breeds in the UK.
| Sequence type | Identification in Nasir et al. (2007) | GenBank no. | Cattle sample(s) | Equine samples | Total | |||||||
| S. Africa | Ethiopia | Australia | Brazil | Italy | Switzerland | Austria | UK | |||||
| 1 | KJ577553 | 3 | 3 | |||||||||
| 2 | KJ577554 | 5 | 1 | 1 | 7 | |||||||
| 3 | KJ577555 | 5 | 5 | |||||||||
| 4 | KJ577556 | 2 | 2 | |||||||||
| 5 | KJ577557 | 2 | 2 | |||||||||
| 6 | KJ577558 | 2 | 2 | |||||||||
| 7 | KJ577559 | 2 | 2 | |||||||||
| 8 | KJ577560 | 2 | 11 | 13 | ||||||||
| 9 | KJ577561 | 2 | 2 | |||||||||
| 10 | KJ577562 | 1 | 1 | |||||||||
| 11 | KJ577563 | 1 | 1 | |||||||||
| 12 | KJ577564 | 1 | 1 | |||||||||
| 13 | KJ577565 | 3 | 3 | |||||||||
| 14 | KJ577566 | 1 | 1 | 2 | 4 | |||||||
| 15 | I (2) | DQ855065 | 2 | 3 | 5 | |||||||
| 16 | III (1) | DQ855067 | 1 | 1 | ||||||||
| 17 | V (1) | DQ855069 | 1 | 1 | ||||||||
| 18 | IV (8) | DQ855068 | 7 | 2 | 8 | 17 | ||||||
| 19 | KJ577567 | 4 | 1 | 1 | 6 | |||||||
| 20 | II (23) | DQ855066 | 1 | 2 | 1 | 2 | 13 | 23 | 4 | 4 | 50 | |
| BPV-1 reference | X02346 | 3 | 3 | |||||||||
| Total | 15 | 4 | 21 | 12 | 6 | 24 | 26 | 10 | 13 | 131 | ||
We noted a division within the BPV-1 maximum-likelihood (ML) phylogeny into two groups of sequences (Fig. 1, groups A and B). The first group appeared ancestral, as it branched off closer to the root of the tree and was restricted to Africa, Brazil and Australia (SVs 1–3, 6, 8–10 and 13), with the exception of SV 2 (also found in Italy) and SV 17 (Switzerland only) (group A). A second, more recently diverged group (group B) contained almost exclusively European samples (SVs 4, 5, 11, 12, 14–16 and 18–20) with the sole exception of SV 20, which was found in all populations and is discussed below. A χ2 test was conducted in r v.3.0.2 (R Core Team, 2013) to compare the geographical origin (European or non-European) of SVs found in group A (Fig. 1; SVs 1–3, 6–10, 13, 17 and BPV-1 reference) with those in group B (SVs 4, 5, 11, 12, 14–16 and 18–20). This confirmed the significant phylogenetic clustering by geographical origin (P<0.001, χ2 = 80.5).
Fig. 1.

ML phylogenetic tree for the LCR sequence variants. Sequence variants found in cattle (BPV-1 reference and SVs 14, 18 and 19) are surrounded by a rectangle (all cattle samples originated from the UK). Variants found in equids are indicated by circle(s), with circle colour(s) corresponding to country of origin and circle size(s) proportionate to the number of samples of that SV from each country (see Table 1 for absolute numbers). The divergence dates for specified nodes 1–4 (diamonds) and their 95 % confidence intervals were calculated according to the LCR evolutionary rate estimate from Rector et al. (2007). Statistical values indicating node support are shown: the left-hand value indicates bootstrap support for each node in the ML phylogeny and the right-hand value gives the posterior probability of the node where present in the Bayesian phylogeny (Fig. S2). HPD, highest posterior density. Bar, nucleotide substitutions per site.
The Bayesian phylogeny (Fig. S2) had a broadly similar topology to the ML tree with one exception: the clade containing the BPV-1 reference sequence, which also consisted of both European (SV 17) and African (SV 7) sequences, took different positions in the ML and Bayesian phylogenies. Excluding this problematic clade from the χ2 test described above had no effect on the significance of results for geographical structuring (P<0.001, χ2 = 77.4).
Genotypic diversity was highest for the European continent (12 SVs), followed by Africa (nine SVs), Brazil (three SVs) and Australia (two SVs), but Europe also accounted for over 50 % of all samples. To take the sample size variation among continents into account, rarefaction curves were constructed (Fig. 2). These indicate that, whilst the European samples showed the highest SV diversity, there was not sufficient sampling from other continents to define the full heterogeneity of BPV-1 variants in these regions.
Fig. 2.
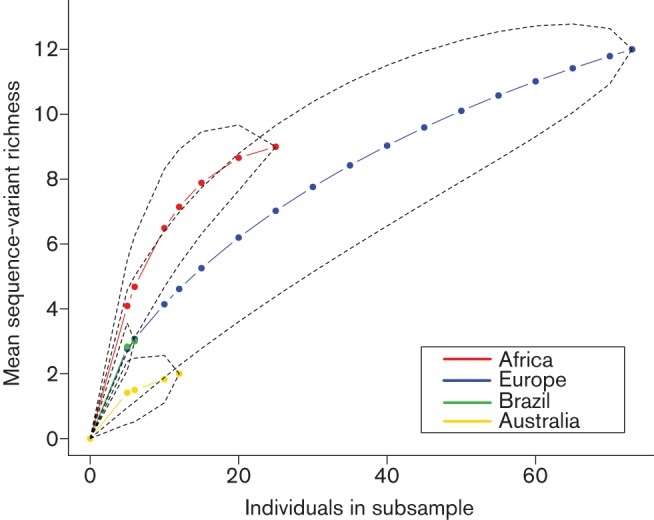
Rarefaction curves showing estimates of sequence-variant richness for each of the different continents sampled whilst taking into account differing sample sizes. Dotted lines give the 95 % confidence intervals around the estimated means.
Divergence times
The substitution rate for the papillomavirus LCR region estimated by Rector et al. (2007) was used to date key nodes within the BPV-1 phylogeny (Fig. 1). The confidence intervals for these estimates were large, reflecting both the comparatively low nucleotide diversity within the viral sequences and the uncertainty in the estimated evolutionary rate used. Despite this limitation, the results clearly suggested that phylogenetic diversification in BPV-1 had occurred tens of thousands of years ago: the most recent divergence event, representing the split of SVs 4, 16 and 20, was dated to 53 thousand years before the present [53 kyear BP; 95 % highest posterior density (HPD) interval: 22–209 kyear BP], whereas the most recent common ancestor (MRCA) of the European clade was estimated at 330 kyear BP (HPD: 151–614 kyear BP). The MRCA of BPV-1 was dated at 628 kyear BP, compared with the split into BPV-1 and -2, estimated to have occurred 1100 kyear BP (95 % HPD: 390–2560 kyear BP).
Applying another papillomavirus substitution rate estimated by Shah et al. (2010) resulted in even older divergence dates, ranging from 188 kyear BP (95 % HPD: 32.4–450 kyear BP) for the most recent to 3700 kyear BP (95 % HPD: 1320–9180 kyear BP) for the MRCA of BPV-1 and -2 (other results not shown).
SV host association and frequency distribution
Of the SVs found in the samples described in this study, one variant (SV 21/BPV-1 reference) was found in cattle only, three variants (SVs 14, 18 and 19) were found in both cattle and equine samples, and all other variants were found in equine samples only (Table 1 gives details of numbers of samples of each SV).
Fifteen bovine samples were included in this dataset, all of which were sampled from Bos taurus cattle in the UK. Of the four different SVs identified within the cattle samples, three variants (SVs 14, 18 and 19) fell within the European clade. No evidence of clustering by host species was apparent within the phylogeny [Fig. 1, SVs found in cattle are surrounded by a rectangle, whilst those found in equids are indicated by coloured circle(s)].
The frequency distribution of SVs from equine sarcoids was highly skewed. One variant, SV 20, was dominant and represented 50 out of all 116 equine samples (43 %). Despite this, it was not found in any of the 15 cattle samples, and a χ2 test conducted in r confirmed that this was a statistically significant difference in host species association (P<0.01, χ2 = 8.7). SV 20 was also the only variant found in all countries sampled and was part of the most recently diverged lineage (SVs 16/20/4, MRCA 53kyear BP), falling within the European clade (Fig. 1).
Discussion
We found substantial genetic variation among BPV-1 sampled worldwide from equine sarcoids, significantly expanding the number of known SVs described previously by Nasir et al. (2007) and facilitating the first phylogenetic examination of the evolutionary history of the virus. Our data revealed a clear geographical segregation between a basal group of sequences (group A) made up of samples from Africa, Brazil and Australia, and a largely European clade derived from the former group (group B). The position and recent divergence of the European clade suggests that BPV-1 did not originate here, and we speculate that the high diversity of SVs found in Africa, despite sampling only two countries (South Africa and Ethiopia), could point to this continent as a possible origin. Clearly, more comprehensive sampling in Africa as well as other continents is needed to properly test this hypothesis.
Divergence dates calculated using papillomavirus substitution rates from the literature (Rector et al., 2007; Shah et al., 2010) suggested that the diversity of the LCR sequences is ancient. Even the most recent divergence (the SV 16/20/4 clade, node 4 in Fig. 1) substantially predated domestication of horses and donkeys (domesticated around 5000 years ago; Kimura et al., 2011; Vilà et al., 2001) and of the two species of cattle (domesticated around 10 000 years ago; Ajmone-Marsan et al., 2010).
The differences in geographical origin of group A and B samples is somewhat surprising, considering the extent of worldwide trade in livestock including cattle. High connectivity should tend to reduce any phylogeographical segregation, especially given the benign nature of BPV infection in cattle, which is unlikely to trigger any action aimed at limiting viral spread. It is interesting to note in this context that domestic cattle consist of two different species, B. taurus and Bos indicus, and that European cattle breeds consist of pure B. taurus stock, whereas African cattle are variable hybrids of the two species, and in Brazil domestic cattle are predominantly B. indicus (Ajmone-Marsan et al., 2010). It is therefore possible that the two geographical groups within our BPV-1 sequences could correspond to the current distribution of different cattle species. Whilst this includes the possibility of co-evolution between BPV-1 and cattle, such an explanation seems unlikely given that all BPV-1 SVs reported here evolved thousands of years ago, predating the large-scale cattle migrations known to have occurred after domestication (Ajmone-Marsan et al., 2010). A more plausible scenario is that pre-existing viral diversity in the respective wild ancestor populations could have been transferred to the two domesticated species and maintained there since. Given that both types of cattle co-exist in many parts of the world, it could be further speculated that the strong geographical segregation is partially maintained by host specificity, in that group A LCR variants have a higher fitness within B. indicus and hybrid breeds compared with European cattle, and vice versa. However, the presence of SV 8 in the majority (11/12) of the Australian sarcoid samples conflicts with this theory. SV 8 segregated within group B, which we hypothesize should be more fit in indicine cattle, whereas the Australian samples originate from New South Wales where the majority of cattle are of B. taurus stock.
Despite the fact that cattle isolates were only available from the UK and were notably undersampled in this study, bovine-derived LCR sequences were scattered throughout the European clade and showed no sign of phylogenetic clustering. Apart from the BPV-1 reference, all three SVs found in cattle (SVs 14, 18 and 19) were also present in equine samples. This suggests that BPV-1 is generally shared between both host species, and that viral transfer between cattle and equine hosts is a repeated and possibly ongoing phenomenon, rather than the product of a single cross-species transmission event at some point in the past.
Although the small number of cattle samples does not allow us to infer the direction of cross-species transmission, the current understanding of the biology of the virus in both species suggests that cattle are more likely to be the origin of infection. Whereas BPV papillomas in cattle are productive infections that generate infectious viral particles, no viral particles have been reported in equids, although virus particle precursors are known to exist (Brandt et al., 2008). Whilst sarcoids can be transmitted between equids (with or without the help of viral particles) (Nasir & Campo, 2008), it appears that BPV-1 is more easily transmissible, and is more likely to be maintained in cattle than in equids. This is supported by transmission studies: Olson & Cook (1951) were able to produce sarcoids in horses from cattle papilloma material with a frequency of 1 in 11, suggesting that the barrier to cross-species transmission of BPV from bovids to equids is not particularly high. However, in the reverse experiment, Ragland et al. (1970) were not able to induce any papillomas in cattle using equine sarcoid material. The nature of BPV infection in cattle and horses is similar to cottontail rabbit papillomavirus, which induces productive infections that rarely progress to cancer in its natural host (wild rabbit), but gives rise to non-productive infections that do become cancerous in domestic rabbits (Campo, 2002).
Given these observations, it seems plausible that BPV-1 is preferentially maintained in the cattle population, with a relatively high occurrence of cross-species transmission to equids. Therefore, we hypothesize that much of the observed genetic diversity of LCR sequences is likely to have evolved within the cattle population, before the virus crossed into equids multiple times from a variety of different (bovine) sources. However, this does not rule out the possibility of evolution also within equid hosts. More samples of BPV-1 from cattle are needed to confirm this theory of host species transfer, with cattle LCR samples from outside Europe being necessary to test whether BPV-1 in cattle segregate with other cattle variants, or with geographical region regardless of host species.
SV 20 may be the exception to the suggested low transmissibility of BPV in equids. This variant was present in 43 % of the equine samples in this study, and in horses and donkeys from all countries sampled, but was not found in any bovine samples. Whilst the low number of cattle samples tested does not rule out the presence of SV 20 in cattle, it was significantly more common in equids than would be expected by chance. This implies that this sequence variant may be transmitting more efficiently through the equine population than other variants. Consistent with this idea, viral DNA with SV 20 LCR (previously classified as variant II) has a significantly higher transcriptional activity than the BPV-1 reference LCR in equine fibroblasts than in bovine fibroblasts (Nasir et al., 2007), and natural equine transmission has been demonstrated for an SV 20 virus (Nasir & Campo, 2008). Further characterization of the behaviour and relative fitness of SV 20-containing BPV-1 is needed to gain a better understanding of the molecular biology of BPV-1 in equine sarcoids, and the specific determinants that underlie establishment and successful transmission in a novel host.
In recent years, several studies have emphasized that the evolution of papillomaviruses is not driven purely through host–pathogen co-evolution, and it is becoming apparent that events such as adaptive radiation and cross-infection between host species have played an important role in shaping the history of this important viral family (Chan et al., 1997; García-Pérez et al., 2014; Gottschling et al., 2007, 2011). In this study, we present what we believe is the first phylogenetic analysis of BPV-1 in equids and bovids, one of only two known ongoing examples of cross-species association of a papillomavirus (the other being BPV-2; Nasir & Campo, 2008). We have presented several novel insights into the evolutionary history of the virus and its association with cattle and equids, adding to our understanding of the mechanisms underlying the diversity and epidemiology of papillomaviruses affecting human and animal health today.
Methods
Samples and DNA isolation.
Tissue samples or paraffin-embedded tissue sections from 131 samples (116 equine sarcoid samples and 15 bovine papilloma samples, Table 1) were obtained from archival material at the University of Glasgow and from samples gathered across four continents over a period of 20 years. Thiry-five of these samples had already been sequence typed in a previous study by Nasir et al. (2007), as indicated in Table 1.
DNA was isolated from tissue samples using standard extraction methods. Briefly, approx. 50 mg tissue was finely dissected and incubated overnight at 37 °C in lysis buffer (5 mM EDTA, 1 % SDS, 50 mM NaCl, 50 mM Tris/HCl, 300 µg proteinase K ml−1). The DNA was then purified using phenol/chloroform, following by precipitation using 3 M sodium acetate. DNA was isolated from paraffin-embedded tissues, using a Qiagen DNeasy kit. All Australian, Italian and Brazilian samples were supplied as paraffin-embedded sections. The samples from Switzerland and the UK were tumour tissues stored at −80 °C and the samples from Austria were supplied as extracted DNA preparations. Samples from Ethiopia and South Africa were supplied as sarcoid cell scrapings from which DNA was isolated essentially as described for frozen tissues.
PCR amplification of BPV-1 LCR.
The LCR regulates replication and transcription of the viral genome. We chose to sequence this region for analysis, as it is has the fastest substitution rate within the papillomavirus genome (Rector et al., 2007) and therefore is expected to be the most informative region for phylogenetic analysis.
The L1 ORF is the most conserved gene within PV genomes and is therefore used for the classification of PV genomes. A difference in nucleotide identity of greater than 10 % in the L1 defines a papillovirus type. Differences of between 2 and 10 % nucleotide identity define a subtype and less than 2 % a variant (Villiers et al., 2004).
In this study, PCR amplification of the LCR was carried out using primers LCRfullF (5′-CAGAAGGTAAGTCAACTG-3′) and LCRfullR (5′-GTGAAAACCGGGGTCTG-3′) corresponding to 7133 and 7150 nt and 74–90 nt, respectively, of BPV-1 GenBank accession number AB626705, which amplify a 796 bp product. Reactions were performed in the presence of either Pfu DNA polymerase (Promega) or Platinum Taq polymerase (Life Technologies), which are both high-fidelity enzymes, according to the manufacturer’s instructions. Cycling conditions were as follows: one cycle of 94 °C for 5 min, followed by 35 cycles of 94 °C for 1 min, 56 °C for 1 min and 72 °C for 2 min, and a final extension of 72 °C for 10 min. In all the PCR experiments, negative controls without a DNA template were also included. In some cases, the amplification product was weak and a second round of PCR was performed using PCR primers BPVLCRF (5′-CGGTACACATCCTGTCCAGCAT-3′) and LCR3Rev (5′-GATGGTGTGATTATTGTTAAC-3′), which amplify a 736 bp product.
Prior to DNA sequencing, PCR products were purified using a Qiaquick PCR clean-up kit or Qiaquick gel extraction kit (Qiagen). Purified products were sequenced using both the forward and reverse primer. DNA sequence analyses were performed using the Applied Biosystems Big Dye terminal sequencing kit and sequencing was performed on an ABI 3100 automated sequencer. Analysis of sequences was performed using the CLC Genomics Workbench (CLC bio) and compared with previously published BPV-1 LCR sequences (Nasir et al., 2007). Where there were difficulties in deciphering the correct sequence, PCR amplification and sequence analyses were repeated. The sequences at the start and end of each sequence were excluded to limit the number of errors. Where the samples were novel and not identical to a previously published sequence, again the PCR and sequencing was repeated. In most cases, a PCR product was detected; however, in a small number of samples, the product was either lost during the clean-up and/or the sequencing reaction failed, or we were not able to confirm/refute specific changes. These samples are not included in this study. The variant loci identified in the BPV-1 LCR sequence alignment are presented in Table S1. Percentage pairwise sequence similarity between BPV LCR SVs is presented in Table S2.
Phylogenetic analysis.
The LCR sequences generated for the bovine and equine samples fell into 21 different SVs, one of which corresponded with the LCR sequence of the BPV-1 reference virus. Of these, SVs 15, 16, 17, 18 and 20 have been described previously (Nasir et al., 2007), and the others were novel to this study (Table 1 summarizes the numbers and origins of samples for each SV and their corresponding GenBank accession numbers). The BPV-2 LCR sequence (GenBank accession no. M20219) was included in this analysis as an outgroup. The 695 bp LCR sequences, located between nt 7252 and 7947, were extracted and then aligned using a global alignment with free end gaps in Geneious v.5.1 (Drummond et al., 2010).
Program jModelTest v.0.1.1 (Posada, 2008) was used to identify a model of nucleotide substitution. The optimum model under the Akaike Inference Criterion was the K80 model (Kimura, 1980) with a proportion of invariable sites and a γ-distributed rate variation among sites, and this model was used for all phylogenetic analyses unless indicated otherwise.
A ML tree was found in PhyML v.3.0 under a heuristic search (Guindon & Gascuel, 2003). Statistical support for individual tree nodes was evaluated based on 1000 non-parametric bootstrap replicates. A Bayesian phylogeny was estimated in MrBayes v.3.1 (Huelsenbeck et al., 2001; Ronquist & Huelsenbeck, 2003) using 107 generations of two simultaneous Markov chain Monte Carlo (MCMC) chains with a sampling frequency of 100 and a burn-in of 2500.
The program beast v.1.6.0 (Drummond & Rambaut, 2007) was used to estimate the divergence times for the LCR phylogeny under a relaxed molecular clock with uncorrelated branch-specific rates drawn from a log-normal distribution (Drummond et al., 2006). Because papillomaviruses are characteristically slowly evolving, differences in sampling dates should be irrelevant to sequence divergence from the MRCA. The program Path-O-Gen (available at http://tree.bio.ed.ac.uk/software/pathogen) was used to test this assumption. As expected, no evidence was found of a positive relationship between root-to-tip divergence and sampling date, confirming that divergence time estimation in beast would require the provision of an external substitution rate (results not shown). Two previous studies have quantified the substitution rate of papillomaviruses, and both were used here to estimate divergence dates. Rector et al. (2007) estimated a mean substitution rate of 2.69×10−8 nucleotide substitutions per site per year for the feline papillomavirus LCR, and this rate was used in beast to impose a normally distributed prior mean for the branch-specific clock rate, with a sd of 5.1×10−9. Shah et al. (2010) estimated a considerably slower evolutionary rate for papillomaviruses based on two coding regions (the E1 and the L1 genes, with substitution rates of 7.10×10−9 and 9.57×10−9 substitutions per site per year, respectively), and we used these two estimates as the upper and lower bound of a uniform mean rate prior. The first of these rate estimates is likely to be more appropriate to this analysis, as it was calculated for the LCR specifically and the depth of phylogeny over which they generated their estimate was closer to the depth we were assessing here (see Ho & Larson, 2006), and therefore we focused on the results from this.
For all beast analyses, the MCMC simulation was run for a chain length of 107 with a sampling frequency of 1000 and a burn-in of 106 under a HKY nucleotide substitution model and a Bayesian skyline model as a flexible demographic prior (Drummond et al., 2005).
Rarefaction.
In order to estimate the SV richness on different continents, whilst accounting for the likelihood that an increased sample size will increase the chance of detecting higher numbers of variants, rarefaction curves were calculated in r using code for individual-based rarefaction downloaded from http://www.jennajacobs.org/R/rarefaction.html. Plots were altered to show 95 % confidence intervals rather than se.
Acknowledgements
H. T. was funded for this work by a BBSRC Doctoral Training Grant. R. B. is supported by the RAPIDD programme of the Science and Technology Directorate of the Department of Homeland Security and NIH Fogarty International Center. We would also like to acknowledge the Horse Race Betting Levy Board for supporting part of this work.
Footnotes
Two supplementary figures and two tables are available with the online Supplementary Material.
References
- Ajmone-Marsan P., Garcia J. F., Lenstra J. A., The Globaldiv Consortium (2010). On the origin of cattle: how aurochs became cattle and colonized the world. Evol Anthropol 19, 148–157 10.1002/evan.20267 [DOI] [Google Scholar]
- Amtmann E., Muller H., Sauer G. (1980). Equine connective tissue tumors contain unintegrated bovine papillomavirus DNA. J Virol 35, 962–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard H.-U. (2013). Taxonomy and phylogeny of papillomaviruses: an overview and recent developments. Infect Genet Evol 18, 357–361. 10.1016/j.meegid.2013.03.011 [DOI] [PubMed] [Google Scholar]
- Bernard H.-U., Calleja-Macias I. E., Dunn S. T. (2006). Genome variation of human papillomavirus types: phylogenetic and medical implications. Int J Cancer 118, 1071–1076. 10.1002/ijc.21655 [DOI] [PubMed] [Google Scholar]
- Bogaert L., Van Poucke M., De Baere C., Dewulf J., Peelman L., Ducatelle R., Gasthuys F., Martens A. (2007). Bovine papillomavirus load and mRNA expression, cell proliferation and p53 expression in four clinical types of equine sarcoid. J Gen Virol 88, 2155–2161. 10.1099/vir.0.82876-0 [DOI] [PubMed] [Google Scholar]
- Boiron M., Levy J. P., Thomas M., Friedmann J. C., Bernard J. (1964). Some properties of Bovine Papilloma Virus. Nature 201, 423–424. [DOI] [PubMed] [Google Scholar]
- Brandt S., Haralambus R., Shafti-Keramat S., Steinborn R., Stanek C., Kirnbauer R. (2008). A subset of equine sarcoids harbours BPV-1 DNA in a complex with L1 major capsid protein. Virology 375, 433–441. 10.1016/j.virol.2008.02.014 [DOI] [PubMed] [Google Scholar]
- Campo M. S. (2002). Animal models of papilloma virus pathogenesis. Virus Res 89, 249–261. [DOI] [PubMed] [Google Scholar]
- Campo M. S. (ed.) (2006). Papillomavirus Research: From Natural History to Vaccines and Beyond. Wymondham, UK: Caister Academic Press. [Google Scholar]
- Chan S.-Y., Bernard H.-U., Ong C.-K., Chan S.-P., Hofmann B., Delius H. (1992). Phylogenetic analysis of 48 papillomavirus types and 28 subtypes and variants: a showcase for the molecular evolution of DNA viruses. J Virol 66, 5714–5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S. Y., Ostrow R. S., Faras A. J., Bernard H. U. (1997). Genital papillomaviruses (PVs) and epidermodysplasia verruciformis PVs occur in the same monkey species: implications for PV evolution. Virology 228, 213–217. 10.1006/viro.1996.8400 [DOI] [PubMed] [Google Scholar]
- Drummond A. J., Rambaut A. (2007). beast: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7, 214. 10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J., Rambaut A., Shapiro B., Pybus O. G. (2005). Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol 22, 1185–1192. 10.1093/molbev/msi103 [DOI] [PubMed] [Google Scholar]
- Drummond A. J., Ho S. Y. W., Phillips M. J., Rambaut A. (2006). Relaxed phylogenetics and dating with confidence. PLoS Biol 4, e88. 10.1371/journal.pbio.0040088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, A., Ashton, B., Buxton, S., Cheung, M., Cooper, A., Heled, J., Kearse, M., Moir, R., Stones-Havas, S. & other authors. (2010). Geneious v5.5. http://www.geneious.com.
- Finlay M., Yuan Z. Q., Burden F., Trawford A., Morgan I. M., Campo M. S., Nasir L. (2009). The detection of Bovine Papillomavirus type 1 DNA in flies. Virus Res 144, 315–317. 10.1016/j.virusres.2009.04.015 [DOI] [PubMed] [Google Scholar]
- García-Pérez R., Ibáñez C., Godínez J. M., Aréchiga N., Garin I., Pérez-Suárez G., de Paz O., Juste J., Echevarría J. E., Bravo I. G. (2014). Novel papillomaviruses in free-ranging Iberian bats: no virus-host co-evolution, no strict host specificity, and hints for recombination. Genome Biol Evol 6, 94–104. 10.1093/gbe/evt211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschling M., Stamatakis A., Nindl I., Stockfleth E., Alonso A., Bravo I. G. (2007). Multiple evolutionary mechanisms drive papillomavirus diversification. Mol Biol Evol 24, 1242–1258. 10.1093/molbev/msm039 [DOI] [PubMed] [Google Scholar]
- Gottschling M., Göker M., Stamatakis A., Bininda-Emonds O. R. P., Nindl I., Bravo I. G. (2011). Quantifying the phylodynamic forces driving papillomavirus evolution. Mol Biol Evol 28, 2101–2113. 10.1093/molbev/msr030 [DOI] [PubMed] [Google Scholar]
- Guindon S., Gascuel O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52, 696–704. 10.1080/10635150390235520 [DOI] [PubMed] [Google Scholar]
- Halpern A. L. (2000). Comparison of papillomavirus and immunodeficiency virus evolutionary patterns in the context of a papillomavirus vaccine. J Clin Virol 19, 43–56. 10.1016/S1386-6532(00)00127-X [DOI] [PubMed] [Google Scholar]
- Hartl B., Hainisch E. K., Shafti-Keramat S., Kirnbauer R., Corteggio A., Borzacchiello G., Tober R., Kainzbauer C., Pratscher B., Brandt S. (2011). Inoculation of young horses with bovine papillomavirus type 1 virions leads to early infection of PBMCs prior to pseudo-sarcoid formation. J Gen Virol 92, 2437–2445. 10.1099/vir.0.033670-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S. Y. W., Larson G. (2006). Molecular clocks: when times are a-changin’. Trends Genet 22, 79–83. 10.1016/j.tig.2005.11.006 [DOI] [PubMed] [Google Scholar]
- Huelsenbeck J. P., Ronquist F., Nielsen R., Bollback J. P. (2001). Bayesian inference of phylogeny and its impact on evolutionary biology. Science 294, 2310–2314. 10.1126/science.1065889 [DOI] [PubMed] [Google Scholar]
- Jackson C. (1936). The incidence and pathology of tumours in domesticated animals in South Africa. Oonderspoort J Vet Sci 6, 375–385. [Google Scholar]
- Kidney B. A., Berrocal A. (2008). Sarcoids in two captive tapirs (Tapirus bairdii): clinical, pathological and molecular study. Vet Dermatol 19, 380–384. 10.1111/j.1365-3164.2008.00698.x [DOI] [PubMed] [Google Scholar]
- Kimura M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16, 111–120. 10.1007/BF01731581 [DOI] [PubMed] [Google Scholar]
- Kimura B., Marshall F. B., Chen S., Rosenbom S., Moehlman P. D., Tuross N., Sabin R. C., Peters J., Barich B. & other authors (2011). Ancient DNA from Nubian and Somali wild ass provides insights into donkey ancestry and domestication. Proc Biol Sci 278, 50–57. 10.1098/rspb.2010.0708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knottenbelt D. (2005). A suggested clinical classification for the equine sarcoid. Clin Tech Equine Pract 4, 278–295 10.1053/j.ctep.2005.10.008 [DOI] [Google Scholar]
- Marais H. J., Page P. C. (2011). Treatment of equine sarcoid in seven Cape mountain zebra (Equus zebra zebra). J Wildl Dis 47, 917–924. 10.7589/0090-3558-47.4.917 [DOI] [PubMed] [Google Scholar]
- Nasir L., Brandt S. (2013). Papillomavirus associated diseases of the horse. Vet Microbiol 167, 159–167. 10.1016/j.vetmic.2013.08.003 [DOI] [PubMed] [Google Scholar]
- Nasir L., Campo M. S. (2008). Bovine papillomaviruses: their role in the aetiology of cutaneous tumours of bovids and equids. Vet Dermatol 19, 243–254. 10.1111/j.1365-3164.2008.00683.x [DOI] [PubMed] [Google Scholar]
- Nasir L., Gault E., Morgan I. M., Chambers G., Ellsmore V., Campo M. S. (2007). Identification and functional analysis of sequence variants in the long control region and the E2 open reading frame of bovine papillomavirus type 1 isolated from equine sarcoids. Virology 364, 355–361. 10.1016/j.virol.2007.02.019 [DOI] [PubMed] [Google Scholar]
- Olson C., Jr, Cook R. H. (1951). Cutaneous sarcoma-like lesions of the horse caused by the agent of bovine papilloma. Proc Soc Exp Biol Med 77, 281–284. 10.3181/00379727-77-18750 [DOI] [PubMed] [Google Scholar]
- Posada D. (2008). jModelTest: phylogenetic model averaging. Mol Biol Evol 25, 1253–1256. 10.1093/molbev/msn083 [DOI] [PubMed] [Google Scholar]
- R Core Team (2013). r: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Ragland W. L., McLaughlin C. A., Spencer G. R. (1970). Attempts to relate bovine papilloma virus to the cause of equine sarcoid: horses, donkeys and calves inoculated with equine sarcoid extracts. Equine Vet J 2, 168–172 10.1111/j.2042-3306.1970.tb04180.x [DOI] [Google Scholar]
- Rector A., Lemey P., Tachezy R., Mostmans S., Ghim S. J., Van Doorslaer K., Roelke M., Bush M., Montali R. J. & other authors (2007). Ancient papillomavirus-host co-speciation in Felidae. Genome Biol 8, R57. 10.1186/gb-2007-8-4-r57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid S. W., Smith K. T., Jarrett W. F. (1994). Detection, cloning and characterisation of papillomaviral DNA present in sarcoid tumours of Equus asinus. Vet Rec 135, 430–432. 10.1136/vr.135.18.430 [DOI] [PubMed] [Google Scholar]
- Robl M. G., Olson C. (1968). Oncogenic action of Bovine Papilloma Virus in hamsters. Cancer Res 28, 1596–1604. [PubMed] [Google Scholar]
- Ronquist F., Huelsenbeck J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. 10.1093/bioinformatics/btg180 [DOI] [PubMed] [Google Scholar]
- Roperto S., Russo V., Ozkul A., Corteggio A., Sepici-Dincel A., Catoi C., Esposito I., Riccardi M. G., Urraro C. & other authors (2013). Productive infection of bovine papillomavirus type 2 in the urothelial cells of naturally occurring urinary bladder tumors in cattle and water buffaloes. PLoS ONE 8, e62227. 10.1371/journal.pone.0062227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadan F. F., Villarreal L. P. (1993). Coevolution of persistently infecting small DNA viruses and their hosts linked to host-interactive regulatory domains. Proc Natl Acad Sci U S A 90, 4117–4121. 10.1073/pnas.90.9.4117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S. D., Doorbar J., Goldstein R. A. (2010). Analysis of host-parasite incongruence in papillomavirus evolution using importance sampling. Mol Biol Evol 27, 1301–1314. 10.1093/molbev/msq015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestre O., Borzacchiello G., Nava D., Iovane G., Russo V., Vecchio D., D’Ausilio F., Gault E. A., Campo M. S., Paciello O. (2009). Bovine papillomavirus type 1 DNA and E5 oncoprotein expression in water buffalo fibropapillomas. Vet Pathol 46, 636–641. 10.1354/vp.08-VP-0222-P-FL [DOI] [PubMed] [Google Scholar]
- Tomita Y., Literak I., Ogawa T., Jin Z., Shirasawa H. (2007). Complete genomes and phylogenetic positions of bovine papillomavirus type 8 and a variant type from a European bison. Virus Genes 35, 243–249. 10.1007/s11262-006-0055-y [DOI] [PubMed] [Google Scholar]
- Vilà C., Leonard J. A., Gotherstrom A., Marklund S., Sandberg K., Liden K., Wayne R. K., Ellegren H. (2001). Widespread origins of domestic horse lineages. Science 291, 474–477. 10.1126/science.291.5503.474 [DOI] [PubMed] [Google Scholar]
- Villiers E.-M., Fauquet C., Broker T. R., Bernard H.-U., zur Hausen H. (2004). Classification of papillomaviruses. Virology 324, 17–27. [DOI] [PubMed] [Google Scholar]
- Yuan Z., Gallagher A., Gault E. A., Campo M. S., Nasir L. (2007). Bovine papillomavirus infection in equine sarcoids and in bovine bladder cancers. Vet J 174, 599–604. 10.1016/j.tvjl.2006.10.012 [DOI] [PubMed] [Google Scholar]
- Yuan Z. Q., Gault E. A., Gobeil P., Nixon C., Campo M. S., Nasir L. (2008). Establishment and characterization of equine fibroblast cell lines transformed in vivo and in vitro by BPV-1: model systems for equine sarcoids. Virology 373, 352–361. 10.1016/j.virol.2007.11.037 [DOI] [PubMed] [Google Scholar]