

Abstract
Human immunodeficiency virus type 2 (HIV-2) infects about two million people worldwide. HIV-2 has fewer treatment options than HIV-1, yet may evolve drug resistance more quickly. We have analysed several novel drugs for anti-HIV-2 activity. It was observed that 5-azacytidine, clofarabine, gemcitabine and resveratrol have potent anti-HIV-2 activity. The EC50 values for 5-azacytidine, clofarabine and resveratrol were found to be significantly lower with HIV-2 than with HIV-1. A time-of-addition assay was used to analyse the ability of these drugs to interfere with HIV-2 replication. Reverse transcription was the likely target for antiretroviral activity. Taken together, several novel drugs have been discovered to have activity against HIV-2. Based upon their known activities, these drugs may elicit enhanced HIV-2 mutagenesis and therefore be useful for inducing HIV-2 lethal mutagenesis. In addition, the data are consistent with HIV-2 reverse transcriptase being more sensitive than HIV-1 reverse transcriptase to dNTP pool alterations.
Individuals infected with human immunodeficiency virus type 2 (HIV-2) are primarily of West African descent and many of the HIV-2 cases worldwide are attributed to immigrant populations of West Africans living abroad (Campbell-Yesufu & Gandhi, 2011; Costarelli et al., 2008; Rey et al., 1989; van der Ende et al., 1990). The lower prevalence of HIV-2 than HIV-1 is attributed to its low infectivity. HIV-2 is considered a naturally attenuated infection and HIV-2-infected individuals are more likely to have lower viral RNA levels and are less likely to progress to AIDS than those infected with HIV-1 (Bourée et al., 1995; Campbell-Yesufu & Gandhi, 2011; Costarelli et al., 2008; Soares et al., 2011). However, when HIV-2 infection induces AIDS, there are fewer treatment options than for those infected with HIV-1, because not all anti-HIV-1 drugs inhibit replication of HIV-2. First generation non-nucleoside reverse transcriptase (RT) inhibitors, the fusion inhibitor enfuvirtide, and several protease inhibitors are known to be ineffective against HIV-2, while the clinical efficacy of the entry inhibitor maraviroc is unknown (Menéndez-Arias & Alvarez, 2014). Furthermore, HIV-2 has been reported to have lower genetic barriers to the evolution of multidrug resistance than HIV-1, further narrowing the already-limited HIV-2 drug treatment options (Gottlieb et al., 2009; MacNeil et al., 2007; Menéndez-Arias & Alvarez, 2014; Rodés et al., 2000; Smith et al., 2009; Witvrouw et al., 2004).
Previous clinical trials to treat HIV-1 infection using drugs or drug candidates that enhance HIV-1 mutagenesis were associated with setbacks. For example, hydroxyurea (an inhibitor of cellular ribonucleotide reductase that has also been shown to enhance HIV-1 mutagenesis) has been tested clinically, but was found to have significant side effects (Biron et al., 1995; Frank et al., 2004; Lori et al., 1997). KP-1212 acts as a viral mutagen by being incorporated into viral DNA during reverse transcription and causing mispairing via tautomerization (Harris et al., 2005). However, a prodrug version (KP-1461) was not able to significantly reduce viral loads in clinical studies – though an altered spectrum of mutations was observed (Mullins et al., 2011).
Several significant advancements have been made recently to raise renewed enthusiasm to therapeutic approaches that seek to reduce HIV-1 infectivity by enhancing the viral mutation rate via lethal mutagenesis in cell culture (Dapp et al., 2013). First, 5-azacytidine was shown to induce HIV-1 lethal mutagenesis by the specific induction of G-to-C transversion mutations (Dapp et al., 2009). The combination of 5-aza-2′-deoxycytidine (decitabine) and gemcitabine was found to synergistically reduce viral infectivity by enhanced viral mutagenesis (Clouser et al., 2010). Decitabine and gemcitabine, both alone and in combination, were found to reduce viral loads in an AIDS mouse model in the absence of toxicity; stable prodrug derivatives have been identified to aid in clinical translation of these drugs for treatment of HIV-1 infection (Clouser et al., 2011, 2012b). Resveratrol, a phytoalexin, has been shown to enhance viral mutation (likely via inhibition of ribonucleotide reductase), and has been shown to potentiate the activity of KP-1212 – which could enhance the likelihood of the clinical utility of KP-1212 in the treatment of HIV-1 infection (Clouser et al., 2012b; Rawson et al., 2013). In this study, we examined the ability of the ribonucleoside mutagen 5-azacytidine and the ribonucleotide reductase inhibitors gemcitabine, resveratrol and clofarabine to reduce HIV-2 infectivity, as compared with HIV-1. A time-of-addition drug assay was utilized in order to identify the step(s) in the HIV-2 life cycle in which viral replication was perturbed.
An HIV-2 vector, pROD-MIG, was used in the drug susceptibility assays. This HIV-2 vector is an envelope-minus vector in which env gene sequences were deleted and a gene cassette composed of the mCherry gene, an internal ribosomal entry site (IRES) and the green fluorescence protein (gfp) gene were cloned into the remaining env gene sequence. As a control, an HIV-1 vector, pNL43-MIG (env-minus HIV-1 vector with an mCherry-IRES-GFP expression cassette), was used in parallel to access HIV-1 infectivity (Rawson et al., 2013). The molecular clone pROD-MIG was created by exchanging the mCherry gene from pHIV-1 MIG with the mouse heat stable antigen gene from pROD10-HIG (Rawson et al., unpublished data). Briefly, a fragment of pHIV-2 HIG spanning env, hsa and IRES was amplified and ligated into pGEM-T (Promega). The resulting construct and pHIV-1 MIG were digested with BamHI and AvrII (New England Biolabs). The appropriate fragments were purified and ligated together using T4 DNA ligase (New England Biolabs), thus exchanging hsa for mCherry within the pGEM-T subclone. The pGEM-T subclone and pHIV-2 HIG were then digested with PmlI, cleaving within env and IRES. The vector was treated with Antarctic phosphatase (New England Biolabs), and the appropriate fragments were ligated together using T4 DNA ligase. Like pNL43-MIG, pROD-MIG expresses mCherry and EGFP and all viral proteins except Env and Nef.
When treating cells with drugs (Fig. 1), vesicular stomatitis virus G glycoprotein (VSV-G) pseudotyped HIV-1 and HIV-2 vector virus stocks were used to infect 10 000 Magi-U373-CXCR4CEM cells per well in a 96-well plate format that had been pre-treated for 2 h with increasing concentrations of drugs or with vehicle, DMSO. Flow cytometry was used to quantify infectivity and EC50 values were calculated in GraphPad Prism6. Table 1 shows the EC50 values calculated for each drug under study along with several drugs that have known mechanisms of action (i.e. raltegravir, tenofovir, zidovudine, nevirapine). Dose–response curves and EC50 values were obtained for each independent experiment using non-linear regression models that generated non-ambiguous EC50 values and that gave acceptable fits in a combined replicates test. These replicate EC50 values were subjected to an unpaired two-tailed t-test to generate a compiled EC50 value for each drug and to compare whether the EC50 values differed for HIV-1 and HIV-2. All drugs tested, except nevirapine, possessed potent anti-HIV-1 and HIV-2 activity. Consistent with previous publications, nevirapine inhibited HIV-1, but not HIV-2, replication (Balzarini, 2004). Resveratrol, clofarabine and 5-azacytidine showed statistically different EC50 values, as indicated by a P value of 0.05 or less. In contrast, zidovudine, tenofovir and raltegravir did not show any significant differences in their EC50 values between HIV-1 and HIV-2, in agreement with previously published reports (Smith et al., 2008, 2011; Witvrouw et al., 2004). This is, to our knowledge, the first report of clofarabine, a known inhibitor of ribonucleotide reductase, having activity against both HIV-1 and HIV-2.
Fig. 1.
Drugs investigated for activity against HIV-2. Drug structures are indicated.
Table 1. EC50 values of drugs under study for HIV-1 and HIV-2 infection.
| Drug | HIV-1 EC50 (μM)[95 % confidence interval]* | HIV-2 EC50 (μM)[95 % confidence interval]* | Significant difference in EC50 values (P<0.05)† | EC50 ratio, HIV-1/HIV-2 |
| Gemcitabine‡ | 1.3×10−3 [2.28×10−4–2.81×10−3] | 6.36×10−4 [3.95×10−4−1.67×10−3] | N | 2.04 |
| Clofarabine§ | 1.12×10−1 [3.06×10−2−1.94×10−1] | 3.21×10−2 [9.52×10−4−6.32×10−2] | Y | 3.78 |
| Resveratrol‡ | 25.6 [20.1–31.0] | 6.08 [4.49–7.68] | Y | 4.21 |
| 5-Azacytidine§ | 20.2 [18.7–21.8] | 14.5 [10.3–18.8] | Y | 1.39 |
| Raltegravir‖‖ | 1.72×10−3 [8.47×10−4–4.30×10−3] | 1.85×10−3 [4.43×10−3−4.15×10−2] | N | 0.93 |
| Tenofovir‖‖ | 1.9×10−1 [49.8×10−2−3.31×10−1] | 1.30×10−1 [1.40×10−2−2.73×10−1] | N | 1.46 |
| Zidovudine‡ | 3.67×10−2 [2.72×10−2−4.62×10−2] | 4.46×10−2 [2.73×10−2−6.19×10−2] | N | 0.823 |
| Nevirapine‡ | 6.2 [5.31–7.12] | No activity | – | – |
Curve fitting was performed using log(inhibitor) vs normalized response. If the goodness of fit was not appropriate as determined by the replicates test and residuals, the curves were fitted using either log(inhibitor) vs response or log(inhibitor) vs response (variable slope) as indicated.
EC50 values for each replicate were used to perform an unpaired two-tailed t-test to determine differences between HIV-1 and HIV-2. Experiments were performed in parallel with HIV-1 and HIV-2 for each drug at least four times.
log(inhibitor) versus normalized response was used to determine EC50.
log response (inhibitor) versus response (variable slope) was used to determine EC50.
‖‖log(inhibitor) versus response (three parameters) was used to determine EC50.
Previous studies have examined the cytotoxicity of 5-azacytidine (Dapp et al., 2009), resveratrol, gemcitabine (Clouser et al., 2010, 2012a; Rawson et al., 2013) and clofarabine (L. B. Beach et al., unpublished data) using the same cell line as reported here. Based on these previous studies, the observed antiviral activity cannot be attributed to cytotoxic effects of these drugs. In support of this, microscopic inspection of the cells as well as forward and side scatter by flow cytometric analysis did not reveal any evidence of increased cell death or abnormalities with any of the drug treatments used. The finding that nevirapine inhibits HIV-1, but not HIV-2, suggests that the antiviral activity is specific and not due to cell cytotoxicity.
The finding of similar antiviral efficacy for zidovudine, tenofovir and raltegravir, but not for nevirapine, correlates well with previously reported findings that compared the anti-HIV-1 and anti-HIV-2 activities of these drugs (Andreatta et al., 2013; Roquebert et al., 2008; Witvrouw et al., 2004). Though some of the confidence intervals were relatively large, Table 1 indicates that HIV-2 is more susceptible to gemcitabine, clofarabine and resveratrol than is HIV-1 by approximately twofold, 3.5-fold and 4.2-fold, respectively. Since all three drugs are known to be inhibitors of ribonucleotide reductase, these observations provide one line of evidence for HIV-2 being more sensitive to ribonucleotide reductase inhibitors than is HIV-1 in cell culture. Intriguingly, only resveratrol and clofarabine showed a statistically significant decrease in EC50 values, but all showed a general trend of inhibiting HIV-2 with greater potency than with HIV-1. It was also observed that HIV-2 was approximately 1.4-fold more sensitive than HIV-1 to 5-azacytidine. Taken together, these data also suggest HIV-2 may be more sensitive than HIV-1 to viral mutagens that can induce lethal mutagenesis.
Time-of-addition drug assays were next done in order to identify the phase in the HIV-2 life cycle perturbed by each drug (Fig. 2). Infections of permissive target cells were done as described above, except that rather than pre-treating with drugs for 2 h before infection, drugs were added to cell culture medium immediately at the time of infection, or at varying time points post-infection. Cell culture medium was changed 24 h after each drug treatment for each time point. Cells were incubated at drug concentrations that could extinguish viral infectivity. The infectivity observed at the 0 h time point for each drug was subtracted and infectivity was subsequently normalized to 24 h no drug infectivity.
Fig. 2.
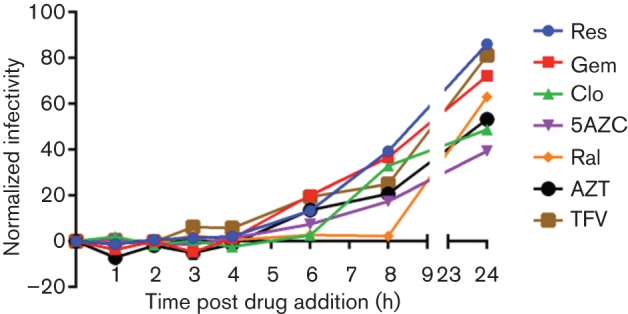
Time-of-addition drug assay. Magi cells were seeded into 96-well plates and were then infected with VSV-G pseudotyped HIV-2 vector virus. At 0, 1, 2, 3, 4, 6, 8 and 24 h post-infection, cells were treated with no drug (DMSO only) or with the IC100 values of clofarabine (Clo), zidovudine (AZT), tenofovir (TFV), resveratrol (Res), raltegravir (Ral), 5-azacytidine (5AZC), or gemcitabine (Gem). Infections were performed in triplicate, background infectivity at the 0 h time point was subtracted, and infectivity for drug treatments was normalized to no drug (DMSO only) infectivity at the 24 h time point (which was 55 %). Values represent the means of three independent experiments, with each treatment at each time point treated in duplicate. Nevirapine was not analysed due to the lack of antiviral activity against HIV-2.
The last time point when a drug was observed to consistently extinguish viral replication was interpreted to correlate with the step of the viral life cycle with which the drug likely interferes (Daelemans et al., 2011). Drugs that interfered with replication at or before 1 h were defined as targeting entry or fusion; at 3–4 h were defined as interfering with reverse transcription; and after 6 h were defined as interfering with integration (Daelemans et al., 2011). Similarly to previous publications, we qualitatively defined the time-of-drug inhibition in our assays as the time point after which infectivity was consistently detected above baseline (Daelemans et al., 2011). For an inhibitor of ribonucleotide reductase, it was hypothesized that, depending on when dNTP pool imbalances occur, the inhibition of viral infectivity could occur after fusion but prior to completion of reverse transcription.
At the 6 h time point, gemcitabine, resveratrol and clofarabine were found to reduce HIV-2 replication, which was interpreted as likely influencing the reverse transcription phase of the virus life cycle (Fig. 2). It was observed that gemcitabine and resveratrol typically lost their antiviral efficacy at earlier time points than clofarabine, but within a similar time frame to the loss of inhibition of tenofovir and zidovudine. This suggests that 2–4 h was the window of time when an antiviral that affects reverse transcription would extinguish viral replication. The size of this window for interfering with viral replication (i.e. 2 h) could be a reflection of the different timing of cellular uptake and phosphorylation of nucleoside analogues in the Magi-U373-CXCR4CEM cell line. This could also be true for the nucleoside analogues analysed in this study (i.e. 5-azacytidine, gemcitabine, clofarabine). The timing of RT perturbation was distinct from the inhibition of integrase in our assay. The integrase inhibitor raltegravir inhibited HIV-2 through 8 h (Fig. 2), the penultimate time point tested. Raltegravir was the only drug tested that inhibited HIV-2 replication beyond 6 h.
Several lines of evidence suggest that HIV-2 RT may be more sensitive than HIV-1 RT to alterations in dNTP pools. First, HIV-2 RT is less processive than HIV-1 RT (Boyer et al., 2012; MacNeil et al., 2007; Post et al., 2003), and this effect is enhanced under reduced dNTP pools (Boyer et al., 2012). Thus, dNTP pool depletion by ribonucleotide reductase inhibitors could more greatly affect RT of HIV-2 than of HIV-1. Second, HIV-2 encodes the Vpx protein (Clavel et al., 1986), which degrades the cellular triphosphorhydrolase SAMHD1 (Ahn et al., 2012). SAMHD1 degradation results in an increase in dNTP pool concentrations, which allows HIV-2 to replicate in cells with low dNTP concentrations, such as macrophages (Baldauf et al., 2012; Kim et al., 2012; Lahouassa et al., 2012; St Gelais et al., 2012). Third, HIV-1 (which does not encode Vpx) can readily replicate in macrophages without counteracting SAMHD1 (Diamond et al., 2004; Nguyen et al., 2014). This argues for an important role of Vpx in maintaining dNTP pool levels for efficient HIV-2 DNA synthesis. Fourth, fidelity differences may exist between HIV-1 RT and HIV-2 RT that could influence the sensitivity to dNTP pool alterations. Taken together, these observations suggest that HIV-2 may be more sensitive to dNTP pool alterations. Therefore, drugs that perturb nucleotide pools could have greater potential for treating HIV-2 rather than HIV-1 infection.
Drug combinations of nucleoside RT inhibitors and ribonucleotide reductase inhibitors may be able to slow the emergence of HIV-2 drug resistance since, unlike HIV-1, HIV-2 RT cannot excise nucleoside RT inhibitors and instead relies only on the exclusion of dNTP analogues to develop antiviral drug resistance (Boyer et al., 2006, 2012; Ntemgwa et al., 2009). Despite a more limited drug repertoire for the treatment of HIV-2 infection due to naturally occurring resistance polymorphisms, and the potential promise of ribonucleotide reductase inhibitors to selectively inhibit HIV-2 replication, no studies have examined the efficacy of ribonucleotide reductase inhibitors (or other classes of viral mutagens) against HIV-2 (Gottlieb et al., 2009; MacNeil et al., 2007; Menéndez-Arias & Alvarez, 2014; Rodés et al., 2000; Smith et al., 2009; Witvrouw et al., 2004).
The observations made in this study indicate the antiviral activities of 5-azacytidine, gemcitabine, resveratrol and clofarabine are selective for the RT phase of HIV-2 replication. These findings also indicate 5-azacytidine, resveratrol and clofarabine have greater antiviral activity against HIV-2 than HIV-1. Nevirapine, as expected, had antiviral activity against HIV-1, but not HIV-2. No statistical difference could be detected in the potency of gemcitabine, zidovudine, tenofovir or raltegravir. Recent reports have demonstrated that HIV-2, but not HIV-1, must degrade the dNTP pool regulator SAMHD1 for successful infection of macrophages and other myeloid cells, which are known to have extremely low dNTP pools (Diamond et al., 2004; Kim et al., 2012). The increased dNTP pools that arise through Vpx-induced SAMHD1 degradation have been hypothesized to be responsible for the rapid emergence of nucleoside RT inhibitor drug resistance in HIV-2-infected individuals (Amie et al., 2013). It is formally possible that treatment of HIV-2 infection with ribonucleotide reductase inhibitors could decrease dNTP pool levels below the Km of HIV-2 RT in macrophages, resting CD4 T-cells, and dendritic cells, which would disrupt the progression of HIV-related pathogenesis and dampen the emergence of drug resistant virus.
Acknowledgements
We thank Christine Clouser for reading and providing constructive comments on the manuscript. We thank Wei-Shau Hu (HIV Drug Resistance Program, NCI, Frederick, MD, USA) for HIV-2 vector constructs. This work was supported by NIH grant R01 GM105876. L. B. B. was supported by NIH grant F31 DA030249 and a fellowship from the University of Minnesota Graduate School. J. M. R. was supported by the Institute for Molecular Virology Training Program (NIH grant T32AI83196) and a Doctoral Dissertation Fellowship from the University of Minnesota Graduate School.
References
- Ahn J., Hao C., Yan J., DeLucia M., Mehrens J., Wang C., Gronenborn A. M., Skowronski J. (2012). HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J Biol Chem 287, 12550–12558. 10.1074/jbc.M112.340711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amie S. M., Daly M. B., Noble E., Schinazi R. F., Bambara R. A., Kim B. (2013). Anti-HIV host factor SAMHD1 regulates viral sensitivity to nucleoside reverse transcriptase inhibitors via modulation of cellular deoxyribonucleoside triphosphate (dNTP) levels. J Biol Chem 288, 20683–20691. 10.1074/jbc.M113.472159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreatta K., Miller M. D., White K. L. (2013). HIV-2 antiviral potency and selection of drug resistance mutations by the integrase strand transfer inhibitor elvitegravir and NRTIs emtricitabine and tenofovir in vitro. J Acquir Immune Defic Syndr 62, 367–374. 10.1097/QAI.0b013e31827b55f1 [DOI] [PubMed] [Google Scholar]
- Baldauf H. M., Pan X., Erikson E., Schmidt S., Daddacha W., Burggraf M., Schenkova K., Ambiel I., Wabnitz G. & other authors (2012). SAMHD1 restricts HIV-1 infection in resting CD4+ T cells. Nat Med 18, 1682–1687. 10.1038/nm.2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzarini J. (2004). Current status of the non-nucleoside reverse transcriptase inhibitors of human immunodeficiency virus type 1. Curr Top Med Chem 4, 921–944. [DOI] [PubMed] [Google Scholar]
- Biron F., Lucht F., Peyramond D., Fresard A., Vallet T., Nugier F., Grange J., Malley S., Hamedi-Sangsari F., Vila J. (1995). Anti-HIV activity of the combination of didanosine and hydroxyurea in HIV-1-infected individuals. J Acquir Immune Defic Syndr Hum Retrovirol 10, 36–40. 10.1097/00042560-199509000-00005 [DOI] [PubMed] [Google Scholar]
- Bourée P., Lamour P., Bisaro F., Didier E. (1995). [Study of an HIV positive, tropical origin population in a refugee centre in France]. Bull Soc Pathol Exot 88, 24–28 (in French). [PubMed] [Google Scholar]
- Boyer P. L., Sarafianos S. G., Clark P. K., Arnold E., Hughes S. H. (2006). Why do HIV-1 and HIV-2 use different pathways to develop AZT resistance? PLoS Pathog 2, e10. 10.1371/journal.ppat.0020010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer P. L., Clark P. K., Hughes S. H. (2012). HIV-1 and HIV-2 reverse transcriptases: different mechanisms of resistance to nucleoside reverse transcriptase inhibitors. J Virol 86, 5885–5894. 10.1128/JVI.06597-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell-Yesufu O. T., Gandhi R. T. (2011). Update on human immunodeficiency virus (HIV)-2 infection. Clin Infect Dis 52, 780–787. 10.1093/cid/ciq248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavel F., Guyader M., Guétard D., Sallé M., Montagnier L., Alizon M. (1986). Molecular cloning and polymorphism of the human immune deficiency virus type 2. Nature 324, 691–695. 10.1038/324691a0 [DOI] [PubMed] [Google Scholar]
- Clouser C. L., Patterson S. E., Mansky L. M. (2010). Exploiting drug repositioning for discovery of a novel HIV combination therapy. J Virol 84, 9301–9309. 10.1128/JVI.01006-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouser C. L., Holtz C. M., Mullett M., Crankshaw D. L., Briggs J. E., Chauhan J., VanHoutan I. M., Patterson S. E., Mansky L. M. (2011). Analysis of the ex vivo and in vivo antiretroviral activity of gemcitabine. PLoS ONE 6, e15840. 10.1371/journal.pone.0015840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouser C. L., Chauhan J., Bess M. A., van Oploo J. L., Zhou D., Dimick-Gray S., Mansky L. M., Patterson S. E. (2012a). Anti-HIV-1 activity of resveratrol derivatives and synergistic inhibition of HIV-1 by the combination of resveratrol and decitabine. Bioorg Med Chem Lett 22, 6642–6646. 10.1016/j.bmcl.2012.08.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouser C. L., Holtz C. M., Mullett M., Crankshaw D. L., Briggs J. E., O’Sullivan M. G., Patterson S. E., Mansky L. M. (2012b). Activity of a novel combined antiretroviral therapy of gemcitabine and decitabine in a mouse model for HIV-1. Antimicrob Agents Chemother 56, 1942–1948. 10.1128/AAC.06161-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costarelli S., Torti C., Rodella A., Baldanti F., Paolucci S., Lapadula G., Manca N., Quiros-Roldan E., Izzo I., Carosi G. (2008). Screening and management of HIV-2-infected individuals in northern Italy. AIDS Patient Care STDS 22, 489–494. 10.1089/apc.2007.0149 [DOI] [PubMed] [Google Scholar]
- Daelemans D., Pauwels R., De Clercq E., Pannecouque C. (2011). A time-of-drug addition approach to target identification of antiviral compounds. Nat Protoc 6, 925–933. 10.1038/nprot.2011.330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dapp M. J., Clouser C. L., Patterson S., Mansky L. M. (2009). 5-Azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. J Virol 83, 11950–11958. 10.1128/JVI.01406-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dapp M. J., Patterson S. E., Mansky L. M. (2013). Back to the future: revisiting HIV-1 lethal mutagenesis. Trends Microbiol 21, 56–62. 10.1016/j.tim.2012.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond T. L., Roshal M., Jamburuthugoda V. K., Reynolds H. M., Merriam A. R., Lee K. Y., Balakrishnan M., Bambara R. A., Planelles V. & other authors (2004). Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J Biol Chem 279, 51545–51553. 10.1074/jbc.M408573200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank I., Bosch R. J., Fiscus S., Valentine F., Flexner C., Segal Y., Ruan P., Gulick R., Wood K. & other authors (2004). Activity, safety, and immunological effects of hydroxyurea added to didanosine in antiretroviral-naive and experienced HIV type 1-infected subjects: a randomized, placebo-controlled trial, ACTG 307. AIDS Res Hum Retroviruses 20, 916–926. 10.1089/aid.2004.20.916 [DOI] [PubMed] [Google Scholar]
- Gottlieb G. S., Badiane N. M., Hawes S. E., Fortes L., Toure M., Ndour C. T., Starling A. K., Traore F., Sall F. & other authors (2009). Emergence of multiclass drug-resistance in HIV-2 in antiretroviral-treated individuals in Senegal: implications for HIV-2 treatment in resouce-limited West Africa. Clin Infect Dis 48, 476–483. 10.1086/596504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris K. S., Brabant W., Styrchak S., Gall A., Daifuku R. (2005). KP-1212/1461, a nucleoside designed for the treatment of HIV by viral mutagenesis. Antiviral Res 67, 1–9. 10.1016/j.antiviral.2005.03.004 [DOI] [PubMed] [Google Scholar]
- Kim B., Nguyen L. A., Daddacha W., Hollenbaugh J. A. (2012). Tight interplay among SAMHD1 protein level, cellular dNTP levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J Biol Chem 287, 21570–21574. 10.1074/jbc.C112.374843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahouassa H., Daddacha W., Hofmann H., Ayinde D., Logue E. C., Dragin L., Bloch N., Maudet C., Bertrand M. & other authors (2012). SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol 13, 223–228. 10.1038/ni.2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lori F., Malykh A. G., Foli A., Maserati R., De Antoni A., Minoli L., Padrini D., Degli Antoni A. M., Barchi E. & other authors (1997). Combination of a drug targeting the cell with a drug targeting the virus controls human immunodeficiency virus type 1 resistance. AIDS Res Hum Retroviruses 13, 1403–1409. 10.1089/aid.1997.13.1403 [DOI] [PubMed] [Google Scholar]
- MacNeil A., Sarr A. D., Sankalé J. L., Meloni S. T., Mboup S., Kanki P. (2007). Direct evidence of lower viral replication rates in vivo in human immunodeficiency virus type 2 (HIV-2) infection than in HIV-1 infection. J Virol 81, 5325–5330. 10.1128/JVI.02625-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menéndez-Arias L., Alvarez M. (2014). Antiretroviral therapy and drug resistance in human immunodeficiency virus type 2 infection. Antiviral Res 102, 70–86. 10.1016/j.antiviral.2013.12.001 [DOI] [PubMed] [Google Scholar]
- Mullins J. I., Heath L., Hughes J. P., Kicha J., Styrchak S., Wong K. G., Rao U., Hansen A., Harris K. S. & other authors (2011). Mutation of HIV-1 genomes in a clinical population treated with the mutagenic nucleoside KP1461. PLoS ONE 6, e15135. 10.1371/journal.pone.0015135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L. A., Kim D. H., Daly M. B., Allan K. C., Kim B. (2014). Host SAMHD1 protein promotes HIV-1 recombination in macrophages. J Biol Chem 289, 2489–2496. 10.1074/jbc.C113.522326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntemgwa M. L., d’Aquin Toni T., Brenner B. G., Camacho R. J., Wainberg M. A. (2009). Antiretroviral drug resistance in human immunodeficiency virus type 2. Antimicrob Agents Chemother 53, 3611–3619. 10.1128/AAC.00154-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post K., Guo J., Howard K. J., Powell M. D., Miller J. T., Hizi A., Le Grice S. F., Levin J. G. (2003). Human immunodeficiency virus type 2 reverse transcriptase activity in model systems that mimic steps in reverse transcription. J Virol 77, 7623–7634. 10.1128/JVI.77.13.7623-7634.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson J. M., Heineman R. H., Beach L. B., Martin J. L., Schnettler E. K., Dapp M. J., Patterson S. E., Mansky L. M. (2013). 5,6-Dihydro-5-aza-2′-deoxycytidine potentiates the anti-HIV-1 activity of ribonucleotide reductase inhibitors. Bioorg Med Chem 21, 7222–7228. 10.1016/j.bmc.2013.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey M. A., Krust B., Laurent A. G., Guétard D., Montagnier L., Hovanessian A. G. (1989). Characterization of an HIV-2-related virus with a smaller sized extracellular envelope glycoprotein. Virology 173, 258–267. 10.1016/0042-6822(89)90242-0 [DOI] [PubMed] [Google Scholar]
- Rodés B., Holguín A., Soriano V., Dourana M., Mansinho K., Antunes F., González-Lahoz J. (2000). Emergence of drug resistance mutations in human immunodeficiency virus type 2-infected subjects undergoing antiretroviral therapy. J Clin Microbiol 38, 1370–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roquebert B., Damond F., Collin G., Matheron S., Peytavin G., Bénard A., Campa P., Chêne G., Brun-Vézinet F. & other authors (2008). HIV-2 integrase gene polymorphism and phenotypic susceptibility of HIV-2 clinical isolates to the integrase inhibitors raltegravir and elvitegravir in vitro. J Antimicrob Chemother 62, 914–920. 10.1093/jac/dkn335 [DOI] [PubMed] [Google Scholar]
- Smith R. A., Gottlieb G. S., Anderson D. J., Pyrak C. L., Preston B. D. (2008). Human immunodeficiency virus types 1 and 2 exhibit comparable sensitivities to Zidovudine and other nucleoside analog inhibitors in vitro. Antimicrob Agents Chemother 52, 329–332. 10.1128/AAC.01004-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R. A., Anderson D. J., Pyrak C. L., Preston B. D., Gottlieb G. S. (2009). Antiretroviral drug resistance in HIV-2: three amino acid changes are sufficient for classwide nucleoside analogue resistance. J Infect Dis 199, 1323–1326. 10.1086/597802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R. A., Raugi D. N., Kiviat N. B., Hawes S. E., Mullins J. I., Sow P. S., Gottlieb G. S., University of Washington-Dakar HIV-2 Study Group (2011). Phenotypic susceptibility of HIV-2 to raltegravir: integrase mutations Q148R and N155H confer raltegravir resistance. AIDS 25, 2235–2241. 10.1097/QAD.0b013e32834d8e52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares R. S., Tendeiro R., Foxall R. B., Baptista A. P., Cavaleiro R., Gomes P., Camacho R., Valadas E., Doroana M. & other authors (2011). Cell-associated viral burden provides evidence of ongoing viral replication in aviremic HIV-2-infected patients. J Virol 85, 2429–2438. 10.1128/JVI.01921-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Gelais C., de Silva S., Amie S. M., Coleman C. M., Hoy H., Hollenbaugh J. A., Kim B., Wu L. (2012). SAMHD1 restricts HIV-1 infection in dendritic cells (DCs) by dNTP depletion, but its expression in DCs and primary CD4+ T-lymphocytes cannot be upregulated by interferons. Retrovirology 9, 105. 10.1186/1742-4690-9-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ende M. E., Kroes A. C., Buitenwerf J., van der Poel C. L. (1990). [Aids caused by HIV-2 in The Netherlands]. Ned Tijdschr Geneeskd 134, 495–497 (in Dutch). [PubMed] [Google Scholar]
- Witvrouw M., Pannecouque C., Switzer W. M., Folks T. M., De Clercq E., Heneine W. (2004). Susceptibility of HIV-2, SIV and SHIV to various anti-HIV-1 compounds: implications for treatment and postexposure prophylaxis. Antivir Ther 9, 57–65. [PubMed] [Google Scholar]