

Abstract
Prostaglandin E2 (PGE2) is a natural lipid-derived molecule that is involved in important physiological functions. Abnormal PGE2 signalling has been associated with pathologies of the nervous system. Previous studies provide evidence for the interaction of PGE2 and canonical Wnt signalling pathways in non-neuronal cells. Since the Wnt pathway is crucial in the development and organization of the brain, the main goal of this study is to determine whether collaboration between these pathways exists in neuronal cell types. We report that PGE2 interacts with canonical Wnt signalling through PKA and PI-3K in neuroectodermal (NE-4C) stem cells. We used time-lapse microscopy to determine that PGE2 increases the final distance from origin, path length travelled, and the average speed of migration in Wnt-activated cells. Furthermore, PGE2 alters distinct cellular phenotypes that are characteristic of Wnt-induced NE-4C cells, which corresponds to the modified splitting behaviour of the cells. We also found that in Wnt-induced cells the level of β-catenin protein was increased and the expression levels of Wnt-target genes (Ctnnb1, Ptgs2, Ccnd1, Mmp9) was significantly upregulated in response to PGE2 treatment. This confirms that PGE2 activated the canonical Wnt signalling pathway. Furthermore, the upregulated genes have been previously associated with ASD. Our findings show, for the first time, evidence for cross-talk between PGE2 and Wnt signalling in neuronal cells, where PKA and PI-3K might act as mediators between the two pathways. Given the importance of PGE2 and Wnt signalling in prenatal development of the nervous system, our study provides insight into how interaction between these two pathways may influence neurodevelopment.
Keywords: Prostaglandin E2, Wnt signalling, Neuroectodermal stem cells, Cell motility, Proliferation, Autism
Background
The plasma membrane phospholipids play a fundamental role in the nervous system and act as a reservoir for second messenger molecules important for the development and normal functioning of the brain. Prostaglandin E2 (PGE2) is a bioactive fatty acid that is derived from arachidonic acid, a major structural component of plasma membrane phospholipids, through the enzymatic metabolism of cyclooxygenases −1 and −2 (COX-1,-2) and then prostaglandin synthases [1]. Extracellular stimuli such as immunological and infectious agents [2-4], environmental toxins such as mercury and lead [5], and exposure to drugs including misoprostol and valproic acid [6] can trigger the local production of PGE2 via specific biosynthetic pathways, resulting in altered cell signal transmission that modulates biological functions such as sleep, fever, inflammation, and pain [7].
The diverse action of PGE2 is achieved through the activation of 4 different G-protein coupled E-prostanoid receptors (EP1 through 4) [8,9]. The divergent role of PGE2 is amplified by the variety of different kinase-mediated signalling cascades that can be activated through its EP receptors, such as the protein kinase A (PKA), phosphatidylinositide 3-kinases (PI-3K), and protein kinase C (PKC) pathways [10].
During the early stages of pregnancy, there are elevated levels of COX-2 and PGE synthases, enzymes responsible for the production of PGE2, which is indicative of the involvement of PGE2 in prenatal development [11]. We have previously shown that the expression profiles of EP receptors during mouse embryonic development changes depending on the embryonic stage, with EP receptor expression highest during E7 (Embryonic day 7) and E15, which corresponds to peak periods of neurogenesis [12]. It has been shown that PGE2 plays a regulatory role in membrane excitability and synaptic transmission in neurons [13]. PGE2 increases the dendritic length and complexity of Purkinje neurons, and can also alter neuronal firing activity in the developing brain [14]. PGE2 is involved in synaptic plasticity and neuroprotection [15], and can also be involved in neuronal cell death and apoptosis [16,17]. Prostaglandins have also been reported to induce the differentiation of neuronal cells [18]. Moreover, the inhibition of COX-2, can suppress neurogenesis and proliferation of neural progenitor cells [19]. These studies show the important role PGE2 can play during normal development of the nervous system. Furthermore, previous research found that PGE2 can exert various effects on cell development, proliferation, and migration in a diversity of cell lines. It has been shown that PGE2 stimulates cell growth and motility in osteoblasts [20], prostate cancer cells [21], and pancreatic stellate cells [22]. The migration of vascular smooth muscle cells [23], intestinal subepithelial myofibroblasts [24], dendritic cells [25], hepatocellular carcinoma cells [26], and mesangial cells [27] can all be regulated by PGE2. However, the effects of PGE2 on neural stem cell behaviour and movement are not well characterized. Our previous studies provide some insight into the molecular mechanisms of abnormal PGE2 signalling on neuronal cells. We have found that exposure to PGE2 results in the retraction of neurites and the elevation of calcium amplitude fluctuations in growth cones of differentiated Neuro-2A cells [12,28].
Abnormal fatty acid metabolism through the PGE2 pathway may contribute to the pathology of neurodevelopmental disorders such as Autism Spectrum Disorders (ASD) [29]. Abnormal levels of PGE2 and other fatty acid metabolites have been identified as potential biomarkers for ASD [30]. PGE2 can act as an endogenous modulator for cerebellar development in the rat brain affecting social interaction and sensory behaviour, which are characteristic behaviours altered in ASD [31]. A clinical study showed that maternal exposure to the drug misoprostol (prostaglandin E analogue), has been associated with the development of Moebius syndrome and autistic-like symptoms [32-34].
Current literature also provides evidence that PGE2 signalling interacts with another crucial developmental pathway, the canonical Wnt (wingless-related MMTV integration site) signalling pathway in various non-neuronal cells [35] such as osteocytes [36], prostate and colon cancer cells [37], hematopoietic stem cells [38], and mesenchymal stem cells [39]. Wnt signalling is tightly regulated in early development and is required for the formation of the nervous system [40]. The canonical Wnt signalling pathway is composed of a network of proteins that modify cell communication and interactions with other cells. Wnt proteins bind to cell surface Frizzled (FZD) receptors, where the signal is then transduced to β-catenin, activating the transcription of Wnt target genes. The Wnt molecules are vital to embryonic development since they can moderate cell proliferation and differentiation by participating in the determination of cell fates [41]. Previous literature shows that convergence of PGE2-dependent signalling with the canonical Wnt pathway can occur at the level of β-catenin through EP1-4 receptors, including the association of the Gαs subunit with Axin, the stimulation of the cAMP/PKA pathway, or the phosphorylation of GSK-3β by PI-3K [42]. However, the interaction of PGE2 and Wnt signalling is not well-characterized in the nervous system. To activate and study canonical Wnt signalling in an in vitro model system, Wnt Agonist (WntA), 2-amino-4-[3,4-(methylenedioxy)benzylamino]-6-(3-methoxyphenyl) pyrimidine, can serve as a useful reagent. WntA is a cell-permeable pyrimidine compound that mimics the effects of Wnt by functioning through the canonical pathway via upregulating TCF-activity without inhibiting the activity of GSK-3β [43]. This is important because GSK-3β plays a regulatory role in many signalling pathways other than Wnt so an agonist that blocks GSK-3β could have disparate effects in cellular models.
This study investigates the effects of PGE2 interaction with the Wnt signalling pathway on the behaviour of murine neuroectodermal (NE-4C) stem cells. We demonstrate that PGE2 modifies canonical Wnt signalling in NE-4C stem cells by altering components of cell motility such as final distance travelled, path length travelled, average speed of migration, as well as cell splitting behaviour. We also reveal that PGE2 can alter the protein expression of non-phospho (active) β-catenin (Ser33/37/Thr41), as well as the expression of specific Wnt-target genes. Interestingly, the genes implicated in our study have been previously associated with ASD. To our knowledge, we show for the first time, that PGE2 signalling interacts with the Wnt pathway in neural stem cells to affect cell behaviour and gene transcription. Our study furthers our understanding of the possible mechanisms by which intracellular cross-talk between PGE2 and Wnt signalling may contribute to cell migration and proliferation during prenatal development of the nervous system.
Results
Expression of EP1-4 receptors in NE-4C cells
To determine whether NE-4C cells endogenously express the receptors of PGE2, we performed real-time quantitative PCR assay, Western blot analysis, and immunocytochemistry. Our results show that in NE-4C cells, EP2 had the highest mRNA expression followed by EP3γ and EP4 receptors. Endogenous EP1 and EP3β receptor expression was considerably low in NE-4C cells, while the EP3α transcript level was nearly absent and may be considered negligible. The relative quantity (RQ = 1) values of EP1, EP2, EP3α, EP3β, EP3γ, and EP4 transcripts expression were 3, 542, 0, 1, 391, and 15, respectively (Figure 1A). Western blot results confirm the expression of all four EP receptors in NE-4C cells (Figure 1B). The localization of the EP receptors in NE-4C cells was also detected with immunocytochemistry using EP1-4 specific antibodies along with antibodies against various cellular organelles including the nuclear envelope, Golgi apparatus, the endoplasmic reticulum, and β-Actin (Figure 1C). Our results show that EP1 receptors were localized in the ER membrane, EP2 receptors were uniformly expressed around the nucleus and co-localized with the nuclear envelope marker, EP3 receptors were located at the plasma membrane, and EP4 receptors at the Golgi apparatus. Hence, NE-4C cells can act as an appropriate experimental model to study PGE2 signalling.
Figure 1.
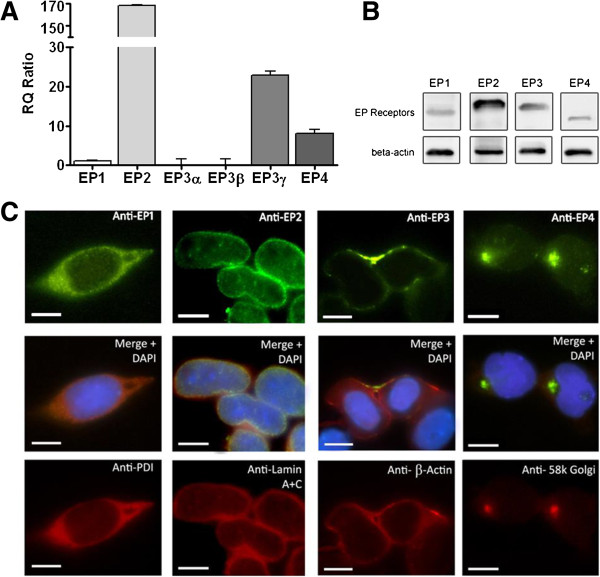
Expression of EP receptors’ mRNA and protein in NE-4C cells. (A) Real-time PCR was used to determine the RQ value for EP1, EP2, EP3α, EP3β, EP3γ and EP4 receptors, which was found to be 2, 16, 1, 2, 46 and 46 respectively. The error bars represent + SEM. (B) Western blot analysis of the EP1, EP2, EP3 and EP4 receptors expression (65, 68, 62 and 53 kDa, respectively). β-Actin was used to indicate equal loading. (C) Immunocytochemistry revealed the subcellular localization of EP1-4 receptors with specific organelles visualized through the use of anti-PDI endoplasmic reticulum marker, anti-Lamin A + C nuclear envelope marker, β-Actin cell membrane marker, and anti-58 K Golgi marker. The scale bar represents 10 μm.
Prostaglandin E2 increases the cell motility of Wnt-induced NE-4C cell migration
The effect of PGE2 on Wnt-dependent migration of NE-4C cells was determined using Nikon Eclipse Ti-E microscope with NIS Elements time-lapse tracking software over a 24 hour period. Final distance, path length, and average speed were quantified after exposure to 1 μM PGE2, 2 μM Wnt Agonist (WntA), or 2 μM WntA with the addition of 1 μM PGE2. The final distance was defined as the distance between the initial and final positions of the cell, represented as a straight line distance. The path length was the total distance travelled from the initial to the final cell position. The average speed of a cell was calculated by dividing the total distance travelled by the time it took to travel between the two positions.
The results show that untreated NE-4C cells moved an average final distance of 65.6 μm following a 24 hour period (Figure 2A). The addition of PGE2 to the cells resulted in a final distance of 56.2 μm which was not significantly different from the untreated control (65.6 μm). WntA only treatment resulted in a significant decrease in final distance of 21.3 μm (p = 0.00242) when compared to the control. The addition of PGE2 to WntA-treated cells resulted in a final distance of 45.0 μm, which is an increase by 23.6 μm (p = 0.04371), as compared to WntA only-treated cells. It represents a 211% increase from the WntA-regulated movement. Visualization of final distance through dispersion XY position plots clearly illustrates that PGE2 signalling restores the Wnt-regulated suppression of cell movement (Figure 2B, WntA + PGE2).
Figure 2.

PGE2-dependent effect on final distance travelled from origin. (A) Final distance from origin was 65.6, 56.2, 21.3, 45.0 μm, respectively. The error bars represent + SEM and values were considered significant at *p < 0.05, **p < 0.01. (B) The Dispersion XY position plots illustrate the effect of PGE2 on Wnt-induced behaviour, where addition of PGE2 to Wnt-activated cells increased the final distance. Addition of H89 (PKA blocker) and Wort (PI-3K) blocker reduces the effect PGE2. Measurements represent an average of 150 cells from three independent experiments (N = 3).
The quantification of path length (Figure 3A) revealed the same pattern. The path length of untreated cells was 458.9 μm. As compared to untreated cells, PGE2 only treatment did not result in a significant change (408.6 μm), but WntA treatment significantly decreased the path length to 103.3 μm (p = 0.00189). Addition of PGE2 to WntA-treated cells led to a path length of 362.1 μm. This is a 350% increase (p = 0.00928) compared to WntA only-treated cells.
Figure 3.
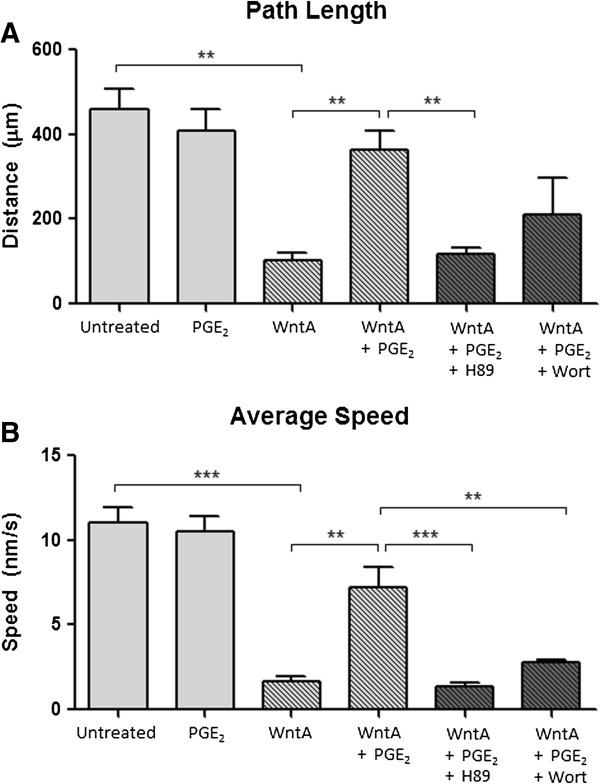
PGE2-dependent effect on path length and average speed. (A) Path length travelled was 459, 409, 103, 362 μm, respectively. (B) Average speed of migration was 11.0, 10.5, 1.7, 7.2 nm/s, respectively. The error bars represent + SEM, **p < 0.01, ***p < 0.001. Results represent an average of 150 cells from three independent experiments (N = 3).
Quantification of average speed showed that PGE2 treated cells travelled at a speed of 10.5 nm/s, which was not significantly different from untreated NE-4C cells that moved at a speed of 11.0 nm/s (Figure 3B). WntA only treatment resulted in a decreased average cell speed of 1.65 nm/s (p = 0.00065). Addition of PGE2 to WntA-treated cells resulted in an average speed of 7.34 nm/s. This suggests that addition of PGE2 elevated the average speed by 439%; an increase of 5.59 nm/s (p = 0.00946) when compared to WntA only-treatment.
In summary, administration of PGE2 treatment leads to significant changes in WntA-regulated cell behaviours such as final distance, path length, and average speed. PGE2 treatment significantly restored the cell kinematic measures which were suppressed by WntA treatment.
Prostaglandin E2 modulates Wnt-induced cell behaviour through PKA and PI-3K kinases
Previous studies in embryonic kidney and colon cancer cells determined that the convergence of PGE2 signalling on the Wnt pathway occurred through the activation of PKA or PI-3K [44-46]. To determine whether PGE2 treatment alters Wnt-induced cell migration behaviour via these kinases in NE-4C cells, we used dihydrochloride hydrate (H89) to block PKA and Wortmannin (Wort) to block PI-3K. Our results show a trend across final distance, path length, and average speed (Figures 2 and 3). With the addition of H89 to WntA + PGE2 treated cells, all cell motility measures significantly decreased compared to the WntA + PGE2 treated cells, resulting in movement profiles that were not statistically different from the WntA-only condition. Specifically, H89-treated cells travelled a final distance of 20.32 μm from the origin (p = 0.02477), path length of 116.01 μm (p = 0.00567), and at an average speed of 1.37 nm/s (p = 0.00073) (Figure 2A and 2B).
With the addition of Wort to WntA + PGE2 treated cells, there was a decreasing trend in final distance and path length but it was not significantly different from PGE2 + WntA treated cells. Only average speed significantly decreased to 2.76 nm/s (N = 3; p = 0.00422) compared to the WntA + PGE2 treatment. Post hoc Dunnett t-test revealed that measurements from the H89 and Wort conditions were not significantly different from the WntA-only treatment, indicating that H89 and Wort significantly diminished the effect of PGE2 on WntA-treated cells. This indicates that PGE2 likely acts through PKA and PI-3K to elicit effects on the WntA-dependent cell motility. However, it appears that H89 may have had a greater effect, suggesting that PGE2 may predominately act through PKA.
Prostaglandin E2 alters cell proliferation behaviour of NE-4C cells induced by Wnt agonist treatment
Previous literature reveals that PGE2 may also affect cell proliferation via the Wnt signalling pathway in prostate and colon cancer cells [37] and hematopoietic [38] and mesenchymal [39] stem cells. We studied the effects of PGE2 on NE-4C cell proliferation using NIS Elements software. The cells were exposed to 1 μM PGE2, 2 μM WntA, or 2 μM WntA with the addition of 1 μM PGE2. Furthermore, H89 or Wort was added to PGE2 + WntA treated cells to determine the effective role of these kinases. The initial number of cells was compared to the final number of cells following 24 hours treatment. PGE2 treatment led to an increase in cell number by 4.60-fold, which was not significantly different from the untreated cells that proliferated by a 4.59 fold-increase (Figure 4A). Administration of WntA resulted in a fold-change of 0.86 (p < 0.001) which was significantly lower than untreated cells. Addition of PGE2 to WntA-treated cells (WntA + PGE2) resulted in a fold-change of 1.03, which was not significant from the WntA only treated condition. Although we observed lower levels of proliferation in the WntA, WntA + PGE2 and WntA + PGE2 + Blocker conditions, we confirmed no change in cell viability between the conditions tested (Figure 4B).
Figure 4.
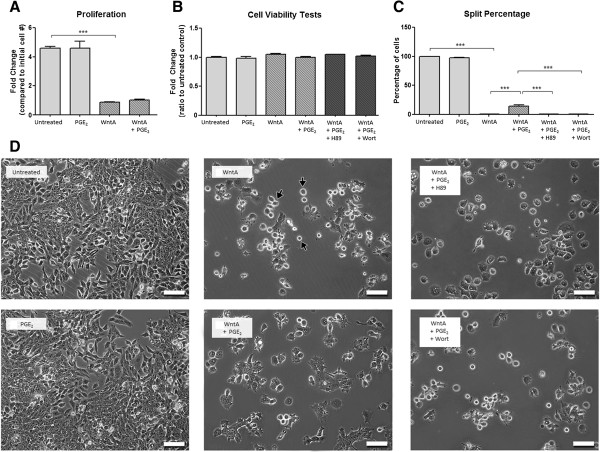
PGE2-dependent effect on proliferation behaviour. (A) Over the experimental duration of 24 hours, the number of cells changed by a fold of 4.60, 4.59, 0.86, 1.03, respectively. (B) Cell viability across treatment conditions was not significantly different. (C) Percentage of successful split ratio was 100%, 98%, 0%, 15%, 0%, and 0% respectively. The error bars represent + SEM, ***p < 0.001. Measurements represent an average of 150 cells from three independent experiments (N = 3). (D) WntA treatment resulted in an arrested state indicted by the black arrows and corresponded with a significant decrease in cell proliferation (***p < 0.001). Scale bar represents 100 μm.
However, we observed distinct differences in cell phenotype between the WntA, WntA + PGE2 and WntA + PGE2 with H89 or Wort treatment. A majority of the cells treated with WntA adopted a shiny circular shape (indicated by black arrows, Figure 4D). This was not as prevalent in the WntA + PGE2 condition. However, the cells treated with WntA + PGE2 and Wort blocker, adopted the shiny circular phenotype seen in the WntA condition. Cells treated with WntA + PGE2 and H89 blocker adopted a circular appearance as well but a smaller population of these round cells were shiny.
Our experiments showed that cell viability was not affected but a distinct shiny circular cell appearance was observed, which is characteristic of a cell just prior to splitting into two daughter cells. Therefore, we also quantified the split percentage, defined as the percentage of cells that successfully divided into two daughter cells during the recorded time period. As expected, the NE-4C untreated cells demonstrated a split percentage of 100% (Figure 4C), indicating that all cells entering a mitotic phase resulted in cell division. A similar pattern was seen in PGE2-treated cells (97.5%). However, treatment of WntA resulted in a significant decrease of split percentage to 0% (p < 0.001), where mitotic cells appeared to become arrested in a round stage denoted in Figure 4D (WntA Image) with black arrows. The addition of 1 μM PGE2 to WntA-treated cells produced a significant increase in split percentage to 14.7% (p < 0.001, Figure 4C) as compared to WntA only treatment. The cells appear to resume their flat morphology. These results suggest that PGE2 treatment can modify Wnt-induced proliferation behaviour such as split percentage. Following treatment with either H89 or Wort, cells returned to a split percentage of 0% as seen with WntA only treatment (Figure 4C, D). This again indicates that PGE2 likely acts on the Wnt pathway through PKA and PI-3K to modify cell proliferation.
To further confirm our results of the cell splitting behaviour, we measured the level of Phospho-Histone H3 (Ser10) (Figure 5) since phosphorylation at Ser10 is tightly associated with chromosome condensation and segregation that occurs during mitosis [47-49]. Compared to untreated cells, PGE2 only-treated cells did not display a significant difference. However, when compared to untreated NE-4C cells, cells treated with WntA, WntA + PGE2 and WntA + PGE2 with H89 or Wort treatment led to a significance increase in Phospho-Histone H3 (Ser10) expression. RQ values were 1.35 (p = 0.033), 1.52 (p = 0.001), 1.36 (p = 0.027), and 1.58 (p = 0.005), respectively. This revealed that although cell numbers were lower under these conditions, the relative expression of Phospho-Histone H3 (Ser10) was significantly higher, indicating that a greater percentage of cells were undergoing mitosis when exposed to these treatments compared to untreated cells. This correlates with our finding that a larger proportion of cells under these conditions adopts and seems to be arrested in a round stage characteristic of cells undergoing mitosis.
Figure 5.
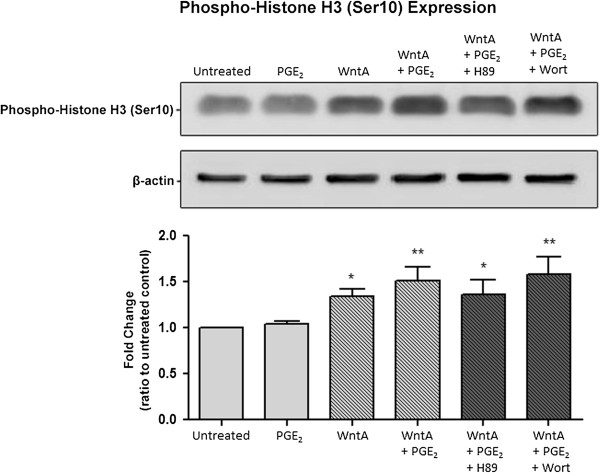
PGE2-dependent effect on phospho-histone H3 (Ser10) expression. Western blot analysis was used to determine Phospho-Histone H3 (Ser10) protein (17 kDa). The expression of Phospho-Histone H3 (Ser10) represented in fold change was 1, 1.04, 1.35, 1.52, 1.36, and 1.58, respectively. The error bars represent + SEM and values were considered significantly different from untreated *p < 0.05, **p < 0.01. Average measurements represent protein from three independent experiments (N = 3). β-Actin was used to indicate equal loading.
Prostaglandin E2 increases active β-catenin expression in Wnt-induced NE-4C cells
β-catenin is a key effector in the canonical Wnt signalling pathway that regulates downstream gene transcription [50]. β-catenin levels can be intricately regulated at multiple phosphorylation sites. Phosphorylation at Ser33, Ser37, and Thr41 leads to its destabilization and primes it for degradation [51], while phosphorylation at Ser552 has been correlated with β-catenin nuclear accumulation [52,53]. We tested the levels of non-phospho-(the active form) β-catenin (Ser33/37/Thr41) and phospho-β-catenin (Ser552). The addition of PGE2 only to NE-4C cells did not significantly change the levels of either form of β-catenin (Figure 6A and B). However, adding PGE2 to WntA-induced NE-4C cells lead to a significant 2.1 fold increase in non-phospho-(active) β-catenin (Ser33/37/Thr41) levels compared to the WntA only treated condition (Figure 7A). There was no significant difference in Phospho-β-catenin (Ser552) levels between the sample conditions (Figure 7B), suggesting that phosphorylation of β-catenin at Ser552 is likely not involved with the behavioural differences in NE-4C cells described earlier. These results indicate that PGE2 may interact with the canonical Wnt signalling pathway by regulation of non-phospho-(active) β-catenin (Ser33/37/Thr41) levels.
Figure 6.
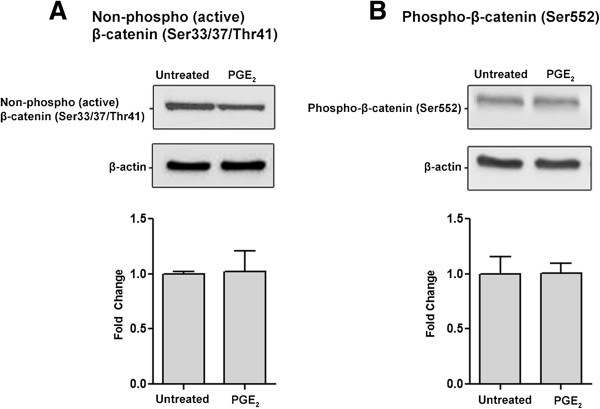
PGE2-dependent effect on β-catenin expression in NE-4C cells. Western blot analysis was used to determine two forms of active β-catenin: (A) non-phospho-(active) β-catenin (Ser33/37/Thr41) and (B) phospho-β-catenin (Ser552) (92 kDa). Addition of PGE2 to NE-4C cells did not yield a significant difference in levels of either active form of β-catenin compared to control. The error bars represent + SEM and values were considered significantly different from control at *p < 0.05, **p < 0.01. Average measurements represent protein from three independent experiments (N = 3). β-Actin was used to indicate equal loading.
Figure 7.
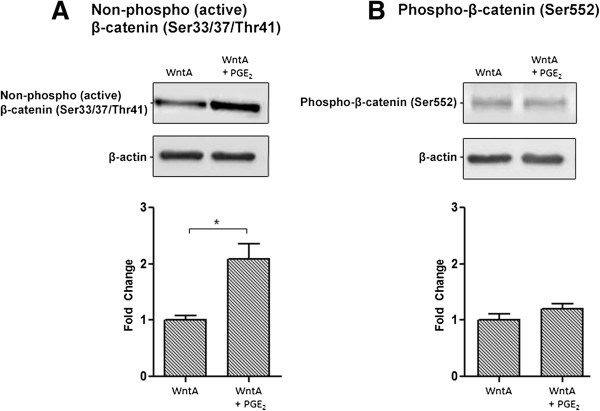
PGE2-dependent effect on β-catenin expression in Wnt-activated NE-4C cells. Western blot analysis was used to determine two forms of active β-catenin: non-phospho-(active) β-catenin (Ser33/37/Thr41) and phospho-β-catenin (Ser552) (92 kDa). (A) The expression of active β-catenin represented in fold change was 1, 2.09, 1.61, and 1.98, respectively. The error bars represent + SEM and values were considered significantly different from control at *p < 0.05. Only PGE2 + WntA condition was significantly different from WntA only condition. (B) There was no significant difference in phospho-β-catenin (Ser552) expression between the conditions. Average measurements represent protein from three independent experiments (N = 3). β-Actin was used to indicate equal loading.
Prostaglandin E2 regulates expression of Wnt-target genes in Wnt-induced NE-4C cells
To investigate whether the addition of PGE2 can influence gene transcription relevant to the canonical Wnt pathway, we screened 29 target genes using Custom TaqMan® Array Plates. We found that Ctnnb1, Ptgs2, Ccnd1, and Mmp9 were differentially regulated (data not shown). Their expression was confirmed with real-time PCR using RNA derived from the same treatment conditions used for behavioural analyses, which includes 1 μM PGE2, 2 μM Wnt Agonist (WntA), or 2 μM WntA with the addition of 1 μM PGE2. Kinase blockers (H89 or Wort) were added to PGE2 + WntA treated cells to determine the potential contribution of PKA and PI3K activity via PGE2 signalling. Our real-time PCR results indicate that PGE2 affects the expression levels of all Wnt-target genes tested (Figure 8).
Figure 8.
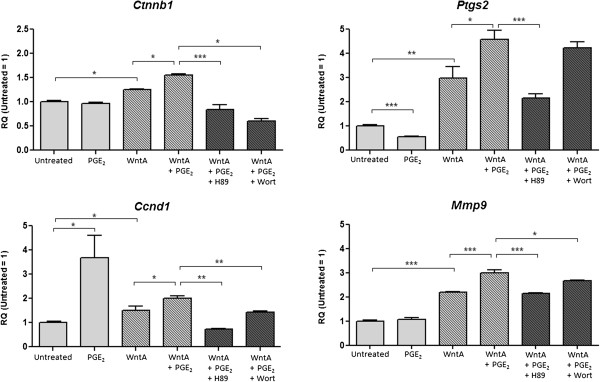
PGE2-dependent effect on Wnt-target genes. Real-time PCR was used to determine the RQ value for Ctnnb1, Ptgs2, Ccnd1, and Mmp9. The expression of Ctnnb1 represented in fold change was 1, 0.97, 1.25, 1.55, 0.84, and 0.60, respectively. The fold change expression of Ptgs2 was 1, 0.56, 2.99, 4.59, 2.16, and 4.22. The fold change expression of Ccnd1 was 1, 3.68, 1.50, 1.99, 0.74, and 1.42. Mmp9 fold change expression was 1, 1.08, 2.19, 3.00, 2.16, and 2.68, respectively. The error bars represent + SEM and values were considered significantly different from control at *p < 0.05, **p < 0.01, and ***p < 0.001. Average measurements are from three independent experiments (N = 3).
Ctnnb1 (beta-catenin) levels were not altered with the addition of PGE2 when compared to untreated NE-4C cells, but cells treated with WntA showed a significant increase of RQ value 1.25 (p = 0.0372). Addition of PGE2 to WntA-induced cells led to a further increase of Ctnnb1 level to an RQ value of 1.55, which was significantly different from the WntA-only condition (p = 0.0131). This pattern was consistent with the expression of phospho (active) β-catenin (Ser33/37/Thr41) protein quantified earlier using Western blot analysis. Addition of H89 or Wort to PGE2 + WntA treated cells resulted in RQ values to 0.83 and 0.60, respectively, compared to untreated cells which was a significant decrease compared to the PGE2 + WntA condition (p > 0.001, p > 0.001). The PKA and PI3K blockers, H89 and Wort, appeared to remove the effect of PGE2 on Ctnnb1 expression in WntA-induced cells, while also reversing the influence on Ctnnb1 levels from WntA-only treatment. This suggests that PKA and PI3K signalling may modify Ctnnb1 expression through PGE2 signalling.
NE-4C cells treated with PGE2 alone had a significant decrease in Ptgs2 (prostaglandin-endoperoxide synthase 2; gene encoding COX-2) mRNA levels compared to untreated cells (RQ = 0.56, p < 0.001), while cells treated with WntA had a significant increase of RQ value 2.99 (p = 0.00286). In contrast, when PGE2 was added to WntA-induced NE-4C cells, Ptgs2 expression was further elevated with an RQ value of 4.59 compared to untreated. This value was significantly different from the PGE2 + WntA condition (p = 0.015). Addition of H89 or Wort to PGE2 + WntA treated cells resulted in RQ values of 2.16 and 4.22, but only the H89 treatment was significantly different from the PGE2 + WntA condition (p < 0.001). This suggests that the effect of PGE2 on WntA-induced cells may be through PKA.
Expression of Ccnd1 (cyclin D1) was also altered. Administration of PGE2 treatment to NE-4C cells correlated with a significant increase of an RQ value to 3.68 (p = 0.045) compared to untreated cells, while WntA-treated cells had a significant increase of RQ value to 1.50 (p = 0.048). Addition of PGE2 to WntA-activated cells was associated with a further increase of Ccnd1 expression, with an RQ value 1.99 compared to untreated cells, which was significantly different from WntA-only treated cells (p = 0.047). H89 or Wort added to PGE2 + WntA treated cells had RQ values of 0.74 and 1.42, respectively, which was significantly different from the PGE2 + WntA condition (p = 0.0054, p = 0.0078). The blockers, H89 and Wort, seemed to attenuate the increase of Ccnd1 levels associated with the addition of PGE2 to WntA-induced cells.
In comparison to untreated NE-4C cells, PGE2 treatment did not change levels of Mmp9 (matrix metalloproteinase 9). However, when compared to WntA-induced NE-4C cells, addition of PGE2 treatment to WntA-treated cells caused a significant increase in expression level (p < 0.001). Specifically, with WntA treatment, Mmp9 expression was significantly elevated to an RQ value of 2.19 (p < 0.001) compared to untreated cells, but addition of PGE2 to WntA-induced cells resulted in a further rise of Mmp9 expression with an RQ value of 3.00. H89 and Wort were added to PGE2 + WntA treated cells and RQ values for Mmp9 were 2.16 and 2.68, respectively, compared to the untreated condition. These values were significantly different from the PGE2 + WntA condition. This indicates that the use of H89 and Wort diminished the increase in Mmp9 expression as a result of PGE2 treatment on WntA-induced cells.
Overall, these results demonstrate that PGE2 can raise the expression of Wnt-target genes, specifically, Ctnnb1, Ptgs2, Ccnd1, and Mmp9, in WntA-induced NE-4C cells. Since H89 and Wort attenuated the changes caused by PGE2, PKA and PI3K likely serve as a molecular link for the interaction between the PGE2 and canonical Wnt signalling pathways.
Discussion
Cell migration and proliferation are crucial components of neural development. Previous studies have shown that elevated levels of PGE2 can result in increased cell motility and proliferation in various non-neuronal cells [46,54-56]. Recent evidence indicates that abnormalities in cell behaviour can result from the interaction between PGE2 with Wnt signalling pathways [44,57]. Our current study provides evidence, for the first time, for the cross-talk between these two pathways in neural stem cells. We report that PGE2 treatment elicits changes in cell behaviour such as an increase in components of cell motility and proliferation, as well as expression of Wnt-target genes, in Wnt-activated NE-4C stem cells. Moreover, the stimulatory effects of PGE2 are subdued through the inhibition of downstream pathway kinases, PKA and PI-3K, suggesting that PGE2 acts through these particular kinases to converge with the Wnt pathway.
Previous studies have shown that PGE2 can increase or decrease the activity of canonical Wnt signalling. PGE2 activates several components of the canonical Wnt pathway in colorectal cancer cells (reviewed in [42]). Specifically in these cells, PGE2 stimulated a significant increase in the activity of Wnt transcription factors, T cell factor-4 (Tcf-4), as well as elevated protein levels of Wnt-target genes [58]. PGE2 acted through its EP2 receptor to modulate β-catenin activity of the Wnt pathway, promoting the growth of colon cancer cells [44]. Wnt activation induced by PGE2 also contributed to abnormal proliferation resulting in enhanced gastric tumorigenesis [57]. Furthermore, PGE2–regulated Wnt signalling had a hepatoprotective effect, aiding in liver regeneration [59]. In pre-osteoblastic cells, concentration-dependent treatment of PGE2 modulated Wnt signalling by altering protein expression of pathway activators, β-catenin and low-density lipoprotein receptor-related protein 5/6 (LRP 5/6), as well as Wnt inhibitor, dickkopf-1 (DKK-1); low doses of PGE2 promoted the Wnt pathway while high doses inhibited it [37]. PGE2 also modified Tcf-luciferase activity of Wnt signalling through the same dose effect [37]. Additionally, in human colorectal adenoma and carcinoma cells, PGE2 treatment up-regulated the protein expression of the Wnt target gene, leucine-rich G-protein coupled receptor 5 (LGR5), which internalizes FZD co-receptor LRP6 and decreases Wnt activity [60]. Altogether, these studies reveal that the interaction between PGE2 and Wnt signalling can have different effects depending on the dose of PGE2 administered and the specific cell type.
We reveal that PGE2 increases the final distance and path length travelled, as well as the average speed of migration in Wnt-activated neuroectodermal stem NE-4C cells. We also show that PGE2 alters the phenotype of Wnt-treated cells, which corresponds to an increase in split percentage. Aberrations in cell motility and proliferation behaviour could have detrimental effects to early development of the nervous system. This is because proper neural development requires an orchestrated system of cellular events, such as migration and proliferation, to occur over particular windows of time [61]. Careful control of these crucial neurobiological processes during prenatal development is required for the formation of complex layered structures in the brain like the cerebral cortex, hippocampus, and cerebellum [62,63].
Our study adds to the current body of research by showing that PGE2 interferes with the Wnt pathway by attenuating Wnt-dependent cell behaviour in NE-4C cells. This is important because Wnt signalling is involved in a myriad of regulatory processes important for the development and organization of the nervous system [64]. It is thoroughly established that Wnt signalling is instrumental to normal anterior-posterior patterning of the embryo [65]. Wnt proteins are key regulators for the formation of the neural tube, as well as neuronal migration and differentiation [40,64]. Wnt signalling also modulates neurite outgrowth [66], axon growth and guidance [67-70], dendritic development and arborization [71,72], radial migration [73], and synapse formation and plasticity [74,75]. Moreover, Wnt signalling is crucial in neuronal fate determination, particularly in the specification and differentiation of neuronal precursors in the midbrain [76] and forebrain [77,78]. Furthermore, epithelial stem cells require Wnt/β-catenin signalling for proliferation and quiescent division [79] and the balance between re-entry and exit of the cell cycle can be altered by Wnt/β-catenin signalling [80]. Additionally, aberrant cortical progenitor cell proliferation patterns and defective hippocampus development can result due to abnormal Wnt signalling [81]. Interestingly, recent findings provide evidence that defective Wnt signalling could contribute to the pathogenesis of psychiatric disorders like schizophrenia and ASD [82-84]. Specifically, Wnt2, located in the putative speech and language region at chromosome 7q31-33, has been identified as a susceptibility gene for autism. [85,86]. Given the importance of Wnt signalling in prenatal development and the existing interaction between Wnt and PGE2 pathways in NE-4C stem cells, alterations in levels of PGE2 via endogenous and exogenous means may have profound effects on nervous system development.
In addition to quantifying cell behaviour, we also demonstrate that PGE2 can affect the expression of non-phospho (active) β-catenin (Ser33/37/Thr41). Wnt/β-catenin signalling occurs through a complex, highly regulated pathway that involves the phosphorylation of multiple sites on β-catenin, which may promote its degradation or activation and subsequent nuclear internalization. For instance, the phosphorylation of sites Ser33, 37, and Thr41 targets β-catenin for ubiquitination and proteasomal degradation [87,88]. Quantification of β-catenin that is non-phosphorylated at these sites has become a common measurement for active or stabilized β-catenin expression. Phosphorylation of β-catenin at the site Ser552 has also been correlated with increased β-catenin/TCF mediated transcriptional activity [89,90]. We found that PGE2 treatment administered to Wnt-activated cells increased the expression of non-phospho (active) β-catenin (Ser33/37/Thr41) protein. In contrast, the phospho- β-catenin (Ser552) levels remained unchanged. It has been established that the regulation of glycogen synthase kinase 3 beta (GSK3β) activity may control stabilization of β-catenin and increased levels of non-phospho (active) β-catenin (Ser33/37/Thr41) protein [91]. It is possible that PGE2 signalling may modify GSK3β activity but this remains to be determined. Nonetheless, the increased levels of non-phospho (active) β-catenin (Ser33/37/Thr41) quantified were in line with our gene expression results that also showed an increase in Ctnnb1 expression as well as other Wnt-target genes. Ctnnb1 encodes for the β-catenin protein, which can regulate cell growth and adhesion and is also a key downstream component of the canonical Wnt pathway. It has also been shown to regulate cortical size; enlarged cortices with increased cortical folds were observed in Ctnnb1 transgenic mice [80]. Interestingly, brain overgrowth and abnormal excess in number of neurons was measured in children with autism [92]. Gene expression of Ctnnb1 was altered in both young and adult autistic cases [93]. Furthermore, de novo mutations of this gene and its relevant network have been ranked significantly as potential autism candidate genes [94,95]. Within the canonical Wnt pathway, the β-catenin/TCF complex can promote the transcription of target genes including Ptgs2[96], Ccnd1[97,98], and Mmp9[99,100]. Expression of these genes was increased as an effect of elevated PGE2 signalling in our study, and interestingly, previous studies have reported a link between these genes and ASD as described below.
Ptgs2, also known as COX-2, is the key enzyme in prostaglandin biosynthesis, including the production of PGE2. COX-2 is a crucial mediator of inflammation and prostanoid signalling [101,102]. Polymorphism of Ptgs2 has been associated with ASD [103]. A recent clinical study proved the efficacy of a COX-2 inhibitor drug, celecoxib, as an adjunctive therapy in the treatment of autism: the treatment was superior for treating irritability, social withdrawal, and stereotypy of children with autism [104].
Another gene affected was Ccnd1. This gene encodes for a protein in the cyclin family, which are important regulators in cell cycle progression, transcription, and neuronal function [105,106]. The increased levels of Ccnd1, as a result of added PGE2, may be involved with the altered proliferation behaviour visualized in this study. Aberrant Ccnd1 levels have also been associated with ASD. In autistic rat pups (model encompassed administration of valproic acid), Ccnd1 expression was atypical in the cerebellum compared to controls [107]. Another study showed that the dysregulation of Ccnd1 lead to abnormal cell cycle and proliferation, neuronal and network excitability and behaviour, and revealed its potential link to human neuro-cardio-facial-cutaneous and related syndromes, which are associated with developmental abnormalities, cognitive deficits, and autism [108]. Diminished expression of 22q11 genes, which disrupts cortical neurogenesis and cell migration, led to alterations in Ccnd1 levels [109]. The authors explain that a developmental disruption, as such, may alter cortical circuitry and establish vulnerability for developmental disorders, including schizophrenia and autism.
Mmp9 is a membrane of the matrix metalloproteinase (MMP) family, which can target many extracellular proteins including proteases, growth factors, and adhesion molecules [110] and are involved with the breakdown of the extracellular matrix in normal physiological processes such as embryonic development and tissue remodelling [111]. MMPs are also important in neuronal development, plasticity, and maintenance of neuronal health [112]. Mmp9 has also been shown to regulate the proliferation and migration of embryonic neural stem cells [99] and participate in neuronal differentiation by regulating neurite elongation and neuronal cell migration [113-115]. Therefore, altered Mmp9 expression may contribute to the deviant behaviour observed in our study. Mmp9 has also been associated with ASD [116]. Elevated levels of MMP9 protein were found in the amniotic fluid of ASD cases compared to controls [117]. Findings from this study provide evidence that molecular and physiological abnormalities in ASD may begin prenatally. Mmp9 has also been implicated in Fragile X syndrome (FXS) [118], which is characterized by behaviours at the extreme of the autistic spectrum. Using in a mouse model of fragile x (Fmr1 KO), levels of MMP9 was found to be elevated in the hippocampus of Fmr1 KO mice [119]. Furthermore, Minocycline, a drug that inhibits MMP9 activity, has been shown to promote dendrite spine maturation and improve behavioural performance in Fmr1 KO mice [119]. These researchers continued their work in human trials and found that Minocycline taken as a daily dose for 8 weeks led to behavioural improvements in FXS patients. This was consistent with their fmr1 KO mouse model results, indicating that MMP9 activity alters underlying neural defects that contribute to behavioural abnormalities seen in ASD [120].
Taken altogether, our gene expression results not only show a potential interaction of the PGE2 and canonical Wnt pathway in the nervous system, but also provide further evidence for a link to ASD.
We show that PGE2 interacts with canonical Wnt signalling through PKA and PI-3K to produce the reported behavioural changes in cell motility and proliferation, as well as gene expression. Specifically, we found that inhibiting these PGE2 downstream pathway kinases, PKA and PI-3K with H89 and Wort respectively, reduced the effect of PGE2. This is in line with previous literature, which found that the convergence of PGE2-dependent effects and the Wnt pathway can occur through the stimulation of PKA or PI-3K in embryonic kidney cells and colon cancer cells [44-46]. Moreover, similar stimulatory effects on cell migration induced by PGE2 in Wnt-activated NE-4C cells from our study were also exhibited in prostate cancer cells through the activation of PI-3K [121]. Our results revealed that H89 had a stronger effect than Wort, suggesting that PGE2 may predominately act through PKA; but future studies are needed to determine which EP receptors are involved. A proposed model is provided in Figure 9.
Figure 9.
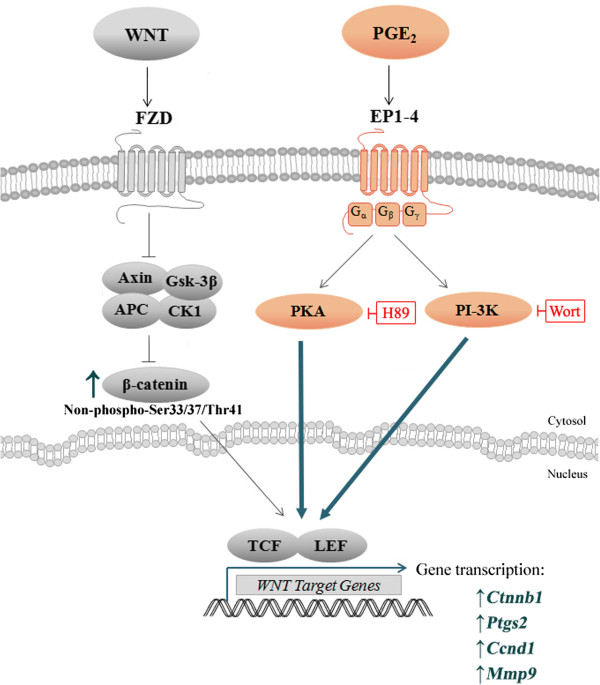
A proposed model for PGE2-Wnt interactions in Wnt-induced NE-4C cells. From the compilation of our results (bolded) and other studies, a schematic model is drawn of the mechanism by which PGE2 might interact with the canonical Wnt pathway.
Increasing evidence for the contribution of environmental factors in the etiology of neurodevelopmental disorders like ASD has prompted urgency to reveal their potential exogenous causes and underlying mechanisms [122]. Environmental factors like exposure to drugs, toxins or infectious agents cause disruptions in PGE2 signalling by increasing the levels of oxidative stress, consequent lipid peroxidation, and the immunological response; these factors and consequences that disturb normal PGE2 signalling have all been linked to ASD [123]. We show that increased PGE2 signalling can modify cell migration, proliferation behaviour, and gene expression in Wnt-activated NE-4C stem cells. Aberrant cell migration and proliferation are pathophysiologic mechanisms that impact the brain broadly, and could be possible factors that contribute to the origination of neurodevelopment disorders. Abnormalities in structure, organization, and connectivity of the brain are all indicators of irregular neural cell migration and proliferation. Local distortions in neural cytoarchitecture, dysplasia, and hypoplasia have been described in brains of autistic subjects [124]. Moreover, structural abnormalities and atypical connectivity of the brain in ASD has been identified by a number of research groups [123,125-128]. Noteworthy, areas of the brain that would be most impacted by dysregulation in neuronal migration and proliferation—that is the cerebellum, cerebral cortex, and hippocampus—are also implicated in ASD [124,129-131]. Despite the assumptions that can be made from our in vitro results, in vivo models must be employed to further describe the possible effects of PGE2 and its interaction with morphogenic signalling pathways, such as Wnt, during prenatal development.
Conclusions
PGE2 is an important bioactive lipid signalling molecule and its interaction with Wnt signalling pathway could have significant effects on prenatal development. Our study shows for the first time that PGE2 can affect Wnt-dependent cell behaviours and gene expression in neuroectodermal stem cells through PKA and PI-3K. Aberrant PGE2 and Wnt signalling have been linked to ASD. Moreover, altered migration and proliferation due to irregular gene expression during embryonic development in ASD have been suggested in previous studies. Our in vitro study provided further evidence that these aberrations may be potential mechanisms in the genesis of neurodevelopment disorders like ASD.
Methods
Cell culture
Mouse NE-4C cells were obtained from American Tissue Culture Collection (ATCC) and grown in Minimum Essential Medium (MEM) supplemented with 10% fetal bovine serum, 2 mM glutamine, 1 X penicillin-streptomycin mixture (Invitrogen). Cells were maintained in an incubator containing 5% CO2 at 95% humidity 37°C. Cells were plated on 0.01% poly-L-lysine (Sigma) coated 100 mm culture plates (BD Falcon) and were subcultured at a 1:10 ratio. Supplemented MEM was changed every 2–3 days.
Cell culture-treatments
NE-4C cells (ATCC) were dissociated with 0.05% trypsin-EDTA, pelleted and resuspended in complete medium as described above. The cells were plated on poly-L-lysine 0.01% (Sigma, MW 70000–150000 kDa) on 35 mm tissue culture dishes (Sarstedt). Plated cells were incubated in 5% CO2 at 95% humidity 37°C overnight before treatment with Wnt Agonist (WntA, Calbiochem), prostaglandin E2 (PGE2, Sigma) and/or blockers. WntA (2 μM), PGE2 (1 μM), H89 dihydrochloride hydrate (H89, 10 μM, Sigma), Wortmannin (WORT, 1 μM, Sigma) or an equivalent volume of vehicle were added to each well. Cells were treated for 24 hours.
Reverse transcription and real-time PCR
Total RNA was extracted from NE-4C cells using the NucleoSpin RNA/Protein Kit (Macherey-Nagel) and was reverse-transcribed into cDNA using MMuLV reverse transcriptase (New England Biolabs) according to manufacturer’s instructions. Primer3 Input software (v. 0.4.0) was used to design forward and reverse primers for EP receptors and have been previously noted [12]. Selection of Wnt-target genes was determined using Custom TaqMan® Array Plates (Life Technologies) as a screening tool (data not shown). Genes that had a greater than 1.8 fold-change were selected for further validation and forward and reverse primers were designed (Table 1). Real-time PCR was performed using the 7500 Fast Real-time PCR system (Applied Biosystems) and the ΔΔCT method was applied to calculate the expression of transcripts. Hypoxanthine phosphoribosyl transferase (HPRT) and Phosphoglycerate Kinase 1 (PGK1) served as endogenous controls. The relative quantification (RQ) ratios were determined from the average of three technical replicates from three biological replicates.
Table 1.
Forward and reverse primer sequences for real-time-PCR
| Gene | Primer sequences (5′ → 3′) | Amplicon size (base pair) |
|---|---|---|
|
Hprt |
F: TCCATTCCTATGACTGTAGATTTTATCAG |
75 |
| R: AACTTTTATGTCCCCCGTTGACT | ||
|
Pgkl |
F: CAGTTGCTGCTGAACTCAAATCTC |
65 |
| R: GCCCACACAATCCTTCAAGAA | ||
|
Ptgs2 |
F: CAGCCAGGCAGCAAATCC |
81 |
| R: TTATACTGGTCAAATCCTGTGCTCAT | ||
|
Ctnnbl |
F: GGACGTTCACAACCGGATTG |
71 |
| R: GAGAATAAAGCAACTG CACAAACAA | ||
|
Ccndl |
F: GCACTTTCTTTCCAGAGTCATCAA |
79 |
| R: CTCCAGAAGGGCTTCAATCTGT | ||
| Mmp9 | F: TCGCGTGGATAAGGAGTTCTCT |
73 |
| R: ATAGGCTTTGTCTTGGTACTGGAAGA |
Western blot analysis
Total protein was extracted from NE-4C cells using the NucleoSpin RNA/Protein Kit (Macherey-Nagel). Samples were separated by polyacrylamide gel electrophoresis (PAGE). Primary antibodies used for EP expression levels include rabbit polyclonal anti-EP1, −EP2, −EP3, −EP4 (1:200; Santa Cruz Biotechnology). Detection of rabbit monoclonal anti-Phospho-Histone H3 (Ser10) (1:1000; Cell Signaling) was used as a measure of cell splitting behaviour. Primary antibodies used for β-catenin expression levels were rabbit monoclonal anti-non-phospho (Active) β-catenin (Ser33/37/Thr41) and rabbit polyclonal anti-phospho-β-catenin (Ser552) (1:1000; Cell Signaling). Blots were reprobed with mouse monoclonal anti-β-Actin (1:10,000; Abcam). Visualization of bound anti-rabbit and anti-mouse horseradish peroxidise-conjugated secondary antibodies was achieved by incubation with ECL Prime Western Blotting Detection Reagent (GE Healthcare) and detection by Geliance 600 Imaging System (Perkin Elmer).
Immunocytochemistry
NE-4C cells were seeded onto 35 mm culture plates containing poly-L-lysine coated coverslips and grown overnight at 37°C. The cells were fixed with 50% acetone and 50% methanol for 20 minutes at −20°C and washed with phosphate buffered saline (4.3 mM Na2HPO4, 137 mM NaCl, 2.7 mM KCl, 1.4 mM KH2PO4). Cells were then incubated with primary antibodies in PBS with 0.3% Triton-X 100 and 2% Normal Goat Serum. Cellular localization of the EP receptors was determined by incubation with anti-EP primary antibodies as described above along with mouse monoclonal anti-Lamin A + C nuclear envelope marker (1:200; Abcam), anti-58 K Golgi marker [58 K-9] (1:100; Abcam), anti-PDI endoplasmic reticulum marker [RL90] (1:100; Abcam) or β-Actin (1:1000; Abcam) at room temperature for 1 hour. Following primary antibody incubation, cells were washed three times with PBS-T for 15 min and incubated with secondary antibodies in PBS-T and 2% NGS for 1 hour at room temperature in the dark. Secondary antibodies used were anti-rabbit fluorescein isothiocyanate (FITC) (1:100; Jackson ImmunoResearch Laboratories) and anti-mouse Texas Red (1:200; Jackson ImmunoResearch Laboratories). Cells were then washed twice with PBS-T for 10 min, followed by a 20 minute incubation of 4′,6-diamidino-2-phenylindole (DAPI) (1:2000; Molecular Probes) at room temperature. Cells were washed twice with PBS-T for 5 min and coverslips were mounted on glass microscope slides with mounting media (Vectashield). The staining was visualized and captured using an Eclipse 80i upright fluorescent microscope with DS-5MC camera (Nikon).
Time-lapse imaging and analysis
Cell behaviour was recorded using Nikon Eclipse Ti-E microscope. Three biological replicates of each treatment condition were performed (N = 3), where an average of 150 cells were tracked. Micrographs were automatically captured every 10 minutes for a 24 hour period from a minimum of three fields. To maintain conditions physiologically suitable for the cells, an enclosed chamber was mounted to the microscope, which was equipped with CO2 supply and temperature thermostat. Cells were kept at 5% CO2, 95% humidity, 37°C. Measurements were completed using NIS Elements software, including a specialized tracking module. Final distance from origin, path length, and average speed were tracked and calculated from an average of 150 cells per treatment condition. Initial and final cell counts were used to determine fold change as a measurement of proliferation. Split percentage was quantified as a measurement of proliferation behaviour. Split percentage was defined as the percentage of cells that fulfilled the complete cell cycle, which was evaluated based on whether the parent cell could successfully split into two daughter cells.
Cell viability analysis
Cells were disassociated and diluted with equal volumes of trypan blue dye (4%). Cell count averages were taken from a minimum of four hemacytometer squares to determine cell number and viability.
Statistical analysis
All numerical data were presented as mean + SEM of three individual experiments. Statistical analysis was performed using student t-test or one-way analysis of variance (ANOVA) followed by Tukey post-hoc comparisons or Dunnett t-test (2-sided). Differences were considered statistically significant at *p < 0.05, **p < 0.01, or ***p < 0.001.
Abbreviations
ASD: Autism spectrum disorders; COX-1,-2: Cyclooxygenases −1 and −2; Ccnd1: Cyclin D1; Ctnnb1: Beta-catenin; EP1-4: E-prostanoid 1 through 4; H89: Dihydrochloride hydrate; Mmp9: Matrix metalloproteinase 9; NE-4C stem cell: Neuroectrodermal stem cells; PGE2: Prostaglandin E2; PI-3K: Phosphatidylinositide 3-kinases; PKA: Protein kinase A; PKC: Protein kinase C; Ptgs2: Prostaglandin-endoperoxide synthase 2; Wnt: Wingless-related MMTV integration site; WntA: Wnt agonist; Wort: Wortmannin.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CTW designed and performed experiments, acquisition and analysis of data, and wrote the manuscript. EA performed experiments and acquisition of data. HYL assisted with research and discussions. DAC participated in the design and coordination of the study, and was involved in drafting the manuscript. All authors read and approved the final manuscript.
Contributor Information
Christine T Wong, Email: cwong87@yorku.ca.
Eizaaz Ahmad, Email: eizaaz@yorku.ca.
Hongyan Li, Email: hongyan@yorku.ca.
Dorota A Crawford, Email: dakc@yorku.ca.
Acknowledgements
This research work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC).
References
- Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol. 2012;72(10):1272–1276. doi: 10.1002/dneu.22024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker-Athill EC, Tan J. Maternal immune activation and autism spectrum disorder: interleukin-6 signaling as a key mechanistic pathway. Neurosignals. 2010;18(2):113–128. doi: 10.1159/000319828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson PH. Maternal infection and immune involvement in autism. Trends Mol Med. 2011;17(7):389–394. doi: 10.1016/j.molmed.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean P, Landrigan PJ. Developmental neurotoxicity of industrial chemicals. Lancet. 2006;368(9553):2167–2178. doi: 10.1016/S0140-6736(06)69665-7. [DOI] [PubMed] [Google Scholar]
- Arndt TL, Stodgell CJ, Rodier PM. The teratology of autism. Int J Dev Neurosci. 2005;23(2–3):189–199. doi: 10.1016/j.ijdevneu.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282(16):11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46(2):205–229. [PubMed] [Google Scholar]
- Furuyashiki T, Narumiya S. Stress responses: the contribution of prostaglandin E(2) and its receptors. Nat Rev Endocrinol. 2011;7(3):163–175. doi: 10.1038/nrendo.2010.194. [DOI] [PubMed] [Google Scholar]
- Andreasson K. Emerging roles of PGE2 receptors in models of neurological disease. Prostaglandins Other Lipid Mediat. 2010;91(3–4):104–112. doi: 10.1016/j.prostaglandins.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Dizier M, Guyader-Joly C, Charpigny G, Grimard B, Humblot P, Ponter AA. Expression of enzymes involved in the synthesis of prostaglandin E2 in bovine in vitro-produced embryos. Zygote. 2011;19(3):277–283. doi: 10.1017/S0967199410000596. [DOI] [PubMed] [Google Scholar]
- Tamiji J, Crawford DA. Prostaglandin E(2) and misoprostol induce neurite retraction in Neuro-2a cells. Biochem Biophys Res Commun. 2010;398(3):450–456. doi: 10.1016/j.bbrc.2010.06.098. [DOI] [PubMed] [Google Scholar]
- Chen C, Bazan NG. Endogenous PGE2 regulates membrane excitability and synaptic transmission in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2005;93(2):929–941. doi: 10.1152/jn.00696.2004. [DOI] [PubMed] [Google Scholar]
- Dean SL, Wright CL, Hoffman JF, Wang M, Alger BE, McCarthy MM. Prostaglandin E2 stimulates estradiol synthesis in the cerebellum postnatally with associated effects on Purkinje neuron dendritic arbor and electrophysiological properties. Endocrinology. 2012;153(11):5415–5427. doi: 10.1210/en.2012-1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Bazan NG. Lipid signaling: sleep, synaptic plasticity, and neuroprotection. Prostaglandins Other Lipid Mediat. 2005;77(1–4):65–76. doi: 10.1016/j.prostaglandins.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Shimamura M, Zhou P, Casolla B, Qian L, Capone C, Kurinami H, Iadecola C, Anrather J. Prostaglandin E type 1 receptors contribute to neuronal apoptosis after transient forebrain ischemia. J Cereb Blood Flow Metab. 2013;33(8):1207–1214. doi: 10.1038/jcbfm.2013.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagishi H, Kosuge Y, Yoneoka Y, Ozone M, Endo M, Osada N, Ishige K, Kusama-Eguchi K, Ito Y. Prostaglandin E2-induced cell death is mediated by activation of EP2 receptors in motor neuron-like NSC-34 cells. J Pharmacol Sci. 2013;121(4):347–350. doi: 10.1254/jphs.12274SC. [DOI] [PubMed] [Google Scholar]
- Choi SY, Choi BH, Suh BC, Chae HD, Kim JS, Shin MJ, Kang SS, Negishi M, Kim KT. Potentiation of PGE(2)-mediated cAMP production during neuronal differentiation of human neuroblastoma SK-N-BE(2)C cells. J Neurochem. 2001;79(2):303–310. doi: 10.1046/j.1471-4159.2001.00577.x. [DOI] [PubMed] [Google Scholar]
- Goncalves MB, Williams EJ, Yip P, Yanez-Munoz RJ, Williams G, Doherty P. The COX-2 inhibitors, meloxicam and nimesulide, suppress neurogenesis in the adult mouse brain. Br J Pharmacol. 2010;159(5):1118–1125. doi: 10.1111/j.1476-5381.2009.00618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raisz LG, Pilbeam CC, Fall PM. Prostaglandins: mechanisms of action and regulation of production in bone. Osteoporos Int. 1993;3(Suppl 1):136–140. doi: 10.1007/BF01621888. [DOI] [PubMed] [Google Scholar]
- Tjandrawinata RR, Dahiya R, Hughes-Fulford M. Induction of cyclo-oxygenase-2 mRNA by prostaglandin E2 in human prostatic carcinoma cells. Br J Cancer. 1997;75(8):1111–1118. doi: 10.1038/bjc.1997.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charo C, Holla V, Arumugam T, Hwang R, Yang P, Dubois RN, Menter DG, Logsdon CD, Ramachandran V. Prostaglandin E2 regulates pancreatic stellate cell activity via the EP4 receptor. Pancreas. 2013;42(3):467–474. doi: 10.1097/MPA.0b013e318264d0f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CC, Lin WN, Cheng SE, Tung WH, Wang HH, Yang CM. Transactivation of EGFR/PI3K/Akt involved in ATP-induced inflammatory protein expression and cell motility. J Cell Physiol. 2012;227(4):1628–1638. doi: 10.1002/jcp.22880. [DOI] [PubMed] [Google Scholar]
- Iwanaga K, Okada M, Murata T, Hori M, Ozaki H. Prostaglandin E2 promotes wound-induced migration of intestinal subepithelial myofibroblasts via EP2, EP3, and EP4 prostanoid receptor activation. J Pharmacol Exp Ther. 2012;340(3):604–611. doi: 10.1124/jpet.111.189845. [DOI] [PubMed] [Google Scholar]
- Joo HJ, Kim HS, Choi YS, Kim H, Kim SJ, Moon WK. Detection of prostaglandin E2-induced dendritic cell migration into the lymph nodes of mice using a 1.5 T clinical MR scanner. NMR Biomed. 2012;25(4):570–579. doi: 10.1002/nbm.1774. [DOI] [PubMed] [Google Scholar]
- Mayoral R, Fernandez-Martinez A, Bosca L, Martin-Sanz P. Prostaglandin E2 promotes migration and adhesion in hepatocellular carcinoma cells. Carcinogenesis. 2005;26(4):753–761. doi: 10.1093/carcin/bgi022. [DOI] [PubMed] [Google Scholar]
- Jaffer S, Mattana J, Singhal PC. Effects of prostaglandin E2 on mesangial cell migration. Am J Nephrol. 1995;15(4):300–305. doi: 10.1159/000168853. [DOI] [PubMed] [Google Scholar]
- Tamiji J, Crawford DA. Misoprostol elevates intracellular calcium in Neuro-2a cells via protein kinase A. Biochem Biophys Res Commun. 2010;399(4):565–570. doi: 10.1016/j.bbrc.2010.07.112. [DOI] [PubMed] [Google Scholar]
- Tamiji J, Crawford DA. The neurobiology of lipid metabolism in autism spectrum disorders. Neurosignals. 2010;18(2):98–112. doi: 10.1159/000323189. [DOI] [PubMed] [Google Scholar]
- El-Ansary A, Al-Ayadhi L. Lipid mediators in plasma of autism spectrum disorders. Lipids Health Dis. 2012;11:160. doi: 10.1186/1476-511X-11-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean SL, Knutson JF, Krebs-Kraft DL, McCarthy MM. Prostaglandin E2 is an endogenous modulator of cerebellar development and complex behavior during a sensitive postnatal period. Eur J Neurosci. 2012;35(8):1218–1229. doi: 10.1111/j.1460-9568.2012.08032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandim JM, Ventura LO, Miller MT, Almeida HC, Costa AE. Autism and Mobius sequence: an exploratory study of children in northeastern Brazil. Arq Neuropsiquiatr. 2003;61(2A):181–185. doi: 10.1590/S0004-282X2003000200004. [DOI] [PubMed] [Google Scholar]
- Miller MT, Stromland K, Ventura L, Johansson M, Bandim JM, Gillberg C. Autism associated with conditions characterized by developmental errors in early embryogenesis: a mini review. Int J Dev Neurosci. 2005;23(2–3):201–219. doi: 10.1016/j.ijdevneu.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Miller G. Neurological disorders. The mystery of the missing smile. Science. 2007;316(5826):826–827. doi: 10.1126/science.316.5826.826. [DOI] [PubMed] [Google Scholar]
- Wong C, Crawford D. In: Comprehensive Guide to Autism. Patel VB, Preedy VR, Martin CR, editor. New York: Springer; 2014. Lipid Signalling in the Pathology of Autism Spectrum Disorders; pp. 1259–1283. [Google Scholar]
- Kitase Y, Barragan L, Qing H, Kondoh S, Jiang JX, Johnson ML, Bonewald LF. Mechanical induction of PGE2 in osteocytes blocks glucocorticoid-induced apoptosis through both the beta-catenin and PKA pathways. J Bone Miner Res. 2010;25(12):2657–2668. doi: 10.1002/jbmr.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XH, Kirschenbaum A, Weinstein BM, Zaidi M, Yao S, Levine AC. Prostaglandin E2 modulates components of the Wnt signaling system in bone and prostate cancer cells. Biochem Biophys Res Commun. 2010;394(3):715–720. doi: 10.1016/j.bbrc.2010.03.057. [DOI] [PubMed] [Google Scholar]
- Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, Weidinger G, Puder M, Daley GQ, Moon RT, Zon LI. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. 2009;136(6):1136–1147. doi: 10.1016/j.cell.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiveland CR, Kassem M, Lea T. Human mesenchymal stem cell proliferation is regulated by PGE2 through differential activation of cAMP-dependent protein kinase isoforms. Exp Cell Res. 2008;314(8):1831–1838. doi: 10.1016/j.yexcr.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Ciani L, Salinas PC. WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nat Rev Neurosci. 2005;6(5):351–362. doi: 10.1038/nrn1665. [DOI] [PubMed] [Google Scholar]
- Buechling T, Boutros M. Wnt signaling signaling at and above the receptor level. Curr Top Dev Biol. 2011;97:21–53. doi: 10.1016/B978-0-12-385975-4.00008-5. [DOI] [PubMed] [Google Scholar]
- Buchanan FG, DuBois RN. Connecting COX-2 and Wnt in cancer. Cancer Cell. 2006;9(1):6–8. doi: 10.1016/j.ccr.2005.12.029. [DOI] [PubMed] [Google Scholar]
- Liu J, Wu X, Mitchell B, Kintner C, Ding S, Schultz PG. A small-molecule agonist of the Wnt signaling pathway. Angew Chem Int Ed Engl. 2005;44(13):1987–1990. doi: 10.1002/anie.200462552. [DOI] [PubMed] [Google Scholar]
- Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310(5753):1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- Fujino H, West KA, Regan JW. Phosphorylation of glycogen synthase kinase-3 and stimulation of T-cell factor signaling following activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. J Biol Chem. 2002;277(4):2614–2619. doi: 10.1074/jbc.M109440200. [DOI] [PubMed] [Google Scholar]
- Sheng H, Shao J, Washington MK, DuBois RN. Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J Biol Chem. 2001;276(21):18075–18081. doi: 10.1074/jbc.M009689200. [DOI] [PubMed] [Google Scholar]
- Goto H, Tomono Y, Ajiro K, Kosako H, Fujita M, Sakurai M, Okawa K, Iwamatsu A, Okigaki T, Takahashi T, Inagaki M. Identification of a novel phosphorylation site on histone H3 coupled with mitotic chromosome condensation. J Biol Chem. 1999;274(36):25543–25549. doi: 10.1074/jbc.274.36.25543. [DOI] [PubMed] [Google Scholar]
- Liokatis S, Stutzer A, Elsasser SJ, Theillet FX, Klingberg R, van Rossum B, Schwarzer D, Allis CD, Fischle W, Selenko P. Phosphorylation of histone H3 Ser10 establishes a hierarchy for subsequent intramolecular modification events. Nat Struct Mol Biol. 2012;19(8):819–823. doi: 10.1038/nsmb.2310. [DOI] [PubMed] [Google Scholar]
- Nowak SJ, Corces VG. Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet. 2004;20(4):214–220. doi: 10.1016/j.tig.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11(24):3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- Wu G, He X. Threonine 41 in beta-catenin serves as a key phosphorylation relay residue in beta-catenin degradation. Biochemistry. 2006;45(16):5319–5323. doi: 10.1021/bi0601149. [DOI] [PubMed] [Google Scholar]
- Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu Z. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem. 2007;282(15):11221–11229. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XC, Yin T, Grindley JC, Tian Q, Sato T, Tao WA, Dirisina R, Porter-Westpfahl KS, Hembree M, Johnson T, Wiedemann LM, Barrett TA, Hood L, Wu H, Li L. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat Genet. 2007;39(2):189–198. doi: 10.1038/ng1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aso H, Ito S, Mori A, Suganuma N, Morioka M, Takahara N, Kondo M, Hasegawa Y. Differential regulation of airway smooth muscle cell migration by E-prostanoid receptor subtypes. Am J Respir Cell Mol Biol. 2013;48(3):322–329. doi: 10.1165/rcmb.2012-0158OC. [DOI] [PubMed] [Google Scholar]
- Bai XM, Zhang W, Liu NB, Jiang H, Lou KX, Peng T, Ma J, Zhang L, Zhang H, Leng J. Focal adhesion kinase: important to prostaglandin E2-mediated adhesion, migration and invasion in hepatocellular carcinoma cells. Oncol Rep. 2009;21(1):129–136. [PubMed] [Google Scholar]
- Yen JH, Khayrullina T, Ganea D. PGE2-induced metalloproteinase-9 is essential for dendritic cell migration. Blood. 2008;111(1):260–270. doi: 10.1182/blood-2007-05-090613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima H, Oshima M. The role of PGE2-associated inflammatory responses in gastric cancer development. Semin Immunopathol. 2013;35(2):139–150. doi: 10.1007/s00281-012-0353-5. [DOI] [PubMed] [Google Scholar]
- Shao J, Jung C, Liu C, Sheng H. Prostaglandin E2 Stimulates the beta-catenin/T cell factor-dependent transcription in colon cancer. J Biol Chem. 2005;280(28):26565–26572. doi: 10.1074/jbc.M413056200. [DOI] [PubMed] [Google Scholar]
- North TE, Babu IR, Vedder LM, Lord AM, Wishnok JS, Tannenbaum SR, Zon LI, Goessling W. PGE2-regulated wnt signaling and N-acetylcysteine are synergistically hepatoprotective in zebrafish acetaminophen injury. Proc Natl Acad Sci U S A. 2010;107(40):17315–17320. doi: 10.1073/pnas.1008209107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Kharusi MR, Smartt HJ, Greenhough A, Collard TJ, Emery ED, Williams AC, Paraskeva C. LGR5 promotes survival in human colorectal adenoma cells and is upregulated by PGE2: implications for targeting adenoma stem cells with NSAIDs. Carcinogenesis. 2013;34(5):1150–1157. doi: 10.1093/carcin/bgt020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles J, Jernigan TL. The basics of brain development. Neuropsychol Rev. 2010;20(4):327–348. doi: 10.1007/s11065-010-9148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evsyukova I, Plestant C, Anton ES. Integrative mechanisms of oriented neuronal migration in the developing brain. Annu Rev Cell Dev Biol. 2013;29:299–353. doi: 10.1146/annurev-cellbio-101512-122400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatten ME. Central nervous system neuronal migration. Annu Rev Neurosci. 1999;22:511–539. doi: 10.1146/annurev.neuro.22.1.511. [DOI] [PubMed] [Google Scholar]
- Ille F, Sommer L. Wnt signaling: multiple functions in neural development. Cell Mol Life Sci. 2005;62(10):1100–1108. doi: 10.1007/s00018-005-4552-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkell RM, Fossat N, Tam PP. Wnt signalling in mouse gastrulation and anterior development: new players in the pathway and signal output. Curr Opin Genet Dev. 2013;23(4):454–460. doi: 10.1016/j.gde.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Lu W, Yamamoto V, Ortega B, Baltimore D. Mammalian Ryk is a Wnt coreceptor required for stimulation of neurite outgrowth. Cell. 2004;119(1):97–108. doi: 10.1016/j.cell.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Bovolenta P, Rodriguez J, Esteve P. Frizzled/RYK mediated signalling in axon guidance. Development. 2006;133(22):4399–4408. doi: 10.1242/dev.02592. [DOI] [PubMed] [Google Scholar]
- Lyuksyutova AI, Lu CC, Milanesio N, King LA, Guo N, Wang Y, Nathans J, Tessier-Lavigne M, Zou Y. Anterior-posterior guidance of commissural axons by Wnt-frizzled signaling. Science. 2003;302(5652):1984–1988. doi: 10.1126/science.1089610. [DOI] [PubMed] [Google Scholar]
- Zou Y. Wnt signaling in axon guidance. Trends Neurosci. 2004;27(9):528–532. doi: 10.1016/j.tins.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Sanchez-Camacho C, Rodriguez J, Ruiz JM, Trousse F, Bovolenta P. Morphogens as growth cone signalling molecules. Brain Res Brain Res Rev. 2005;49(2):242–252. doi: 10.1016/j.brainresrev.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Rosso SB, Sussman D, Wynshaw-Boris A, Salinas PC. Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat Neurosci. 2005;8(1):34–42. doi: 10.1038/nn1374. [DOI] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron. 2006;50(6):897–909. doi: 10.1016/j.neuron.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Zhou CJ, Zhao C, Pleasure SJ. Wnt signaling mutants have decreased dentate granule cell production and radial glial scaffolding abnormalities. J Neurosci. 2004;24(1):121–126. doi: 10.1523/JNEUROSCI.4071-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Park CS, Tang SJ. Activity-dependent synaptic Wnt release regulates hippocampal long term potentiation. J Biol Chem. 2006;281(17):11910–11916. doi: 10.1074/jbc.M511920200. [DOI] [PubMed] [Google Scholar]
- Gogolla N, Galimberti I, Deguchi Y, Caroni P. Wnt signaling mediates experience-related regulation of synapse numbers and mossy fiber connectivities in the adult hippocampus. Neuron. 2009;62(4):510–525. doi: 10.1016/j.neuron.2009.04.022. [DOI] [PubMed] [Google Scholar]
- Prakash N, Brodski C, Naserke T, Puelles E, Gogoi R, Hall A, Panhuysen M, Echevarria D, Sussel L, Weisenhorn DM, Martinez S, Arenas E, Simeone A, Wurst W. A Wnt1-regulated genetic network controls the identity and fate of midbrain-dopaminergic progenitors in vivo. Development. 2006;133(1):89–98. doi: 10.1242/dev.02181. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Gotoh Y. Stage-dependent fate determination of neural precursor cells in mouse forebrain. Neurosci Res. 2005;51(4):331–336. doi: 10.1016/j.neures.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Zhou CJ, Borello U, Rubenstein JL, Pleasure SJ. Neuronal production and precursor proliferation defects in the neocortex of mice with loss of function in the canonical Wnt signaling pathway. Neuroscience. 2006;142(4):1119–1131. doi: 10.1016/j.neuroscience.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Lowry WE, Blanpain C, Nowak JA, Guasch G, Lewis L, Fuchs E. Defining the impact of beta-catenin/Tcf transactivation on epithelial stem cells. Genes Dev. 2005;19(13):1596–1611. doi: 10.1101/gad.1324905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297(5580):365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- Lee SM, Tole S, Grove E, McMahon AP. A local Wnt-3a signal is required for development of the mammalian hippocampus. Development. 2000;127(3):457–467. doi: 10.1242/dev.127.3.457. [DOI] [PubMed] [Google Scholar]
- Kalkman HO. A review of the evidence for the canonical Wnt pathway in autism spectrum disorders. Mol Autism. 2012;3(1):10. doi: 10.1186/2040-2392-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okerlund ND, Cheyette BN. Synaptic Wnt signaling-a contributor to major psychiatric disorders? J Neurodev Disord. 2011;3(2):162–174. doi: 10.1007/s11689-011-9083-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter D, Kerwin R, Al-Sarraji S, Brion JP, Chadwich A, Lovestone S, Anderton B, Everall I. Abnormalities of Wnt signalling in schizophrenia--evidence for neurodevelopmental abnormality. Neuroreport. 1998;9(7):1379–1383. doi: 10.1097/00001756-199805110-00024. [DOI] [PubMed] [Google Scholar]
- Lin PI, Chien YL, Wu YY, Chen CH, Gau SS, Huang YS, Liu SK, Tsai WC, Chiu YN. The WNT2 gene polymorphism associated with speech delay inherent to autism. Res Dev Disabil. 2012;33(5):1533–1540. doi: 10.1016/j.ridd.2012.03.004. [DOI] [PubMed] [Google Scholar]
- Wassink TH, Piven J, Vieland VJ, Huang J, Swiderski RE, Pietila J, Braun T, Beck G, Folstein SE, Haines JL, Sheffield VC. Evidence supporting WNT2 as an autism susceptibility gene. Am J Med Genet. 2001;105(5):406–413. doi: 10.1002/ajmg.1401. [DOI] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108(6):837–847. doi: 10.1016/S0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- Kimelman D, Xu W. Beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene. 2006;25(57):7482–7491. doi: 10.1038/sj.onc.1210055. [DOI] [PubMed] [Google Scholar]
- Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. J Biol Chem. 2006;281(15):9971–9976. doi: 10.1074/jbc.M508778200. [DOI] [PubMed] [Google Scholar]
- Zhao J, Yue W, Zhu MJ, Sreejayan N, Du M. AMP-activated protein kinase (AMPK) cross-talks with canonical Wnt signaling via phosphorylation of beta-catenin at Ser 552. Biochem Biophys Res Commun. 2010;395(1):146–151. doi: 10.1016/j.bbrc.2010.03.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Chen G, Romero G, Moschos S, Xu X, Hu J. Induction of Gsk3beta-beta-TrCP Interaction Is Required for Late Phase Stabilization of beta-catenin in Canonical Wnt Signaling. J Biol Chem. 2014. Epub ahead of print. [DOI] [PMC free article] [PubMed]
- Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ, Barnes CC, Pierce K. Neuron number and size in prefrontal cortex of children with autism. JAMA. 2011;306(18):2001–2010. doi: 10.1001/jama.2011.1638. [DOI] [PubMed] [Google Scholar]
- Chow ML, Pramparo T, Winn ME, Barnes CC, Li HR, Weiss L, Fan JB, Murray S, April C, Belinson H, Fu XD, Wynshaw-Boris A, Schork NJ, Courchesne E. Age-dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genet. 2012;8(3):e1002592. doi: 10.1371/journal.pgen.1002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm N, O’Roak BJ, Shendure J, Eichler EE. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014;37(2):95–105. doi: 10.1016/j.tins.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485(7397):246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez F, Bravo S, Cruzat F, Montecino M, De Ferrari GV. Wnt/beta-catenin signaling enhances cyclooxygenase-2 (COX2) transcriptional activity in gastric cancer cells. PLoS One. 2011;6(4):e18562. doi: 10.1371/journal.pone.0018562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein EA, Assoian RK. Transcriptional regulation of the cyclin D1 gene at a glance. J Cell Sci. 2008;121(Pt 23):3853–3857. doi: 10.1242/jcs.039131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96(10):5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingraham CA, Park GC, Makarenkova HP, Crossin KL. Matrix metalloproteinase (MMP)-9 induced by Wnt signaling increases the proliferation and migration of embryonic neural stem cells at low O2 levels. J Biol Chem. 2011;286(20):17649–17657. doi: 10.1074/jbc.M111.229427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Crampton SP, Hughes CC. Wnt signaling induces matrix metalloproteinase expression and regulates T cell transmigration. Immunity. 2007;26(2):227–239. doi: 10.1016/j.immuni.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. COX-2’s new role in inflammation. Nat Chem Biol. 2010;6(6):401–402. doi: 10.1038/nchembio.375. [DOI] [PubMed] [Google Scholar]
- Seibert K, Masferrer JL. Role of inducible cyclooxygenase (COX-2) in inflammation. Receptor. 1994;4(1):17–23. [PubMed] [Google Scholar]
- Yoo HJ, Cho IH, Park M, Cho E, Cho SC, Kim BN, Kim JW, Kim SA. Association between PTGS2 polymorphism and autism spectrum disorders in Korean trios. Neurosci Res. 2008;62(1):66–69. doi: 10.1016/j.neures.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Asadabadi M, Mohammadi MR, Ghanizadeh A, Modabbernia A, Ashrafi M, Hassanzadeh E, Forghani S, Akhondzadeh S. Celecoxib as adjunctive treatment to risperidone in children with autistic disorder: a randomized, double-blind, placebo-controlled trial. Psychopharmacology (Berl) 2013;225(1):51–59. doi: 10.1007/s00213-012-2796-8. [DOI] [PubMed] [Google Scholar]
- Bloom J, Cross FR. Multiple levels of cyclin specificity in cell-cycle control. Nat Rev Mol Cell Biol. 2007;8(2):149–160. doi: 10.1038/nrm2105. [DOI] [PubMed] [Google Scholar]
- Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140(15):3079–3093. doi: 10.1242/dev.091744. [DOI] [PubMed] [Google Scholar]
- Kim JE, Shin MS, Seo TB, Ji ES, Baek SS, Lee SJ, Park JK, Kim CJ. Treadmill exercise ameliorates motor disturbance through inhibition of apoptosis in the cerebellum of valproic acid-induced autistic rat pups. Mol Med Rep. 2013;8(2):327–334. doi: 10.3892/mmr.2013.1518. [DOI] [PubMed] [Google Scholar]
- Pucilowska J, Puzerey PA, Karlo JC, Galan RF, Landreth GE. Disrupted ERK signaling during cortical development leads to abnormal progenitor proliferation, neuronal and network excitability and behavior, modeling human neuro-cardio-facial-cutaneous and related syndromes. J Neurosci. 2012;32(25):8663–8677. doi: 10.1523/JNEUROSCI.1107-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meechan DW, Tucker ES, Maynard TM, LaMantia AS. Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome. Proc Natl Acad Sci U S A. 2009;106(38):16434–16445. doi: 10.1073/pnas.0905696106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCawley LJ, Matrisian LM. Matrix metalloproteinases: they’re not just for matrix anymore! Curr Opin Cell Biol. 2001;13(5):534–540. doi: 10.1016/S0955-0674(00)00248-9. [DOI] [PubMed] [Google Scholar]
- Vu TH, Werb Z. Matrix metalloproteinases: effectors of development and normal physiology. Genes Dev. 2000;14(17):2123–2133. doi: 10.1101/gad.815400. [DOI] [PubMed] [Google Scholar]
- Fujioka H, Dairyo Y, Yasunaga K, Emoto K. Neural functions of matrix metalloproteinases: plasticity, neurogenesis, and disease. Biochem Res Int. 2012;2012:789083. doi: 10.1155/2012/789083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambaut-Guerin AM, Herigault S, Rouet-Benzineb P, Rouher C, Lafuma C. Induction of matrix metalloproteinase MMP-9 (92-kDa gelatinase) by retinoic acid in human neuroblastoma SKNBE cells: relevance to neuronal differentiation. J Neurochem. 2000;74(2):508–517. doi: 10.1046/j.1471-4159.2000.740508.x. [DOI] [PubMed] [Google Scholar]
- Ferguson TA, Muir D. MMP-2 and MMP-9 increase the neurite-promoting potential of schwann cell basal laminae and are upregulated in degenerated nerve. Mol Cell Neurosci. 2000;16(2):157–167. doi: 10.1006/mcne.2000.0859. [DOI] [PubMed] [Google Scholar]
- Shubayev VI, Myers RR. Matrix metalloproteinase-9 promotes nerve growth factor-induced neurite elongation but not new sprout formation in vitro. J Neurosci Res. 2004;77(2):229–239. doi: 10.1002/jnr.20160. [DOI] [PubMed] [Google Scholar]
- Abdallah M, Michel T. Matrix metalloproteinases in autism spectrum disorders. J Mol Psychiatry. 2013;1(1):1–5. doi: 10.1186/2049-9256-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdallah MW, Pearce BD, Larsen N, Greaves-Lord K, Norgaard-Pedersen B, Hougaard DM, Mortensen EL, Grove J. Amniotic fluid MMP-9 and neurotrophins in autism spectrum disorders: an exploratory study. Autism Res. 2012;5(6):428–433. doi: 10.1002/aur.1254. [DOI] [PubMed] [Google Scholar]
- Janusz A, Milek J, Perycz M, Pacini L, Bagni C, Kaczmarek L, Dziembowska M. The Fragile X mental retardation protein regulates matrix metalloproteinase 9 mRNA at synapses. J Neurosci. 2013;33(46):18234–18241. doi: 10.1523/JNEUROSCI.2207-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilousova TV, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW, Ethell IM. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet. 2009;46(2):94–102. doi: 10.1136/jmg.2008.061796. [DOI] [PubMed] [Google Scholar]
- Paribello C, Tao L, Folino A, Berry-Kravis E, Tranfaglia M, Ethell IM, Ethell DW. Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol. 2010;10:91. doi: 10.1186/1471-2377-10-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo BT, Morton D Jr, Komaragiri S, Millena AC, Leath C, Khan SA. TGF-beta effects on prostate cancer cell migration and invasion are mediated by PGE2 through activation of PI3K/AKT/mTOR pathway. Endocrinology. 2013;154(5):1768–1779. doi: 10.1210/en.2012-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrigan PJ, Lambertini L, Birnbaum LS. A research strategy to discover the environmental causes of autism and neurodevelopmental disabilities. Environ Health Perspect. 2012;120(7):a258–a260. doi: 10.1289/ehp.1104285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecker C, Ronan L, Feng Y, Daly E, Murphy C, Ginestet CE, Brammer M, Fletcher PC, Bullmore ET, Suckling J, Baron-Cohen S, Williams S, Loth E, Consortium MRCAIMS, Murphy DG. Intrinsic gray-matter connectivity of the brain in adults with autism spectrum disorder. Proc Natl Acad Sci U S A. 2013;110(32):13222–13227. doi: 10.1073/pnas.1221880110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel J, Kuchna I, Nowicki K, Imaki H, Wegiel J, Marchi E, Ma SY, Chauhan A, Chauhan V, Bobrowicz TW, de Leon M, Louis LA, Cohen IL, London E, Brown WT, Wisniewski T. The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010;119(6):755–770. doi: 10.1007/s00401-010-0655-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrams DA, Lynch CJ, Cheng KM, Phillips J, Supekar K, Ryali S, Uddin LQ, Menon V. Underconnectivity between voice-selective cortex and reward circuitry in children with autism. Proc Natl Acad Sci U S A. 2013;110(29):12060–12065. doi: 10.1073/pnas.1302982110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Abe O, Nippashi Y, Yamasue H. Comparison of white matter integrity between autism spectrum disorder subjects and typically developing individuals: a meta-analysis of diffusion tensor imaging tractography studies. Mol Autism. 2013;4(1):25. doi: 10.1186/2040-2392-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmonte MK, Allen G, Beckel-Mitchener A, Boulanger LM, Carper RA, Webb SJ. Autism and abnormal development of brain connectivity. J Neurosci. 2004;24(42):9228–9231. doi: 10.1523/JNEUROSCI.3340-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshew NJ, Williams DL. The new neurobiology of autism: cortex, connectivity, and neuronal organization. Arch Neurol. 2007;64(7):945–950. doi: 10.1001/archneur.64.7.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Tayama M, Murakawa K, Yoshimoto T, Miyazaki M, Harada M, Kuroda Y. Development of the brainstem and cerebellum in autistic patients. J Autism Dev Disord. 1995;25(1):1–18. doi: 10.1007/BF02178163. [DOI] [PubMed] [Google Scholar]
- Penzes P, Buonanno A, Passafaro M, Sala C, Sweet RA. Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. J Neurochem. 2013;126(2):165–182. doi: 10.1111/jnc.12261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauda F, Geda E, Sacco K, D’Agata F, Duca S, Geminiani G, Keller R. Grey matter abnormality in autism spectrum disorder: an activation likelihood estimation meta-analysis study. Neurol Neurosurg Psychiatry. 2011;82(12):1304–1313. doi: 10.1136/jnnp.2010.239111. [DOI] [PubMed] [Google Scholar]