

Abstract
Metabolic acidosis is a common complication of chronic kidney disease. Accumulating evidence identifies acidosis not only as a consequence of, but as a contributor to, kidney disease progression. Several mechanistic pathways have been identified in this regard. The dietary acid load, even in the absence of overt acidosis, may have deleterious effects. Several small trials now suggest that the treatment of acidosis with oral alkali can slow the progression of kidney disease.
Keywords: Bicarbonate, Dietary acid, Net endogenous acid production, Sodium bicarbonate, Alkali, Ammonia, Complement, Endothelin, Aldosterone
Review
Metabolic acidosis is a common complication of chronic kidney disease (CKD). Based on a cross-sectional analysis of the National Health and Nutrition Examination Survey, an estimated 26 million adults in the United States have CKD, and approximately 700,000 individuals have an estimated glomerular filtration rate (eGFR) less than 30 mL/min/1.73 m2[1]. As 30-50% of those with eGFR <30 mL/min/1.73 m2 have metabolic acidosis [2-4], approximately 200,000 to 350,000 individuals with CKD stage 4 and 5 have chronic metabolic acidosis in the United States.
Chronic metabolic acidosis may have various adverse effects in patients with CKD, including altered skeletal metabolism [5], insulin resistance [6], protein-energy wasting [7-9], and accelerated progression of kidney disease. In epidemiologic studies, low serum bicarbonate levels have been associated with high mortality (Table 1). In a study of 1,240 male patients with non-dialysis dependent CKD, the lowest mortality was observed among those with baseline serum bicarbonate levels of 26–29 mEq/L, whereas patients with levels <22 mEq/L had a 43% higher risk of mortality [10]. Using data from the African American Study of Kidney Disease and Hypertension (AASK) trial, Raphael et al. showed that higher bicarbonate levels within the range of 20 to 30 mEq/L were associated with reduced risk of the clinical composite outcome of death, dialysis or worsening renal function [11]. This review focuses specifically on the effect of metabolic acidosis on the progression of kidney disease.
Table 1.
Observational studies of serum bicarbonate and long-term outcomes in persons without end-stage renal disease
| Study | Population | Main outcome(s) | Findings | pH or pCO 2 available | Strengths | Limitations |
|---|---|---|---|---|---|---|
| Shah et al. 2009 [4] |
5,422 outpatients in the Bronx, NY; 9% with eGFR < 60 mL/min/1.73 m2 |
Kidney disease progression, defined as 50% decrease in eGFR or eGFR < 15 mL/min/1.73 m2 |
HR 1.54 (95% CI 1.13-2.09) for progression, for serum bicarbonate ≤22 mEq/L compared with 25–26 mEq/L |
No |
• Ethnically diverse cohort |
• Single measure of serum bicarbonate |
| • Data derived from clinical and administrative dataset | ||||||
| Menon et al. 2010 [12] |
1,781 participants (839 randomized, 942 non-randomized) from the MDRD study |
(1) ESRD (need for dialysis or transplantation); (2) all-cause mortality; (3) composite of 1 and 2 |
HR 1.05 (0.87-1.28), 0.99 (0.75-1.13), 1.04 (0.87-1.24) for need for kidney failure, all-cause mortality, and composite outcome, respectively, for serum bicarbonate 11–20 compared with 26–40 mEq/L |
No |
• Well-characterized cohort |
• Single measure of serum bicarbonate |
| • Adjustment for measured GFR | ||||||
| Raphael et al. 2011 [11] |
1,090 participants of the AASK trial |
Composite outcome of death, ESRD (dialysis or transplantation), or GFR event (defined as a GFR reduction by 50% or by 25 ml/min/1.73 m2 from baseline) |
HR 0.960 (0.924-0.998) for composite outcome, per mEq/L higher baseline serum bicarbonate |
No |
• Well-characterized cohort |
• Single measure of serum bicarbonate |
| • Adjustment for measured GFR | ||||||
| • Adjustment for errors in measurement of GFR and proteinuria | ||||||
| Kovesdy et al. 2009 [10] |
1,240 adults at a Veterans Affairs Medical Center; 87% with CKD stages 3 and 4 |
(1) All-cause mortality; (2) composite of predialysis mortality and initiation of dialysis |
U-shaped association; HR for mortality 1.43 (1.10-1.87) for serum bicarbonate <22 compared with 26–29 mEq/L; similar results for composite outcome |
No |
• Adjustment for time-varying serum bicarbonate levels |
• Data derived from clinical and administrative dataset |
| Navaneethan et al. 2011 [13] |
41,749 outpatients with eGFR < 60 mL/min/1.73 m2 in Cleveland, OH |
All-cause mortality |
U-shaped association; HR 1.23 (1.16-1.31) for bicarbonate <23 compared with 23–32 mEq/L; HR 1.59 (1.49-1.69) for reaching bicarbonate <23 mEq/L |
No |
• Examination of temporal change in serum bicarbonate |
• Data derived from clinical and administrative dataset |
| • Large sample size | ||||||
| Dobre et al. 2013 [14] |
3,939 participants from the CRIC study |
(1) Renal outcome, defined as 50% decrease in eGFR or ESRD (dialysis or transplantation); (2) atherosclerotic events; (3) CHF events; (4) all-cause mortality |
Per mEq/L higher serum bicarbonate, HR 0.97 (0.94-0.99) for renal outcome; 0.99 (0.95-1.03) for atherosclerotic event; 1.14 (1.03-1.26) for CHF for serum bicarbonate ≥24 mEq/L; 0.98 (0.95-1.02) for mortality |
No |
• Well-characterized cohort |
• Single measure of serum bicarbonate |
| Kanda et al. 2013 [15] |
113 Japanese patients ≥60 years old with eGFR < 60 mL/min/1.73 m2 |
Kidney disease progression, defined as 25% decrease in eGFR or initiation of dialysis |
HR 0.791 (0.684-0.914) for progression, per mEq/L higher serum bicarbonate |
No |
• Focus on elderly cohort |
• Single measure of serum bicarbonate |
| • Small sample size | ||||||
| Raphael et al. 2013 [16] | 15,836 participants of NHANES III | All-cause mortality | HR 1.75 (1.12-2.74), 1.56 (0.78-3.09), and 2.56 (1.49-4.38) for total population, non-CKD, and CKD subgroups, respectively, for serum bicarbonate <22 mEq/L compared with 26–30 mEq/L | No | • Nationally representative cohort |
• Single measure of serum bicarbonate |
| • Compared CKD and non-CKD subgroups |
Abbreviations: HR, hazard ratio; CI, confidence interval; GFR, glomerular filtration rate; MDRD, Modification of Diet in Renal Disease; AASK, African American Study of Kidney Disease and Hypertension; ESRD, end-stage renal disease; CKD, chronic kidney disease; CRIC, Chronic Renal Insufficiency Cohort; CHF, congestive heart failure; NHANES, National Health and Nutrition Examination Survey.
Pathophysiology leading to acidosis in CKD
Under normal conditions, the extracellular H+ concentration is tightly regulated and varies little from the normal value of approximately 40 nanomol/L. Regulation of acid–base homeostasis involves three basic steps: chemical buffering by extracellular and intracellular buffers, alteration of alveolar ventilation, and changes in renal H+ excretion [17]. The kidneys regulate H+ excretion by reabsorbing filtered HCO3− and generating new HCO3− in response to various stimuli. Secreted H+ combine with urinary buffers such as HPO42− and ammonia. In CKD the reduction in functioning nephrons causes defects in renal excretion of acid, mainly in the form of ammonium [18]. A fraction of patients may have inappropriate bicarbonate wasting in the urine [19].
The acidosis in CKD usually remains relatively stable. In uncomplicated acidosis, the serum bicarbonate levels typically are >12 mEq/L and blood pH is >7.2 [19,20]. There are two general possibilities for this stabilization of serum bicarbonate levels. One is that after an initial period of acid retention, the excretion and production of acid equalize. The other possibility is that acidosis evokes extrarenal mechanisms that dispose of the endogenous acid not excreted in the urine. The major source of such buffering would be base derived from bone. Carefully conducted balance studies in CKD patients with acidosis found a positive acid balance of approximately 10–20 mEq per day and negative calcium balance that improved with correction of acidosis [21,22]. However, the degree of acid retention has been debated, with the suggestion that these findings resulted from systematic measurement error [23]. While the magnitude of acid retention may be less than reported, it seems likely that acid production does exceed excretion in the setting of chronic renal acidosis [24].
The acidosis in mild to moderate CKD is predominantly hyperchloremic, with an increased anion gap observed variably and generally only with advanced CKD [25-27]. However, accounting for changes in serum albumin and other electrolytes in the anion gap calculation reveals a slightly more nuanced view. This maneuver reveals minor elevations in the anion gap, which are associated with decrements in serum bicarbonate, even in the CKD stage 2 eGFR range [28]. Although small, these differences in anion gap are independently associated with mortality [28]. Thus, there may be low-level accumulation of organic solutes even when the GFR is relatively preserved that partially accounts for changes in the serum bicarbonate level.
Metabolic acidosis and progression of kidney disease
Epidemiologic studies have demonstrated that lower serum bicarbonate is associated with an increased risk of kidney disease progression (Table 1). In a single center retrospective cohort study involving patients with and without kidney disease, the risk of CKD progression was 54% (95% confidence interval (CI) 13-109%) higher for patients with bicarbonate levels <22 mEq/L compared with bicarbonate levels of 25 to 26 mEq/L [4]. Among 1,781 participants with CKD stages 2–4 from the Modification of Diet in Renal Disease Study, compared with serum bicarbonate levels ≥26 mEq/L, levels ≤20 mEq/L were associated with a higher risk of kidney failure (hazard ratio (HR) 2.22 (95% CI, 1.83-2.68)) while adjusting for demographic and cardiovascular disease factors, serum albumin, proteinuria, and cause of kidney disease [12]. However, the association was substantially attenuated and non-significant after adjustment for GFR, measured using iothalamate clearance (HR 1.05 (95% CI, 0.87-1.28)). The implication was that lower serum bicarbonate was a marker for more advanced kidney disease, and that analyses adjusting for estimated GFR were limited by residual confounding. However, Raphael et al. performed a similar analysis using data from 1,094 African American participants of the AASK trial with GFR measured by iothalamate clearance [11]. After controlling for measured GFR, higher serum bicarbonate in the 20 to 30 mEq/L range was associated with a lower risk of dialysis or worsening kidney function (HR 0.932 (95% CI, 0.881-0.986) per 1 mEq/L serum bicarbonate). Most recently, in 3,939 participants with CKD stages 2–4 in the Chronic Renal Insufficiency Cohort, every 1 mEq/L higher serum bicarbonate level was associated with a 3% (95% CI, 1-6%) lower risk of developing end-stage renal disease or a 50% decline in eGFR during follow-up [14].
Several factors have been implicated in the effect of metabolic acidosis on the progression of kidney disease. These include ammonia-induced complement activation and increased production of endothelin and aldosterone (Figure 1). Although total ammonium excretion decreases with progressive CKD, ammonia generation per nephron actually increases [29]. This adaptive response may be deleterious for the surviving nephrons [30]. Nath et al. examined the role of ammonia in the pathogenesis of tubulo-interstitial injury using the rat remnant kidney model [31]. Chronic sodium bicarbonate supplementation lowered renal vein total ammonia concentrations, reduced tubular deposition of complement components C3 and C5b-9, and ameliorated structural and functional evidence of tubulo-interstitial damage. The authors proposed that ammonia, as a nitrogen nucleophile, reacted biochemically with C3 to trigger the alternative complement pathway. Therefore, the compensatory increase in single-nephron ammoniagenesis observed in CKD could further instigate progressive kidney injury.
Figure 1.
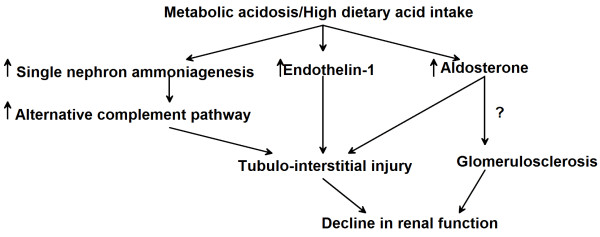
Pathogenesis of kidney disease progression due to chronic metabolic acidosis. Metabolic acidosis and/or a high dietary acid load may contribute to progressive kidney disease through multiple mechanisms, including increased ammonia generation per nephron leading to activation of the alternative complement pathway and increased endothelin-1 and aldosterone levels in the kidney. Each of these factors may cause tubule-interstitial injury leading to a decline in kidney function. Hyperaldosteronism could also accelerate glomerulosclerosis, although glomerular injury due to metabolic acidosis has not been reported in animal models.
Acidosis has been shown to increase endothelin (ET)-mediated tubulo-interstitial injury as well. ET is an endothelial cell-derived peptide with 3 mammalian isoforms: ET-1, ET-2, and ET-3. The kidneys produce ET-1 in relatively high amounts and contain abundant ET receptors, particularly in the vasculature and the medulla [32]. In vitro evidence has demonstrated that ET-1 promotes the synthesis of fibronectin and collagen [33]. Several in vivo studies have correlated renal ET-1 gene expression and urinary excretion with the degree of proteinuria and glomerular and tubulo-interstitial damage [34]. ET regulates multiple renal functional parameters, one of which is acid–base handling. Specifically, ET-1 mediates increased renal acid excretion in response to a systemic acid challenge. Rats given a dietary acid load as (NH4)2SO4 increased ET-1 addition to renal interstitial fluid, and pharmacological inhibition of ET receptors using bosentan blunted distal tubule acidification [35]. Using a remnant kidney model, Wesson and colleagues demonstrated that dietary protein-induced tubulo-interstitial injury and GFR decline were mediated by ET, and that alkali supplementation prevented these effects [36,37].
Excess aldosterone could also mediate the decline in GFR caused by acidosis, via its hemodynamic effects and its pro-fibrotic actions [38]. Hyperaldosteronism in the setting of reduced nephron mass contributes to hypertension, proteinuria and glomerulosclerosis in the remnant kidney model [39,40]. Chronic aldosterone treatment plus a high salt diet produced hypertension and severe glomerular and tubulo-interstitial injury in non-nephrectomized rats [41,42]. Schambelan et al. examined the effect of chronic metabolic acidosis on adrenocortical hormone production by administering NH4Cl for 5 days to four normal subjects, and found that chronic metabolic acidosis induced a sustained stimulation of aldosterone secretion [43]. Aldosterone mediates increased distal nephron acidification in the setting of metabolic acidosis, and prevention of kidney disease with alkali in the rat remnant kidney model was associated with reduced aldosterone production in kidney cortex [44,45].
More recently, the effect of acidosis on kidney injury has been explored using gene expression analyses. Raj et al. examined the effect of exposure of Madin-Darby canine kidney cells (an immortalized cell line derived from the dog distal renal tubule) to medium with pH 7.4 and 7.0 for 24 hours, and found that acid stress upregulated the expression of genes that might participate in the pro-inflammatory process leading to glomerulosclerosis and fibrosis [46].
Dietary acid, acid retention and progression of kidney disease
The net dietary acid load is determined by the balance of acid-forming and base-forming precursors in the diet. Common foods that provide a high dietary acid load include cheese, meat, eggs and grains, whereas fruits and vegetables are rich in alkali precursors [47,48]. Generally speaking, diets high in animal protein produce high levels of net endogenous acid production (NEAP), whereas vegan and vegetarian diets result in low, or even negative, NEAP. Low NEAP can be achieved even while preserving the proportion of energy derived from dietary protein. This was demonstrated in the Dietary Approaches to Stop Hypertension (DASH) trial, where a diet incorporating increased fruit and vegetable intake produced substantially lower NEAP than the typical American diet [47].
The usual diet consumed in the Western world is quite different from that which our ancestors ate. Ancestral human diets were largely plant-based, and were characterized by much lower levels of refined carbohydrates and sodium, and much higher levels of fiber and potassium, than contemporary diets [49,50]. Sebastian et al. estimated the net acid load from retrojected ancestral pre-agricultural diets and compared it with contemporary diets [51]. They found that 87% of 159 retrojected pre-agricultural diets were net base-producing with a mean NEAP of −88 mEq per day, compared with +48 mEq per day for the average American diet. Similarly, Strohle et al. examined the dietary net acid load for 229 worldwide historically-studied hunter-gatherer societies, and confirmed that the NEAP became progressively more positive as the dietary plant-to-animal ratio declined [52]. The historical shift from negative to positive NEAP, or from net base-producing to acid-producing, is thought due to the displacement of high-bicarbonate-yielding plant foods in the ancestral diet by cereal grains and energy-dense, nutrition-poor foods in the contemporary diet. It has been postulated that on an evolutionary scale this change has occurred rapidly, in a time span too brief for adequate genetic adaptation [53-55]. The resultant mismatch between the modern Western diet and a core metabolic machinery that was selected for 50,000 to 100,000 years ago may therefore contribute to chronic disease [56].
The high dietary acid load of contemporary diets could impair kidney function by inducing metabolic acidosis or even subclinical acid retention. In rats, acid loading increased blood and renal cortical acid content measured by microdialysis, consistent with net acid retention [57]. Despite no difference in blood pH or serum bicarbonate, rats with reduced nephron mass had higher tissue acid content compared with control animals. Dietary acid accelerated GFR decline, which was ameliorated by dietary alkali [58]. The decline in GFR caused by dietary acid could be mediated by increased kidney ET and aldosterone production [45,59] (Figure 1).
In humans, higher NEAP has been associated with lower serum bicarbonate in persons with kidney disease and in the general population [60,61]. Furthermore, this association is strongest among those with more advanced CKD and among middle-aged and older individuals, populations in whom the capacity to excrete an acid load is relatively impaired [60,61]. Among 632 participants from the AASK cohort study, higher NEAP was associated with a faster decline in GFR over a median follow up of 3.2 years [62]. Thus, animal studies and epidemiologic data link dietary acid load with the pathogenesis of progressive CKD.
Treatment
Several single-center clinical studies have examined the effect of alkali therapy on the progression of kidney disease. In a 2-year, open-label, randomized trial, 134 patients with CKD stage 4 and serum bicarbonate levels of 16 to 20 mEq/L were assigned to receive oral sodium bicarbonate or to continue routine care [63]. Sodium bicarbonate was provided as 600 mg tablets dosed thrice daily and increased as necessary to maintain bicarbonate levels ≥23 mEq/L. Compared with the control group, bicarbonate-supplemented patients experienced a slower decline in creatinine clearance (5.93 vs. 1.88 mL/min/1.73 m2; p < 0.0001) and fewer developed ESRD. This was followed by another prospective interventional study of 59 patients with a clinical diagnosis of hypertensive nephropathy, all treated with regimens including an angiotensin-converting enzyme (ACE) inhibitor, who had an eGFR 20–59 mL/min/1.73 m2 and a serum bicarbonate <22 mEq/L [64]. Thirty patients prescribed sodium citrate were compared with the remaining 29 patients who were unable or unwilling to take sodium citrate or bicarbonate. After 24 months, patients taking sodium citrate had lesser eGFR decline and significantly lower urinary ET-1 excretion and tubulo-interstitial injury, as measured by urinary N-acetyl-β-D-glucosaminidase (NAG). In a second study by the same group, 120 patients with hypertensive nephropathy and CKD stage 2, all taking ACE inhibitors, were randomized in a blinded fashion to receive 0.5 mEq/kg/day sodium bicarbonate, sodium chloride, or matching placebo. After 5 years, the rate of eGFR decline was slowest in the patients randomized to sodium bicarbonate, and they had lower urine ET-1 and NAG compared with the other groups [65]. Of note, the mean serum bicarbonate at study entry among these 120 participants was 26.2 mEq/L, well within the normal range.
More recently, dietary modification has been examined by Goraya and colleagues [66,67]. In patients with CKD stage 2 due to hypertensive nephropathy, 30 days of increased fruit and vegetable consumption produced similar reductions in urinary NAG and albuminuria as did oral sodium bicarbonate [66]. Similar results were found in 71 patients with stage 4 CKD and serum bicarbonate <22 mEq/L who were randomized to 1 year of sodium bicarbonate at 1.0 mEq/kg per day or increased fruits and vegetables to reduce dietary acid by half [67]. The serum bicarbonate increased with the dietary intervention, although less than in the bicarbonate group, whose alkali dose would be expected to nearly completely neutralize the dietary acid load. The aforementioned markers of kidney injury declined similarly in the 2 groups. It is encouraging that there were no complications from hyperkalemia, but it should be noted that only patients with plasma K+ ≤4.6 mEq/L were enrolled in the study.
Thus, accumulating evidence suggests that treatment of chronic metabolic acidosis could slow the progression of CKD. However, the evidence base is not yet definitive. A systemic review was performed examining the published literature regarding alkali therapy through July 2011 [68]. The authors concluded that although oral alkali might afford a long-term benefit in slowing the progression of CKD, differences in study protocols and small sample sizes precluded definitive conclusions. Additional, more nuanced questions also remain unanswered. If alkali therapy does retard progression, what serum bicarbonate level should be targeted? Is there a risk to overcorrection of acidosis? Higher serum bicarbonate was associated with increased risk of congestive heart failure in the CRIC study [14], but to date interventional studies have not demonstrated significant metabolic alkalosis or fluid overload with even high-dose alkali. Is serum bicarbonate even the right target, given evidence of acid retention and deleterious effects within the normal range of serum bicarbonate? To what extent should dietary interventions to reduce the dietary acid load be employed in lieu of, or in addition to, oral alkali? There are 3 ongoing randomized controlled trials of alkali therapy in patients with CKD that have the potential to answer some of these questions and to impact clinical practice (NCT01640119 [69]; NCT01452412 [70]; EUDRACT Number 2012-001824-36 [71]). A summary of these trials is provided in Table 2. Several smaller studies are examining other effects of base, including changes in urinary TGF-β1 (NCT01574157), vascular endothelial function (NCT02031770), and its role in sickle cell disease (NCT01894594).
Table 2.
Summary of on-going randomized clinical trials of alkali therapy in patients with chronic kidney disease
| Title | Correction of Metabolic Acidosis with Use of Bicarbonate in Chronic Renal Insufficiency (NCT01640119)[69] | Alkali Therapy in Chronic Kidney Disease (NCT01452412)[70] ^ | Oral Sodium Bicarbonate Supplementation in Patients with Chronic Metabolic Acidosis and Chronic Kidney Disease (EUDRACT Number 2012-001824-36)[71] |
|---|---|---|---|
| Estimated primary completion date |
12/2013 |
1/2015 |
Not available# |
| Anticipated sample size |
728 |
150 |
200 |
| CKD stage |
Stage 3b & 4 |
Stage 3 & 4 |
Stage 3 & 4 |
| Serum bicarbonate levels at randomization |
≥18 mEq/L |
20-26 mEq/L |
<21 mEq/L |
| Study design |
Randomized, open label |
Randomized, placebo-controlled, double blind |
Randomized, open label |
| Intervention |
Bicarbonate administration to keep bicarbonate levels between 24–28 mEq/L |
Sodium bicarbonate 0.4 mEq / kg ideal body weight per day |
Sodium bicarbonate with target bicarbonate levels of 24 ± 1 mEq/L |
| Control |
No intervention, partial correction if bicarbonate <18 mEq/L (up to 22 mEq/L) |
Placebo |
Rescue therapy of sodium bicarbonate with target bicarbonate level of 20 ± 1 mEq/L |
| Locations |
Multiple centers in Italy |
2 centers in the United States (Bronx, NY and Cleveland, OH) |
Single center in Vienna, Austria |
| Follow up length |
36 months |
24 months |
24 months |
| Primary outcome |
Doubling of Cr |
HOMA-IR, sit to stand to sit speed, DEXA of wrist, urinary NGAL & KIM-1 |
Means of eGFR, calculated using the 4-variable-MDRD Study equation |
| Secondary outcome measures | All-cause death, start of dialysis | Glucose disposal rate by euglycemic hyperinsulinemic clamp, hand-grip strength, serum calcium, phosphate, 1,25-dihydroxyvitamin D, PTH, Cr, cystatin C, urinary albumin/Cr ratio, urinary cystatin | Death, need for renal replacement therapy, change in markers of bone metabolism |
Abbreviations: CKD, chronic kidney disease; Cr, creatinine; HOMA-IR, homeostasis model assessment-estimated insulin resistance; DEXA, dual energy X-ray absorptiometry; NGAL, urinary neutrophil gelatinase-associated lipocalin; KIM-1, kidney injury molecule-1; PTH, parathyroid hormone; eGFR, estimated glomerular filtrate rate; MDRD, modification of diet in renal disease; NY, New York; OH, Ohio.
^Our institution is one of the centers for the study (NCT01452412) and one of us (MKA) is a co-investigator. The information presented is updated from clinicaltrials.gov.
#Recruitment was planned to start in October 2013.
Conclusion
Patients with CKD develop metabolic acidosis due to reduced kidney mass and defects in renal acid excretion. Chronic metabolic acidosis is a common complication of CKD and appears to contribute to the progression of kidney disease. High dietary acid intake has also been demonstrated to worsen kidney function by induction of metabolic acidosis and/or subclinical acid retention. The factors that have been implicated in this effect of acidosis on CKD progression include ammonia-induced activation of the alternative complement system, as well as increased endothelin and aldosterone production. Existing evidence from clinical trials suggests that alkali therapy could retard the progression of CKD. Increased fruit and vegetable consumption appears to be a reasonable alternative to alkali therapy for patients with mild metabolic acidosis and without hyperkalemia. However, definitive evidence is lacking for optimal evidence-based practice guidelines. Ongoing trials will hopefully facilitate more evidence-based treatment of metabolic acidosis in the future.
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
WC drafted the manuscript and both WC and MKA revised it and approved the final manuscript. Both authors have read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Wei Chen, Email: wchen@montefiore.org.
Matthew K Abramowitz, Email: matthew.abramowitz@einstein.yu.edu.
Acknowledgements
This research was supported by an American Society of Nephrology Carl W. Gottschalk Research Scholar Grant, National Institutes of Health (NIH) grant T32 DK007110, and by Clinical and Translational Science Award (CTSA) grants 1 UL1 TR001073-01, 1 TL1 TR001072-01, 1 KL2 TR001071-01 from the National Center for Advancing Translational Sciences (NCATS), a component of the NIH. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
References
- Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, Van Lente F, Levey AS. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298(17):2038–2047. doi: 10.1001/jama.298.17.2038. [DOI] [PubMed] [Google Scholar]
- Moranne O, Froissart M, Rossert J, Gauci C, Boffa JJ, Haymann JP, M’Rad MB, Jacquot C, Houillier P, Stengel B, Fouqueray B. the NephroTest Study Group. Timing of onset of CKD-related metabolic complications. J Am Soc Nephrol. 2009;20(1):164–171. doi: 10.1681/ASN.2008020159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eustace JA, Astor B, Muntner PM, Ikizler TA, Coresh J. Prevalence of acidosis and inflammation and their association with low serum albumin in chronic kidney disease. Kidney Int. 2004;65(3):1031–1040. doi: 10.1111/j.1523-1755.2004.00481.x. [DOI] [PubMed] [Google Scholar]
- Shah SN, Abramowitz M, Hostetter TH, Melamed ML. Serum bicarbonate levels and the progression of kidney disease: a cohort study. Am J Kidney Dis. 2009;54(2):270–277. doi: 10.1053/j.ajkd.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraut JA, Mishler DR, Singer FR, Goodman WG. The effects of metabolic acidosis on bone formation and bone resorption in the rat. Kidney Int. 1986;30(5):694–700. doi: 10.1038/ki.1986.242. [DOI] [PubMed] [Google Scholar]
- Mak RH. Effect of metabolic acidosis on insulin action and secretion in uremia. Kidney Int. 1998;54(2):603–607. doi: 10.1046/j.1523-1755.1998.00023.x. [DOI] [PubMed] [Google Scholar]
- Bailey JL, Wang X, England BK, Price SR, Ding X, Mitch WE. The acidosis of chronic renal failure activates muscle proteolysis in rats by augmenting transcription of genes encoding proteins of the ATP-dependent ubiquitin-proteasome pathway. J Clin Invest. 1996;97(6):1447–1453. doi: 10.1172/JCI118566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Gao XL, Hirschberg R, Vadgama JV, Kopple JD. Impaired actions of insulin-like growth factor 1 on protein Synthesis and degradation in skeletal muscle of rats with chronic renal failure. Evidence for a postreceptor defect. J Clin Invest. 1996;97(4):1064–1075. doi: 10.1172/JCI118499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballmer PE, McNurlan MA, Hulter HN, Anderson SE, Garlick PJ, Krapf R. Chronic metabolic acidosis decreases albumin synthesis and induces negative nitrogen balance in humans. J Clin Invest. 1995;95(1):39–45. doi: 10.1172/JCI117668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovesdy CP, Anderson JE, Kalantar-Zadeh K. Association of serum bicarbonate levels with mortality in patients with non-dialysis-dependent CKD. Nephrol Dial Transplant. 2009;24(4):1232–1237. doi: 10.1093/ndt/gfn633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphael KL, Wei G, Baird BC, Greene T, Beddhu S. Higher serum bicarbonate levels within the normal range are associated with better survival and renal outcomes in African Americans. Kidney Int. 2011;79(3):356–362. doi: 10.1038/ki.2010.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon V, Tighiouart H, Vaughn NS, Beck GJ, Kusek JW, Collins AJ, Greene T, Sarnak MJ. Serum bicarbonate and long-term outcomes in CKD. Am J Kidney Dis. 2010;56(5):907–914. doi: 10.1053/j.ajkd.2010.03.023. [DOI] [PubMed] [Google Scholar]
- Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Wehbe E, Raina R, Simon JF, Srinivas TR, Jain A, Schreiber MJ Jr, Nally JV Jr. Serum bicarbonate and mortality in stage 3 and stage 4 chronic kidney disease. Clin J Am Soc Nephrol. 2011;6(10):2395–2402. doi: 10.2215/CJN.03730411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobre M, Yang W, Chen J, Drawz P, Hamm LL, Horwitz E, Hostetter T, Jaar B, Lora CM, Nessel L, Ojo A, Scialla J, Steigerwalt S, Teal V, Wolf M, Rahman M. Association of serum bicarbonate with risk of renal and cardiovascular outcomes in CKD: a report from the Chronic Renal Insufficiency Cohort (CRIC) study. Am J Kidney Dis. 2013;62(4):670–678. doi: 10.1053/j.ajkd.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda E, Ai M, Yoshida M, Kuriyama R, Shiigai T. High serum bicarbonate level within the normal range prevents the progression of chronic kidney disease in elderly chronic kidney disease patients. BMC Nephrol. 2013;14:4. doi: 10.1186/1471-2369-14-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphael KL, Zhang Y, Wei G, Greene T, Cheung AK, Beddhu S, El Nahas M. Serum bicarbonate and mortality in adults in NHANES III. Nephrol Dial Transplant. 2013;28(5):1207–1213. doi: 10.1093/ndt/gfs609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose BD, Post TW. Clinical Physiology of Acid–base and Electrolyte Disorders. Fifth. USA: McGraw-Hill; 2000. [Google Scholar]
- Van Slyke DD, Linder GC, Hiller A, Leiter L, McIntosh JF. The Excretion of Ammonia and Titratable Acid in Nephritis. J Clin Invest. 1926;2(3):255–288. doi: 10.1172/JCI100045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz WB, Hall PW 3rd, Hays RM, Relman AS. On the mechanism of acidosis in chronic renal disease. J Clin Invest. 1959;38(1, Part 1):39–52. doi: 10.1172/JCI103794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relman AS. Renal Acidosis and Renal Excretion of Acid in Health and Disease. Adv Intern Med. 1964;12:295–347. [PubMed] [Google Scholar]
- Goodman AD, Lemann J Jr, Lennon EJ, Relman AS. Production, Excretion, and Net Balance of Fixed Acid in Patients with Renal Acidosis. J Clin Invest. 1965;44:495–506. doi: 10.1172/JCI105163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litzow JR, Lemann J Jr, Lennon EJ. The effect of treatment of acidosis on calcium balance in patients with chronic azotemic renal disease. J Clin Invest. 1967;46(2):280–286. doi: 10.1172/JCI105530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uribarri J, Douyon H, Oh MS. A re-evaluation of the urinary parameters of acid production and excretion in patients with chronic renal acidosis. Kidney Int. 1995;47(2):624–627. doi: 10.1038/ki.1995.79. [DOI] [PubMed] [Google Scholar]
- Gennari FJ. In: Acid–base disorders and their treatment. Gennari FJ, Adrogue HJ, Galla JH, Madias NE, editor. Boca Raton: Taylor & Francis; 2005. Metabolic acidosis in chronic renal insufficiency; pp. 469–485. [Google Scholar]
- Hakim RM, Lazarus JM. Biochemical parameters in chronic renal failure. Am J Kidney Dis. 1988;11(3):238–247. doi: 10.1016/s0272-6386(88)80156-2. [DOI] [PubMed] [Google Scholar]
- Wallia R, Greenberg A, Piraino B, Mitro R, Puschett JB. Serum electrolyte patterns in end-stage renal disease. Am J Kidney Dis. 1986;8(2):98–104. doi: 10.1016/s0272-6386(86)80119-6. [DOI] [PubMed] [Google Scholar]
- Widmer B, Gerhardt RE, Harrington JT, Cohen JJ. Serum electrolyte and acid base composition. The influence of graded degrees of chronic renal failure. Arch Intern Med. 1979;139(10):1099–1102. doi: 10.1001/archinte.1979.03630470021010. [DOI] [PubMed] [Google Scholar]
- Abramowitz MK, Hostetter TH, Melamed ML. The serum anion gap is altered in early kidney disease and associates with mortality. Kidney Int. 2012;82(6):701–709. doi: 10.1038/ki.2012.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson DP. Control of hydrogen ion homeostasis and renal acidosis. Medicine. 1971;50(6):503–541. doi: 10.1097/00005792-197111000-00002. [DOI] [PubMed] [Google Scholar]
- Halperin ML, Ethier JH, Kamel KS. Ammonium excretion in chronic metabolic acidosis: benefits and risks. Am J Kidney Dis. 1989;14(4):267–271. doi: 10.1016/s0272-6386(89)80200-8. [DOI] [PubMed] [Google Scholar]
- Nath KA, Hostetter MK, Hostetter TH. Pathophysiology of chronic tubulo-interstitial disease in rats. Interactions of dietary acid load, ammonia, and complement component C3. J Clin Invest. 1985;76(2):667–675. doi: 10.1172/JCI112020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan DE, Inscho EW, Wesson D, Pollock DM. Physiology of endothelin and the kidney. Compr Physiol. 2011;1(2):883–919. doi: 10.1002/cphy.c100039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Ortega M, Gomez-Garre D, Alcazar R, Palacios I, Bustos C, Gonzalez S, Plaza JJ, Gonzalez E, Egido J. Involvement of angiotensin II and endothelin in matrix protein production and renal sclerosis. J Hypertens Suppl. 1994;12(4):S51–58. [PubMed] [Google Scholar]
- Remuzzi G. Role of endothelin in the development of glomerulosclerosis. Kidney Blood Press Res. 1996;19(3–4):182–183. doi: 10.1159/000174070. [DOI] [PubMed] [Google Scholar]
- Wesson DE. Endogenous endothelins mediate increased distal tubule acidification induced by dietary acid in rats. J Clin Invest. 1997;99(9):2203–2211. doi: 10.1172/JCI119393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesson DE, Nathan T, Rose T, Simoni J, Tran RM. Dietary protein induces endothelin-mediated kidney injury through enhanced intrinsic acid production. Kidney Int. 2007;71(3):210–217. doi: 10.1038/sj.ki.5002036. [DOI] [PubMed] [Google Scholar]
- Phisitkul S, Hacker C, Simoni J, Tran RM, Wesson DE. Dietary protein causes a decline in the glomerular filtration rate of the remnant kidney mediated by metabolic acidosis and endothelin receptors. Kidney Int. 2008;73(2):192–199. doi: 10.1038/sj.ki.5002647. [DOI] [PubMed] [Google Scholar]
- Ponda MP, Hostetter TH. Aldosterone antagonism in chronic kidney disease. Clin J Am Soc Nephrol. 2006;1(4):668–677. doi: 10.2215/CJN.00120106. [DOI] [PubMed] [Google Scholar]
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest. 1996;98(4):1063–1068. doi: 10.1172/JCI118867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldigier JC, Kanjanbuch T, Ma LJ, Brown NJ, Fogo AB. Regression of existing glomerulosclerosis by inhibition of aldosterone. J Am Soc Nephrol. 2005;16(11):3306–3314. doi: 10.1681/ASN.2004090804. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Abe Y. Molecular mechanisms and therapeutic strategies of chronic renal injury: renoprotective effects of aldosterone blockade. J Pharmacol Sci. 2006;100(1):9–16. doi: 10.1254/jphs.FMJ05003X3. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Yao L, Nagai Y, Miyata K, Yoshizumi M, Kagami S, Kondo S, Kiyomoto H, Shokoji T, Kimura S, kohno M, Abe Y. Possible contributions of reactive oxygen species and mitogen-activated protein kinase to renal injury in aldosterone/salt-induced hypertensive rats. Hypertension. 2004;43(4):841–848. doi: 10.1161/01.HYP.0000118519.66430.22. [DOI] [PubMed] [Google Scholar]
- Schambelan M, Sebastian A, Katuna BA, Arteaga E. Adrenocortical hormone secretory response to chronic NH4Cl-induced metabolic acidosis. Am J Physiol. 1987;252(4 Pt 1):E454–460. doi: 10.1152/ajpendo.1987.252.4.E454. [DOI] [PubMed] [Google Scholar]
- Khanna A, Simoni J, Wesson DE. Endothelin-induced increased aldosterone activity mediates augmented distal nephron acidification as a result of dietary protein. J Am Soc Nephrol. 2005;16(7):1929–1935. doi: 10.1681/ASN.2004121054. [DOI] [PubMed] [Google Scholar]
- Wesson DE, Simoni J. Acid retention during kidney failure induces endothelin and aldosterone production which lead to progressive GFR decline, a situation ameliorated by alkali diet. Kidney Int. 2010;78(11):1128–1135. doi: 10.1038/ki.2010.348. [DOI] [PubMed] [Google Scholar]
- Raj S, Scott DR, Nguyen T, Sachs G, Kraut JA. Acid stress increases gene expression of proinflammatory cytokines in Madin-Darby canine kidney cells. Am J Physiol Ren Physiol. 2013;304(1):F41–48. doi: 10.1152/ajprenal.00128.2012. [DOI] [PubMed] [Google Scholar]
- Scialla JJ, Anderson CA. Dietary acid load: a novel nutritional target in chronic kidney disease? Adv Chronic Kidney Dis. 2013;20(2):141–149. doi: 10.1053/j.ackd.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remer T. Influence of diet on acid–base balance. Semin Dial. 2000;13(4):221–226. doi: 10.1046/j.1525-139x.2000.00062.x. [DOI] [PubMed] [Google Scholar]
- Copeland SR. Potential hominin plant foods in northern Tanzania: semi-arid savannas versus savanna chimpanzee sites. J Hum Evol. 2009;57(4):365–378. doi: 10.1016/j.jhevol.2009.06.007. [DOI] [PubMed] [Google Scholar]
- Konner M, Eaton SB. Paleolithic nutrition: twenty-five years later. Nutr Clin Pract. 2010;25(6):594–602. doi: 10.1177/0884533610385702. [DOI] [PubMed] [Google Scholar]
- Sebastian A, Frassetto LA, Sellmeyer DE, Merriam RL, Morris RC Jr. Estimation of the net acid load of the diet of ancestral preagricultural Homo sapiens and their hominid ancestors. Am J Clin Nutr. 2002;76(6):1308–1316. doi: 10.1093/ajcn/76.6.1308. [DOI] [PubMed] [Google Scholar]
- Strohle A, Hahn A, Sebastian A. Estimation of the diet-dependent net acid load in 229 worldwide historically studied hunter-gatherer societies. Am J Clin Nutr. 2010;91(2):406–412. doi: 10.3945/ajcn.2009.28637. [DOI] [PubMed] [Google Scholar]
- Eaton SB, Konner MJ, Cordain L. Diet-dependent acid load, Paleolithic [corrected] nutrition, and evolutionary health promotion. Am J Clin Nutr. 2010;91(2):295–297. doi: 10.3945/ajcn.2009.29058. [DOI] [PubMed] [Google Scholar]
- Smith E, Morowitz HJ. Universality in intermediary metabolism. Proc Natl Acad Sci U S A. 2004;101(36):13168–13173. doi: 10.1073/pnas.0404922101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med. 1985;312(5):283–289. doi: 10.1056/NEJM198501313120505. [DOI] [PubMed] [Google Scholar]
- Cordain L, Eaton SB, Sebastian A, Mann N, Lindeberg S, Watkins BA, O’Keefe JH, Brand-Miller J. Origins and evolution of the Western diet: health implications for the 21st century. Am J Clin Nutr. 2005;81(2):341–354. doi: 10.1093/ajcn.81.2.341. [DOI] [PubMed] [Google Scholar]
- Wesson DE. Dietary acid increases blood and renal cortical acid content in rats. Am J Physiol. 1998;274(1 Pt 2):F97–103. doi: 10.1152/ajprenal.1998.274.1.F97. [DOI] [PubMed] [Google Scholar]
- Wesson DE, Simoni J. Increased tissue acid mediates a progressive decline in the glomerular filtration rate of animals with reduced nephron mass. Kidney Int. 2009;75(9):929–935. doi: 10.1038/ki.2009.6. [DOI] [PubMed] [Google Scholar]
- Wesson DE, Simoni J, Broglio K, Sheather S. Acid retention accompanies reduced GFR in humans and increases plasma levels of endothelin and aldosterone. Am J Physiol Ren physiol. 2011;300(4):F830–837. doi: 10.1152/ajprenal.00587.2010. [DOI] [PubMed] [Google Scholar]
- Amodu A, Abramowitz MK. Dietary Acid, Age, and Serum Bicarbonate Levels among Adults in the United States. Clin J Am Soc Nephrol. 2013;8(12):2034–2042. doi: 10.2215/CJN.03600413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scialla JJ, Appel LJ, Astor BC, Miller ER 3rd, Beddhu S, Woodward M, Parekh RS, Anderson CA. Estimated net endogenous acid production and serum bicarbonate in African Americans with chronic kidney disease. Clin J Am Soc Nephrol. 2011;6(7):1526–1532. doi: 10.2215/CJN.00150111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scialla JJ, Appel LJ, Astor BC, Miller ER 3rd, Beddhu S, Woodward M, Parekh RS, Anderson CA. African American Study of Kidney D, Hypertension Study G. Net endogenous acid production is associated with a faster decline in GFR in African Americans. Kidney Int. 2012;82(1):106–112. doi: 10.1038/ki.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol. 2009;20(9):2075–2084. doi: 10.1681/ASN.2008111205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phisitkul S, Khanna A, Simoni J, Broglio K, Sheather S, Rajab MH, Wesson DE. Amelioration of metabolic acidosis in patients with low GFR reduced kidney endothelin production and kidney injury, and better preserved GFR. Kidney Int. 2010;77(7):617–623. doi: 10.1038/ki.2009.519. [DOI] [PubMed] [Google Scholar]
- Mahajan A, Simoni J, Sheather SJ, Broglio KR, Rajab MH, Wesson DE. Daily oral sodium bicarbonate preserves glomerular filtration rate by slowing its decline in early hypertensive nephropathy. Kidney Int. 2010;78(3):303–309. doi: 10.1038/ki.2010.129. [DOI] [PubMed] [Google Scholar]
- Goraya N, Simoni J, Jo C, Wesson DE. Dietary acid reduction with fruits and vegetables or bicarbonate attenuates kidney injury in patients with a moderately reduced glomerular filtration rate due to hypertensive nephropathy. Kidney Int. 2012;81(1):86–93. doi: 10.1038/ki.2011.313. [DOI] [PubMed] [Google Scholar]
- Goraya N, Simoni J, Jo CH, Wesson DE. A comparison of treating metabolic acidosis in CKD stage 4 hypertensive kidney disease with fruits and vegetables or sodium bicarbonate. Clin J Am Soc Nephrol. 2013;8(3):371–381. doi: 10.2215/CJN.02430312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susantitaphong P, Sewaralthahab K, Balk EM, Jaber BL, Madias NE. Short- and long-term effects of alkali therapy in chronic kidney disease: a systematic review. Am J Nephrol. 2012;35(6):540–547. doi: 10.1159/000339329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correction of Metabolic Acidosis in End Stage Renal Disease. [clinicaltrials.gov/show/ NCT01640119]
- Akali Therapy in Chronic Kidney Disease. [clinicaltrials.gov/show/ NCT01452412]
- Gaggl M, Cejka D, Plischke M, Heinze G, Fraunschiel M, Schmidt A, Horl WH, Sunder-Plassmann G. Effect of oral sodium bicarbonate supplementation on progression of chronic kidney disease in patients with chronic metabolic acidosis: study protocol for a randomized controlled trial (SoBic-Study) Trials. 2013;14(1):196. doi: 10.1186/1745-6215-14-196. [DOI] [PMC free article] [PubMed] [Google Scholar]