

Abstract
Chromatin is vital to normal cells, and its deregulation contributes to a spectrum of human ailments. An emerging concept is that aberrant chromatin regulation culminates in gene expression programs that set the stage for the seemingly diverse pathologies of cancer, developmental disorders and neurological syndromes. However, the mechanisms responsible for such common etiology have been elusive. Recent evidence has implicated lesions affecting chromatin-remodeling proteins in cancer, developmental disorders and neurological syndromes, suggesting a common source for these different pathologies. Here, we focus on the chromodomain helicase DNA binding chromatin-remodeling family and the recent evidence for its deregulation in diverse pathological conditions, providing a new perspective on the underlying mechanisms and their implications for these prevalent human diseases.
Keywords: cancer, CHD proteins, chromatin remodeling, copy number variation, developmental disorders, DNA damage, male infertility, mutation, neurological syndromes
In eukaryotic cells, DNA is packaged into chromatin, the fundamental unit of which is the nucleosome. Nucleosomes contain an octamer consisting of two copies of each core histone protein: H2A, H2B, H3 and H4, around which 146 base pairs of DNA is wrapped, with histone H1 being positioned on the DNA linkers between adjacent nucleosomes. During all DNA template-based cellular processes, chromatin undergoes dynamic remodeling to allow optimal states for efficient transcriptional regulation, DNA replication, DNA recombination and repair, as well as chromosome condensation and segregation. Such dynamic chromatin remodeling is enabled through the interplay of elaborate mechanisms including chromatin remodeling by ATP-dependent chromatin remodelers, covalent histone modifications, exchange of canonical histones with histone variants, DNA methylation and noncoding RNAs. In particular, chromatin remodelers perform critical roles in regulating DNA accessibility and chromatin structure through their ability to mobilize and restructure nucleosomes using the energy derived from ATP hydrolysis. ATP-dependent chromatin remodelers are highly conserved across organisms from yeast to humans, and share an ATPase domain similar to that of the SNF2 (sucrose nonfermenting 2) family of DNA translocases [1]. Based on the presence of additional functional domains, chromatin remodelers are classified into four families: SWI/SNF (switching defective/sucrose non-fermenting), ISWI (imitation SWI), INO80 (inositol requiring 80) and CHD (chromo domain helicase DNA binding) [1]. Members of these chromatin-remodeling families have been shown to play important roles in the regulation of gene transcription, DNA replication, DNA recombination, DNA repair, higher-order chromatin assembly and chromosome segregation, and their perturbations are linked to human diseases including cancer and developmental disorders [1,2]. In this review, we focus specifically on the CHD chromatin remodeler family [3,4], highlighting emerging evidence demonstrating the functional roles of CHD proteins in the pathogenesis of cancer, developmental disorders and neurological syndromes.
The CHD family of chromatin remodelers
The CHD family encompasses nine members (CHD1–9) [3,4] (Figure 1). CHD proteins are highly conserved from yeast to humans, being characterized by a SNF2-like helicase-ATPase domain located in the central region, and signature tandem chromodomains (chromatin organization modifier domains) located in the amino terminal region of the protein [3-6]. The SNF2-like domain confers the ATP-dependent enzymatic activity that enables CHD proteins to mobilize and restructure nucleosomes [4,7-11]. Chromodomains are evolutionarily conserved motifs that mediate interactions with chromatin by binding directly to DNA, RNA or methylated histones [12]. Interestingly, DNA-binding activity by chromodomains is important for the ATPase and transcriptional repression activities of CHD4 [7,13,14]. The two tandem chromodomains of CHD1 co-operate to bind to lysine 4-methylated histone H3 (H3K4me) [15]. The chromodomains of Chd5 directly bind to and are required for maintenance of H3K27me3 on Polycomb group-regulated target genes [16].
Figure 1. The chromodomain helicase DNA binding family of chromatin remodelers.
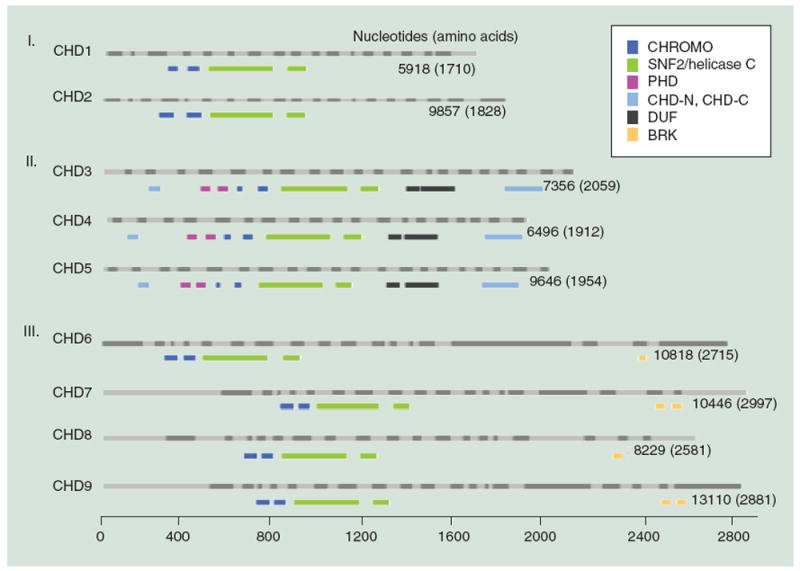
CHD proteins are classified into three subfamilies (Roman numerals) based on their functional motifs (see legend). The human CHD family based on ensemble is drawn to scale, with light and dark gray bars depicting alternating exons (above) and the functional motifs from PFAM (a database of protein families of multiple sequence alignments generated using hidden Markov models) shown in color (below) for each CHD member. The number of nucleotides and amino acid residues for the CHD transcript and protein, respectively, are shown.
BRK: Brahma and Kismet domains; CHD: chromodomain helicase DNA binding; CHROMO: Chromodomain; CHD-N, CHD-C: CHD_N and CHD_C are shown in upstream and downstream region, respectively; DUF: Domain of unknown function; PHD: Plant homeodomain; SNF2/helicase C: SNF2_N and helicase_C are shown in upstream and downstream region, respectively.
Besides these domains that are common to the nine CHD proteins, additional functional domains divide CHD proteins into three subfamilies [3,4] (Figure 1). Subfamily I members (CHD1, CHD2) contain a DNA-binding domain with a preference for binding AT-rich sequences [6]. Subfamily II members (CHD3, CHD4, CHD5) lack a defined DNA-binding domain, but harbor dual plant homeodomain (PHD) zinc finger motifs upstream of the chromodomains. PHD fingers are multifaceted readers of the N-terminal tail of histone H3, including the methylation state of Lys4 (H3K4me2/3 vs. H3K4me0), Arg2 (H3R2me0 vs. H3R2me2), Lys9 (H3K9me3) and Lys36 (H3K36me3), as well as the acetylation state of Lys9 (H3K9ac) and Lys14 (H3K14ac) [17,18]. For example, the two PHD fingers of CHD4 bind to H3 tails with unmodified H3K4 and/or trimethylated H3K9, but not trimethylated H3K4 [19,20]. Furthermore, the PHD fingers are required for the nucleosome remodeling and repressive functions of CHD4 [7,21]. Similarly, our lab discovered that the tandem PHD fingers of CHD5 bind to H3 tails with unmodified H3K4 and this interaction is required for CHD5’s ability to regulate transcription [22]. Interestingly, PHD fingers are also implicated in binding to nonhistone domains such as RNA recognition motif (RRM) and homology domain 1 (HD1) [17]. Subfamily III members (CHD6, CHD7, CHD8, CHD9) contain additional functional motifs in their C-terminal regions, including paired BRK (Brahma and Kismet) domains, a SANT-like (switching-defective protein 3, adaptor 2, nuclear receptor corepressor, transcription factor IIIB) domain, conserved region (CR) motifs and a less-defined DNA-binding domain. SANT domains are suggested to interact primarily with unmodified histone tails, and couple histone binding to ATP catalysis [28], whereas functions of BRK and CR domains are not well understood. The diverse functional domains of CHD proteins suggest that they could have multi-faceted roles as ATP-dependent chromatin remodelers, ‘readers’ of covalent histone modifications and DNA-binding factors, while the specific domains ascribed to different CHD family members could equip distinct CHD proteins with unique biological roles. Numerous studies have revealed roles for CHD proteins in cellular processes such as transcriptional regulation, chromatin assembly, nucleosome remodeling, DNA damage repair, RNA processing, ribosomal RNA biogenesis, cellular proliferation, senescence, apoptosis and differentiation [3,4,29-39]. Increasing evidence also links genomic lesions such as mutations, deletions, translocations and copy number variations (CNVs), in CHD family members to human disease syndromes [3,4].
CHD proteins in cancer
Recent studies have revealed that a number of CHD proteins are potent tumor suppressors and their deficiencies contribute to development of a variety of cancers. Our laboratory identified CHD5 as a tumor suppressor gene mapping to human 1p36 [40], a genomic region frequently deleted in a broad range of cancers including neural, epithelial and hematopoietic malignancies [41]. Chd5 is a tumor suppressor that controls cellular proliferation, senescence and apoptosis by its ability to facilitate transcriptional activation of Cdkn2a, thereby inducing p16/Rb and p19/p53-mediated tumor-suppressive pathways [40]. In addition, Chd5 binds other regions of the genome, showing preferential binding to genomic loci lacking H3K4me3, and its interaction with H3 is crucial for Chd5-mediated transcriptional modulation of genes implicated in cancer, cell cycle control and chromatin regulation [22]. Chd5 binds unmodified H3 via its dual PHDs [22,42]. Mutation of specific conserved residues within the PHDs of Chd5 abrogate the Chd5-H3 interaction, and in contrast to wild-type Chd5, H3-binding defective Chd5 mutants are not able to suppress proliferation or to induce differentiation in cultured cells, and hence are not able to inhibit tumor growth in vivo [22]. In addition, the chromodomains of CHD5 bind H3 tails that are trimethylated at Lys27 (H3K27me3) and the interaction is required for maintenance of H3K27me3 at polycomb-repressive genes required for neuronal differentiation [16]. These findings suggest that CHD5 is capable of interacting with nucleosomes through multiple structural domains with preference for specific histone modifications and potentially via combinatorial recognition of multiple histone modifications.
Since the time that CHD5 was discovered as a tumor suppressor [40], a flurry of studies have reported tumor-suppressive functions for CHD5 and revealed a high frequency of CHD5 lesions (including compromised expression, promoter hypermethylation, mutation and/or deletion) in diverse cancers including neuroblastoma [43-47], glioma [40,48,49], breast cancer [48,50], lung cancer [51], ovarian cancer [52], gastric cancer [53], gallbladder carcinoma [54], colorectal cancer [48,55,56], hepatocellular carcinoma [57], melanoma [58], leukemia [59] and laryngeal squamous cell carcinoma [60]. A number of studies have also defined robust CHD5 expression as an independent prognostic marker for better survival for patients with neuroblastoma [44], glioma [49], ovarian cancer [52] and gallbladder carcinoma [54]. Notably, CHD5 expression has been primarily detected in brain and testis to date [47,61-65]. It is thus intriguing that CHD5 seems to exert tumor-suppressive functions in cell types where it is not normally detected. It is possible that Chd5 affects cell fate choices, such that its perturbation leads to unbridled expansion of a cell type in which Chd5 is not expressed, as recently proposed [66]. Another possibility is that Chd5 expression is induced in response to DNA-damage, but is not normally detected in those tissues, and therefore its loss leads to increased DNA damage and eventually tumorigenesis. In support of this idea, we found that DNA-damaging agents induce Chd5 expression [64]. Another possibility is that Chd5 in an ‘expressing’ cell type has paracrine effects on cell types in which it is not expressed, for example, due to its ability to modulate cancer pathways and/or the DNA damage response [22,64]. Thus, whereas it is clear that CHD5 is a potent tumor suppressor, a better understanding of how it works in different cellular contexts and how its perturbation leads to diverse cancers is needed.
While CHD5 was the first CHD protein to be implicated in cancer [40], tumor-suppressive roles for additional CHD members have also been found. For the most part, these studies have been based on sequencing of human tumors. Kim et al. examined mutational status of CHD1, CHD2, CHD3, CHD4, CHD7, CHD8 and CHD9 in 28 gastric cancers (GCs) with high microsatellite instability (MSI-H), 45 GCs with low MSI (MSI-L), 35 colorectal cancers (CRCs) with MSI-H and 45 CRCs with MSI-L and assessed CHD4 and CHD8 expression in these tumors by immunohistochemistry. CHD1, CHD2, CHD3, CHD4, CHD7, CHD8 and CHD9 mutations were, respectively, found in 7.9% (5/63), 30.2% (19/63), 4.8% (3/63), 7.9% (5/63), 11.1% (7/63), 15.9% (10/63) and 11.1% (7/63) of the high MSI tumors (GC and CRC combined) [67]. Loss of CHD4 expression was observed in 56.4% of GCs and 55.7% of CRCs, and loss of CHD8 was observed in 35.7% of GCs and 28.6% of CRCs [67]. These findings suggest that mutations and deficiency of CHD proteins may be common in cancers of the stomach and colon, and that their disruption could contribute to tumorigenesis. Increasing genetic and functional evidence for individual CHD family members provides further support for this idea.
In addition to the frequent frameshift mutations of CHD7 and CHD8 in cancers of the stomach and colon discovered by Kim et al. [67], Tahara et al. also found that mutations of CHD7 and CHD8 occur in 42% (18/42) of CRCs with CpG island methylator phenotype 1, referred to as CIMP1 CRCs, making CHD7 and CHD8 the most commonly mutated genes in the study [68]. CHD7 and CHD8 mutations are also found in 9% (3/34) CIMP2 CRCs and 6% (2/34) CIMP-negative CRCs [68]. By analyzing expression of CHD8 in 101 cases of GCs and corresponding noncancerous tissues, Sawada et al. found that CHD8 expression is significantly compromised in GCs, and is an independent prognostic marker for biological aggressiveness of this tumor type [69]. Through gene set enrichment analysis, low CHD8 expression was found to be significantly associated with components of the WNT/β-catenin signaling pathway and with cell cycle regulators, and CHD8 knockdown in gastric cancer cells promotes proliferation [69]. These reports are consistent with previous studies showing that CHD8 inhibits β-catenin-mediated gene expression through its direct interaction with β-catenin [9], and cyclin E2 expression through its interaction with RNA polymerase II during transcriptional elongation [70]. These genomic and functional findings indicate a tumor-suppressive role for CHD8 in gastric cancer, and implicate both CHD8 and CHD7 in CRCs.
In addition, Colbert et al. reported that CHD7 expression predicts favorable survival in patients with resected pancreatic ductal adenocarcinoma (PDAC) [71]. CHD7 depletion sensitizes PDAC cells to gemcitabine treatment and delays their growth in tumor xenografts. Immunohistochemical analysis of specimens from 59 patients with resected PDAC receiving adjuvant gemcitabine revealed that low CHD7 expression was associated with increased recurrence-free survival and overall survival [71]. In addition, Pleasance et al. identified rearrangements of the CHD7 locus in three small-cell lung cancer cell lines, two carrying a PVT1-CHD7 fusion gene in the setting of MYC amplification and one carrying a tandem duplication of exons 3–8 of CHD7 [72,73]. These data imply roles for CHD7 in pancreatic and lung cancers.
A number of studies also identified CHD1 as a tumor suppressor mapping to human 5q21, a region frequently deleted in prostate cancer [74-76]. Berger et al. identified splice site mutations and intragenic breakpoints of CHD1 in 42.9% (3/7) of primary prostate tumors analyzed by whole-genome sequencing [77]. Grasso et al. identified focal deletions/mutations of CHD1 in 8% (10/119) of prostate primary tumors analyzed through exome sequencing and array comparative genomic hybridization analysis [78]. Burkhardt et al. found that CHD1 is required for efficient recruitment of the androgen receptor (AR) to promoters of hormone-responsive genes, and that CHD1 regulates expression of AR-responsive tumor suppressor genes, including NKX3–1 (NK3 homeobox 1), FOXO1 (Fork-head box 01) and PPAR-γ [74]. Interestingly, Huang et al. and Liu et al. independently revealed that depletion of CHD1 in prostate cancer cells increases cell invasiveness without altering cellular proliferation [75,76]. Together, these findings strongly indicate tumor-suppressive functions for CHD1 in the prostate and suggest that CHD1 deficiency may contribute to prostate cancer metastasis.
Increasing evidence also reveals important roles for CHD4 in serous tumors and other cancers. By exon sequencing of 52 primary serous endometrial tumors, Le Gallo et al. identified somatic mutations of CHD4 in 17% of the samples, significantly higher than the background mutation rate (q ≤0.0353) [79]. Zhao et al. also identified somatic missense mutations in CHD4, many of which appear to impair CHD4 function, in 27.3% (6/22) uterine serous carcinomas analyzed, which was a significant increase in mutation burden than expected (p < 2.4 × 10-6) [80]. In addition, copy number gain and loss of the region of human chromosome 12 that harbors CHD4 are found in 28% (7/25) and 12% (3/25), respectively, of serous carcinomas [80]. These genomic data provide strong support for a role of CHD4 perturbation in serous tumor development. Functionally, a number of studies have indicated that CHD4 is a key regulator of the DNA-damage response and genome maintenance [29,31,32,81], suggesting that CHD4 is critical for genomic integrity and that its disruption contributes to tumorigenesis. In addition, CHD3 and CHD4 are catalytic components of the NuRD (nucleosome remodeling histone deacetylase) complex, which has been associated with tumorigenesis, epithelial-to-mesenchymal transition and metastasis [82]. For example, CHD4 and CHD3 bind ZGPAT (zinc finger, CCCH-type with G patch domain), a transcriptional repressor of genes encoding modulators of cellular proliferation, survival and migration, thereby inhibiting breast cancer development [83].
Some studies suggest roles for CHD6 in CRC and bladder cancer. Mouradov et al. identified CHD6 mutations in 19.5% (8/41) of CRC cell lines studied [84]. Hassan et al. performed CNV profiling of 64 paired CRC-normal specimens from the same patient, and revealed significant gains of 20q12—a chromosomal region that encompasses CHD6 and seven other genes—in 45.31% of the tumors [85]. In addition, Gui and colleagues detected missense mutations in CHD6 in 7% (7/97) of transitional cell bladder carcinomas [86]. These genomic findings implicate a role for CHD6 in colorectal cancer and bladder cancer, although additional functional data are required to support this hypothesis. Similarly, genomic data for the prevalence of CHD2 lesions in human cancer are currently limited. However, deficiency of Chd2 in mice has been shown to lead to lymphomagenesis [35]. Chd2 mutant cells accumulate higher levels of γ-H2AX and exhibit a defect in repair of DNA damage after γ-irradiation, implying a role for Chd2 in the DNA damage response and maintenance of genomic stability [35]. In addition, cystic endometrial hyperplasia occurs in female mice that are heterozygous for a different Chd2 mutant allele [87].
Together, this accumulating body of literature indicates important roles for CHD family members in a broad range of cancers. Particularly compelling evidence has been shown for the tumor-suppressive roles of CHD5 in a variety of cancers, CHD1 in prostate cancer, CHD4 in serous cancer and CHD8 in gastric cancer. Multiple CHD proteins including CHD2, CHD4, CHD5, CHD6, CHD7 and CHD8 are also implicated in tumorigenesis, particularly in CRCs. These findings imply that perturbation of CHD-mediated chromatin remodeling is a common theme in tumorigenesis, while individual CHD proteins could exert particular functions in specific cancer types. On the other hand, significant variations in mutation frequencies of CHD genes have been reported in different studies, which will require further analyses to clarify. More functional data are also needed to further demonstrate and elucidate the roles of CHD proteins such as CHD6 in cancer development.
CHD proteins in neurological syndromes
Increasing evidence reveals important roles for CHD proteins in neurological syndromes. A battery of independent studies discovered 13 de novo mutations of CHD8 in patients with autism spectrum disorder (ASD) and consistently identified CHD8 as one of the most significant risk genes for autism [88-91]. In addition, McCarthy et al. reported de novo mutations in CHD8 in patients with schizophrenia through whole-exome sequencing of 57 parent–parent–offspring trios [92]. In addition to these recurrent CHD8 mutations, lesions in other CHDs (CHD1, CHD2, CHD3, CHD5 and CHD7) were identified in the same studies [88-91]. Of particular note, mutations in CHD7 cause CHARGE syndrome, a genetic syndrome characterized by a constellation of developmental defects that often includes behavioral abnormalities [93-98]. A large percentage (28–42%) of CHARGE syndrome patients are reported to have autism-like behaviors [99-102], suggesting a role for CHD7 in the pathogenesis of ASD. Jiang and colleagues identified an inherited missense mutation in CHD7 in a family with autism [103]. In addition, Pinto et al. reported distinct de novo deletions within CHD2 in two brothers with ASD, one with deletion of the first six exons of CHD2, and the other with an 83 kbp deletion within CHD2 [104]. In addition, a de novo missense mutation in CHD2 was reported in ASD [89], and a 15q26.1 microdeletion encompassing CHD2 and the flanking gene RGMA (repulsive guidance family molecule a) was reported in a patient with autism-like behavior and epilepsy [105]. Together, these findings suggest that disruption in genes encoding CHD chromatin remodelers such as CHD2, CHD7 and CHD8 play important roles in the pathogenesis of ASD.
An increasing body of evidence also strongly implicates critical roles for CHD2 in epileptic encephalopathy. Targeted sequencing of 46 candidate genes in 500 individuals affected with epileptic encephalopathy—a severe form of epilepsy beginning in infancy—revealed six mutations in CHD2, representing 1.2% of the subjects and identifying CHD2 as the most highly mutated gene [106]. Suls et al. performed whole-exome sequencing of proband–parent trios that included nine patients with Dravet syndrome, a severe form of epilepsy, and identified de novo mutations in CHD2 in 33% (3/9) of the individuals [107]. The authors also showed that knockdown of chd2 in zebrafish caused altered locomotor activity and epileptiform discharges similar to seizures described in affected persons [107]. Lund et al. found mutations in CHD2 in 9% (2/22) of patients with Lennox–Gastaut syndrome, an epileptic encephalopathy with a heterogeneous etiology [108]. De novo mutations in CHD2 in Lennox–Gastaut syndrome were found by exome sequencing of 109 probands and their parents [109]. In addition, a de novo CHD2 frame shift mutation was reported in a patient with intellectual disability [110]. These findings indicate that CHD2 is a risk gene for epileptic encephalopathy, and suggests that CHD2 perturbation may contribute to other neurological disorders as well.
Preliminary evidence also suggests potential roles for several other CHD family members in neurological dysfunctions. For example, several human ataxia syndromes have CNVs on chromosome 20q12 that encompass CHD6 [112-114], and deletion of exon 12 of Chd6 in mice impairs motor co-ordination [115]. A de novo balanced (4; 20)(q33; q12) translocation that disrupts CHD6 and compromises its expression was also reported in a patient with severe mental retardation [111]. In addition, deletion of 1p36.32-p36.22, a chromosome region encompassing CHD5 was reported in patients with developmental delay and intellectual disability [117,118]. We found that Chd5-compromised mice have severe behavioral phenotypes and aberrant dendritic arborization (Mills, unpublished). A de novo mutation in CHD4 was reported in 1/109 patients with epileptic encephalopathy [109], and conditional disruption of Chd4 specifically within Schwann cells leads to abnormal motor co-ordination and hind limb reflexes [116]. While more supporting human genetic data are needed, these functional data in model organisms and preliminary reports in humans suggest that perturbation of CHD4, CHD5 and CHD6 might also play important roles in neurological dysfunction.
Taken together, compelling evidence has established critical roles for CHD2, CHD7 and CHD8 in a number of neurological disorders, in particular ASD and epileptic encephalopathy, while preliminary data implicate roles for CHD4, CHD5 and CHD6 in neurological dysfunction, suggesting common roles for perturbation of CHD chromatin remodelers in the pathogenesis of neurological diseases.
CHD proteins in developmental disorders
CHD proteins are also implicated in development. Most notably, lesions in CHD7 cause CHARGE syndrome, a developmental syndrome named by its constellation of birth defects including ocular coloboma, congenital heart defects, choanal atresia, retardation of growth and development, genital hypoplasia and ear anomalies associated with deafness [93-98]. The diverse developmental anomalies that clinically define CHARGE syndrome suggest that CHD7 plays pleiotropic roles in development, consistent with the fact that CHD7 is expressed in a wide variety of tissues during embryogenesis and in the adult [98,119]. CHD7 binds to H3K4me3 at enhancer regions of numerous genes, and is bound to these regulatory elements in a cell type and stage-specific manner [120]. CHD7 is implicated in neurogenesis, ear morphogenesis, osteoblast formation and somitogenesis [119,121-123]. Nucleosome remodeling is believed to be a key function for CHD7 during development, as CHARGE syndrome mutations impair CHD7’s chromatin-remodeling activity [8]. In addition to CHARGE syndrome, polymorphisms within CHD7 are associated with susceptibility to idiopathic scoliosis, a progressive spine deformity that is often deadly [124].
In addition to CHD7, many studies imply diverse roles for other CHD members in development. Disruption of Chd2 using a gene trap strategy leads to neonatal lethality in homozygous mice and partial lethality in heterozygotes [87]. Heterozygous mice that survive have developmental delay and are runted as adults, with extremely hypoplastic subcutaneous fat and pronounced lordokyphosis by 4 months of age [87]. A small proportion (2/15) of heterozygous mice have eye defects [87]. A distinct Chd2 mutant mouse model with a mutation that abrogates the DNA-binding activity of Chd2 has delayed embryogenesis and perinatal lethality [125,126]. Surviving heterozygous mice have an enhanced susceptibility to damage in many primary organs, in particular the kidney and heart [125,126]. The diverse developmental defects exhibited by Chd2-deficient mice indicate that Chd2 plays important roles in the development of multiple tissues and organs. Human developmental disorders associated with CHD2 dysfunction have not yet been reported, however, a CHD2 breakpoint mutation was found in a patient with scoliosis, consistent with the lordokyphosis phenotype observed in Chd2-deficient mice [87]. In addition, Qin et al. reported an association between a genetic variant of CHD2 and nonobstructive azoospermia [127]. We and others recently discovered that Chd5 is critical for sperm development [64,65]. We defined Chd5 as a master regulator of the histone-to-protamine replacement process during the post-meiotic phase of sperm development [64]. Inactivation of Chd5 in mice results in deficient histone acetylation, nucleosome eviction and increased DNA damage during spermatid maturation, leading to deficient sperm production and defective sperm function, culminating in male infertility [64]. Consistent with the compromised fertility of Chd5-deficient mice, we also found that low CHD5 expression is correlated with low human male fertility [64]. In addition, both our lab and others discovered that Chd5 is induced during neuronal development and is required for neurogenesis [16,63]. CHD5 binds a large cohort of genes and is required for the maintenance of H3K27me3 at these loci and activation of polycomb-repressed neuronal genes [16]. In addtion, Chd4 is essential for polycomb-mediated inhibition of astroglial differentiation through its interaction with Ezh2 and suppression of genes that induce the astrogenic lineage [128]. Chd4 also interacts with Nab corepressors to direct Schwann cell mediated peripheral nerve myelination, and conditional ablation of Chd4 in Schwann cells leads to delayed myelination, radial sorting defects, hypomyelination and deregulation of promyelinating Schwann cells [116]. In addition, Chd4 is required for expression of CD4 (CD4 molecule) and T-cell development [129], and CHD4 forms a complex with GATA3 (GATA-binding protein 3) to simultaneously activate transcription of Th2 cytokines and to repress transcription of the Th1 cytokine IFNG (interferon, γ) to establish T helper 2 cell identity [130]. Chd4 is also required for the establishment of basal keratinocytes within the epidermis of the skin and for the morphogenesis of hair follicles [131]. Consistent with Chd4’s role in skin development, autoantibodies against CHD3 and CHD4 are detected in 10–30% of patients with dermatomyositis [132]. Also, Chd8 is essential for mouse embryogenesis, as Chd8−/− embryos manifest growth retardation at embryonic day 5.5 (E5.5) and developmental arrest accompanied by massive apoptosis at E7.5 [133]. Furthermore, Chd8 recruits histone H1 to suppress p53-mediated apoptosis during early embryogenesis [134]. The Xenopus laevis CHD8 ortholog Duplin encodes a negative regulator of Wnt/β-catenin signaling that inhibits axis formation during embryonic development [135]. Injection of mRNA encoding Duplin into the dorsal axis of the developing embryo leads to defective head development [135]. Mutation of Chd1 causes abnormal wing development, male infertility and female subfertility in Drosophila melanogaster, indicating a multifaceted role for Chd1 during embryogenesis [136]. Chd1 plays a crucial role during ovarian follicle development in the silkmoth Bombyx mori, as it specifically binds and repositions nucleosomes located near promoters of genes encoding chorion proteins, allowing binding of CEBP (CCAAT/enhancer-binding protein) and TBP (TATA box-binding protein) and initiation of transcription [137]. In mouse, Chd1 is essential for maintaining an open chromatin conformation and for pluripotency of embryonic stem cells [138]. Altogether, these findings indicate that CHD proteins are widely implicated in developmental processes. Although human developmental disorders reported to be associated with lesions of CHD genes are currently limited (with the exception of CHD7), the diverse roles demonstrated in development of mouse and other organisms suggest that dysfunction of CHD proteins could also bear severe consequences that contribute to a range of developmental anomalies and syndromes.
Connections
The roles of CHD proteins in cancer, neurological syndromes and developmental disorders not only suggests diverse functions for these chromatin remodelers, but also indicates shared risks and interconnections between the pathogenesis of these major human diseases. Increasing clinical and epidemiologic studies also suggest links between cancer, neurological diseases and developmental disorders. For example, Shavelle et al. analyzed the cause of death for 13,111 patients with autism that had been followed between 1983 and 1997, and found that there was a higher standardized mortality from cancer, ranging from 1.9 in subjects with no or mild intellectual disability, to 2.9 in subjects with moderate, severe or profound intellectual disability, although the absolute number of deaths in both categories was only 6 and 15, respectively [139]. Kao et al. analyzed the prevalence of autism in cancer subjects that were diagnosed in the United States, and found significant correlations between the prevalence of autism and the incidence of in situ breast cancer, but no correlation with the other types of cancers analyzed [140]. Although more studies are needed to validate these findings and to exclude other factors such as environmental effects, these epidemiologic studies suggest a link between certain forms of cancer and autism.
Similarly, a recent study analyzed the medical records of 2,238 infertile men, 451 of whom had azoospermia (i.e., lack of sperm), and found that infertile men were 1.7-times more likely to develop cancer than the general population [141]. Furthermore, men with azoospermia had a 2.9-fold increased risk of developing cancer. This finding is in agreement with a previous study reporting a significant association between subfertility and increased risk of testicular cancer using meta-analysis of seven case–control studies that included 4,954 participants [142], suggesting a common etiology between cancer and male infertility. In line with this study, we discovered that deficiency of Chd5 leads to male infertility, cancer and behavioral anomalies, at least in mice [40,64]. We defined CHD5 as a tumor suppressor mapping to human 1p36 [40], and a trove of later studies identified lesions in CHD5 and demonstrated tumor-suppressive functions for CHD5 that when compromised lead to a variety of human cancers [43-46,48-55,58,60,143]. We discovered that CHD5 deficiency also correlates with male infertility in humans [64]. Together, these findings define perturbation of CHD5-mediated chromatin regulation as one potential common etiology for the three types of diseases. Similarly, CHD8 is both a risk gene for ASD [88-91] and an independent prognostic factor for gastric cancer [69], while it is also essential for embryonic development including axis formation and head development [134,135]. CHD8 negatively regulates WNT/β-catenin signaling [135], which could be a common pathway affected by CHD8 lesions in pathogenesis of all these diseases, as WNT/β-catenin signaling is implicated in neurological syndromes, tumorigenesis and development [144-146]. However, CHD8 also has WNT/β-catenin-independent functions [134], through which CHD8 could affect pathogenesis of the three disease categories. CHD7 mutations are the main cause of CHARGE syndrome, a disease characterized by an array of developmental defects [93-98], with 28–42% of CHARGE syndrome patients having autism-like behaviors [99-102]. CHD7 mutations are also found in ASD patients [103]. In addition, CHD7 is highly mutated in CRCs with the CpG island methylator phenotype 1 [68] and is rearranged in small-cell lung cancer cell lines [72,73]. CHD7 expression also predicts a favorable survival outcome for patients with resected PDAC [71]. These observations indicate that CHD7 is involved in the pathogenesis of developmental and neurological syndromes as well as cancer. Similarly, CHD2 is implicated in pathogenesis of epilepsy and ASD [88-91,104-110], lymphoid cancers [35], as well as being essential for the development of the axial skeleton, kidney and other organs [87,126], suggesting a common role for CHD2 lesions in pathogenesis of these diseases. CHD4 is reported to be among the most frequently mutated gene in serous tumors [79,80], and is also essential for astroglial differentiation [128], T-cell development [129] and skin development [131]. Autoantibodies against CHD4 are detected in 10–30% of human patients with dermatomyositis [132]. While human genetic data implicating CHD4 alterations in neurologic diseases are currently limited, ablation of Chd4 in Schwann cells leads to abnormal motor co-ordination and hind limb reflexes [116], suggesting that CHD4 could affect development of all three types of diseases. Altogether, these findings define deregulation of CHD proteins as a common culprit for three main categories of human diseases, and also indicate shared mechanisms underlying the pathogenesis for the different disease syndromes. In addition to CHD proteins, mutations in other chromatin remodelers, such as BAF250A, BAF250B, BRG1 and BRM, have also been identified and implicated in the pathogenesis of both cancer and neurodevelopmental syndromes, as recently reviewed by Ronan et al. [147], suggesting perturbation of chromatin remodeling as a common pathogenic mechanism.
At the molecular level, how could chromatin-remodeling proteins, more specifically CHD proteins, impact pathogenesis that impinges upon cancer, neurological syndromes and developmental disorders? These human ailments are each characterized by genomic lesions, including mutations and CNVs. Several CHD proteins play critical roles for the DNA damage response and for maintaining genome integrity [29-35]. For example, CHD4 is a critical factor that co-ordinates both checkpoint signaling and repair of DNA damage [29-32]. Depletion of CHD4 disrupts the DNA damage response at the chromatin level, leading to an increase in DNA breaks and rampant genomic instability [29-32]. CHD2 is also important for mediating DNA repair [34,35], and Chd2-deficient cells have a compromise in repair of DNA damage after ionizing or ultraviolet irradiation [34,35]. We also found that Chd5 is required for an efficient DNA damage response in the male germ line and that DNA damage induces Chd5 expression [64]. Given the common roles for CHD proteins in repair of DNA damage and maintenance of genomic stability [29-35] and their shared functions in modulating chromatin structure and gene expression programs in diverse cellular processes [1], we speculate that a compromise in CHD proteins renders chromatin more susceptible to intrinsic and/or extrinsic damage, resulting in a higher extent of genomic lesions. The functional genomic elements jeopardized by these perturbations could have versatile roles in regulating cellular proliferation, tissue development and neural function, thereby culminating in cancer, developmental disorders and neurological syndromes in the same subject. Depending on the specific combination of functional genomic elements jeopardized by these lesions and the compounding consequences, these perturbations could also lead to a specific type of disease or make one disease more pronounced than the others.
Conclusions & future perspectives
The common implications for CHD proteins in cancer, neurological syndromes and developmental disorders and the potential underlying molecular mechanisms linking the pathogenesis of these diseases, exemplify the interconnections between these diseases and the converging roles of chromatin-remodeling proteins in the pathogenesis of multiple diseases. We expect future studies to add increasing evidence for the interconnections between cancer, neurological syndromes and developmental disorders, and to provide a clearer understanding of the underlying molecular mechanisms. Such insight will be valuable for developing better diagnosis, treatment and management for patients affected by these prevalent diseases (Table 1).
Table 1.
Chromo domain helicase DNA binding (CHD) proteins and human disease.
| Gene | Cancer | Developmental diseases | Neurologic diseases |
|---|---|---|---|
| CHD1 | Prostate cancer [74-76], gastric cancer [67], colorectal cancer [67] | Abnormal wing development, male infertility and female subfertility in Drosophila melanogaster [136] ovarian follicle development in the silkmoth Bombyx mori [137] | Autism spectrum disorder [89] |
| CHD2 | Lymphoma [35], cystic endometrial hyperplasia [87] | Scoliosis [87], embryogenesis, growth retardation, kidney, heart, eye and fatty tissue development in mouse [87,125,126], myogenesis [148] | Autism spectrum disorder [89,104,105], epilepsy [104-109], intellectual disability [110] |
| CHD3 | Prostate cancer [149], breast cancer [83] gastric cancer [67], colorectal cancer [67] | Autism spectrum disorder [91] | |
| CHD4 | Serous endometrial tumors [79,80], prostate cancer [149], breast cancer [83] | Astrogenic differentiation [128], Schwann cell-mediated peripheral nerve myelination [116], T-cell development [129], skin development and morphogenesis of hair follicles [131], dermatomyositis [132] | Epileptic encephalopathy [109], abnormal motor co-ordination and hind limb reflexes [116] |
| CHD5 | Neuroblastoma [43-47], glioma [40,48,49], breast cancer [48,50], lung cancer [51], ovarian cancer [52], gastric cancer [53], gallbladder carcinoma [54], colorectal cancer [48,55,56], hepatocellular carcinoma [57], melanoma [58], leukemia [59], squamous cell carcinoma [60] | Sperm development [64,65], neurogenesis [16,63] | Intellectual disability [117,118], hyperactivity and repetitive behaviors (Mills, unpublished) |
| CHD6 | Colorectal cancer [84,85], bladder carcinoma [86] | Mental retardation [111], ataxia syndromes [112-115], | |
| CHD7 | Colorectal cancer [67,68], pancreatic ductal adenocarcinoma [71], small-cell lung cancer [72,73] | CHARGE syndrome [93-98] scoliosis [124] neurogenesis, ear morphogenesis, osteoblast formation and somitogenesis [119,121-123] | Autism spectrum disorder [91,103,99-102] |
| CHD8 | Colorectal cancer [67,68], gastric cancer [69] | Embryogenesis [133] head development [135] | Autism spectrum disorders [88-91], schizophrenia [92] |
Executive summary.
Chromatin-remodeling proteins are classified into four families (SWI/SNF, ISWI, INO80 and chromodomain helicase DNA binding [CHD]) and play important roles in diverse molecular and cellular processes.
Nine CHD chromatin-remodeling proteins (CHD1–9) are divided into three subfamilies based on their distinct functional domains; these proteins share common ATPase domains and tandem chromodomains and are implicated in a number of biological processes and human diseases.
Increasing evidence identifies important roles for CHD proteins in cancer and demonstrates CHD proteins as useful for predicting patient survival.
Emerging evidence is establishing lesions in CHD genes as top risk factors for neurological diseases such as autism spectrum disorder and epileptic encephalopathy.
CHD proteins are essential for diverse developmental processes, and mutations in CHD genes can lead to severe developmental disorders (e.g., CHD7 mutations in CHARGE syndrome).
The common roles for CHD perturbation in cancer, neurological diseases and developmental disorders indicate shared etiology among these diseases, which is supported by increasing epidemiological evidence.
Critical roles of CHD proteins in the DNA damage response and the maintenance of genome integrity may underlie the pathogenesis of cancer, neurological diseases and developmental disorders.
Future studies will add increasing evidence for the interconnections between cancer, neurological diseases and developmental disorders and provide a clearer understanding for the molecular mechanisms that are responsible.
Acknowledgments
Work discussed in this review was supported in part by NIH awards R01CA190997 and R21OD018332 to AAM.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
-
•
of interest;
-
••
of considerable interest
- 1.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 2.Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, part II: ATP-dependent chromatin remodeling. Trends Mol Med. 2007;13(9):373–380. doi: 10.1016/j.molmed.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marfella CG, Imbalzano AN. The Chd family of chromatin remodelers. Mutat Res. 2007;618(1–2):30–40. doi: 10.1016/j.mrfmmm.2006.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4•.Hall JA, Georgel PT. CHD proteins: a diverse family with strong ties. Biochem Cell Biol. 2007;85(4):463–476. doi: 10.1139/O07-063. This review provides a detailed summary of the domain structure and molecular functions of chromodomain helicase DNA binding (CHD) proteins. [DOI] [PubMed] [Google Scholar]
- 5.Woodage T, Basrai MA, Baxevanis AD, Hieter P, Collins FS. Characterization of the CHD family of proteins. Proc Natl Acad Sci USA. 1997;94(21):11472–11477. doi: 10.1073/pnas.94.21.11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delmas V, Stokes DG, Perry RP. A mammalian DNA-binding protein that contains a chromodomain and an SNF2/SWI2-like helicase domain. Proc Natl Acad Sci USA. 1993;90(6):2414–2418. doi: 10.1073/pnas.90.6.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Watson AA, Mahajan P, Mertens HD, et al. The PHD and chromo domains regulate the ATPase activity of the human chromatin remodeler CHD4. J Mol Biol. 2012;422(1):3–17. doi: 10.1016/j.jmb.2012.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bouazoune K, Kingston RE. Chromatin remodeling by the CHD7 protein is impaired by mutations that cause human developmental disorders. Proc Natl Acad Sci USA. 2012;109(47):19238–19243. doi: 10.1073/pnas.1213825109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson BA, Tremblay V, Lin G, Bochar DA. CHD8 is an ATP-dependent chromatin remodeling factor that regulates beta-catenin target genes. Mol Cell Biol. 2008;28(12):3894–3904. doi: 10.1128/MCB.00322-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lutz T, Stoger R, Nieto A. CHD6 is a DNA-dependent ATPase and localizes at nuclear sites of mRNA synthesis. FEBS Lett. 2006;580(25):5851–5857. doi: 10.1016/j.febslet.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 11.Lusser A, Urwin DL, Kadonaga JT. Distinct activities of CHD1 and ACF in ATP-dependent chromatin assembly. Nat Struct Mol Biol. 2005;12(2):160–166. doi: 10.1038/nsmb884. [DOI] [PubMed] [Google Scholar]
- 12.Eissenberg JC. Structural biology of the chromodomain: form and function. Gene. 2012;496(2):69–78. doi: 10.1016/j.gene.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 13.Bouazoune K, Mitterweger A, Langst G, et al. The dMi-2 chromodomains are DNA binding modules important for ATP-dependent nucleosome mobilization. EMBO J. 2002;21(10):2430–2440. doi: 10.1093/emboj/21.10.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramirez J, Dege C, Kutateladze TG, Hagman J. MBD2 and multiple domains of CHD4 are required for transcriptional repression by Mi-2/NuRD complexes. Mol Cell Biol. 2012;32(24):5078–5088. doi: 10.1128/MCB.00819-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flanagan JF, Mi LZ, Chruszcz M, et al. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438(7071):1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- 16.Egan CM, Nyman U, Skotte J, et al. CHD5 is required for neurogenesis and has a dual role in facilitating gene expression and polycomb gene repression. Dev Cell. 2013;26(3):223–236. doi: 10.1016/j.devcel.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 17.Musselman CA, Kutateladze TG. Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res. 2011;39(21):9061–9071. doi: 10.1093/nar/gkr613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanchez R, Zhou MM. The PHD finger: a versatile epigenome reader. Trends Biochem Sci. 2011;36(7):364–372. doi: 10.1016/j.tibs.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mansfield RE, Musselman CA, Kwan AH, et al. Plant homeodomain (PHD) fingers of CHD4 are histone H3-binding modules with preference for unmodified H3K4 and methylated H3K9. J Biol Chem. 2011;286(13):11779–11791. doi: 10.1074/jbc.M110.208207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Musselman CA, Mansfield RE, Garske AL, et al. Binding of the CHD4 PHD2 finger to histone H3 is modulated by covalent modifications. Biochem J. 2009;423(2):179–187. doi: 10.1042/BJ20090870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Musselman CA, Ramirez J, Sims JK, et al. Bivalent recognition of nucleosomes by the tandem PHD fingers of the CHD4 ATPase is required for CHD4-mediated repression. Proc Natl Acad Sci USA. 2012;109(3):787–792. doi: 10.1073/pnas.1113655109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paul S, Kuo A, Schalch T, et al. Chd5 requires PHD-mediated histone 3 binding for tumor suppression. Cell Rep. 2013;3(1):92–102. doi: 10.1016/j.celrep.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ooi SKT, Qiu C, Bernstein E, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448(7154):714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li HT, Ilin S, Wang WK, et al. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442(7098):91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pena PV, Davrazou F, Shi XB, et al. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442(7098):100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wysocka J, Swigut T, Xiao H, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442(7098):86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 27.Lan F, Collins RE, De Cegli R, et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448(7154):718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyer LA, Latek RR, Peterson CL. The SANT domain: a unique histone-tail-binding module? Nat Rev Mol Cell Biol. 2004;5(2):158–163. doi: 10.1038/nrm1314. [DOI] [PubMed] [Google Scholar]
- 29.Urquhart AJ, Gatei M, Richard DJ, Khanna KK. ATM mediated phosphorylation of CHD4 contributes to genome maintenance. Genome Integr. 2011;2(1):1. doi: 10.1186/2041-9414-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’shaughnessy A, Hendrich B. CHD4 in the DNA-damage response and cell cycle progression: not so NuRDy now. Biochem Soc Trans. 2013;41(3):777–782. doi: 10.1042/BST20130027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larsen DH, Poinsignon C, Gudjonsson T, et al. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J Cell Biol. 2010;190(5):731–740. doi: 10.1083/jcb.200912135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan MR, Hsieh HJ, Dai H, et al. Chromodomain helicase DNA-binding protein 4 (CHD4) regulates homologous recombination DNA repair, and its deficiency sensitizes cells to poly(ADP-ribose) polymerase (PARP) inhibitor treatment. J Biol Chem. 2012;287(9):6764–6772. doi: 10.1074/jbc.M111.287037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goodarzi AA, Kurka T, Jeggo PA. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat Struct Mol Biol. 2011;18(7):831–839. doi: 10.1038/nsmb.2077. [DOI] [PubMed] [Google Scholar]
- 34.Rajagopalan S, Nepa J, Venkatachalam S. Chromodomain helicase DNA-binding protein 2 affects the repair of X-ray and UV-induced DNA damage. Environ Mol Mutagen. 2012;53(1):44–50. doi: 10.1002/em.20674. [DOI] [PubMed] [Google Scholar]
- 35.Nagarajan P, Onami TM, Rajagopalan S, Kania S, Donnell R, Venkatachalam S. Role of chromodomain helicase DNA-binding protein 2 in DNA damage response signaling and tumorigenesis. Oncogene. 2009;28(8):1053–1062. doi: 10.1038/onc.2008.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ling T, Xie W, Luo M, et al. CHD4/NuRD maintains demethylation state of rDNA promoters through inhibiting the expression of the rDNA methyltransferase recruiter TIP5. Biochem Biophys Res Commun. 2013;437(1):101–107. doi: 10.1016/j.bbrc.2013.06.045. [DOI] [PubMed] [Google Scholar]
- 37.Zentner GE, Hurd EA, Schnetz MP, et al. CHD7 functions in the nucleolus as a positive regulator of ribosomal RNA biogenesis. Hum Mol Genet. 2010;19(18):3491–3501. doi: 10.1093/hmg/ddq265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuan CC, Zhao X, Florens L, Swanson SK, Washburn MP, Hernandez N. CHD8 associates with human Staf and contributes to efficient U6 RNA polymerase III transcription. Mol Cell Biol. 2007;27(24):8729–8738. doi: 10.1128/MCB.00846-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39•.Stanley FK, Moore S, Goodarzi AA. CHD chromatin remodelling enzymes and the DNA damage response. Mutat Res. 2013;750(1–2):31–44. doi: 10.1016/j.mrfmmm.2013.07.008. Provides a detailed review of the established and emerging roles for CHD proteins in DNA repair, the oxidative stress response, the maintenance of genomic stability and cancer prevention. [DOI] [PubMed] [Google Scholar]
- 40••.Bagchi A, Papazoglu C, Wu Y, et al. CHD5 is a tumor suppressor at human 1p36. Cell. 2007;128(3):459–475. doi: 10.1016/j.cell.2006.11.052. This publication was the first to identify CHD5 as a tumor suppressor mapping to human 1p36, and was also the first to implicate a CHD family member in human cancer, triggering a flurry of studies on CHD proteins in oncogenesis. [DOI] [PubMed] [Google Scholar]
- 41.Bagchi A, Mills AA. The quest for the 1p36 tumor suppressor. Cancer Res. 2008;68(8):2551–2556. doi: 10.1158/0008-5472.CAN-07-2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oliver SS, Musselman CA, Srinivasan R, Svaren JP, Kutateladze TG, Denu JM. Multivalent recognition of histone tails by the PHD fingers of CHD5. Biochemistry. 2012;51(33):6534–6544. doi: 10.1021/bi3006972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fujita T, Igarashi J, Okawa ER, et al. CHD5, a tumor suppressor gene deleted from 1p36.31 in neuroblastomas. J Natl Cancer Inst. 2008;100(13):940–949. doi: 10.1093/jnci/djn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garcia I, Mayol G, Rodriguez E, et al. Expression of the neuron-specific protein CHD5 is an independent marker of outcome in neuroblastoma. Mol Cancer. 2010;9:277. doi: 10.1186/1476-4598-9-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Xu W, Huang Y, Huang X, Xu L, Lv Z. Genistein demethylates the promoter of CHD5 and inhibits neuroblastoma growth in vivo. Int J Mol Med. 2012;30(5):1081–1086. doi: 10.3892/ijmm.2012.1118. [DOI] [PubMed] [Google Scholar]
- 46.Koyama H, Zhuang T, Light JE, et al. Mechanisms of CHD5 Inactivation in neuroblastomas. Clin Cancer Res. 2012;18(6):1588–1597. doi: 10.1158/1078-0432.CCR-11-2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson PM, Gotoh T, Kok M, White PS, Brodeur GM. CHD5, a new member of the chromodomain gene family, is preferentially expressed in the nervous system. Oncogene. 2003;22(7):1002–1011. doi: 10.1038/sj.onc.1206211. [DOI] [PubMed] [Google Scholar]
- 48.Mulero-Navarro S, Esteller M. Chromatin remodeling factor CHD5 is silenced by promoter CpG island hypermethylation in human cancer. Epigenetics. 2008;3(4):210–215. doi: 10.4161/epi.3.4.6610. [DOI] [PubMed] [Google Scholar]
- 49.Wang L, He S, Tu Y, et al. Downregulation of chromatin remodeling factor CHD5 is associated with a poor prognosis in human glioma. J Clin Neurosci. 2013;20(7):958–963. doi: 10.1016/j.jocn.2012.07.021. [DOI] [PubMed] [Google Scholar]
- 50.Wu X, Zhu Z, Li W, et al. Chromodomain helicase DNA binding protein 5 plays a tumor suppressor role in human breast cancer. Breast Cancer Res. 2012;14(3):R73. doi: 10.1186/bcr3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao R, Yan Q, Lv J, et al. CHD5, a tumor suppressor that is epigenetically silenced in lung cancer. Lung Cancer. 2012;76(3):324–331. doi: 10.1016/j.lungcan.2011.11.019. [DOI] [PubMed] [Google Scholar]
- 52.Wong RR, Chan LK, Tsang TP, et al. CHD5 downregulation associated with poor prognosis in epithelial ovarian cancer. Gynecol Obstet Invest. 2011;72(3):203–207. doi: 10.1159/000323883. [DOI] [PubMed] [Google Scholar]
- 53.Wang X, Lau KK, So LK, Lam YW. CHD5 is down-regulated through promoter hypermethylation in gastric cancer. J Biomed Sci. 2009;16:95. doi: 10.1186/1423-0127-16-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Du X, Wu T, Lu J, et al. Decreased expression of chromodomain helicase DNA-binding protein 5 is an unfavorable prognostic marker in patients with primary gallbladder carcinoma. Clin Transl Oncol. 2013;15(3):198–204. doi: 10.1007/s12094-012-0903-2. [DOI] [PubMed] [Google Scholar]
- 55.Cai C, Ashktorab H, Pang X, et al. MicroRNA-211 expression promotes colorectal cancer cell growth in vitro and in vivo by targeting tumor suppressor CHD5. PloS One. 2012;7(1):e29750. doi: 10.1371/journal.pone.0029750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fatemi M, Paul TA, Brodeur GM, Shokrani B, Brim H, Ashktorab H. Epigenetic silencing of CHD5, a novel tumor-suppressor gene, occurs in early colorectal cancer stages. Cancer. 2014;120(2):172–180. doi: 10.1002/cncr.28316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao R, Wang N, Huang H, Ma W, Yan Q. CHD5, a tumor suppressor is epigenetically silenced in hepatocellular carcinoma. Liver Int. 2014;34(6):e151–160. doi: 10.1111/liv.12503. [DOI] [PubMed] [Google Scholar]
- 58.Lang J, Tobias ES, Mackie R. Preliminary evidence for involvement of the tumour suppressor gene CHD5 in a family with cutaneous melanoma. Br J Dermatol. 2011;164(5):1010–1016. doi: 10.1111/j.1365-2133.2011.10223.x. [DOI] [PubMed] [Google Scholar]
- 59.Zhao R, Meng F, Wang N, Ma W, Yan Q. Silencing of CHD5 gene by promoter methylation in leukemia. PloS One. 2014;9(1):e85172. doi: 10.1371/journal.pone.0085172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang J, Chen H, Fu S, Xu ZM, Sun KL, Fu WN. The involvement of CHD5 hypermethylation in laryngeal squamous cell carcinoma. Oral Oncol. 2011;47(7):601–608. doi: 10.1016/j.oraloncology.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 61.Egan CM, Skotte Julie, Streubel Gundula, et al. CHD5 is required for neurogenesis and has a dual role in facilitating gene expression and polycomb gene repression. Dev Cell. 2013;26(3):223–236. doi: 10.1016/j.devcel.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 62.Potts RC, Zhang P, Wurster AL, et al. CHD5, a brain-specific paralog of Mi2 chromatin remodeling enzymes, regulates expression of neuronal genes. PloS One. 2011;6(9):e24515. doi: 10.1371/journal.pone.0024515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vestin A, Mills AA. The tumor suppressor Chd5 is induced during neuronal differentiation in the developing mouse brain. Gene Expr Patterns. 2013;13(8):482–489. doi: 10.1016/j.gep.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li W, Wu J, Kim SY, et al. Chd5 orchestrates chromatin remodelling during sperm development. Nat Commun. 2014;5:3812. doi: 10.1038/ncomms4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhuang T, Hess RA, Kolla V, Higashi M, Raabe TD, Brodeur GM. CHD5 is required for spermiogenesis and chromatin condensation. Mech Dev. 2014;131:35–46. doi: 10.1016/j.mod.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vestin A, Mills AA. The tumor suppressor Chd5 is induced during neuronal differentiation in the developing mouse brain. Gene Expr Patterns. 2013;13(8):482–489. doi: 10.1016/j.gep.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67•.Kim MS, Chung NG, Kang MR, Yoo NJ, Lee SH. Genetic and expressional alterations of CHD genes in gastric and colorectal cancers. Histopathology. 2011;58(5):660–668. doi: 10.1111/j.1365-2559.2011.03819.x. Investigated the mutation status of most CHD genes in gastric and colorectal cancers and found that they are commonly mutated. [DOI] [PubMed] [Google Scholar]
- 68.Tahara T, Yamamoto E, Madireddi P, et al. Colorectal carcinomas with CpG island methylator phenotype 1 frequently contain mutations in chromatin regulators. Gastroenterology. 2014;146(2):530–538 e535. doi: 10.1053/j.gastro.2013.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sawada G, Ueo H, Matsumura T, et al. CHD8 is an independent prognostic indicator that regulates Wnt/beta-catenin signaling and the cell cycle in gastric cancer. Oncol Rep. 2013;30(3):1137–1142. doi: 10.3892/or.2013.2597. [DOI] [PubMed] [Google Scholar]
- 70.Rodriguez-Paredes M, Ceballos-Chavez M, Esteller M, Garcia-Dominguez M, Reyes JC. The chromatin remodeling factor CHD8 interacts with elongating RNA polymerase II and controls expression of the cyclin E2 gene. Nucleic Acids Res. 2009;37(8):2449–2460. doi: 10.1093/nar/gkp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Colbert LE, Petrova AV, Fisher SB, et al. CHD7 expression predicts survival outcomes in patients with resected pancreatic cancer. Cancer Res. 2014;74(10):2677–2687. doi: 10.1158/0008-5472.CAN-13-1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pleasance ED, Stephens PJ, O’meara S, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463(7278):184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Campbell PJ, Stephens PJ, Pleasance ED, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40(6):722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Burkhardt L, Fuchs S, Krohn A, et al. CHD1 is a 5q21 tumor suppressor required for ERG rearrangement in prostate cancer. Cancer Res. 2013;73(9):2795–2805. doi: 10.1158/0008-5472.CAN-12-1342. [DOI] [PubMed] [Google Scholar]
- 75.Liu W, Lindberg J, Sui G, et al. Identification of novel CHD1-associated collaborative alterations of genomic structure and functional assessment of CHD1 in prostate cancer. Oncogene. 2012;31(35):3939–3948. doi: 10.1038/onc.2011.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang S, Gulzar ZG, Salari K, Lapointe J, Brooks JD, Pollack JR. Recurrent deletion of CHD1 in prostate cancer with relevance to cell invasiveness. Oncogene. 2012;31(37):4164–4170. doi: 10.1038/onc.2011.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Berger MF, Lawrence MS, Demichelis F, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470(7333):214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Le Gallo M, O’hara AJ, Rudd ML, et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet. 2012;44(12):1310–1315. doi: 10.1038/ng.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao S, Choi M, Overton JD, et al. Landscape of somatic single-nucleotide and copy-number. Proc Natl Acad Sci USA. 2013;110(8):2916–2921. doi: 10.1073/pnas.1222577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010;29(18):3130–3139. doi: 10.1038/emboj.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer. 2011;11(8):588–596. doi: 10.1038/nrc3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li R, Zhang H, Yu W, et al. ZIP: a novel transcription repressor, represses EGFR oncogene and suppresses breast carcinogenesis. EMBO J. 2009;28(18):2763–2776. doi: 10.1038/emboj.2009.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mouradov D, Sloggett C, Jorissen RN, et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014;74(12):3238–3247. doi: 10.1158/0008-5472.CAN-14-0013. [DOI] [PubMed] [Google Scholar]
- 85.Ali Hassan NZ, Mokhtar NM, Kok Sin T, et al. Integrated analysis of copy number variation and genome-wide expression profiling in colorectal cancer tissues. PloS One. 2014;9(4):e92553. doi: 10.1371/journal.pone.0092553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gui Y, Guo G, Huang Y, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43(9):875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kulkarni S, Nagarajan P, Wall J, et al. Disruption of chromodomain helicase DNA binding protein 2 (CHD2) causes scoliosis. Am J Med Genet A. 2008;146A(9):1117–1127. doi: 10.1002/ajmg.a.32178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.O’roak BJ, Vives L, Fu W, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338(6114):1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Neale BM, Kou Y, Liu L, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Talkowski ME, Rosenfeld JA, Blumenthal I, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149(3):525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91•.O’roak BJ, Vives L, Girirajan S, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485(7397):246–250. doi: 10.1038/nature10989. Reports recurrent de novo mutations in CHD8 as a top risk gene ASD, and also identified de novo mutations in CHD3 and CHD7 in patients with autism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mccarthy SE, Gillis J, Kramer M, et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry. 2014;19(6):652–658. doi: 10.1038/mp.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, Van Ravenswaaij-Arts CM. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet. 2011;48(5):334–342. doi: 10.1136/jmg.2010.087106. [DOI] [PubMed] [Google Scholar]
- 94.De Arriba Munoz A, Monge Galindo L, Lopez Pison J, et al. CHARGE syndrome and CHD7 gene mutation. Neurologia. 2011;26(4):255. doi: 10.1016/j.nrl.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 95.Jongmans MC, Admiraal RJ, Van Der Donk KP, et al. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet. 2006;43(4):306–314. doi: 10.1136/jmg.2005.036061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wincent J, Holmberg E, Stromland K, et al. CHD7 mutation spectrum in 28 Swedish patients diagnosed with CHARGE syndrome. Clin Genet. 2008;74(1):31–38. doi: 10.1111/j.1399-0004.2008.01014.x. [DOI] [PubMed] [Google Scholar]
- 97.Martin DM. Chromatin remodeling in development and disease: focus on CHD7. PLoS Genet. 2010;6(7):e1001010. doi: 10.1371/journal.pgen.1001010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Layman WS, Hurd EA, Martin DM. Chromodomain proteins in development: lessons from CHARGE syndrome. Clin Genet. 2010;78(1):11–20. doi: 10.1111/j.1399-0004.2010.01446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Johansson M, Gillberg C, Rastam M. Autism spectrum conditions in individuals with Mobius sequence, CHARGE syndrome and oculo-auriculo-vertebral spectrum: diagnostic aspects. Res Dev Disabil. 2010;31(1):9–24. doi: 10.1016/j.ridd.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 100.Vervloed MP, Hoevenaars-Van Den Boom MA, Knoors H, Van Ravenswaaij CM, Admiraal RJ. CHARGE syndrome: relations between behavioral characteristics and medical conditions. Am J Med Genet A. 2006;140(8):851–862. doi: 10.1002/ajmg.a.31193. [DOI] [PubMed] [Google Scholar]
- 101.Hartshorne TS, Grialou TL, Parker KR. Autistic-like behavior in CHARGE syndrome. Am J Med Genet A. 2005;133A(3):257–261. doi: 10.1002/ajmg.a.30545. [DOI] [PubMed] [Google Scholar]
- 102.Smith IM, Nichols SL, Issekutz K, Blake K. Canadian paediatric surveillance P: behavioral profiles and symptoms of autism in CHARGE syndrome: preliminary Canadian epidemiological data. Am J Med Genet A. 2005;133A(3):248–256. doi: 10.1002/ajmg.a.30544. [DOI] [PubMed] [Google Scholar]
- 103.Patient CMDIA, Jiang YH, Yuen RK, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet. 2013;93(2):249–263. doi: 10.1016/j.ajhg.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pinto D, Delaby E, Merico D, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94(5):677–694. doi: 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Capelli LP, Krepischi AC, Gurgel-Giannetti J, et al. Deletion of the RMGA and CHD2 genes in a child with epilepsy and mental deficiency. Eur J Med Genet. 2012;55(2):132–134. doi: 10.1016/j.ejmg.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 106.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45(7):825–830. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Suls A, Jaehn JA, Kecskes A, et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet. 2013;93(5):967–975. doi: 10.1016/j.ajhg.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lund C, Brodtkorb E, Oye AM, Rosby O, Selmer KK. CHD2 mutations in Lennox–Gastaut syndrome. Epilepsy Behav. 2014;33:18–21. doi: 10.1016/j.yebeh.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 109.Epi KC, Allen AS, et al. Epilepsy Phenome/Genome P. De novo mutations in epileptic encephalopathies. Nature. 2013;501(7466):217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rauch A, Wieczorek D, Graf E, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380(9854):1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 111.Yamada K, Fukushi D, Ono T, et al. Characterization of a de novo balanced t(4;20)(q33;q12) translocation in a patient with mental retardation. Am J Med Genet A. 2010;152A(12):3057–3067. doi: 10.1002/ajmg.a.33174. [DOI] [PubMed] [Google Scholar]
- 112.Bennetts JS, Rendtorff ND, Simpson F, Tranebjaerg L, Wicking C. The coding region of TP53INP2, a gene expressed in the developing nervous system, is not altered in a family with autosomal recessive non-progressive infantile ataxia on chromosome 20q11-q13. Dev Dyn. 2007;236(3):843–852. doi: 10.1002/dvdy.21064. [DOI] [PubMed] [Google Scholar]
- 113.Hertz JM, Sivertsen B, Silahtaroglu A, et al. Early onset, non-progressive, mild cerebellar ataxia co-segregating with a familial balanced translocation t(8;20)(p22;q13) J Med Genet. 2004;41(3):e25. doi: 10.1136/jmg.2003.011510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tranebjaerg L, Teslovich TM, Jones M, et al. Genome-wide homozygosity mapping localizes a gene for autosomal recessive non-progressive infantile ataxia to 20q11-q13. Hum Genet. 2003;113(3):293–295. doi: 10.1007/s00439-003-0967-8. [DOI] [PubMed] [Google Scholar]
- 115.Lathrop MJ, Chakrabarti L, Eng J, et al. Deletion of the Chd6 exon 12 affects motor coordination. Mamm Genome. 2010;21(3–4):130–142. doi: 10.1007/s00335-010-9248-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hung H, Kohnken R, Svaren J. The nucleosome remodeling and deacetylase chromatin remodeling (NuRD) complex is required for peripheral nerve myelination. J Neurosci. 2012;32(5):1517–1527. doi: 10.1523/JNEUROSCI.2895-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Iourov IY, Vorsanova SG, Kurinnaia OS, Zelenova MA, Silvanovich AP, Yurov YB. Molecular karyotyping by array CGH in a Russian cohort of children with intellectual disability, autism, epilepsy and congenital anomalies. Mol Cytogenet. 2012;5(1):46. doi: 10.1186/1755-8166-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kaminsky EB, Kaul V, Paschall J, et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med. 2011;13(9):777–784. doi: 10.1097/GIM.0b013e31822c79f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Janssen N, Bergman JE, Swertz MA, et al. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat. 2012;33(8):1149–1160. doi: 10.1002/humu.22086. [DOI] [PubMed] [Google Scholar]
- 120.Schnetz MP, Bartels CF, Shastri K, et al. Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res. 2009;19(4):590–601. doi: 10.1101/gr.086983.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jacobs-Mcdaniels NL, Albertson RC. Chd7 plays a critical role in controlling left-right symmetry during zebrafish somitogenesis. Dev Dyn. 2011;240(10):2272–2280. doi: 10.1002/dvdy.22722. [DOI] [PubMed] [Google Scholar]
- 122.Hurd EA, Capers PL, Blauwkamp MN, et al. Loss of Chd7 function in gene-trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mamm Genome. 2007;18(2):94–104. doi: 10.1007/s00335-006-0107-6. [DOI] [PubMed] [Google Scholar]
- 123.Feng W, Khan MA, Bellvis P, et al. The chromatin remodeler CHD7 regulates adult neurogenesis via activation of soxc transcription factors. Cell Stem Cell. 2013;13(1):62–72. doi: 10.1016/j.stem.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 124.Gao X, Gordon D, Zhang D, et al. CHD7 gene polymorphisms are associated with susceptibility to idiopathic scoliosis. Am J Hum Genet. 2007;80(5):957–965. doi: 10.1086/513571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Marfella CG, Henninger N, Leblanc SE, et al. A mutation in the mouse Chd2 chromatin remodeling enzyme results in a complex renal phenotype. Kidney Blood Press Res. 2008;31(6):421–432. doi: 10.1159/000190788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Marfella CG, Ohkawa Y, Coles AH, Garlick DS, Jones SN, Imbalzano AN. Mutation of the SNF2 family member Chd2 affects mouse development and survival. J Cell Physiol. 2006;209(1):162–171. doi: 10.1002/jcp.20718. [DOI] [PubMed] [Google Scholar]
- 127.Qin Y, Ji J, Du G, et al. Comprehensive pathway-based analysis identifies associations of BCL2, GNAO1 and CHD2 with non-obstructive azoospermia risk. Hum Reprod. 2014;29(4):860–866. doi: 10.1093/humrep/deu013. [DOI] [PubMed] [Google Scholar]
- 128.Sparmann A, Xie Y, Verhoeven E, et al. The chromodomain helicase Chd4 is required for Polycomb-mediated inhibition of astroglial differentiation. EMBO J. 2013;32(11):1598–1612. doi: 10.1038/emboj.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Williams CJ, Naito T, Arco PGD, et al. The chromatin remodeler Mi-2 beta is required for CD4 expression and T cell development. Immunity. 2004;20(6):719–733. doi: 10.1016/j.immuni.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 130.Hosokawa H, Tanaka T, Suzuki Y, et al. Functionally distinct Gata3/Chd4 complexes coordinately establish T helper 2 (Th2) cell identity. Proc Natl Acad Sci USA. 2013;110(12):4691–4696. doi: 10.1073/pnas.1220865110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kashiwagi M, Morgan BA, Georgopoulos K. The chromatin remodeler Mi-2beta is required for establishment of the basal epidermis and normal differentiation of its progeny. Development. 2007;134(8):1571–1582. doi: 10.1242/dev.001750. [DOI] [PubMed] [Google Scholar]
- 132.Targoff IN, Reichlin M. The association between Mi-2 antibodies and dermatomyositis. Arthritis Rheum. 1985;28(7):796–803. doi: 10.1002/art.1780280711. [DOI] [PubMed] [Google Scholar]
- 133.Nishiyama M, Nakayama K, Tsunematsu R, Tsukiyama T, Kikuchi A, Nakayama KI. Early embryonic death in mice lacking the beta-catenin-binding protein Duplin. Mol Cell Biol. 2004;24(19):8386–8394. doi: 10.1128/MCB.24.19.8386-8394.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nishiyama M, Oshikawa K, Tsukada Y, et al. CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat Cell Biol. 2009;11(2):172–182. doi: 10.1038/ncb1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sakamoto I, Kishida S, Fukui A, et al. A novel beta-catenin-binding protein inhibits beta-catenin-dependent Tcf activation and axis formation. J Biol Chem. 2000;275(42):32871–32878. doi: 10.1074/jbc.M004089200. [DOI] [PubMed] [Google Scholar]
- 136.Mcdaniel IE, Lee JM, Berger MS, Hanagami CK, Armstrong JA. Investigations of CHD1 function in transcription and development of Drosophila melanogaster. Genetics. 2008;178(1):583–587. doi: 10.1534/genetics.107.079038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Papantonis A, Tsatsarounos S, Vanden Broeck J, Lecanidou R. CHD1 assumes a central role during follicle development. J Mol Biol. 2008;383(5):957–969. doi: 10.1016/j.jmb.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 138.Gaspar-Maia A, Alajem A, Polesso F, et al. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature. 2009;460(7257):863–868. doi: 10.1038/nature08212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139•.Shavelle RM, Strauss DJ, Pickett J. Causes of death in autism. J Autism Dev Disord. 2001;31(6):569–576. doi: 10.1023/a:1013247011483. Revealed an association between autism and cancer. It examined the cause of death for 13,111 ambulatory patients with autism and identified a higher standardized mortality from cancer in subjects with moderate, severe or profound intellectual disability. [DOI] [PubMed] [Google Scholar]
- 140.Kao HT, Buka SL, Kelsey KT, Gruber DF, Porton B. The correlation between rates of cancer and autism: an exploratory ecological investigation. PLoS One. 2010;5(2):e9372. doi: 10.1371/journal.pone.0009372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141•.Eisenberg ML, Betts P, Herder D, Lamb DJ, Lipshultz LI. Increased risk of cancer among azoospermic men. Fertil Steril. 2013;100(3):681–685. doi: 10.1016/j.fertnstert.2013.05.022. Revealed an association between fertility and cancer. It examined medical records of 2238 infertile men and found that infertile men were 1.7-times more likely to develop cancer than the general population. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Peng X, Zeng X, Peng S, Deng D, Zhang J. The association risk of male subfertility and testicular cancer: a systematic review. PloS One. 2009;4(5):e5591. doi: 10.1371/journal.pone.0005591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mallette FA, Richard S. JMJD2A promotes cellular transformation by blocking cellular senescence through transcriptional repression of the tumor suppressor CHD5. Cell Rep. 2012;2(5):1233–1243. doi: 10.1016/j.celrep.2012.09.033. [DOI] [PubMed] [Google Scholar]
- 144.Macdonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Inestrosa NC, Toledo EM. The role of Wnt signaling in neuronal dysfunction in Alzheimer’s Disease. Mol Neurodegener. 2008;3:9. doi: 10.1186/1750-1326-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.De Ferrari GV, Moon RT. The ups and downs of Wnt signaling in prevalent neurological disorders. Oncogene. 2006;25(57):7545–7553. doi: 10.1038/sj.onc.1210064. [DOI] [PubMed] [Google Scholar]
- 147.Ronan JL, Wu W, Crabtree GR. From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet. 2013;14(5):347–359. doi: 10.1038/nrg3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Harada A, Okada S, Konno D, et al. Chd2 interacts with H3.3 to determine myogenic cell fate. EMBO J. 2012;31(13):2994–3007. doi: 10.1038/emboj.2012.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Srinivasan R, Mager GM, Ward RM, Mayer J, Svaren J. NAB2 represses transcription by interacting with the CHD4 subunit of the nucleosome remodeling and deacetylase (NuRD) complex. J Biol Chem. 2006;281(22):15129–15137. doi: 10.1074/jbc.M600775200. [DOI] [PubMed] [Google Scholar]