

Abstract
Arylboronic acids and their derivatives have been widely exploited as important synthetic precursors in organic synthesis, materials science, and pharmaceutical development. In addition to numerous applications in transition-metal-mediated cross-coupling reactions, transition-metal-free transformations involving arylboronic acids and derivatives have recently received a surge of attention for converting the C-B bond to C-C, C-N, C-O, and many other C-X bonds. Consequently, a wide range of useful compounds, e.g., phenols, anilines, nitroarenes, and haloarenes, have been readily synthesized. Amongst these efforts, many versatile reagents have been developed and a lot of practical approaches demonstrated. The research in this promising field is summarized in the current review and organized on the basis of the type of bonds being formed.
Keywords: Arylboronic acid, Hydroxylation, Amination, Halogenation, Transition-metal-free
1 Introduction
During the past few decades, arylboronic acids and their derivatives (boronate esters, trifluoroborates and boroxines) have found broad utility in organic synthesis, materials science, and pharmaceutical development.[1] As a consequence of their many intrinsic merits, such as high reactivity, low toxicity, and extensive commercial availability, arylboronic acids have become one of the most prevalent feedstocks for the introduction of aryl substituents into diverse molecules. Since the first report of palladium-catalyzed cross-coupling of organoboranes with carbon halides by Suzuki in 1979,[2] the transition-metal-mediated functionalization of arylboronic acids has burgeoned.[3] In the presence of transition metals, arylboronic acids are prone to undergo transmetallation, via which the carbon-boron (C-B) bond is converted into a carbon-carbon (C-C) bond or carbon-heteroatom (C-N, C-O, etc.) bond. Despite the robust catalytic capability of transition metals, some drawbacks have been gradually realized in transition-metal-mediated reactions. For instance, metal catalysts are often air- and/or moisture-sensitive, expensive, and residual transition metals can be difficult as well as costly to remove, especially in pharmaceutical applications. Consequently, transition-metal-free processes have emerged as valuable alternatives to more conventional transition-metal-catalyzed reactions.
In organoboronic acids, a vacant p-orbital causes the sp2-hybridized boron to behave as a Lewis acid. Addition of a nucleophile (X-LG) or Lewis base (Scheme 1) generates a tetravalent boron “ate” complex and induces rehybridization to generate a sp3-boron. The heightened electron density on the boron and increased steric crowding in the “ate” complex facilitates R-B bond dissociation and subsequent alkyl/aryl migration to the adjacent acceptor atom with retention of configuration. The overall process of ipso-substitution is achieved without the assistance of transition metals (Scheme 1). To better understand this unique migratory characteristic, Brown and Matteson conducted the seminal studies on the transformations of alkylboranes.[4] Later, Aggarwal and others contributed many remarkable studies into the conversion of chiral alkylboranes.[5] Complementary to the aforementioned transformations of alkylboranes, considerable progress has been made recently in transition-metal-free transformations of arylboronic acids for the construction of C-C, C-N, C-O and many other C-X bonds. These efforts will be summarized herein and organized on the basis of the type of bonds being formed. This review is focussed upon an unique viewpoint for the transformation of arylboronic acids, and complementary to existing reviews on transition-metal-catalyzed transformations.[3]
Scheme 1.
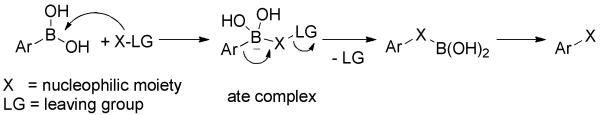
General Pathway for Transition-Metal-Free Functionalization of Organoboronic Acids.
2 Carbon-Oxygen Bond Formation
Phenols are a privileged structural moiety in numerous natural products, pharmaceuticals and polymers.[6] They also frequently serve as versatile synthetic intermediates for the construction of complex molecules. Thus, it is of great utility to develop practical access to polyfunctionalized phenols. In this regard, transition-metal-free ipso-hydroxylation of arylboronic acids offers the opportunity to produce phenols in a mild and efficient manner which otherwise might be difficult to achieve by other means.
The seminal work, reported by Ainley and Challenger[7] and later Hawthorne,[8] converted phenylboronic acids into phenols using alkaline hydrogen peroxide and hydrogen peroxide, respectively. Only a few examples were provided in low to moderate yields. Nearly a half century later, this chemistry was extensively studied and numerous practical modifications were introduced to improved functional group compatibility.
In 1995, Webb and Levy reported the oxidation of arylboronic acids to phenols using potassium monopersulfate, available commercially as a stable triple salt (2KHSO5·KHSO4·K2SO4) under the trademark Oxone®.[9] Both electron-rich and deficient phenylboronic acids gave good yields (Scheme 2). Phenylboronate bis-ester was also a suitable substrate. The reaction was performed at 2 °C in 10-15% aqueous acetone. Lower yields were obtained in the absence of co-solvent acetone, suggesting that in situ generated dimethyldioxirane competed with Oxone® in the oxidation. All of the reactions required careful control of reaction time (< 5 min) and temperature. Subsequently, KHSO5, the putative oxidant in Oxone®, was isolated and employed in the oxidation of phenylboronic acid. The transformation gave a comparable yield of phenol as did Oxone® itself.[10]
Scheme 2.

Hydroxylation of Arylboronic Acids by Oxone®.
The oxidation of phenylboronic acids with hydrogen peroxide (H2O2) was systematically studied by Prakash, Petasis and co-workers.[11] In the presence of 30% H2O2, a variety of phenylboronic acids bearing different substituents were readily converted to phenols in good yields (Scheme 3). This oxidation along with the following coupling furnished symmetrical diaryl ethers in one pot. The kinetics of the reaction between phenylboronic acid and H2O2 were investigated in detail by Kuivila and co-workers.[12]
Scheme 3.

Hydroxylation of Arylboronic Acids by H2O2.
A modified procedure employing PVD-H2O2 and PVP-H2O2 complex was then reported from the same group (Scheme 4).[13] These solid-H2O2 equivalents were found to be more efficient for the ipso-hydroxylation of phenylboronic acids to phenols in terms of chemical yields and reaction rate. The complex can be reused for three runs without compromising the chemical yields.
Scheme 4.

Hydroxylation of Arylboronic Acids by Solid-H2O2 Complex.
Other variations based on H2O2 oxidation were also developed for the ipso-hydroxylation of arylboronic acids. For example, the addition of catalytic amounts of iodine or recyclable Amberlite IR-120 resin promoted the reaction to generate a variety of phenols in good yields under mild reaction conditions.[14]
Other oxidants, such as mCPBA and TBHP, could also accomplish the transformation efficiently. In aqueous EtOH, mCPBA furnished the corresponding phenols in good yields,[15] whereas TBHP in water accelerated the reaction and were complete within 10 min.[16]
An interesting example was the hydroxylation of arylboronic acids with hydroxylamine.[17] The reaction of arylboronic acids or their pinacol esters with in situ generated hydroxylamine from hydroxylammonium chloride and sodium hydroxide gave the corresponding phenols in moderate to good yields at room temperature (Scheme 5). The electronic property of substituents appreciably influenced the reaction outcome. For electron-deficient substrates, the yields were lower and the reaction failed completely in the case of p-acetyl. The conversion was much more sluggish, generally 1-2 days, than the reactions using Oxone® or H2O2.
Scheme 5.

Hydroxylation of Arylboronic Acids by NH2OH.
Recently, Zhu and Falck disclosed a highly efficient ipso-hydroxylation of arylboronic acids by means of tertiary amine N-oxide.[18] The transformation was mild, rapid, and had broad functional group tolerance. As shown in Scheme 6, regardless of the electronic or steric character of the substrate, all arylboronic acids afforded phenols in excellent chemical yields. The examples bearing oxidation-sensitive functionality (4-CHO) and halogens (I, Br), which could not survive exposure to peroxide or during transition-metal catalysis, are noteworthy. The reaction conditions were also applicable to heteroarylboronic acids. All oxygen-, nitrogen-, and sulfur-containing substrates led to the corresponding hydroxylated products in useful yields. In addition, other boronic acid surrogates such as boronate esters and trifluoroborate were apt substrates.
Scheme 6.

Hydroxylation of Aryl/Heteroarylboronic Acids by N-Oxide.
A mechanism of leaving group-enhanced 1,2-aryl migration was proposed for the transformation (Scheme 7). Initially, nucleophilic attack of N-oxide at boron forms an “ate” complex intermediate. Migration of the aryl from boron to the oxygen of the N-oxide with simultaneous internal displacement (SN2) ruptures the ammonium-O bond and is an important driving force for the reaction. Standard aqueous isolation or direct chromatographic purification on silica gel hydrolyzes the resultant boronate ester to furnish the corresponding phenol.
Scheme 7.
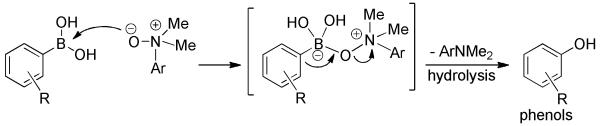
Proposed Mechanism for N-Oxide-mediated Hydroxylation of Arylboronic Acids
An environmentally appealing aerobic oxidation of arylboronic acids promoted by quinone[19] or thiols has been found.[20] In the presence of catalytic quinone or stoichiometric aminothiophenol, arylboronic acids were quantitatively converted into phenols under air. However, the substrate scope was limited.
Under an oxygen or air atmosphere, two similar studies from Fuchigami et al. and Jørgensen et al. revealed that the transformation of arylboronic acids to phenols can also take place by means of electrochemical hydroxylation (Scheme 8).[21] A variety of arylboronic acids were converted into corresponding phenols in good yields. A pathway involving a superoxide anion intermediate, generated via one-electron reduction of oxygen at the cathode, was proposed by both groups.
Scheme 8.

Electrochemical Hydroxylation of Arylboronic Acids.
Recently, Huang and co-workers developed a copper-promoted electrochemical hydroxylation of arylboronic acids (Scheme 9).[22] A variety of phenols from arylboronic acids in aqueous ammonia were generated efficiently in an undivided cell. The formation of phenols versus anilines could be controlled with good chemoselectivity by simply changing the aqueous ammonia concentration and the anode potential.
Scheme 9.
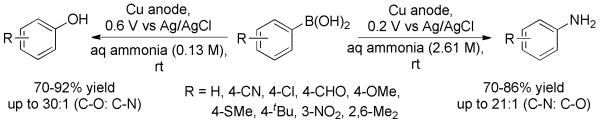
Chemoselective Electrochemical Hydroxylation versus Amination of Arylboronic Acids.
Alcohols labelled with 18O are highly valued in biological studies. Most recently, Rozen and co-workers developed a general method to access phenols containing a heavy oxygen isotope in excellent yields at room temperature.[23] The 18O-labelled phenols were synthesized by the treatment of arylboronic acids with H18OF·CH3CN complex (Scheme 10). Both electron-rich and -deficient substituents were well tolerated. The origin of the 18O isotope is H218O; the H18OF·CH3CN complex is prepared by bubbling dilute F2 through acetonitrile and H218O. The use of fluorine gas requires special precautions and, thus, detracts from the general utility of the method.
Scheme 10.
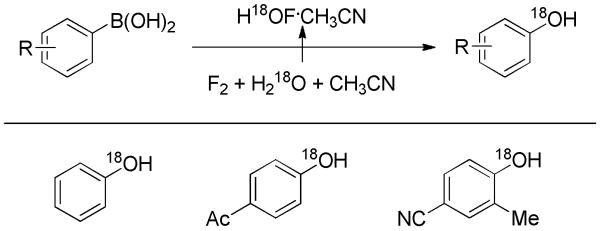
Preparation of 18O-Labelled Phenols.
The transformation of potassium aryltrifluoroborates to phenols was investigated by Molander and Cavalcanti.[24] Upon treatment with Oxone® in acetone/water, a wide range of aryltrifluoroborates were efficiently converted to the respective phenols within a few minutes (Scheme All electron-rich, -deficient, and neutral substrates afforded excellent chemical yields. Remarkably, heteroaryl trifluoroborates were also compatible with the reaction conditions, giving the corresponding heterocycles in high yields.
3 Carbon-Nitrogen Bond Formation
3.1. Amination of Arylboronic Acids and Derivatives
The versatility of aromatic amines renders them of broad utility in pharmaceuticals, agrochemicals, polymers, and dyes.[25] Apart from the conventional preparations via metal-mediated processes or at high temperature and pressure, transition-metal-free amination of arylboronic acids and derivatives provides ready access to aniline derivatives with susceptible functional groups. In this context, a few examples have been disclosed recently.
Yu and co-workers described the formation of secondary anilines via transition-metal-free C-N bond formation between arylboronic acids and organic azides.[26] By just heating the two components in xylene, the corresponding products were obtained in moderate to good yields (Scheme 12). Electron-deficient arylboronic acids gave better yields than electron-rich ones. Both alkyl and aryl azides were competent substrates. However, the harsh reaction conditions, i.e., 140 °C and the use of potentially explosive azides, obviously limit its attractiveness and scope.
Scheme 12.

Amination of Arylboronic Acids with Organic Azides.
Afterward, the transition-metal-free formation of tertiary anilines through amination of arylboroxines was developed by Wang and co-workers.[27] O-Benzoyl hydroxylamine was employed as aminating reagent. A number of tertiary anilines, as well as a few secondary anilines, were readily generated at 130 °C (Scheme 13). A variety of functional groups such as halogen, ester, amide, and nitrile were well tolerated.
Scheme 13.

Amination of Arylboroxines with O-Benzoyl Hydroxylamine.
Very recently, two tactically related studies aimed at the direct primary amination of aromatic organoboronates were revealed. Attributing the lack of reactivity between the aminating agent and organoboron to their ineffective association, Morken and co-workers manipulated the nucleophilicity of methoxyamine by adding 3 equiv nBuLi to the reaction.[28] This enabled the lithiated methoxyamine to incorporate with pinacol boronate, forming an active “ate” complex which ultimately led to the primary amination product. The reaction proved to be efficient with electron-rich substrates, but electron-deficient ones generally resulted in low yields (Scheme 14). Heterocyclic substrates failed to react. Additionally, the amination was demonstrated with alkyl boronates. When chiral boronates were subjected to the reaction conditions, the corresponding amines were stereospecifically furnished with retention of configuration at the carbon atom.
Scheme 14.

Primary Amination of Pinacol Boronates with MeONH2.
A variation for primary amination of arylboronic acids with broader substrate scope was developed by Zhu et al.[29] Evaluation of several potential aminating agents revealed O-(2,4-dinitrophenyl)hydroxylamine (DPH) was the only suitable one for the amination of arylboronic acids under mild conditions. A diverse array of arylboronic acids was readily transformed into the corresponding primary anilines in useful yields using only 1.1 equiv DPH (Scheme 15). Both electron-rich and -deficient substrates were well tolerated. Notably, some halogenated anilines (I, Br), which might be labile under transition-metal-mediated procedures, could be made without complications. In a few cases, the addition of 1.2 equiv Cs2CO3 promoted the amination reaction rates and improved the yields. The authors also demonstrated the reaction could be scaled up to the multigram-scale without attenuating the chemical yield. The methodology, however, failed with nitrogen- and sulfur-containing heterocyclic boronic acids, albeit dibenzofuran boronic acid gave the expected product in satisfactory yield.
Scheme 15.

Primary Amination of Arylboronic Acids with DPH.
As shown in Scheme 9, primary anilines could also be prepared through electrochemical synthesis depending upon the reaction conditions and electrode potential. A range of functionalized anilines were obtained in good yields and chemoselectivities.
3.2. Nitration/Nitrosation of Arylboronic Acids and Derivatives
Nitroarenes are important building blocks widely used in both fundamental organic chemistry research and by industry.[30] They are generally produced by electrophilic aromatic nitration, though this process often employs strong nitrating agent, highly acidic reaction conditions, and often leads to over-oxidation and/or mixtures of isomers.[30b] Hence, a mild, chemoselective, and regioselective nitration approach is much in demand.
In 2000, Prakash, Petasis, Olah, and co-workers reported the transition-metal-free formation of nitroarenes via ipso-nitration of arylboronic acids.[31] In the presence of Crivello’s reagent (mixture of ammonium nitrate/trifluoroacetic anhydride), arylboronic acids were converted to the corresponding nitroarenes in moderate yields (Scheme 16). Despite carefully regulating the reaction temperature at −35 °C, the by-product from undesired di-nitration could not be suppressed.
Scheme 16.
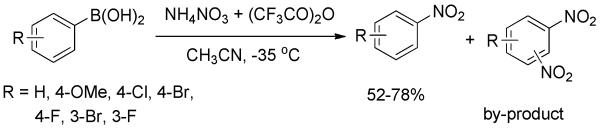
Nitration of Arylboronic Acids by Crivello’s Reagent.
Afterward, this procedure was improved via modulation of the nitrating agent.[32] NH4NO3 (sometimes AgNO3) in combination with TMSCl, in lieu of (CF3CO)2O, generated an active nitrating agent TMS-O-NO2 capable of regioselective ipso-nitration of arylboronic acids. The proposed mechanistic pathway is shown in Scheme 17. The reaction was carried out at room temperature for 1-3 days, and a variety of nitroarenes were readily afforded in good yields (Scheme 18). It should be noted that in the case of substrates bearing a strong deactivating group, e.g., CF3 and NO2, yields for the electrophilic nitration decreased significantly.
Scheme 17.
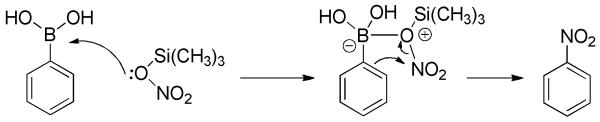
Proposed Mechanism for Nitration of Arylboronic Acids.
Scheme 18.

Nitration of Arylboronic Acids by TMSCl/Nitrate Salts.
Direct ipso-nitration of arylboronic acids using a single reagent, i.e., tert-butyl nitrite, was recently described by Wu, Beller and co-workers.[33] Both electron-rich and deficient substrates were well tolerated, furnishing the corresponding nitroarenes in moderate to good yields (Scheme 19). Regarding the mechanism, it was suggested reaction of the phenylboronic acids with the nitriting agent initially resulted in nitrosobenzenes, which were concomitantly oxidized in situ by air to nitrobenzenes. Compared to the above approaches, this one was operationally simple; however, excessive nitrating agent (10 equiv) was required for complete conversion.
Scheme 19.

Nitration of Arylboronic Acid with tert-Butyl Nitrite.
An efficient nitration of arylboronic acids was reported by Maiti and co-workers, in which Bi(NO3)3·5H2O/K2S2O8 was employed as nitrating agent.[34] This protocol demonstrated a broader substrate scope than previous reports. A series of nitroarenes possessing electron-rich and -deficient substituents were synthesized in moderate to good yields (Scheme 20). Moreover, heteroaryls such as dibenzofuranyl, dibenzothienyl, and quinolinyl boronic acids were also suitable substrates. Unlike the common 1,2-aryl migration pathway, an aryl radical mechanism was proposed for the transformation. It was verified by control experiments in the presence of radical scavengers. With the addition of TEMPO or hydroquinone to the reaction, the chemical yield dropped significantly.
Scheme 20.

Nitration of Aryl/Heteroaryl Boronic Acids with Bismuth Nitrate and Perdisulfate.
Nitrosoarenes comprise a class of highly reactive synthetic intermediates for which numerous chemical transformations have been reported, inter alia, cycloadditions and aldol reactions.[35] However, satisfactory access to functionally diverse nitrosoarenes has been a challenge. Very recently, based on the strategy of transition-metal-free nitration of aryltrifluoroborates, Molander and Cavalcanti disclosed an efficient synthesis of variously functionalized nitrosoarenes.[36] Nitrosonium tetrafluoroborate (NOBF4) served as nitrosating agent and demonstrated a remarkably high reactivity in the transformation. In the case of electron-rich aryltrifluoroborates, the reaction was completed in just 30 seconds at room temperature; electron-deficient substrates were likewise fully converted within 1 minute under the same reaction conditions. All the reactions resulted in good chemical yields, and displayed broad functional group tolerance (Scheme 21). Impressively, many heteroaryl trifluoroborates including dibenzofuranyl, dibenzothienyl, benzothienyl, indolyl, pyrimidinyl, and pydidinyl derivatives were compatible, readily generating the corresponding nitrosoheteroaryls in useful yields.
Scheme 21.

Nitrosation of Aryl/Heteroaryl Trifluoro-borates with NOBF4.
4 Carbon-Carbon Bond Formation
Carbon-carbon (C-C) bond formation represents one of the most fundamental transformations in organic synthesis. Along with the booming development of cross-coupling reactions over the last several decades, organoboron compounds have been mostly commonly applied for transition-metal-catalyzed C-C bond creation such as the Suzuki-Miyaura reaction. On the other hand, there has been a growing interest in transition-metal-free functionalization of organoboronates, which offers a powerful tool for the construction of C-C bond as illustrated by the Petasis-Borono Mannich reaction (PBM). Considering that there are already many outstanding comprehensive reviews with respect to PBM and relevant nucleophilic additions of organoborons to unsaturated bonds,[37] those reactions will not be discussed herein. In this section, we wish to focus on recent examples in which C-C bonds are constructed via a 1,2-aryl migration pathway.
Barluenga and co-workers recently reported an efficient transition-metal-free C-C bond formation between arylboronic acids and tosylhydrazones.[38] The latter were used as an in situ source of diazo compounds in the presence of base. After a survey of solvents and bases, the optimized reaction conditions (dioxane, K2CO3, 110 °C) were used to determine the substrate scope. Importantly, the functional group tolerance was extremely broad. All transformations readily provided the corresponding coupled products regardless of the electronic properties of the substituents on both arylboronic acids and hydrazones (Scheme 22). The reaction could be accomplished with hydrazones derived from aldehydes or ketones, with aryl, heteroaryl, or alkyl substituents. These could be coupled not only with aryl, but also heteroaryl and alkyl boronic acids. Additional noteworthy features highlighted were: a) no inert atmosphere or dry solvents were required, a major advantage vis-a-vis most transition-metal-mediated couplings; b) the reaction can be conducted in one pot directly from the carbonyl compound without isolating the intermediate tosylhydrazone.
Scheme 22.
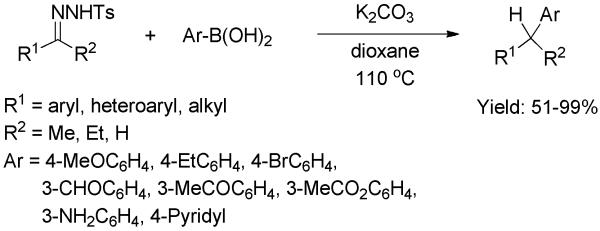
C-C bond formation between Tosylhydrazones and Arylboronic Acids.
The overall transformation is initiated by the thermolysis of tosylhydrazone I in the presence of base. This results in the in situ generation of diazo compound II, which is the actual reactive species in the subsequent conversion to benzylboronic acid VI. This assumption is unambiguously supported by the following control experiments. When ethyl diazoacetate (or phenyl diazomethane) in lieu of its hydrazone precursor is treated with p-methoxyphenyl boronic acid under the standard reaction conditions, the same product VI is obtained. Then, protodeboronation of VI gives rise to the final product VII. However, the question remained as to which of two pathways could possibly lead to VI starting from II. The diazo compound II could react with boronic acid through intermediate III to give VI (top pathway, Scheme 23). This one is a commonly accepted mechanistic route via 1,2-aryl migration. Alternatively, carbene IV might be generated from II followed by insertion into the C-B bond of the boronic acid forming zwitterion V, which could also result in VI (bottom pathway, Scheme 23).
Scheme 23.

Possible Mechanistic Pathways for C-C bond formation between Tosylhydrazones with Boronic Acids.
A similar study involving the C-C bond formation between diazo compounds with arylboroxines was disclosed by Wang and co-workers.[39] The addition of diisopropylamine was needed to neutralize the reaction milieu, thus preventing decomposition of the diazo substrates. The diazocarbonyl substrates ranged from diazoesters to diazoamides and diazoketones (Scheme 24). The substituents on the arylboroxines could be electron-rich or -deficient. A mechanistic pathway initiated by nucleophilic attack of the diazo carbon upon the boroxine, consistent with the one shown in the top of Scheme 23, was suggested.
Scheme 24.
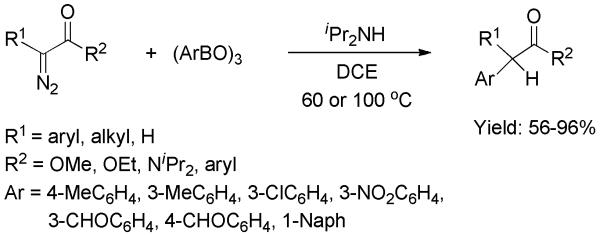
C-C bond formation between Diazocarbonyl Compounds and Arylboroxines.
Based on a similar mechanism as above, a stepwise reaction of boronate esters with diazocarbonyl compounds was developed (Scheme 25).[40] Dimethyl boronate ester was first converted into CF3-substituted borate I by treatment with TMSCF3 and KF. Salt I was then converted to the corresponding borane II, thus enhancing its electrophilicity towards diazocarbonyls. The subsequent addition gave rise to enol ether intermediate III which could be either quenched by a proton or trapped with an imine.
Scheme 25.

Stepwise Reaction of Arylboronate Esters and Diazocarbonyl Compounds.
As shown in Scheme 25, the α-diazocarbonyl, generated in situ as a consequence of the 1,2-aryl migration, suffers deboronation via formation of a boron enolate intermediate. On the other hand, one might anticipate that retention of the boron at the α-position would be even more useful as it could be further applied in C-C bond formation or other functionalizations.
This aim was achieved very recently by Molander and co-workers with the use of trifluorodiazoethane.[41] Trifluorodiazoethane was prepared in situ from the corresponding ammonium salt. It reacted with arylboronic acids, leading to the α-trifluoromethylated pinacol boronates after quenching the reaction mixture with pinacol. Although detectable by 1H NMR, the products were unstable and prone to oxidation during purification. The use of aryltrifluoroborates rendered the process practical as the corresponding products were now more stable. Treatment with TMSCl or p-tolylSiCl3 as fluorophiles converted aryltrifluoroborates into dihaloboranes, which were highly reactive towards trifluorodiazoethane. As shown in Scheme 26, the reaction gave the isolable α-trifluoromethylated organoboronates in good yields. Both electron-rich and -deficient substituents were compatible. Heterocyclic substrates likewise proceeded smoothly without affecting the reaction efficiency. Significantly, the α-trifluoromethylated organoboronates proved to be extremely useful building blocks for incorporating the CF3 unit at sp3-carbons.
Scheme 26.

Synthesis of Stable α-Trifluoromethylated Organoboronates.
Kusama and co-workers described a photochemical-promoted transition-metal-free C-C bond formation between acylsilanes with arylboronic esters to afford ketones at room temperature.[42] The reaction was irradiated by a 500-W super high-pressure Hg lamp. The authors hypothesized a carbene-involved pathway. Acylsilane I undergoes a photo-induced 1,2-silyl shift to generate siloxycarbene II, which is able to insert into the C-B bond of the boronic ester. The resultant intermediate III then undergoes 1,2-migration of R3 to form boronate IV. Quenching using an acidic medium releases the corresponding ketone V (Scheme 27).
Scheme 27.
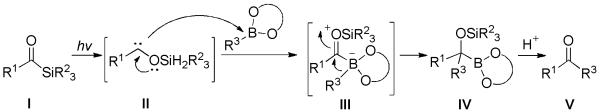
Postulated Mechanism for C-C bond formation between Acylsilanes with Boronic Esters.
Conclusions from optimization of reaction parameters: a) the nonpolar solvent hexane gave a better outcome than other common solvents; b) TMS-containing substrates are ideal; c) conversions using neopentyl glycol ester boronates are faster than other boronates; and d) the chemical yield was further improved by addition of 4Å molecular sieves. A wide range of acylsilanes and boronic esters were compatible in the transformation (Scheme 28). Apart from many applications with diazo compounds, this was a rare example in which acylsilanes were efficiently applied in a transition-metal-free C-C bond-formation reaction.
Scheme 28.
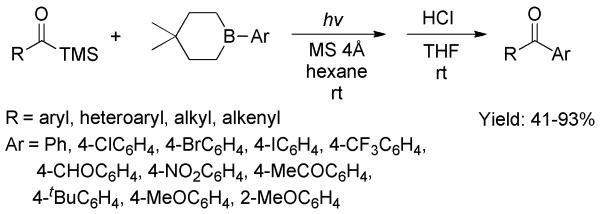
Photocatalytic C-C bond formation between Acylsilanes and Arylboronic Esters.
5 Carbon-Halogen (F, Cl, Br, I) Bond Formation
Aryl halides are ubiquitous and have been extensively employed as versatile synthetic precursors for the formation of carbon-carbon and carbon-heteroatom bonds.[43] Thus, the chemo-/regio-selective introduction of aryl-halogen bonds into complex polyfunctional molecules is an important and still challenging subject.
The transition-metal-free ipso-fluorination of arylboronic acids and derivatives was initiated by Widdowson and co-workers.[44] Cesium fluoroxysulphate (CsSO4F, abbreviated as CFS) was employed as fluorinating agent. In the reaction, boronic acids were firstly transformed into their diethanolamine esters as the latter were more stable and generally gave slightly better yields than the former. Then, treatment of the boronate esters with CFS generated aryl fluorides in low to moderate yields (Scheme 29). Electron-deficient substrates resulted in low yields in this electrophilic fluorination. Catalytic amounts of 1,3-dinitrobenzene were added to the reaction to suppress competitive SET or radical reactions. The direct fluorination of arylboronic acids could also be accomplished using MeOH as solvent.[45] However, this preliminary research was still limited by low chemical yields and a narrow substrate scope.
Scheme 29.
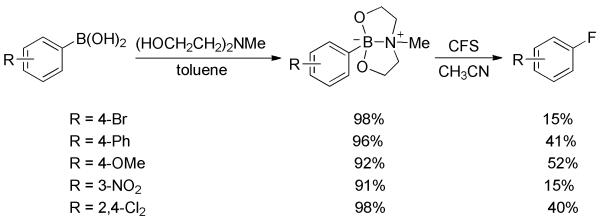
Fluorination of Arylboronic Acids with CFS.
Inspired by the pioneering research on electrophilic fluorination of alkenyl trifluoroborates from Petasis, Prakash, and Olah,[46] Lemaire and co-workers recently developed the Selectfluor™-mediated ipso-fluorination of arylboronic acids and trifluoroborates.[47] Using Selectfluor™ as fluorinating agent, a few arylboronic acids and trifluoroborates were converted to the corresponding fluoroarenes in moderate to good yields at room temperature (Scheme 30). Inexplicably, the example with a 3-BnO substituent failed. Moreover, only electron-rich substrates were tolerated. Accordingly, a broadly applicable transition-metal-free fluorination of arylboronic acids and derivatives still remains a challenge.
Scheme 30.

Fluorination of Arylboronic Acids and Trifluoroborates with Selectfluor™.
The mild ipso-bromination and iodination of arylboronic acids with N-halosuccinimides were achieved by Prakash, Petasis, and Olah.[48] In the presence of NIS, a wide range of arylboronic acids were transformed into iodoarenes in moderate to good yields (Scheme 31). Both electron-donating and -withdrawing functional groups were tolerated. Sulfur-containing heterocycles such as thienyl and benzothienyl boronic acids were also readily converted to the corresponding products. Similarly, a series of bromoarenes were obtained from arylboronic acids using NBS. The presence of the strongly deactivating nitro substituent resulted in low yields for bromination and iodination.
Scheme 31.

Bromination and Iodination of Arylboronic Acids with NBS and NIS.
Later, an efficient access to bromoarenes and chloroarenes via ipso-halogenation of arylboronic acids was disclosed.[49] By use of 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) as brominating agent and catalytic amounts of sodium methoxide, arylboronic acids were converted to bromoarenes with excellent yields (Scheme 32). In contrast to the above example, nitro-substituted boronic acid gave a 96% yield. This protocol was then applied to the chlorination of arylboronic acids using the analogous 1,3-chloro-5,5-dimethylhydantoin (DCDMH), although yields were lower and more variable.
Scheme 32.

Bromination and Chlorination of Arylboronic Acids with DBDMH and DCDMH.
The transformation of aryltrifluoroborates into aryl bromides and iodides was accomplished by Kabalka and Mereddy.[50] The iodinating (or brominating) agent was prepared in situ from a mixture of chloramine-T and sodium iodide (or sodium bromide). For ipso-iodination, a variety of aryltrifluoroborates including electron-rich and -deficient substrates were converted at room temperature, generating iodoarenes in good yields (Scheme 33). Thienyl and benzothienyl iodides were also obtained. Under similar brominating conditions, lots of aryl, heteroaryl, alkenyl, and alkynyltrifluoroborates were readily reacted, producing the corresponding bromides in good yields.
Scheme 33.

Bromination and Iodination of Aryltrifluoroborates with Chloramine-T.
The ipso-bromination of aryltrifluoroborates could also be achieved using tetrabutylammonium tribromide (TBATB) as the brominating agent.[51] A variety of arylbromides were obtained in high yields. Oxygen- and nitrogen-containing heterocyclic substrates were readily brominated without significant dibromination.
The iodinating conditions, NaI/chloramine-T, can also be applied to the ipso-iodination of other arylboronic acid derivatives. For instance, aromatic neopentylboronates underwent iodination to afford the desired aryl iodides in modest yields (Scheme 34).[52] Another example is the iodination of aryl triolborate, a water-soluble complex of arylboronic acid.[53] Both electron-rich and -deficient derivatives, as well as heterocyclic substrates led to a series of iodides in good yields (Scheme 35).
Scheme 34.
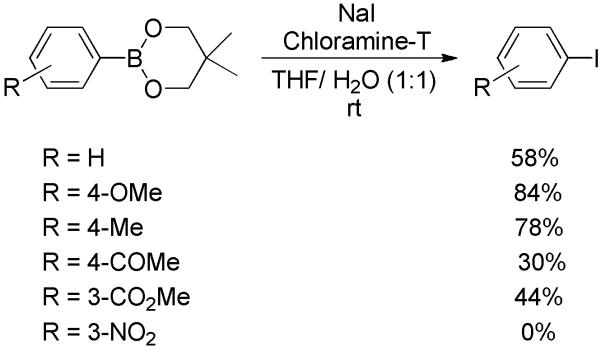
Iodination of Aryl Neopentylboronates.
Scheme 35.

Iodination of Aryltriolbororate Complex.
The ipso-chlorination of aryltrifluoroborates was explored by Molander and Cavalcanti.[54] Several chlorination conditions were competent for the transformation of electron-rich aryltrifluoroborates to the corresponding chloroarenes. For example, in the presence of NaOCl or NaCl/chloramine-T, an assortment of chloroarenes was conveniently prepared from precursors bearing electron-donating groups. However, electron-deficient substrates failed to work. This problem was solved by utilizing trichloroisocyanuric acid (TCICA) as chlorinating agent (Scheme 36). Not only electron-rich but - deficient aryltrifluoroborates were readily converted into chloroarenes in excellent yields. Significantly, a diverse array of heteroaryl chlorides was obtained under mild conditions through this practical approach. With pyridyl and benzofuryl trifluoroborates, the reactions gave rise to the dichlorinated products. Recently, Mayr and co-workers investigated the kinetics and mechanism of transition-metal-free electrophilic substitution of heteroaryltrifluroborates.[55] It was believed that the BF3K group often directed electrophiles to adjacent or remote position, and substitutions at CH positions were always faster than ipso-substitutions of the BF3K group. Hence, in the dichlorination process, the introduction of chlorine via electrophilic chlorination probably occurred before ipso-chlorination of the BF3K group.
Scheme 36.

Chlorination of Aryl/Heteroaryl Trifluoroborates.
Since the electrophilic halogenating agents were employed in the above transformations, these reactions likely proceed mechanistically via an electrophilic halogenation pathway. This transition-metal-free halogenation process is mild and enjoys a broad substrate scope. It can be envisioned that these processes may be particularly useful for the preparation of radiolabelled haloarenes, which have found wide applications as research tools in diagnoses and therapies. For example, 125I labelled arenes could be easily prepared at the late stage using commercially available and comparatively inexpensive Na125I.
A transition-metal-free synthesis of diaryliodonium sulfonates from arylboronic acids was disclosed by Widdowson and co-workers.[56] Treatment of electron-rich arylboronic acids with PhI(OAc)2·2TfOH in CH2Cl2 at room temperature produced a set of diphenyliodonium triflates in moderate to good yields (Scheme 37). The reaction conditions were not applicable to electron-deficient substrates and heteroarylboronic acids. However, in an analogous application of PhI(OH)(OTs) (Koser’s reagent), heteroarylboronic acids were efficiently transformed into the corresponding heteroarylphenyliodonium tosylates in useful yields.
Scheme 37.

Synthesis of Diaryliodonium Sulfonates from Arylboronic Acids.
The carbon-sulfur bond could also be constructed via the transition-metal-free sulfuration of phenyltrifluoroborate.[57] In the presence of S2Cl2 or (PhSO2)2S, phenylsulfide or phenyldisulfide were formed, respectively, in high chemical yields (Scheme 38). Although only phenyltrifluoroborate was studied therein, this study shed light on the concept of building C-S bonds through transition-metal-free ipso-sulfuration of arylboronic acids and derivatives. This patently underexploited field warrants further investigation.
Scheme 38.
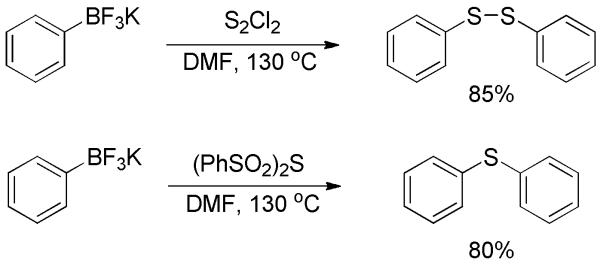
Sulfuration of Phenyltrifluoroborate.
6. Conclusion
Organoboron compounds have been widely used as synthetic intermediates in fields ranging from organic and pharmaceutical chemistry to materials science. Despite the dominance of transition-metal-mediated cross-coupling reactions, recent years have witnessed considerable progress in transition-metal-free transformations of arylboronic acids and their derivatives for the construction of C-C, C-N, C-O and many other chemical bonds. As a consequence, a vast number of useful products were afforded, some for the first time. The current review summarizes the recent developments in this area, especially the production of phenols (C-O bond), anilines (C-N bond), nitroarenes (C-N bond) and haloarenes (C-F, C-Cl, C-Br, C-I bond). In addition, reactions involving C-C bond and C-S bond formation were also briefly discussed. Amongst these works, many versatile reagents have been developed and a lot of practical approaches validated.
transition-metal-free processes possess many advantages including the avoidance of metals, thus obviating metal-contamination of the reaction products and the environment, and good tolerance of functional groups which are otherwise incompatible with transition metals. Further significant endeavors, such as applications in the synthesis of more complex molecules, should be anticipated.
Scheme 11.

Hydroxylation of Aryl/Heteroaryl Trifluoroborates.
Acknowledgements
C.Z. thanks the financial support from Soochow University and The Project of Scientific and Technologic Infrastructure of Suzhou (SZS201207). J.R.F. received support from the Robert A. Welch Foundation (GL625910) and NIH GM31278.
Biographies
Author Biographies
Chen Zhu received a BS degree from Xiamen University in 2003, and a PhD degree from Shanghai Institute of Organic Chemistry in 2008 under the supervision of Prof. Guo-Qiang Lin. After postdoctoral research in Gakushuin University, Japan with Prof. Takahiko Akiyama, he moved to the University of Texas Southwestern Medical Center, working with Prof. John R. Falck and Prof. Chuo Chen. He was appointed as professor at Soochow University, China in Dec. 2013. His current research interests include transition-metal-free transformations and their applications in the construction of natural products and biologically active compounds.

J. R. Falck earned his B.Sc. and Ph.D. from Colorado State University in 1970 and 1973, respectively, followed by a D.I.C. in 1974. He pursued advanced training in the laboratories of Sir Derek H. R. Barton (Imperial College, England), Theodore Cohen (Univ. Pittsburgh), and E. J. Corey (Harvard Univ.) before joining the faculty at UT Southwestern where he occupies the Robert A. Welch Distinguished Chair in Chemistry. He has contributed extensively to synthetic methodology, organometallic chemistry, and natural products, especially eicosanoids and other signal transduction molecules.

References
- [1].Hall DG, editor. Boronic Acids. 2nd ed. Wiley-VCH; Weinheim: 2011. [Google Scholar]
- [2].a) Miyaura N, Yamada K, Suzuki A. Tetrahedron Lett. 1979;20:3437–3440. [Google Scholar]; b) Miyaura N, Suzuki A. Chem. Commun. 1979:866–867. [Google Scholar]
- [3].a) Suzuki A. In: Metal-Catalyzed Cross-Coupling Reactions. Diederich F, Stang PJ, editors. Wiley-VCH; Weinheim: 1998. pp. 49–98. [Google Scholar]; b) Suzuki A. Pure Appl. Chem. 1991;63:419–422. [Google Scholar]; c) Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457–2483. [Google Scholar]; d) Suzuki A. J. Organomet. Chem. 1999;576:147–168. [Google Scholar]; e) Suzuki A. Angew. Chem. Int. Ed. 2011;50:6722–6737. doi: 10.1002/anie.201101379. [DOI] [PubMed] [Google Scholar]
- [4].a) Matteson DS. In: Boronic Acids. 1st ed. Hall DG, editor. Wiley-VCH; Weinheim: 2005. pp. 305–342. [Google Scholar]; b) Matteson DS. Acc. Chem. Res. 1970;3:186–193. [Google Scholar]; c) Brown HC, Midland MM. Angew. Chem. Int. Ed. 1972;11:692–700. [Google Scholar]; d) Brown HC, Jadhav PK, Mandal AK. Tetrahedron. 1981;37:3547–3587. [Google Scholar]; e) Brown HC, Singaram B. Acc. Chem. Res. 1988;21:287–293. [Google Scholar]; f) Matteson DS. Acc. Chem. Res. 1988;21:294–300. [Google Scholar]; g) Matteson DS. Chem. Rev. 1989;89:1935–1551. [Google Scholar]; h) Brown HC, Ramachandran PV. Acc. Chem. Res. 1992;25:16–24. [Google Scholar]
- [5].a) Scott HK, Aggarwal VK. Chem. Eur. J. 2011;17:13124–13132. doi: 10.1002/chem.201102581. [DOI] [PubMed] [Google Scholar]; b) Partridge BM, Chausset-Boissarie L, Burns M, Pulis AP, Aggarwal VK. Angew. Chem. Int. Ed. 2012;51:11795–11799. doi: 10.1002/anie.201203198. [DOI] [PubMed] [Google Scholar]; c) Watso CG, Aggarwal VK. Org. Lett. 2013;15:1346–1349. doi: 10.1021/ol400289v. [DOI] [PubMed] [Google Scholar]; d) Pulis AP, Blair DJ, Torres E, Aggarwal VK. J. Am. Chem. Soc. 2013;135:16054–16057. doi: 10.1021/ja409100y. [DOI] [PubMed] [Google Scholar]
- [6].a) Tyman JHP, editor. Synthetic and Natural Phenols. Elsevier; New York: 1996. [Google Scholar]; b) Rappoport Z, editor. The Chemistry of Phenols. Wiley-VCH; Weinheim: 2003. [Google Scholar]
- [7].Ainley AD, Challenger F. J. Chem. Soc. 1930:2171–2180. [Google Scholar]
- [8].Hawthorne MF. J. Org. Chem. 1957;22:1001. [Google Scholar]
- [9].Webb KS, Levy D. Tetrahedron Lett. 1995;36:5117–5118. [Google Scholar]
- [10].Travis BR, Ciaramitaro BP, Borhan B. Eur. J. Org. Chem. 2002:3429–3434. [Google Scholar]
- [11].Simon J, Salzbrunn S, Prakash GKS, Petasis NA, Olah GA. J. Org. Chem. 2001;66:633–634. doi: 10.1021/jo0015873. [DOI] [PubMed] [Google Scholar]
- [12].a) Kuivila HG. J. Am. Chem. Soc. 1954;76:870–874. [Google Scholar]; b) Kuivila HG. J. Am. Chem. Soc. 1955;77:4014–4016. [Google Scholar]; c) Kuivila HG, Wiles RA. J. Am. Chem. Soc. 1955;77:4830–4834. [Google Scholar]; d) Kuivila HG, Armour AG. J. Am. Chem. Soc. 1957;79:5659–5662. [Google Scholar]
- [13].Prakashi GKS, Chacko S, Panja C, Thomas TE, Gurung L, Rasul G, Mathew T, Olah GA. Adv. Synth. Catal. 2009;351:1567–1574. [Google Scholar]
- [14].a) Gogoi A, Bora U. Synlett. 2012;23:1079–1081. [Google Scholar]; b) Mulakayala N, Kumar KM, Rapolu RK, Kandagatla B, Rao P, Oruganti S, Pal M. Tetrahedron Lett. 2012;53:6004–6007. [Google Scholar]
- [15].Chen D-S, Huang J-M. Synlett. 2013;24:499–501. [Google Scholar]
- [16].Guo S, Lu L, Cai H. Synlett. 2013;24:1712–1714. [Google Scholar]
- [17].Kianmehr E, Yahyaee M, Tabatabai K. Tetrahedron Lett. 2007;48:2713–2715. [Google Scholar]
- [18].Zhu C, Wang R, Falck JR. Org. Lett. 2012;14:3494–3497. doi: 10.1021/ol301463c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cammidge AN, Goddard VHM, Schubert CPJ, Gopee H, Hughes DL, Gonzalez-Lucas D. Org. Lett. 2011;13:6034–6034. doi: 10.1021/ol202500z. [DOI] [PubMed] [Google Scholar]
- [20].Kaewmati P, Somsook E, Dhital RN, Sakurai H. Tetrahedron Lett. 2012;53:6104–6106. [Google Scholar]
- [21].a) Hosoi K, Kuriyama Y, Inagi S, Fuchigami T. Chem. Commun. 2010;46:1284–1286. doi: 10.1039/b914093j. [DOI] [PubMed] [Google Scholar]; b) Jiang H, Lykke L, Pedersen SU, Xiao W-J, Jørgensen KA. Chem. Commun. 2012;48:7203–7205. doi: 10.1039/c2cc32711b. [DOI] [PubMed] [Google Scholar]
- [22].Qi H-L, Chen D-S, Ye J-S, Huang J-M. J. Org. Chem. 2013;78:7482–7487. doi: 10.1021/jo400981f. [DOI] [PubMed] [Google Scholar]
- [23].Gatenyo J, Vints I, Rozen S. Chem. Commun. 2013;49:7379–7381. doi: 10.1039/c3cc42337a. [DOI] [PubMed] [Google Scholar]
- [24].Molander GA, Cavalcanti LN. J. Org. Chem. 2011;76:623–630. doi: 10.1021/jo102208d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Lawrence SA, editor. Amines: Synthesis, Properties and Applications. Cambridge University Press; Cambridge: 2004. [Google Scholar]; b) Rappoport Z, editor. The Chemistry of Anilines, Parts 1 and 2. John Wiley & Sons; New York: 2007. [Google Scholar]; c) Ricci A, editor. Amino Group Chemistry: From Synthesis to the Life Science. Wiley-VCH; Weinheim: 2008. [Google Scholar]
- [26].Ou L, Shao J, Zhang G, Yu Y. Tetrahedron Lett. 2011;52:1430–1431. [Google Scholar]
- [27].Xiao Q, Tian L, Tan R, Xia Y, Qiu D, Zhang Y, Wang J. Org. Lett. 2012;14:4230–4233. doi: 10.1021/ol301912a. [DOI] [PubMed] [Google Scholar]
- [28].Mlynarski SN, Karns AS, Morken JP. J. Am. Chem. Soc. 2012;134:16449–16451. doi: 10.1021/ja305448w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhu C, Li G, Ess DH, Falck JR, Kurti L. J. Am. Chem. Soc. 2012;134:18253–18256. doi: 10.1021/ja309637r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].a) Ono N, editor. The Nitro Group in Organic Synthesis. Wiley-VCH; New York: 2001. [Google Scholar]; b) Olah GA, Malhotra R, Narang SC, editors. Nitration: Methods and Mechanisms. VCH; New York: 1989. [Google Scholar]
- [31].Salzbrunn S, Simon J, Prakash GKS, Petasis NA, Olah GA. Synlett. 2000:1485–1487. [Google Scholar]
- [32].Prakash GKS, Panja C, Mathew T, Surampudi V, Petasis MA, Olah GA. Org. Lett. 2004;6:2205–2207. doi: 10.1021/ol0493249. [DOI] [PubMed] [Google Scholar]
- [33].Wu X-F, Schranck J, Neumann H, Beller M. Chem. Commun. 2011;47:12462–12463. doi: 10.1039/c1cc15484b. [DOI] [PubMed] [Google Scholar]
- [34].Manna S, Maity S, Rana S, Agasti S, Maiti D. Org. Lett. 2012;14:1736–1739. doi: 10.1021/ol300325t. [DOI] [PubMed] [Google Scholar]
- [35].a) Patai S, editor. The Chemistry of Amino, Nitroso, Nitro and Related Groups. Wiley-VCH; Weinheim: 1996. [Google Scholar]; b) Momiyama N, Yamamoto H. Chem. Commun. 2005:3514–3525. doi: 10.1039/b503212c. [DOI] [PubMed] [Google Scholar]
- [36].Molander GA, Cavalcanti LN. J. Org. Chem. 2012;77:4402–4413. doi: 10.1021/jo300551m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].a) Ramadhar TR, Batey RA. In: Boronic Acids. 2nd ed. Hall DG, editor. Wiley-VCH; Weinheim: 2011. pp. 427–477. [Google Scholar]; b) Petasis NA. In: Multicomponent Reactions. Zhu J, Bienaymé H, editors. Wiley-VCH; Weinheim: 2005. pp. 199–223. [Google Scholar]; c) Candeias NR, Montalbano F, Cal PMSD, Gois PMP. Chem. Rev. 2010;110:6169–6193. doi: 10.1021/cr100108k. [DOI] [PubMed] [Google Scholar]; d) Yu T, Li H, Wu X, Yang J. Chin. J. Org. Chem. 2012;32:1836–1845. [Google Scholar]
- [38].Barluenga J, Tomás-Gamasa M, Aznar F, Valdés C. Nature Chem. 2009;1:494. doi: 10.1038/nchem.328. [DOI] [PubMed] [Google Scholar]
- [39].Peng C, Zhang W, Yan G, Wang J. Org. Lett. 2009;11:1667. doi: 10.1021/ol900362d. [DOI] [PubMed] [Google Scholar]
- [40].a) Elkin PK, Levin VV, Dilman AD, Struchkova MI, Belyakov PA, Arkhipov DE, Korlyukov AA, Tartakovsky VA. Tetrahedron Lett. 2011;52:5259–5263. [Google Scholar]; b) Elkin PK, Levin VV, Dilman AD, Struchkova MI, Arkhipov DE, Korlyukov AA. Tetrahedron Lett. 2012;53:6216–6218. [Google Scholar]
- [41].Argintaru OA, Ryu D, Aron I, Molander GA. Angew. Chem. Int. Ed. 2013;52:13656–13660. doi: 10.1002/anie.201308036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ito K, Tamashima H, Iwasawa N, Kusama H. J. Am. Chem. Soc. 2011;133:3716. doi: 10.1021/ja1102597. [DOI] [PubMed] [Google Scholar]
- [43].Diederich F, Stang PJ, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; Weinheim: 1998. [Google Scholar]
- [44].Clough JM, Diorazio LJ, Widdowson DA. Synlett. 1990:761–762. [Google Scholar]
- [45].Diorazio LJ, Widdowson DA, Clough JM. Tetrahedron. 1992;48:8073–8088. [Google Scholar]
- [46].Petasis NA, Yudin AK, Zavialov IA, Prakash GKS, Olah GA. Synlett. 1997:606–608. [Google Scholar]
- [47].Cazorla C, Métay E, Andrioletti B, Lemaire M. Tetrahedron Lett. 2009;50:3936–3938. [Google Scholar]
- [48].Thiebes C, Prakash GKS, Petasis NA, Olah GA. Synlett. 1998:141–142. [Google Scholar]
- [49].Szumigala RH, Jr., Devine PN, Gauthier DR, Jr., Volante RP. J. Org. Chem. 2004;69:566–569. doi: 10.1021/jo035184p. [DOI] [PubMed] [Google Scholar]
- [50].a) Kabalka GW, Mereddy AR. Tetrahedron Lett. 2004;45:343–345. [Google Scholar]; b) Kabalka GW, Mereddy AR. Organometallics. 2004;23:4519–4521. [Google Scholar]
- [51].Yao M-L, Reddy MS, Yong L, Walfish I, Blevins DW, Kabalka GW. Org. Lett. 2010;12:700–703. doi: 10.1021/ol9027144. [DOI] [PubMed] [Google Scholar]
- [52].Thompson ALS, Kabalka GW, Akula MR, Huffman JW. Synthesis. 2005:547–550. [Google Scholar]
- [53].Akula MR, Yao M-L, Kabalka GW. Tetrahedron Lett. 2010;51:1170–1171. [Google Scholar]
- [54].Molander GA, Cavalcanti LN. J. Org. Chem. 2011;76:7195–7203. doi: 10.1021/jo201313a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Berionni G, Morozova V, Heininger M, Mayer P, Knochel P, Mayr H. J. Am. Chem. Soc. 2013;135:6317–6324. doi: 10.1021/ja4017655. [DOI] [PubMed] [Google Scholar]
- [56].Carroll MA, Pike VW, Widdowson DA. Tetrahedron Lett. 2000;41:5393–5396. [Google Scholar]
- [57].Kerverdo S, Gingras M. Tetrahedron Lett. 2000;41:6053–6057. [Google Scholar]