

Abstract
Comparative analysis of a number of studies in drought‐stressed maize (Zea mays L.) reporting quantitative trait loci (QTLs) for abscisic acid concentration, root characteristics, other morpho‐physiological traits (MPTs) and grain yield (GY) reveals their complex genetic basis and the influence of the genetic background and the environment on QTL effects. Chromosome regions (e.g. near umc11 on chromosome 1 and near csu133 on chromosome 2) with QTLs controlling a number of MPTs and GY across populations and conditions of different water supply have been identified. Examples are presented on the use of QTL information to elucidate the genetic and physiological bases of the association among MPTs and GY. The QTL approach allows us to develop hypotheses accounting for these associations which can be further tested by developing near isogenic lines (NILs) differing for the QTL alleles. NILs also allow for a more accurate assessment of the breeding value of MPTs and, in some cases, may allow for the map‐based cloning of the gene(s) underlying the QTL. Although QTL analysis is still time‐consuming and resource‐demanding, its integration with genomics and post‐genomics approaches (e.g. transcriptome, proteome and metabolome analyses) will play an increasingly important role for the identification and validation of candidate genes affecting MPTs and GY.
Key words: Review, quantitative trait locus, QTL, drought stress, maize, genomics, ABA, roots, grain yield, near isogenic lines, NILs, Zea mays
INTRODUCTION
In the past decades, considerable effort has been devoted to investigating the relationships between genetic variation in morpho‐physiological traits (MPTs) and adaptation of crop plants to drought conditions (Blum, 1988; Richards, 1988; Ludlow and Muchow, 1990; McDonald and Davies, 1996; Passioura, 1996). More recently, this enduring challenge has laid more emphasis on the effects of drought at the cellular and molecular levels (Bray, 1997; Bajaj et al., 1999; Zhang et al., 2000; Bohnert and Bressan, 2001; Knight and Knight, 2001). Although the wealth of information produced by these studies has considerably enhanced our understanding of how plants can adapt to water‐stressed conditions, the improvement in crop performance has seldom benefited from this knowledge. Due to the extremely complex genetic basis of yield, improving and stabilizing crop performance under drought‐stressed conditions remains a slow, laborious and mostly empirical process.
Progress in increasing yield and its stability under water‐limited conditions through a direct selection has been hampered by the low heritability of yield, particularly under drought, and by its large ‘genotype × environment’ interaction (Blum, 1988; Ceccarelli and Grando, 1996). As an alternative to a direct selection for yield under drought conditions, MPTs genetically correlated with yield have been targeted in selection programmes pursued in collaboration between physiologists and breeders (Crosbie, 1982; Blum, 1988). The successful application of this strategy requires MPTs that are cheap and easy to score, characterized by a high genetic correlation with yield and heritability higher than that of yield. Unfortunately, only a very small number of MPTs meet these prerequisites, which is why only a few ‘success’ stories have so far been reported for enhancing yield under drought by applying an indirect selection for MPTs (Richards, 1996; Ribaut et al., 1997a). Additionally, it remains difficult to identify accessions (genotypes) with as many as possible ‘favourable’ (in terms of effects on yield) alleles at the loci governing the expression of the desired MPT.
The expression of MPTs is usually governed by several loci and thus favourable alleles of all such loci are likely to be dispersed in different combinations in the accessions of each crop. Therefore, even when crossing parental lines carrying the desirable alleles at most of the major loci regulating the targeted MPT, the probability of recovering at least one recombinant genotype carrying favourable alleles at all the major loci for the trait under selection is extremely low. For example, if an F2 population segregates for ten unlinked and functionally polymorphic genes controlling a particular trait, then more than 2·4 × 106 F2 plants should be considered to retrieve, at a probability level (P) of 0·90, at least one F2 plant homozygous for the favourable alleles at all ten loci, according to the formula (1 – (1/4)10)n = 0·10, where ‘n’ is the number of plants to be considered. The underlying but clearly unrealistic assumption is the possibility of correctly identifying such genotypes based solely on the phenotype. This goal might possibly be achieved for highly heritable traits (e.g. flowering time) by evaluating in replicated experiments the selfed progenies (e.g. F3 or, preferably, later generations) of such plants or, for species which can be vegetatively propagated, a number of clones. However, the costs associated with this approach would clearly be prohibitive.
The traditional methods applied to investigate the genetic control of multigenic (quantitative) traits such as MPTs and yield in a segregating population (Falconer, 1981; Hallauer and Miranda Fo, 1988), although valuable, are insufficient to provide us with information on: (1) the chromosome regions regulating the variation of each trait; (2) the simultaneous effects of each chromosome region on other traits and the genetic basis (pleiotropy and/or linkage) of such associated effects; and (3) the interpretation of possible cause–effect relationships among traits. Some of these constraints can now be partially overcome by using molecular markers which not only allow for the identification of the quantitative trait loci (QTLs) controlling the chosen MPT, but also enable us to assess the effects of the same QTL region on other traits (Tanksley, 1993; Prioul et al., 1997) and, more importantly, on yield (Stuber et al., 1987, 1999).
The main objectives of this article are to: (1) summarize the basic principles of QTL analysis; (2) illustrate how QTL analysis can help in elucidating the genetic basis of traits association; (3) review the published information in maize (Zea mays L.) on QTLs for abscisic acid (ABA) concentration and root traits, both of which are involved in the adaptive response of maize to drought; (4) analyse and interpret the co‐location of the QTLs for ABA concentration and root traits with QTLs for other MPTs (e.g. anthesis–silking interval, stomatal conductance, etc.) and for grain yield in maize; (5) critically analyse the limitations and future perspectives of QTL analysis; (6) present alternative approaches to uncover the presence of QTLs; and (7) illustrate how QTLs can lead to gene discovery. The current understanding of the breeding value of the MPTs herein considered is good for the anthesis–silking interval only (Bolaños and Edmeades, 1996, 1997; Ribaut et al., 1997a), and is otherwise poor, as in the case of root traits, or even controversial, as in the case of ABA concentration (Blum, 1988; Quarrie, 1993, 1996; Landi et al., 1995, 2001b; Setter, 1997; Sanguineti et al., 1999; Mugo et al., 2000). For the sake of clarity, we will adopt the terminology of Ludlow and Muchow (1990), which distinguishes traits providing drought escape and drought resistance, with the former further subdivided in terms of dehydration avoidance and dehydration tolerance.
PRINCIPLES OF QTL ANALYSIS
The ultimate goals of QTL analysis are to dissect the complex inheritance of quantitative traits into ‘Mendelian‐like’ factors amenable to selection through the analysis of the flanking molecular markers and to clone the genes underlying the QTLs. The dissection of a quantitative trait into its discrete genetic components is generally made possible through the production of an experimental population (e.g. F2 plants, F3 families, recombinant inbred lines, double haploids, etc.) starting from an F1 cross between two inbred lines showing genetic variation for the trait(s) of interest (Tanksley, 1993; Lee, 1995; Quarrie, 1996; Prioul et al., 1997). The number of plants or progenies considered for a QTL study usually varies from approx. 100 (Lebreton et al., 1995; Agrama and Moussa, 1996; Guingo et al., 1998; Tuberosa et al., 1998b) to over 400 (Openshaw and Frascaroli, 1997; Melchinger et al., 1998).
Typically, a QTL study includes an accurate phenotypic evaluation of an adequately large mapping population, its molecular profiling and a statistical analysis to test the association between a phenotype and a marker genotype. For most species, an adequate coverage of the genome can be achieved with approx. 100–150 marker loci evenly spaced along the chromosomes. Once a marker density of approx. one marker/20 cM is reached, then it becomes more profitable to increase the number of progenies rather than the number of markers to increase the accuracy of QTL detection (Darvasi et al., 1993).
At its simplest, QTL analysis relies on one‐way ANOVA testing whether the phenotypic means of the possible genotype classes at a specific chromosomal position are significantly different. If a significant difference is found, then it can be concluded that a QTL is probably linked to the chromosomal position under investigation. A major limitation of ANOVA is that it does not provide information on the distance of the QTL from the associated marker; furthermore, it may not be possible to ascertain whether the detected effect is due to a minor QTL tightly linked to the marker, or to a major, but more distant, QTL. These limitations can be overcome through the use of statistical approaches based on information of multiple markers which provide greater accuracy for QTL mapping (Martinez and Curnow, 1992; Zeng, 1994; Utz and Melchinger, 1996; Liu, 1998; Kao et al., 1999; Korol et al., 2001; Sen and Churchill, 2001). In this case, a frequently used output‐statistic to describe the results is the LOD (logarithm of the odds ratio) score; the LOD value at a particular chromosome position is computed as log10 of the ratio between the chance of a real QTL being present given the effect measured at that position divided by the chance of having a similar effect with no QTL being present. The most likely position of the QTL is thus at the peak of the LOD profile. The graphical output can also provide us with a confidence interval around the QTL peak, thus delineating the range of the most likely QTL position moving away from each side of the peak. To avoid declaring ‘false‐positive’ QTLs (i.e. declaring the presence of a QTL when the QTL is absent), the threshold value of the LOD score should be set reasonably high (usually >2·0).
The large volume of literature available on the diverse statistical approaches, experimental designs and other general features of QTL analysis will not be reviewed in detail here because several authors have already thoroughly addressed such aspects (Jansen, 1993; Tanksley, 1993; Jansen and Stam, 1994; Lee, 1995; Beavis, 1998; Lynch and Walsh, 1998; Churchill and Doerge, 1999; Flint and Mott, 2001; Hackett, 2002; Mauricio, 2001). However, a limited number of related issues will be briefly addressed as necessary.
Although the principles of QTL analysis were first outlined and successfully applied in the early 1920s to map a QTL for seed size in bean tightly linked to a gene controlling seed pigmentation (Sax, 1923), a wide‐scale application of QTL analysis was not possible at that time due to the paucity of genetic markers available. The systematic identification and characterization of QTLs was finally made possible more than 50 years later, following the introduction of the first class of molecular markers (restriction fragment length polymorphisms, RFLPs) suitable for an adequately detailed genome‐wide survey (Botstein et al., 1980). In maize, the first linkage map based on molecular markers was established by Helentjaris et al. (1986). The time‐consuming process of map construction was considerably shortened with the introduction of the PCR‐based microsatellite (SSR, simple sequence repeat) markers, particularly suited for mapping purposes due to their high level of polymorphism (Taramino and Tingey, 1996). From the mid‐1990s, the addition of AFLPs (amplified fragment length polymorphisms; Vos et al., 1995) has provided an unprecedented level of map saturation (Vuylsteke et al., 1999). Also, sequenced cDNAs (also known as ESTs, expressed sequence tags) became a source of molecular markers and have now been integrated into maize genetic maps (Causse et al., 1996; Davis et al., 1999; Sharopova et al., 2002).
The comparative analysis between QTL data of two or more mapping populations is made possible by directly using the information provided by markers (usually RFLPs and/or SSRs) common to the maps being compared and/or indirectly by referring each mapping population to a reference map. A widely‐used reference map for maize is the UMC map (Davis et al., 1999), which contains more than 1700 markers. To facilitate the cross‐referring to QTL studies in maize or other cereals, the UMC map has been subdivided into 100 sectors (bins) marked by reliable RFLP markers. The bin framework has been shown to be extremely useful for comparison of QTL positions across experiments (Lin et al., 1995) and it will also be exploited throughout this review for comparing QTLs for MPTs and grain yield across experiments and genetic backgrounds. It is noteworthy that the average genetic length of bins (approx. 17 cM in the map reported by Davis et al., 1998) is roughly similar to the average chromosome interval supporting a QTL peak. Additionally, the UMC map allows us to compare the map position of mutants (Neuffer et al., 1997) with that of QTLs, thus contributing relevant information for the identification of possible candidate genes for a particular trait. Robertson (1985) even postulated that a mutant phenotype at a particular locus may be caused by an allele whose effect is more drastic than that of the QTL alleles at the same locus. In maize, Robertson’s hypothesis has been validated for plant height when a QTL for this trait was found to co‐localize with the map position of the mutant dwarf3 (Touzet et al., 1995; Winkler and Helentjaris, 1995), and for plant architecture when a QTL controlling the level of branching was shown to coincide with the mutant tb1 (Doebley et al., 1995).
In principle, once QTLs have been identified, introgression of the favourable alleles and their pyramiding into elite germplasm (e.g. parental lines, populations, etc.) becomes possible through marker‐assisted selection (MAS; Ribaut and Hoisington, 1998; Stuber, 1999; Young, 1999). However, only a few successful applications of MAS for the improvement of quantitative traits have been described to date (Hu et al., 1997; Ragot et al., 2000; Ribaut et al., 2000) due mainly to weak associations (in terms of genetic distance) between markers and target QTLs and/or the high costs of MAS (Stuber et al., 1999; Salvi et al., 2001). Certainly, a more rosy picture for MAS emerges considering single‐gene traits such as disease resistance (Bus et al., 2000; Witcombe and Hash, 2000).
QTL ANALYSIS AS A MEANS TO INVESTIGATE ASSOCIATIONS BETWEEN TRAITS
A number of approaches can be utilized to analyse the physiological and genetic basis of the associations between traits. Physiological relationships can be established by analysing correlated changes occurring in time or following treatments influencing the target traits (Quarrie and Jones, 1977); these studies can be conducted using a single accession (genotype). The presence of a genetic association between traits can be investigated by testing a number of either unrelated genotypes differing in their mean value for the traits of interest (Tuberosa et al., 1994), or related families derived by divergent selection started from a population segregating for the target trait (Innes et al., 1984; Tuberosa et al., 1986; Landi et al., 2001a, b). Each of these approaches has flaws and is prone to biases, so rarely allows us to ascertain with reasonable precision the extent to which the association is due to pleiotropy and/or linkage. Alternatively, QTL analysis enables us to investigate with greater precision the genetic basis of trait association merely by looking for co‐location on the genetic map of the corresponding QTLs. Consequently, QTL studies provide us with useful information to elucidate the causal pathways linking two or more traits (Simko et al., 1997), as is the case of MPTs and yield under drought (Lebreton et al., 1995; Prioul et al., 1997; Sanguineti et al., 1999). Co‐location of QTL peaks for two traits can result from a number of alternative scenarios as shown in Fig. 1 (redrawn after Lebreton et al., 1995): (A) two tightly linked genes modulating the expression of separate traits, but not separated by the statistical tests adopted; (B) one gene with a single function which leads to a sequence of causally related events; (C) one gene with an effect on two or more traits independent of each other; or (D) two tightly linked genes with effects on the same two or more traits. Linkage, namely the first scenario, can be distinguished from pleiotropy, the second and third scenarios, whenever an improved mapping resolution (e.g. increasing the number of tested progenies and/or more markers mapped in the region of interest) leads to the identification of recombinant progenies for the two associated traits, i.e. the parental alleles of the linked genes controlling the two traits separate in one or more recombinant progenies. The opposite finding (i.e. no recombinant progeny) cannot prove with certainty that pleiotropy is the cause of the association; however, pleiotropy becomes increasingly plausible when co‐segregation of the two traits is maintained when an increasing number of segregating individuals are considered. The final proof for pleiotropy can be obtained through the cloning and manipulation of the gene/s underlying the QTL in question. Further complexity can be added to the models presented in Fig. 1A–C because co‐location of QTLs could also be due to the combined effects of linked genes with possible pleiotropic effects as shown in Fig. 1D.
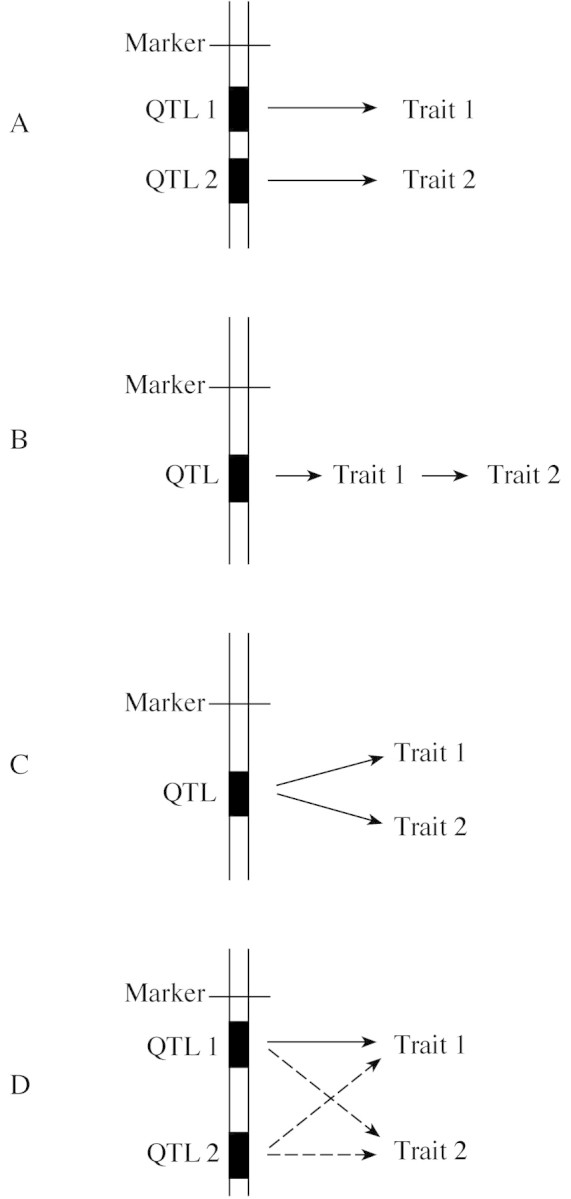
Fig. 1. (redrawn after Lebreton et al., 1995) Four models for the possible association between two traits whose expression is influenced by the same chromosome region. A, Linkage. Traits 1 and 2 controlled independently by two distinct QTLs which are closely linked but not separated by the statistical tests adopted. B, Pleiotropy. Two traits causally associated and regulated by a QTL acting directly on one trait only. C, Pleiotropy. Traits 1 and 2 controlled independently by a single gene. D, Linkage and pleiotropy. Traits 1 and 2 controlled independently by two distinct QTLs which also have pleiotropic effects (see text for an example).
For an example of pleiotropic effects as depicted in Fig. 1C and pertinent to this article, we will consider leaf ABA concentration and root growth (corresponding to traits 1 and 2, respectively, in Fig. 1). Under drought conditions, the scenario presented in Fig. 1C could be reconciled with either (1) a gene influencing the signalling cascade (e.g. gene encoding a signal receptor) leading from the perception of the drought stress to the activation of the genes independently governing leaf ABA concentration and root growth; or (2) a regulatory gene encoding a protein (e.g. transcription factor) with a DNA‐binding domain capable of promoting the expression of the independent genes influencing leaf ABA concentration and root growth. Eventually, the scenario described in Fig. 1B, which implies a causal effect of ABA on root growth, can be validated if a QTL for ABA concentration in root tips is also mapped in the same region. However, it should be noted that, given the complex relationships between ABA concentration and root growth, the opposite explanation (i.e. root growth influencing ABA concentration) is also plausible, particularly at a later stage of development (e.g. near flowering). Therefore, when considering traits such as ABA concentration and root size whose cause–effect relationships may switch according to the growth stage (i.e. from ABA having a greater influence on root size at an early stage, to root size influencing ABA at a later stage; see also Sanguineti et al., 1999), the interpretation of the results becomes more complex.
When considering two or more complex traits for which several QTLs have been mapped, co‐location of the QTL peaks can occur by mere chance due to linkage. For example, in maize, where the map has been subdivided into bins of approximately similar genetic length, if a number (N) of QTLs for traits A and B have been mapped to NA and NB chromosome bins, respectively, then the percentage of bins expected to harbour, by mere chance, QTLs for both traits should be equal to [(NA × NB)/NT2] × 100, where NT indicates the total number of chromosome bins covered by the map used to identify the QTLs. The application of this formula assumes: (1) the possibility of correctly assigning the position of the QTL peak to a single chromosome bin, an assumption not always met with QTLs characterized by wide support intervals and maps with low marker density; and (2) a uniform distribution of the QTLs among bins, which is also a rather unlikely assumption in maize as already shown by the work of Khavkin and Coe (1997). As more data on QTL distribution become available for maize, the extent of the bias introduced by the non‐uniform distribution of QTLs among chromosome bins could be accounted for in the formula including appropriate coefficients for each bin. On the same line, the formula will underestimate the degree of overlap whenever the QTLs are only 5–10 cM apart but are assigned to adjacent bins.
Another indirect and empirical way of evaluating whether linkage or pleiotropy causes trait association at a number ‘N’ of regions where the QTLs for the traits in question overlap is provided by the analysis of the sign of the additive effects of the QTLs (computed as half of the difference between the average values of the parental QTL alleles) at all ‘N’ regions. Although genetically linked genes may account for the association between two traits at any particular QTL, it is very unlikely that linkage is the cause of the association between the same traits at a number of different QTLs when the sign of the association (positive or negative) between the genetic effects remains the same at all QTLs. In fact, if linkage causes the association, the chances of finding alleles linked in coupling (i.e. ‘plus’ or ‘minus’ alleles at both loci controlling the two traits) equal the chances of finding alleles linked in repulsion (i.e. one ‘plus’ and one ‘minus’ allele at each one of the two loci), unless selection has favoured one vs. the other, an event rather unlikely to occur at a number of independent loci. Conversely, if pleiotropy is the primary cause of the association between two traits, then it is more likely that a similar relationship is found at most, if not all, of the QTLs where the overlap occurs. Therefore, the probability of finding the same linkage phase (coupling or repulsion) by chance alone at a number of ‘N’ QTL regions equals 0·5N. However, the presence of contrasting associations at regions showing co‐location of QTLs does not disprove pleiotropy, since the sign of the association between traits could well vary according to the growth stage and the environmental conditions present when the gene(s) underlying that particular QTL was expressed, as previously indicated for the association between ABA concentration and root size (see also Sanguineti et al., 1999).
The first attempt to utilize QTL data to test for possible causal relationships among MPTs in drought‐stressed maize was carried out by Lebreton et al. (1995) who suggested that if trait A has a regulatory role on trait B (e.g. xylem ABA controlling stomatal conductance), the most significant QTLs for trait A should have a measurable effect at the same QTL regions on trait B. Conversely, if trait B (e.g. stomatal conductance) has a more complex genetic basis as compared with trait A (e.g. xylem ABA), then trait B will probably have significant QTLs not coincident with those for trait A.
LITERATURE REVIEW FOR QTLS FOR ABA CONCENTRATION AND ROOT TRAITS
QTLs for ABA concentration
An increase in ABA concentration is a universal and firmly established response of plants subjected to drought and other abiotic stresses (Larqué‐Saavedra and Wain, 1976; Davies and Zhang, 1991; Quarrie, 1991; Ribaut and Pilet, 1991). Extensive evidence indicates the pivotal role of ABA in regulating several processes at the molecular, cellular, organ and whole plant levels under conditions of water deficit (Zeevaart and Creelman, 1988; Saab et al., 1995; Sharp, 1996; Bray, 1997; Netting, 2000). ABA modulates the expression of a large number of genes whose products may protect the cell from the harmful effects of an excessive water loss (Close, 1996, 1997; Ingram and Bartels, 1996; Bray, 1997). In maize seedlings subjected to artificially induced conditions of water deficit (Sharp, 1996), an increased ABA concentration increased the root to shoot ratio, an adaptive change which could be important for avoiding dehydration under conditions of low water availability in the upper layer of the soil profile. It has been suggested that the role of ABA in maintaining root cell elongation at low water potential may involve enhanced expression or activity of cell wall‐yielding factors through an interaction with ethylene production (Wu et al., 1996; Spollen et al., 2000). It has also been shown that ABA affects proline accumulation throughout maize primary roots (Ober and Sharp, 1994); this suggests that ABA may play a regulatory role in maintaining the osmotic status of the roots at low water potential (McDonald and Davies, 1996). The most widely recognized adaptive role of an increased ABA concentration under water‐limited conditions is the reduction of stomatal conductance and water lost by transpiration (Tardieu et al., 1992, 1993; Tuberosa et al., 1994; Trejo et al., 1995; Li et al., 2000). It has also been shown that ABA facilitates water uptake into maize roots as soil starts drying, particularly under non‐transpiring conditions, when the apoplastic path of water transport is largely excluded (Hose et al., 2000). More recent work carried out in arabidopsis has shown that ABA also plays an important role in mediating the stimulation of lateral root elongation by local NO3– applications (Signora et al., 2001).
A literature search of the bibliographic databases indicates that QTLs for leaf ABA (L‐ABA) concentration in maize have been investigated in three different populations (Lebreton et al., 1995; Tuberosa et al., 1998a, b). One of these studies has also reported QTLs for the concentration of ABA in the xylem sap (X‐ABA; Lebreton et al., 1995). Figure 2 reports a tentative allocation of the QTLs for L‐ABA and X‐ABA of the three populations to the bins of the UMC reference map. The large number of QTLs governing variation in ABA concentration in these populations should be related to their different genetic backgrounds as well as to differences in the growing conditions (e.g. glasshouse or field), dynamics (e.g. duration, timing and intensity) of the water‐stress episodes and in the growth stage at which plants were subjected to drought. Altogether, these results confirm and expand the complexity of the genetic basis of ABA concentration in maize which had already been postulated in earlier studies based on more conventional genetic approaches (Ivanovic et al., 1992; Sanguineti et al., 1996).

Fig. 2. Bin allocation on the maize map of the QTLs for root traits in hydroponics and in the field, ABA concentration in the leaf and in the xylem sap, anthesis–silking interval (ASI) and grain yield (GY) in 11 maize populations (a–k). Coloured bars indicate QTLs assigned to a single bin; within each bin, bar position does not reflect the most likely position of the QTL peak. Vertical parentheses spanning two bins indicate QTLs that could not be assigned to a single bin. According to the population considered, numbers in parentheses indicate the following: number of root characteristics for which a QTL was detected in hydroponics (populations a and b) or in the field (populations a, c and d); number of samplings and/or environments in which a QTL was detected for leaf ABA concentration (populations c, e and f); ASI populations a, b, e, g and h); and grain yield (GY; populations a, b, c, e, g, h, i, j and k). Asterisks indicate QTLs for the concentration of ABA in the xylem sap. QTLs for root traits and ABA concentration are reported to the left of each chromosome, while QTLs for ASI and GY are reported to the right of each chromosome. For the sake of simplicity, the QTLs for GY of the B73 × H99 population tested under low‐ and high‐nitrogen conditions have been indicated as obtained under water‐stressed and well‐watered conditions, respectively.
A more detailed analysis of the results of the above‐mentioned experiments indicates that the highest number (16) of QTLs for L‐ABA was reported in the Os420 × IABO78 background (Tuberosa et al., 1998c). The high number of QTLs identified in this 2‐year study investigating ABA at two growth stages (at stem elongation and tasseling) is most likely to be the result of the increased chances of detecting QTLs in at least one of the four samplings analysed, the large variation in L‐ABA among the 80 F4 families (from 264 to 680 ng ABA g–1 d. wt) and the high heritability of L‐ABA (from 0·68 to 0·88). Of the 16 QTLs identified, only four significantly influenced mean L‐ABA values across samplings. At all these four primary QTLs, the effect of the parent alleles was consistent when analysed in each individual sampling; the alleles which increased L‐ABA were contributed by Os420, the high‐ABA parent line. The most important and consistent QTL mapped on bin 2·04 near csu133. The effects associated with the QTL near csu133 were more pronounced near flowering. In the same region, a QTL for L‐ABA has also been described in Polj17 × F‐2 at two growth stages (Lebreton et al., 1995); also in this case, the QTL showed a stronger effect near anthesis. Additionally, preliminary results have indicated that in the population described in Ribaut et al. (1996, 1997c) the QTL likelihood profile for ABA concentration in the ear peaks near csu133 (M. Ribaut and T. Setter, pers. comm.).
The presence of a QTL for L‐ABA near csu133 has been validated through a divergent selection for L‐ABA started from 480 (Os420 × IABO78) F2 plants (Sanguineti et al., 1996; Landi et al., 2001a). Following the completion of two cycles of selection (from F2 to F4) for high or low L‐ABA under conditions of moderate drought stress in the field, the csu133 profiles of the eight F4 families with the highest ABA values and the eight F4 families with the lowest ABA values were investigated. The csu133 allele associated with the low L‐ABA parental allele was fixed in all eight low L‐ABA families, while the csu133 allele associated with the high L‐ABA parental allele was fixed in seven high L‐ABA families, with only one F4 family being heterozygous (Salvi et al., 1997). These results provided convincing evidence that the chromosome region near csu133 harbours one or more genes with a strong effect on L‐ABA. The consistency of the LOD values (up to 7·1) and the size of the additive effects (up to 86 ng ABA g–1 d. wt) shown by this QTL on L‐ABA coupled with its narrow support interval (approx. 5 cM) in Os420 × IABO78 (Tuberosa et al., 1998c) prompted the derivation of backcross‐derived lines (BDLs) in the Os420 × IABO78 genetic background (Landi et al., 2002). Six parallel cycles of backcrossing allowed us to obtain BDLs homozygous for the high‐ABA allele or for the low‐ABA allele of both parents (Os420 and IABO78). The backcross procedure was aided by using the two closely linked RFLP loci flanking (approx. 9 cM apart) the target QTL. Preliminary results obtained under conditions of moderate drought in the field have shown a significant difference between the mean value of BDLs homozygous for the allele increasing ABA (provided by Os420) and BDLs homozygous for the allele decreasing ABA (provided by IABO78). In particular, the additive effect of the QTL averaged across BDLs was equal to 31 ng ABA g–1 d. wt, corresponding to 12·4 % of the overall mean (Landi et al., 2002). In our previous investigation conducted on F4 families derived from the same cross (Tuberosa et al., 1998c), the additive effect at this QTL was estimated to be equal to 49 ng ABA g–1 d. wt, corresponding to 12·3 % of the overall mean. These results thus indicate that the target QTL was successfully transferred by the marker‐assisted backcross, and further confirm the stability and the relative importance of its additive effect.
As compared with the previous study, a smaller number (seven; see Fig. 2) of QTLs was found to influence L‐ABA at two growth stages (early‐ and mid‐phases of stem elongation) in the 151 F3 families derived from the cross A662 × B73 which were tested under rainfed conditions in two locations (Tuberosa et al., 1998a). The limited number of QTLs for L‐ABA evidenced in this study was probably a consequence of the low level of water stress experienced by the plants at the time of sampling, as indicated by the low average value of L‐ABA in all four samplings (from 192 to 241 ng ABA g–1 d. wt), and/or the small difference in L‐ABA value of the parental lines (235 and 266 ng ABA g–1 d. wt in A662 and B73, respectively, across samplings; unpubl. res.). Two QTLs (on bins 5·06–5·07 and 8·07) influenced L‐ABA in both locations, while no overlap was evidenced between growth stages within each location.
A high number of QTLs influenced ABA concentration in leaf and xylem samples collected in 81 F2 plants derived from the cross Polj17 × F‐2 (Fig. 2; Lebreton et al., 1995). Samples were collected 3 weeks before flowering under well‐watered conditions and at flowering under conditions of water deficit. With the exception of chromosome 8, which was poorly represented (only two markers), all other chromosomes were found to harbour QTLs influencing ABA concentration. Of these regions, one on bin 1·06 and one on bin 2·04 near csu133 significantly influenced L‐ABA at both water regimes. The QTL with the strongest effect (LOD = 8·0) under drought conditions was located on bin 3·06 near umc39b. The QTLs for X‐ABA showed a poor overlap with those for L‐ABA, a result in keeping with the low correlation found between these two traits in maize (Zhang and Davies, 1990; Tardieu et al., 1991; Tuberosa et al., 1994). Only two of the four QTLs which influenced leaf water potential (LWP) were located in regions near QTLs for X‐ABA or L‐ABA, a result which led Lebreton et al. (1995) to suggest that it was unlikely that variation in L‐ABA was consequent to variation in the degree of drought stress. In this case, no data were collected on leaf relative water content (LRWC), another indicator of the water status of the plant.
A fairly extensive overlap among QTLs for L‐ABA and QTLs for LRWC was found in Os420 × IABO78: of the 16 QTLs that significantly affected L‐ABA, seven concomitantly influenced LRWC in at least one of the two growth stages considered (Sanguineti et al., 1999). At all QTLs but one (mapped on bin 10·04), the corresponding additive effects for LRWC and L‐ABA were negatively associated, a finding that suggests that in this case L‐ABA mainly represented an indicator of the level of drought stress experienced by the plant at sampling, contrary to the results reported by Lebreton et al. (1995). It is most likely that this apparent discrepancy is due to the different traits considered to survey the water status of the plant and/or to the different genetic backgrounds of the material evaluated in the two studies. Additionally, the differences in the expected inbreeding levels of the two populations (0·875 in the F4 families of Os420 × IABO78 vs. 0·500 in the F2 plants in Polj17 × F‐2) may have influenced their vigour and thus may have magnified the effects on L‐ABA of differences in the water status of the plants.
Tuberosa et al. (1998c) investigated whether known mutants affected in ABA biosynthesis might be possible candidates for the QTLs controlling L‐ABA. Based on information provided by RFLP markers common to the UMC map and the Os420 × IABO78 map, it was shown that the map positions of mutants impaired in ABA biosynthesis (e.g. vp5, vp14, etc.) were outside the support intervals of the QTLs influencing L‐ABA. The rate‐determining step for ABA biosynthesis is controlled by vp14 (Schwartz et al., 1997; Milborrow, 2001), which has been mapped to bin 1·08 (Tan et al., 1997). Also, it should be noted that in both A662 × B73 and Polj17 × F‐2, no QTL was found for ABA concentration in bin 1·08. These results, while providing no evidence that the major gene involved in the biosynthesis of ABA might be responsible for the QTLs for L‐ABA, leave the question open as to what sort of genes may underlie these QTLs. Among a number of possibilities, feasible candidates could be the genes influencing the intensity of the transduction signal associated with turgor loss, a major determinant in the regulation of ABA concentration (Jensen et al., 1996), and/or genes controlling MPTs (e.g. root size and architecture, leaf area, leaf angle, osmotic adjustment, etc.) affecting the water balance of the plant, hence its turgor. Interestingly, the results of Lebreton et al. (1995) showed that the QTL for L‐ABA near csu133 (bin 2·04) clearly overlapped with a QTL for root pulling force. Additionally, Quarrie et al. (1999) reported that recurrent selection for grain yield under drought conditions significantly changed allele frequencies at csu133 in two populations developed at the International Maize and Wheat Improvement Center (CIMMYT), namely Tuxpeño Sequia (Bolaños and Edmeades, 1993) and Drought Tolerant Population (Bolaños et al., 1993). Collectively, these results support the importance of this region in controlling drought‐related traits and yield in maize.
QTLs for root traits
Although root characteristics have been shown to be important factors influencing drought avoidance (Sullivan, 1983; O’Toole and Bland, 1987), little is known about their genetic control and their contribution to yield under drought conditions. This is largely due to the difficulty in properly investigating roots in a large number of plants, particularly in field experiments. In field‐grown maize, root pulling force (RPF) is one of the traits which allows for large‐scale investigation of roots below the soil surface (Kevern and Hallauer, 1983; Fincher et al., 1985). It is important to determine the extent to which RPF reflects root characteristics investigated with more precise but much more laborious approaches like the analysis of root density in soil cores. Sanguineti et al. (1998) have shown that maize root density in both the 30–70 cm and in the 70–110 cm layers was positively associated with RPF (r = 0·67 and 0·71, respectively) in a set of inbred lines differing in root size and architecture; in contrast, no significant association with RPF was evident in the 0–30 cm layer. It should also be mentioned that RPF was one of the MPTs significantly affected by eight cycles of recurrent selection for grain yield under drought conditions in tropical maize (Bolaños et al., 1993). In this case, RPF was negatively associated with grain yield, which led the authors to suggest that improved drought tolerance was due to increased partitioning of biomass towards the developing ear during a severe drought stress coinciding with flowering, rather than a change in plant water status.
Lebreton et al. (1995) identified QTLs for root traits in the Polj17 × F‐2 population also investigated for L‐ABA and X‐ABA (see previous section). The phenotypic evaluation focused on seminal root number (SRN), nodal root number at the base of the stem (NRN) and on RPF measured at the end of the season. There were seven QTLs with a LOD value higher than 2·0 for RPF and four for both SRN and NRN. Because QTL data were also available for ABA concentration, the causal relationships between root traits and ABA concentration were analysed by adopting the procedures summarized earlier (see ‘QTL analysis as a means to investigate associations between traits’). The findings supported the hypothesis that ABA concentration was more likely to regulate RPF than vice versa, an interpretation consistent with the positive relationship between the endogenous ABA concentration and primary root growth in maize seedlings grown under artificial conditions of drought stress (Sharp et al., 1994; McDonald and Davies, 1996). At all QTLs but one, the sign of the additive effects on ABA concentration and RPF was similar. This result could be due to one or more genes in this particular QTL region regulating root growth independently from ABA but capable of influencing its concentration via the water status of the plant: in this case, the QTL effects on root growth and ABA concentration are more likely to be negatively correlated. As to NRN, the comparative analysis of the effects at the most important QTLs revealed a striking correlation between nodal root number and X‐ABA (r = 0·84, P < 0·001), which led the authors to suggest that ABA content in the xylem collected from the leaves is largely determined by the nodal roots.
Under field conditions, Guingo et al. (1998) described maize QTLs for root traits in a 2‐year study carried out with 100 test‐crosses obtained from crossing an early dent line (F252) with 100 recombinant inbred lines (RILs) derived from the cross F‐2 × Io (Causse et al., 1996). Among other traits, the number of nodal roots (below soil surface) on internodes 6, 7 and 8 (RI6, RI7 and RI8, respectively) and the mean diameter of roots on internode 7 (Droot7) were considered. Although the heritability of these traits was fairly high (from 0·56 to 0·62), only one QTL was found to significantly influence the variation measured for RI6, RI7 and RI8, while three QTLs were found for Droot7. The small number of QTLs detected for these traits is probably related to the limited number of test‐cross families considered and/or, at least for RI6, RI7 and Droot7, to the rather limited variation reported in the mean values of the test‐crosses. Accordingly, the coefficients of total genetic determination (R2) were low (from 21 to 44 %), a finding which suggests that the majority of QTLs modulating the investigated traits went undetected. Epistasis was another possible reason invoked by Guingo et al. (1998) to account for the small number of QTLs detected in their study. The only QTL that concomitantly influenced two root traits (RI7 and RI8) was on bin 5·05 between SC343B and SC403; from a practical standpoint, the co‐location of QTLs for different root traits indicated that MAS for this QTL region on chromosome 5 could have beneficial effects on root lodging and yield.
The main limitations to field studies of roots in the soil are the large amount of work required and the usually destructive nature of the measurements (Beck et al., 1987). As an alternative to field studies, hydroponics offers a number of noticeable advantages for investigating roots. These include easy access to roots, non‐destructive and subsequent observations on the same plants, coupled with the possibility of measuring a large number of plants in a small space; furthermore, the addition of polyethylene glycol (Nagy et al., 1995) allows us to test plants under predetermined and uniform conditions of water stress, a condition very difficult to achieve otherwise. The most distinct disadvantage of hydroponics is the very unnatural environment in which roots grow. It is important to appreciate that in terms of QTL identification of root traits and practical application of this information (e.g. MAS), what matters is not the absolute values of root traits measured in hydroponics and in the soil, but rather the magnitude and type (‘cross‐over’ or not) of ‘genotype × environment’ interaction for the root traits analysed in the progenies of the mapping populations. Therefore, if QTLs influencing genetic variation of root traits in hydroponics also regulate root growth in the field, it may be possible to identify among such QTLs those with an associated effect on yield, provided, of course, that genetic variability in root traits affects yield. This aspect is particularly relevant in maize in which the importance of the seminal (embryogenic) roots declines as a result of the prevailing role in the adult plant of shoot‐born roots, commonly named ‘adventitious’ nodal roots (Kiesselbach, 1949). Nevertheless, a positive correlation between root traits of maize seedlings and those of mature plants was reported by Nass and Zuber (1971). Furthermore, a relationship was found between seminal root traits of maize seedlings in hydroponics and root lodging in the field, an important trait influencing grain yield (Landi et al., 1998; Sanguineti et al., 1998). Significant, albeit weak, associations have also been reported between seminal root traits in hydroponics and RPF in the field (Landi et al., 2001b).
QTLs for root traits in hydroponics were investigated in 171 F3 families derived from the cross between Lo964 and Lo1016, two lines which were previously shown to differ greatly for root characteristics (Sanguineti et al., 1998). Tuberosa et al. (2002) identified 11, seven, nine and ten QTLs (LOD >2·5) regulating primary root length (R1L), primary root diameter (R1D), primary root weight (R1W) and the weight of the adventitious seminal roots (R2W), respectively. The high LOD value (>5·0) of ten QTLs and their sizeable R2 values (from 14·7 to 32·6 %) suggested the presence of QTLs with major effects. In total, 37 QTLs were grouped and assigned to 24 bins; ten bins concomitantly influenced at least two root traits (Fig. 2). Four of these regions were on chromosome 1 (bins 1·02, 1·06, 1·07–1·08 and 1·11). The QTL with the most sizeable effects (LOD values of 14·7, 6·4 and 8·3 for R1D, R1L and R2W, respectively) was found on bin 1·06 between PGAMCTA205 and php20644. To verify whether some of the QTL regions influencing root traits in hydroponics also modulate root growth in the field, 118 F3 families of the same mapping population were tested for RPF in three replicated field experiments (Giuliani et al., 2000 and unpubl. res.). QTLs were assigned to 19 bins, 11 of which (representing 13·7 % of the 80 bins explored by the Lo964 × Lo1016 map) also harboured a QTL for one or more root traits in hydroponics. In this case, applying the formula described in the section on QTL analysis and considering the 80 bins explored by the map, the percentage of bins expected by chance to harbour QTLs for root traits in hydroponics and in the field equals 7·1 {computed as [(24 × 19)/802] × 100}.
Among the QTLs identified in hydroponics in Lo964 × Lo1016, only one on bin 2·03 overlapped with one of the six root QTLs described in the field study of Guingo et al. (1998). In the Polj17 × F‐2 population, a total of 15 QTLs were detected for RPF, NRN and/or seedling root number; these QTLs were located on 14 different bins, six of which also harboured QTLs for root traits identified in hydroponics and/or in the field with the Lo964 × Lo1016 population.
The same root traits investigated in hydroponics in Lo964 × Lo1016 were also measured by Tuberosa et al. (2000 and unpubl. res.) in 120 RILs of the mapping population developed at CIMMYT from the cross Ac7729 × Ac7643/TZSRW and previously tested as F3 families under drought conditions for yield and other agronomic traits (Ribaut et al., 1996, 1997c). Among the 16 bins which carried a QTL for root traits (R. Tuberosa et al., unpubl. res.), eight were common to both populations (Fig. 2).
Collectively, these results indicate the feasibility of using hydroponics to identify QTLs for root traits, a number of which might also influence variation in root traits under field conditions. However, it is impossible to ascertain at this stage the extent to which these results might be due to random coincidence of a number of closely linked QTLs independently affecting root traits in hydroponics and in the field rather than to the effects of the same set of genes modulating root growth in hydroponics and also in the field. The most noticeable overlap for QTLs influencing root traits in hydroponics and in the field occurred on bin 1·06 which represents the most promising candidate region for a validation study through MAS. In fact, this bin harbours QTLs for root traits in hydroponics in Lo964 × Lo1016 (Tuberosa et al., 2002) and Ac7729 × Ac7643/TZSRW (Tuberosa et al., 2000), and QTLs for root pulling force in both Lo964 × Lo1016 (Giuliani et al., 2000) and Polj17 × F‐2 (Lebreton et al., 1995). Furthermore, this bin also revealed QTLs for L‐ABA in Os420 × IABO78 (Tuberosa et al., 1998c) and in Polj17 × F‐2 (Lebreton et al., 1995), for the ASI in Lo964 × Lo1016 (R. Tuberosa et al., unpubl. res.) and QTLs for grain yield (see next section). Altogether, the sizeable effects of this region on root traits and grain yield warrant the development of near isogenic lines (NILs) differing for the parental segment at this QTL. The availability of NILs for this QTL region would also open the way for undertaking positional cloning of the corresponding gene(s), in which case hydroponics would allow for a quick root phenotyping at an early growth stage.
Another interesting region is located on bin 1·03 near umc11 where a QTL for root traits was found in two of the four populations herein considered. Bin 1·03 also harboured the major QTL for root biomass and leaf growth rate in a drain‐pipe experiment carried out in DTP79 × B73 (S. A. Quarrie, pers. comm.). Furthermore, bin 1·03 also harbours QTLs for ABA concentration in the leaf (in Os420 × IABO78 and A662 × B73) and in the xylem (in Polj17 × F‐2). The root mutant rtcs (rootless for crown and lateral seminal root) has been mapped approx. 15–20 cM away from umc11 (Hochholdinger et al., 1998). Other maize mutants have been described (Doyle, 1978; Wen and Schnable, 1994; Neuffer, 1997; Hochholdinger and Feix, 1998; Hochholdinger et al., 1998; Krebs et al., 1999), but have not been mapped with sufficient precision to allow for a meaningful comparison with the QTL data herein presented. Finally, it is worth mentioning that a comparative analysis based on sinteny relationships between maize and rice presented by Quarrie (1996) indicated that at least five QTL regions for root traits in Polj17 × F‐2 correspond to regions in rice regulating root characteristics (Champoux et al., 1995). The most notable coincidence was again between the region near umc11 on chromosome 1 in maize and the region between RG104A and RG227 on chromosome 3 in rice. This chromosome region of rice also influenced root penetration ability (Price et al., 2000) and RPF (Ali et al., 2000).
ANALYSIS OF THE CO‐LOCATION OF QTLS FOR ABA CONCENTRATION AND ROOT TRAITS WITH QTLS FOR ASI AND GRAIN YIELD UNDER DROUGHT CONDITIONS
The majority of QTL studies aimed at evaluating drought tolerance in crops have been carried out at only one water regime, thus precluding the distinction between the constitutive (per se) and adaptive nature of QTL effects. This type of information would be of great value for applying MAS more effectively to each targeted environment. Frequently, the effects of specific QTL alleles on yield show substantial variation according to the type of environment. Sorting out constitutive from adaptive QTL effects is made possible by the evaluation of the same mapping population at different water regimes (Ribaut et al., 1996b, 1997c; Frova et al., 1999; Pelleschi et al., 1999; Sari‐Gorla et al., 1999; Tuberosa et al., 2002).
In maize, Ribaut et al. (1996, 1997c) reported QTLs for grain yield (GY) and ASI at three water regimes evaluating 240 F3 families derived from the cross Ac7729 × Ac7643/TZSRW (Fig. 2). The main aim of these authors was to evaluate the effects on GY of the degree of synchronization of pollen shed and silk extrusion, also known as ASI. In maize, it is well known that the most critical stage in terms of yield losses due to drought is just before and during flowering (Westgate and Boyer, 1985; Artlip et al., 1995; Jones and Setter, 2000; Saini and Westgate, 2000) and that ASI and GY are negatively associated under drought (Bolaños and Edmeades, 1993, 1997; Agrama and Moussa, 1996; Chapman and Edmeades, 1999). ASI can thus be used as a reliable and an easily scorable indicator of the level of water stress experienced by the maize plant. Although ASI can be selected effectively based on visual observations, it is desirable to identify molecular markers linked to the QTLs for ASI in order to select for this trait more effectively under drought and to continue selecting in the absence of drought at flowering (Ribaut et al., 1997b). Additionally, MAS allows for the early selection of plants carrying the most favourable allelic combination at the QTLs influencing ASI. The results of Ribaut and coworkers highlighted the presence of QTLs with fairly stable effects on ASI across the two drought‐stress treatments: of the seven QTLs evidenced altogether, five were common to both water regimes. Furthermore, three of these QTLs (on bins 1·08, 2·07–2·08 and 6·05) were also evidenced for ASI in well‐watered conditions. A QTL for ASI on bin 1·08 was also found in B73 × H99 (Sari‐Gorla et al., 1999).
The co‐location between QTLs for GY and those for L‐ABA at two growth stages (stem elongation and tassel appearance) and ASI was reported in the 2‐year study carried out with the mapping population derived from Os420 × IABO78 (Sanguineti et al., 1999). The overlap between QTLs affecting both L‐ABA and ASI was more extensive in 1994 (four of the eight QTLs for ASI) compared with 1995 (one of the five QTLs for ASI). With the exception of a QTL on chromosome 2 near csu133 (bin 2·04), at all the other QTLs a high L‐ABA was associated with a longer ASI, a finding that suggests the likely presence of pleiotropic effects. The four and six QTLs identified for GY in 1994 and 1995 accounted for 49·1 and 66·2 % of the genetic variation among F4 families, respectively. Overlap of QTLs for L‐ABA and GY occurred at two of the four QTLs for GY in 1994 and four of the six QTLs for GY in 1995. Two QTLs (on chromosome 1 near umc128, bins 1·07–1·08, and on chromosome 7 near asg8, bin 7·00) significantly influenced GY in both years; the values of the additive effects of these QTLs were the highest and showed opposite directions relative to an increase in L‐ABA. To interpret these contrasting results in terms of pleiotropic effects, the authors suggested a tentative model based on reciprocal cause–effect relationships between ABA and root size according to the growth stage. Although models like this one are highly speculative, they show how QTL information can be utilized to better understand the effects on GY due to genetic variation in MPTs. It should be noted that although root characteristics were not measured in the mapping population derived from Os420 × IABO78, a field experiment indicated that IABO78 (the low‐ABA parental line), as compared with Os420 (the high‐ABA parental line), was characterized by a significantly higher RPF coupled with a higher root density in deeper soil layers (Sanguineti et al., 1998). In general, the QTL results reported in Sanguineti et al. (1999) indicate that L‐ABA mainly represented an indicator of the level of drought stress experienced by plants at sampling. This was, in turn, related to the fact that plants grown under droughted field conditions and differing in MPTs influencing their water status will also differ in L‐ABA, in part independently from their capacity to accumulate ABA at a similar level of drought stress. Based on these considerations, future work considering ABA concentration as a selection criterion should preferably be carried out under conditions which allow for testing segregating progenies under a uniform level of drought stress.
QTLs for GY under well‐watered (WW) and water‐stressed (WS) conditions, together with QTLs for root traits, were reported in Lo964 × Lo1016 (Tuberosa et al., 2002). In the field, seven and eight QTLs were identified for GY‐WW and GY‐WS, respectively, which represented a total of 12 bins. Several overlaps occurred between the QTLs for root traits and QTLs affecting GY. Based on the number of bins harbouring QTLs for root traits in hydroponics (24 bins) and QTLs for GY (12 bins), the percentage of bins that influenced both categories of traits (7·5 %) was higher than that expected by chance (4·5 %). Among the root traits investigated (see previous section), R2W most frequently and consistently overlapped with QTLs for GY‐WW and GY‐WS. At four QTL regions (bins 1·06, 1·08, 10·04 and 10·07), an increase in R2W was positively associated with GY. The consistency of the sign of the additive effects for R2W with those for GY‐WW and GY‐WS at these four QTLs makes it more likely that pleiotropy caused this association in all cases, in accordance with the earlier section on ‘QTL analysis’. A 10 cM interval on bin 1·06 between PGAMCTA205 and php20644 showed the strongest effects on R1L, R1D, R2W, GY‐WW and GY‐WS. At least nine bins in Lo964 × Lo1016 (Tuberosa et al., 2002) which harboured QTLs for GY and for root traits in hydroponics also overlapped with QTLs for RPF and/or brace roots surveyed in three field experiments in which the destructive nature of RPF precluded the collection of data for GY (Giuliani et al., 2000, unpubl. res.).
QTLs for ASI and GY identified by Agrama and Moussa (1996) under conditions of limited water availability in SD34 × SD35 have also been reported in Fig. 2. Three of the five QTLs (LOD >3·0) that significantly affected GY in SD34 × SD35 mapped in bins (1·03, 3·09 and 8·03) also harboured QTLs for root traits in hydroponics of other mapping populations.
An important consideration in drought‐stress experiments is that as a soil dries, both water and nitrogen availability may become limiting (McDonald and Davies, 1996). A perturbation in nitrogen supply can alter the hormonal balance (ABA and cytokinins; Clarkson and Touraine, 1994). A decreased availability of nitrogen or other nutrients has been associated with an increased ABA concentration (McDonald and Davies, 1996). Agrama et al. (1999) tested a mapping population derived from B73 × G79 at two levels of nitrogen availability; this led to the identification of six and five QTLs (LOD >2·5) for GY at low and high nitrogen availability, respectively. Only two regions (bins 9·06–9·07 and 10·04–10·05) harboured QTLs affecting GY at both nutrient levels. In both cases, these regions also affected root traits of other populations tested in hydroponics and in the field (Fig. 2).
More recent work (Hirel et al., 2001) with test‐crosses of RILs derived from F‐2 × Io grown at two nitrogen levels (high and low) identified several QTLs for nitrogen use efficiency. Coincidence of QTLs for nitrogen use efficiency and GY with genes encoding enzymes involved in nitrogen metabolism (e.g. cytosolic glutamine synthetase) was detected on several bins. The most striking coincidence was for a gene (gln4) encoding glutamine synthetase which maps in bin 5·07 (Hirel et al., 2001). It is also worth mentioning that one of the QTLs evidenced at both nitrogen levels can be assigned to bin 1·06, thus confirming the importance of this region for determining GY under conditions of limited water and nutrient supply.
QTLs for invertase activity have been mapped in F‐2 × Io subjected to drought stress (Pelleschi et al., 1999). In this case, water shortage produced an early and strong stimulation of acid‐soluble invertase activity in adult leaves, whereas cell wall invertase activity remained constant. This response was closely related to the mRNA level for only one (Ivr2) of the invertase genes. In total, four QTLs were detected for invertase activity under well‐watered conditions and nine under drought conditions. One QTL common to both treatments was located near Ivr2, which can be assigned to bin 5·03. Other QTLs for invertase activity were found close to carbohydrate QTLs and some of them formed what the authors referred to as ‘stress clusters’. The two main clusters mapped on bins 1·03 and 5·03.
The results herein summarized indicate that a number of chromosome regions have concomitant effects on ABA concentration, root traits, other MPTs and GY in drought‐stressed maize. Among others, the regions corresponding to bins 1·03 and 1·06 warrant further attention to elucidate the genetic basis of such effects and to evaluate the response to MAS for the QTL alleles with a beneficial influence on grain yield. An important trait for which no QTL information is presently available and which could help in interpreting the effects of ABA on other MPTs is sensitivity to ABA. Significant variability among maize lines has been shown for stomatal sensitivity to ABA concentration (Conti et al., 1994) and pollen tube growth at different ABA levels (Frascaroli and Tuberosa, 1993). These findings suggest the possibility of identifying suitable lines for a QTL study targeting sensitivity to ABA. Little information is presently available on the mechanistic basis of variation in sensitivity to ABA (McDonald and Davies, 1996).
UNDERSTANDING AND DEALING WITH THE SHORTCOMINGS OF QTL ANALYSIS
One of the major shortcomings of QTL experiments is the low resolution power in detecting the real number of QTLs regulating the expression of the investigated traits (Charcosset and Gallais, 1996). Sampling variation further reduces the capacity to detect QTLs, particularly with populations of small size (<200 progenies). In a landmark paper, Beavis (1994) applied a Monte Carlo simulation to experimental data and showed that with populations of approx. 100–200 progenies (i.e. the size of a typical QTL experiment), only a modest fraction of QTLs are discovered; additionally, the effects of each single QTL were generally overestimated. A later article showed that with fewer than 500 progenies and independently from marker density, it is very unlikely that QTLs of small effects are uncovered (Beavis, 1998). These predictions were validated in experiments carried out with maize mapping populations sufficiently large (>400 progenies) to allow for meaningful subsamplings (Openshaw and Frascaroli, 1997; Melchinger et al., 1998). The effectiveness of detecting QTLs using the values of single environments and their mean has been evaluated in a number of studies. Jansen et al. (1995) concluded that the chances of detecting a QTL in several environments is small even if no ‘QTL × environment’ interaction is present. Other authors have reported the inconsistency of QTL detection across environments (Paterson et al., 1991; Mather et al., 1997; Wang et al., 1999). Recently, using a simulation approach, Otto and Jones (2000) showed that the number of loci detected in a QTL study is not linearly related with the actual number of underlying QTLs. They also proposed an estimator of the total number of QTLs segregating in experimental populations.
Because of its low resolution power, QTL analysis cannot provide us with a complete molecular dissection of the genetic basis underlying a QTL. As an example, a support interval of approx. 15 cM in a maize map of approx. 1500 cM is expected to contain an average of approx. 400 genes, assuming that approx. 40 000 genes are uniformly distributed along the genome. Indeed, increasing the size of the segregating population also improves the level of map resolution, which can lead to resolving a single QTL into multiple tightly linked loci (Phillips et al., 1992; Graham et al., 1997; Yamamoto et al., 1998; Legare et al., 2000). So far, the cloning of two QTLs for fruit size and sugar content in tomato (Frary et al., 2000; Fridman et al., 2000) and three QTLs for flowering time in rice (Yano et al., 2000; Takahashi et al., 2001) has indicated that in all five cases a single locus was responsible for each QTL. However, more than one gene may underlie a large portion of the QTLs described in the literature (Flint and Mott, 2001). The small number of QTLs which have been cloned to date probably represents a biased sample in terms of the genetic control of QTLs, because it is much easier to clone a single gene rather than two or more tightly clustered genes.
One of the prerequisites for increasing the probability of identifying QTLs is the utilization of mapping populations derived from the cross of two experimental lines diverging widely for the investigated traits, a condition which rarely occurs among elite inbred lines of comparable maturity which are commonly utilized for hybrid production in maize. Despite this, it should be pointed out that QTLs can also be identified in mapping populations obtained from lines with similar values for the investigated traits. It is important to realize that only the QTLs with functionally different alleles segregating in the mapping population will be detected, while important genes for the control of the trait, but for which allelic differences are absent, will go unnoticed. Furthermore, only few QTLs have large effects (Kearsey and Farquhar, 1998). Bost et al. (1999) suggested that this may be particularly frequent with metabolic pathways whose fluxes can be considered as model quantitative traits defined by QTLs controlling the activity or quantity of the enzymes. Major QTLs for metabolic pathways have been described in maize (McMullen et al., 1998; Pelleschi et al., 1999; Hirel et al., 2001). Based on Monte Carlo simulations, Bost et al. (1999) showed that a highly asymmetric (L‐shaped) distribution of the contributions of individual QTLs to the variance of metabolic fluxes is consistently expected in an F2 progeny. Another computer simulation has shown that MAS should be most successful when few loci (e.g. approx. 10) control the trait; indeed, MAS is not expected to be as convenient as phenotypic selection when many loci (e.g. >50) control the trait (Bernardo, 2001), a condition which decreases the precision of the estimates of gene effects.
An additional shortcoming of QTL studies based on the analysis of a mapping population is that the number, location and effects of the identified QTLs vary according to the genetic background of the population. Therefore, if a QTL allele is identified that is particularly beneficial for a target trait and its effects validated in population A, its introgression by means of MAS in population B will not necessarily lead to tangible benefits because, as compared with population A, population B may already have alleles of similar or even greater value at this QTL and/or because of different interactions between the QTL and the two genetic backgrounds. As an alternative to a traditional QTL study involving one or more mapping populations, a linkage disequilibrium study involving a large number (at least approx. 100–200) of unrelated accessions (e.g. inbred lines or hybrids) provides the opportunity to uncover the QTLs most important in regulating the variation within the germplasm of a particular crop (Thornsberry et al., 2001; see also section on ‘Association mapping’, below).
Finally, the greatest limitation of QTL analysis is that it is neither cheap nor fast. The time elapsing from the initial cross to derive a mapping population to the actual identification of the QTLs is seldom less than 3 years, provided that F2 plants or F1‐derived double‐haploids are evaluated; this period can stretch up to 6–8 years when highly homozygous lines (RILs) are obtained through selfing. The costs of a traditional QTL study remain extremely high due to the number of molecular data points required and to the extensive phenotypic evaluation, particularly for traits characterized by low heritability such as yield. Although it is now considerably cheaper and faster to construct a genetic map than a decade ago, in most cases the type of expertise and equipment needed to complete a QTL study are not available, particularly in developing countries. The recent progress achieved in miniaturization and robotics has allowed for the introduction of high‐density oligonucleotide arrays for allele identification, making it feasible to genotype thousands of markers (e.g. SNPs, single nucleotide polymorphisms) in a single experiment (Wang et al., 1998; Cho et al., 1999; Dinakar et al., 2002). A sequence independent microarray format for genotyping has also been developed (Jaccoud et al., 2001). The utilization of these techniques will drastically reduce the time required to collect the data necessary for obtaining a genetic map; unfortunately, the cost will remain high, at least until these technological platforms are applied more routinely.
The shortcomings herein illustrated should not deter the utilization of QTL analysis; an awareness of these shortcomings is nevertheless necessary to fully appreciate the type of questions that can be addressed with QTL analysis, the kind of answers that can be obtained and the associated costs.
ALTERNATIVE APPROACHES TO UNCOVER THE PRESENCE OF QTLS
QTL studies are commonly based on the phenotypic and molecular analysis of single genotypes (individual plants or progenies) of a mapping population most commonly derived from the cross of inbred lines chosen within the cultivated germplasm pool. However, a number of alternative approaches are available whose application can contribute to partially circumvent some of the limitations reviewed in the previous section.
Selective genotyping and bulk segregant analysis
Cheaper and faster alternatives to conventional QTL studies are offered by the application of selective genotyping (Lander and Botstein, 1989) and bulk segregant analysis (BSA; Michelmore et al., 1991). In selective genotyping, only a portion of individuals of the mapping population, typically the uppermost and the lowermost fractions in respect of a quantitative trait, are molecularly scored; consequently, very large populations can be screened, thus allowing for an increase in the power of QTL mapping. However, this approach suffers from possible biased estimates of QTL effects if the process of selection of the individuals to be included in the QTL analysis is not taken into consideration (Darvasi and Soller, 1992). A major limitation of the application of selective genotyping and BSA is that only QTLs controlling a very large proportion of the phenotypic variance are expected to be detected (Wang and Paterson, 1994). Furthermore, QTLs of traits genetically unrelated to the target trait will not be evidenced.
In BSA, originally introduced as a means of improving the marker density in the chromosomal region around a Mendelian trait, the molecular analysis is further reduced to only two DNA samples obtained by pooling the DNA of a small number of individuals or progenies (approx. 10–16) characterized by the lowest and the highest phenotypic values for the target trait in the segregating population. In this case, if a QTL region controls a significant portion of the variability among progenies for the target trait, any polymorphic marker closely linked to such QTL will also show different allele frequencies in the bulks with ‘low’ and ‘high’ phenotypic values; this will be evidenced by the different intensities of the corresponding parental bands in the profile of the two DNA bulks. In contrast, for any polymorphic marker unlinked to the QTL, the two bulks will present the same intensities for the corresponding band(s). The probability that an unlinked dominant marker such as a RAPD (random amplified polymorphic DNA) or an AFLP is found to be polymorphic between two bulks of ten individuals is extremely low (approx. 2 × 10–6; Michelmore et al., 1991). In BSA, while the efforts required for phenotyping will increase more or less linearly with the number of progenies, the molecular work necessary to identify the QTLs will remain the same. Although BSA offers some clear advantages, particularly when dealing with traits characterized by simple or nearly Mendelian inheritance, it has rarely been adopted for identifying QTLs of traits with low heritability, a condition which may lead to bulk DNA of progenies which carries the ‘wrong’ alleles (i.e. inclusion in the ‘high’ bulk of one or more genotypes with a ‘low’ allele and/or vice versa). A major shortcoming of BSA is that no information is provided on the distance of the QTL from the polymorphic marker; therefore, markers obtained in a BSA effort need to be mapped with standard approaches. Analogously to selective genotyping, QTLs of traits genetically unrelated to the target trait will not be evidenced.
In maize, the identification of QTLs for agronomic traits by means of BSA has been reported for flowering time in maize (Tuberosa et al., 1998c). The only application of BSA to locate QTLs regulating yield in drought‐stressed maize has been reported by Quarrie et al. (1999). In this case, BSA was applied in two different genetic backgrounds (Tuxpeño Sequia and Drought Tolerant Population) to investigate changes in allele frequency between the base population and the corresponding population obtained following several cycles of recurrent selection for yield under drought. Because each population comprised several alleles at each marker locus, at least 50 individuals/population were combined in the bulked DNA samples; in this case, codominant and multi‐allelic markers (RFLPs) were utilized. In both populations, a number of markers showed consistent differences in allele frequency in the bulks obtained from the unselected and selected populations. A number of these markers (umc11 on chromosome 1, csu149 and umc126b on chromosome 5, csu94 on chromosome 6) correspond to QTL locations for grain yield and its components also found in other studies (Agrama and Moussa, 1996; Austin and Lee, 1996; Ribaut et al., 1997c; Sanguineti et al., 1999; Tuberosa et al., 2002). Furthermore, Quarrie et al. (1999) noticed that csu94 maps very close to a gene for sucrose phosphate synthase (SPS) and a QTL for adult leaf carbohydrate concentration (glucose and fructose) reported in F‐2 × Io (Causse et al., 1995; Prioul et al., 1999).
Advanced backcross QTL analysis
The objective of advanced backcross QTL analysis (ABQA) is to quickly identify and exploit beneficial QTL alleles by integrating QTL discovery and variety improvement in a single process (Tanksley and Nelson, 1996). The strategy relies on the evaluation of backcross (BC) families between an elite variety used as recurrent parent and a donor accession (often a wild species sexually compatible with the cultivated species). In ABQA, the QTL analysis is delayed until the BC2 or BC3 generation, after selecting against characteristics that may have a negative effect from an agronomic standpoint (e.g. photoperiod sensitivity for maize or ear shattering for barley). The BC lines carrying the favourable QTL alleles will already be rather similar genetically to the elite variety and thus amenable for commercial exploitation. ABQA has already proven its validity for the exploitation of exotic germplasm in tomato (Tanksley et al., 1996; Bernacchi et al., 1998) and rice (Xiao et al., 1998). The applicability of ABQA in maize, starting from crosses with teosinte, is under evaluation (Harjes et al., 1999). Parallel efforts have been undertaken in barley in the attempt to uncover beneficial QTL alleles for drought tolerance from H. spontaneum, the wild progenitor of barley (Forster et al., 2000): preliminary results of three field trials show that at a number of QTLs for GY the allele increasing the value of the trait was contributed by H. spontaneum (R. Tuberosa et al., unpubl. res.).
Association mapping
Recently, QTL identification without the analysis of a mapping population became a reality with the development of analytical approaches that explore the residual linkage disequilibrium between markers and closely linked QTLs present in pools of unrelated accessions (Meuwissen and Goddard, 2000). The underlying principles derive from the so‐called association studies or case‐control tests from human genetics (Jorde, 2000; Cardon and Bell, 2001). Under this approach, marker‐trait association is only expected when a QTL is tightly linked to the marker because the accumulated recombination events that occurred during the development of the lines will prevent the detection of any marker/trait association in any situation where the QTL is not tightly linked to the investigated molecular marker. In maize, Vuylsteke et al. (2000a) successfully searched for genetic effects on GY associated with AFLP markers on a panel of F1 hybrids representative of the major maize heterotic groups. Because of their high level of informativeness, AFLPs are particularly suited to assess and characterize genetic diversity in germplasm collections (Vuylsteke et al., 2000b). Based on a similar principle, but with an analysis carried out at a much higher level of genetic resolution, Thornsberry et al. (2001) identified an association between flowering time in maize and sequence polymorphisms at the dwarf8 locus by evaluating a number of traits and the target gene sequence of 92 inbred lines. Although an estimate of the level of the residual linkage disequilibrium present in the cultivated germplasm is presently unavailable, these association methods may offer powerful alternatives for identifying candidate genes at QTLs, particularly in selfing species.
FROM QTLS TO GENES
QTL analysis, while lifting the ‘statistical fog’ surrounding conventional quantitative genetics (Mauricio, 2001), provides a powerful magnifying lens for deciphering the chromosome regions regulating complex traits. In this context, the introduction of QTL analysis in quantitative genetics can be compared with the introduction of the optical microscope in cell biology. However, the ultimate dissection of a phenotype can only be considered complete with a direct connection with a DNA sequence variation, and this cannot be obtained at present under a simple and unique QTL analysis framework. Several options are available to proceed from a supporting interval delimiting the QTL to the actual gene(s) responsible for the QTL effect.
Presently, positional cloning appears to be the main strategy toward QTL cloning. All the requirements for the positional cloning of Mendelian genes (Wicking and Williamson, 1991; Tanksley et al., 1995) are also needed for the positional cloning of the gene underlying a QTL; additionally, a much larger effort is needed for the phenotypic scoring of the segregating progenies. The prerequisites are: (1) the confining of the QTL to a 2–3 (or preferably shorter) cM region and the availability of at least one pair of NILs carrying opposite alleles at the target region; (2) the availability of a large mapping population (>2000 plants) derived from the cross of the NILs; (3) the presence of a high level of polymorphism between the NILs in order to allow for a level of marker density (usually AFLPs) in the target region suitable to chromosome walking or chromosome landing; (4) a high rate of recombination (i.e. a high ratio between genetic and physical distances) in the target region; (5) the availability of a BAC (bacterial artificial chromosome) and/or YAC (yeast artificial chromosome) genomic library covering the QTL region; and (6) a system (e.g. transformation) for proving the identity and testing the effects of the selected candidate gene.
It is thus evident that the routine implementation of positional cloning QTL in maize is not possible due to the massive efforts required in phenotyping progenies for quantitative traits and for the mass screening of molecular markers. Furthermore, steps (4) and (5) of the cloning process are complicated by the highly complex and repetitive structure of the maize genome. This notwithstanding, an effort is presently underway in maize to attempt the cloning of the gene(s) responsible for a QTL regulating flowering time (Salvi et al., 2002).
A number of parallel approaches which are now being deployed in maize genetics and genomics may ultimately facilitate QTL cloning. One approach is based on the identification and mapping of a massive number of ESTs (expressed sequence tags) whose mapping will provide candidate genes for the QTLs (Davis et al., 1999). A similar approach is the construction of functional maps, where ESTs/genes sharing similarity in structure, function or pathway are mapped with the aim of utilizing them as candidate genes (Schneider et al., 1999). In maize, limited information is available as to sequence variation of drought‐related genes and the functional implications of this variation. Dehydrins are a class of proteins that could provide an interesting starting point for the construction of functional maps related to drought stress in maize. Dehydrins are induced under conditions of water loss, particularly during seed maturation, and have been postulated to play a positive role in the adaptive response of the cell to drought (Close et al., 1989; Close, 1996; Choi et al., 1999).
It is also technically feasible to assemble a physical map of the maize genome by ordering overlapping genomic clones (BACs or YACs), and exploiting the ESTs to cross‐link the physical map to the genetic map. At that stage, QTL cloning will be reduced to fine mapping at the genomic clone level, followed by the sequencing of the genomic clone most likely to contain the gene(s) responsible for the QTL. This procedure has already been successfully exploited for QTL cloning in rice (Takahashi et al., 2001) following the construction of a rice physical map (Kurata et al., 1997).
An increasing amount of information is being obtained on the colinearity relationships between maize and rice chromosomes (Wilson et al., 1999), making the latter the reference species for the identification of candidate genes following the identification of the rice region colinear to the maize QTL region. Eventually, the maize genome will also be sequenced providing the ultimate resource of candidate genes for QTL mapping and cloning.
The possibility of assembling and exploiting colinearity maps between rather distantly related species was also suggested (Paterson et al., 1996), a promising proposition in view of the recent public disclosure of the annotated sequence of the arabidopsis genome (TAGI, 2000). Unfortunately, recent work has shown that the colinearity between arabidopsis and rice (Devos et al., 1999), and between arabidopsis and maize (van Buuren et al., 2002) has been eroded to such a degree that deploying map information of arabidopsis does not seem to help the identification of related genes in cereals. This notwithstanding, arabidopsis could still provide interesting contributions for unravelling the genetic basis of QTLs: its small size and very short life cycle, the small genome, the availability of the annotated genome sequence and a vast number of mutants coupled with a very efficient transformation system, all contribute to make this model species particularly attractive for the identification of QTLs and genes regulating the response to drought and other abiotic stresses. The work carried out in arabidopsis will be of particular value in elucidating the molecular mechanisms involved in the perception of cellular dehydration as well as in the signal transduction pathway leading to ABA accumulation and the activation of the suite of genes involved in the adaptive response to water deficit. Surprisingly little work has been carried out in arabidopsis to identify QTLs for MPTs and biomass production under drought conditions; analogously, no QTLs for ABA accumulation or root characteristics have been described. Recently, the discovery of a number of genes (e.g. CBF or DREB; Jaglo‐Ottosen et al., 1998; Kasuga et al., 1999) quickly triggered by abiotic stresses and encoding for transcription factors controlling the expression of drought‐responsive genes has sparked great interest for investigating their role in the major crops (Riechmann and Ratcliffe, 2000). DREB homologues have been identified in barley (T. Close, pers. comm.) and maize (van Buuren et al., 2002). Once these DREB homologues are mapped, it will be interesting to verify the presence in the same regions of QTLs for yield under drought.
Finally, the application of reverse genetics approaches (i.e. based on gene inactivation due to the insertion of active transposons) is presently not directly applicable to clone maize QTLs because of the difficulty in identifying the effect of the inactivation of a target QTL in the diverse and non‐homogeneous genetic backgrounds utilized to prepare the tagged population.
FUTURE PERSPECTIVES FOR QTL ANALYSIS AND GENE DISCOVERY
The spectacular increase in crop yields achieved in the past century has been equally attributed to plant breeding and to agronomy. In maize, the success of the hybrids most widely grown in the central portion of the ‘U.S. corn belt’ has been attributed to improved tolerance to abiotic and biotic stresses, coupled with maintenance of the ability to maximize yield per plant under non‐stress conditions (Duvick, 1997; Bruce et al., 2002). Unless new sources of genetic variation are tapped and more effective selection schemes are devised, it may not be possible to maintain the linear gains in yield of the 20th century, particularly in view of the increased unpredictability of weather patterns and depletion of water and nutrient supplies. It is thus clear that breeding for abiotic stresses, particularly drought and nutrient availability, will play an increasingly important role for securing a food supply adequate in quantity and quality (Salamini, 1999). New and sophisticated tools promise to vastly improve our ability to manipulate the genome of crop plants in order to improve their adaptability to environmental insults (Bowen and Luedtke, 1997; O’Sullivan et al., 1999; Habben et al., 2000; Cushman and Bohnert, 2001). The real challenge for the scientific community will be assigning functions to genes, and sorting out and exploiting those which are relevant for breeding purposes (Miflin, 2000).
QTL analysis should be viewed as an integral part of functional genomics which can help us to determine the relevance of allelic variation (natural and/or artificially induced) on grain yield. The impressive progress achieved in the mass‐scale profiling of the transcriptome (mRNAs) through microarray analysis (Ruan et al., 1998; Somerville and Somerville, 1999; Lockhart and Winzeler, 2000; Schaffer et al., 2000; Deyholos and Galbraith, 2001; Kawasaki et al., 2001; Zhu et al., 2001; Ozturk et al., 2002) opens the possibility of identifying QTLs for mRNA levels of 1000s of genes or even for the whole genome, as in arabidopsis and rice, whose genome sequences are now available. In maize, a number of microarrays have been made available to the scientific community through a publicly funded project (http://www.zmdb.iastate.edu). Microarrays would allow for the quantitative profiling of mRNAs of an entire mapping population, thus providing the opportunity to identify TQLs (transcript quantity loci) and verify their coincidence with QTLs for traits of interest. However, the present costs to implement this technological platform are still too high to conceive its routine application in QTL studies. Instead, microarrays are very suitable for detailed studies of changes in mRNA profiling of a limited number of genotypes (e.g. transgenics, NILs, BDLs, etc.). The major limitations of microarray‐based studies are: (1) low‐abundant mRNAs may not be represented by the array (except of course when the array includes all the genes of one particular species) and/or detected upon hybridization; (2) the correlation between the level of mRNAs and the products of their translation or their biological effect can be low; and (3) the difficulty, with the current technology, of verifying the level of gene expression in small samples.
As an alternative to close‐ended methods such as microarrays, a number of open‐ended methods for gene expression profiling are available: differential display (Liang and Pardee, 1992); representational difference analysis (Hubank and Schatz, 1994); cDNA‐AFLP (Bachem et al., 1996); SAGE (serial analysis of gene expression, Velculescu et al., 1995; Matsumura et al., 1999); GeneCalling (Shimkets et al., 1999; Bruce et al., 2000); and MPSS (massively parallel signature sequencing, Brenner et al., 2000). Bruce et al. (2001) used the GeneCalling method in whole root tissue of two maize inbred lines characterized by contrasting root‐related traits: out of approx. 13 500 cDNA fragments that were analysed at two growth stages, 69 showed a two‐fold or greater difference between the lines at both samplings, suggesting a relationship between these genes and root anchorage traits. When seven of these 69 fragments were selected for further analysis based on their match to publicly known sequences, five known genes were identified. Also in this case, it will be interesting to verify if maize QTLs for GY under drought map near these genes or near genes encoding for transcription factors regulating their expression level.
Among the emerging approaches, particular attention is being devoted to proteomics (Wilkins et al., 1996; Yates, 1998; Consoli et al., 2002). The rapid improvement in the techniques required to quantify proteins allows for the investigation of hundreds of proteins in the individuals of a mapping population. This, in turn, leads to the application of QTL analysis for mapping genes influencing protein quantity (PQL, protein quantity locus), a concept that was first introduced by Damerval et al. (1994) and further investigated in a number of studies (Touzet et al., 1995; de Vienne et al., 1999; Thiellement et al., 1999; Zivy and de Vienne, 2000; Consoli et al., 2002). Co‐localization of a PQL with its protein‐coding locus would indicate that allelic differences at that locus influence the expression of the protein, while co‐localization between a PQL and a QTL for a different trait would allow us to infer an association between the ‘candidate protein’ and trait variation. This strategy has been described by de Vienne et al. (1999) together with promising examples of its application in maize in order to identify suitable candidate genes conferring drought tolerance. The rapid technical improvements in proteomics and also in metabolomics will probably emphasize the search of QTLs for molecules (e.g. structural proteins, enzymes, metabolites, etc.) determining the structure and functions of the cell, and QTLs controlling key metabolic processes such as the maintenance of the redox potential (Foyer and Noctor, 2000; Bartels, 2001) or the biosynthesis of starch in the developing maize kernel (Zinselmeier et al., 1995, 1999).
In terms of target traits to be considered in future work, a promising approach is to identify QTLs for MPTs characterized by low ‘genotype × environment’ interactions, a source of variation which constantly biases the results of QTL studies and limits their applicability. Tardieu and co‐workers have shown that the elongation rate at the base of a maize leaf shows an invariant pattern during both the steady‐state elongation and the establishment of the elongation zone (Muller et al., 2001). Elongation rate is genotype‐specific and is linearly influenced by environmental variables (e.g. temperature, water availability) whose measurement can thus remove or at least mitigate the confounding effects of uncontrolled variation in such environmental factors. Crossing genotypes differing in elongation rates can thus lead to the identification of the QTLs for this trait (Tardieu et al., 2001). Recent results obtained by manipulating ABA concentration in Nicotiana plumbaginifolia using a transgenic approach have shown that the concentration of xylem ABA can also be linearly related in a genotype‐dependent fashion to predawn leaf water potential (Borel et al., 2001). In this case, however, the collection of the phenotypic data for a QTL analysis would certainly be very daunting. Monitoring large‐scale changes in transcript profiles may eventually allow for the identification of transcript networks accounting for ‘genotype × environment’ interactions in different genotypes.
CONCLUDING REMARKS
Clearly, in the 15 years following the first report of a QTL in maize (Stuber et al., 1987), QTL analysis has improved our knowledge of the genetic basis of grain yield and a number of morpho‐physiological traits involved in the response to drought of this important crop. The information on QTLs for MPTs and GY in maize herein summarized identifies a number of chromosome regions that warrant further studies in order to: (1) verify the validity of using marker‐assisted selection for improving GY under drought; (2) elucidate whether the associated effects observed at the QTL are due to pleiotropy or linkage; and (3) identify the candidate gene(s) underlying the QTL effects and, possibly, clone such gene(s) through positional cloning. A number of possible candidate genes mapping near QTLs regulating important MPTs and GY in maize have already been identified. Further studies based on genetic engineering, association genetics and, from an applicative standpoint, marker‐assisted selection will allow us to validate the role of a particular gene in controlling the variation of a target trait. Functional maps obtained with EST and/or cDNA clones of known function will vastly improve our ability to identify candidate genes.
Based on the QTL results summarized here, models have tentatively been proposed to account for association of traits observed at QTLs influencing traits for which a causal relationship can be established. The challenge is now to find suitable candidate genes for such QTLs and validate their role. The emerging picture for these genes and putatively associated QTLs is that of functional clusters non‐randomly distributed along the maize chromosomes, as shown in the comprehensive survey presented by Khavkin and Coe (1997) who hypothesized that major QTLs are clusters of genes (e.g. homeotic genes and other genes encoding for transcription factors) regulating development and that many plant reactions to abiotic stresses mediated by the growth machinery rely on such gene clusters. Chromosomal domains of gene expression have also been reported for the human genome (Cohen et al., 2000; Caron et al., 2001). In arabidopsis, the completion of genome sequencing and annotation has indicated that approx. 7 % of all open reading frames encode for proteins with significant similarity to known classes of plant transcription factors (TAGI, 2000). Although only a handful of candidate genes for QTLs has been identified in arabidopsis (Alonso‐Blanco and Koornneef, 2000), it will be interesting to verify the percentage of QTLs determined by sequence polymorphism at loci encoding for transcription factors. Our prejudice is that transcription factors, as compared with structural genes, will be over‐represented by QTLs.
The road from QTL analysis to gene discovery will remain a long and winding one, and will be applicable to a limited number of cases. Although the caveats of QTL analysis will probably deter many of us from undertaking this journey, an improved elucidation of the genetic basis of MPTs, made possible by the different QTL approaches herein presented, will significantly contribute not only to improve our understanding of the physiological and morphological bases of yield, but also to a more sound application of such information to plant breeding. Additionally, QTL analysis offers stimulating opportunities of collaboration between the different categories of plant scientists whose common objective is to contribute methods and materials for improving and stabilizing yield under drought. One of the best opportunities is provided by studies involving NILs obtained for a target QTL. NILs are particularly suited for physiological studies and only a decade ago it was stated that their development was thought to be restricted to traits controlled by one or only a few genes (Ludlow and Muchow, 1990). Clearly, NILs are a good example of how QTL analysis can contribute to develop a valuable type of genetic material for a quantitative trait. In conclusion, we believe that QTL analysis is here to stay.
ACKNOWLEDGEMENTS
The authors wish to thank Steve Quarrie for helpful and stimulating discussions. Contribution of the Interdepart mental Centre for Biotechnology, University of Bologna.
Supplementary Material
Received: 4 July 2001; Returned for revision: 3 January 2002; Accepted: 13 February 2002.
References
- AgramaHAS, Moussa ME.1996. Mapping QTLs in breeding for drought tolerance in maize (Zea mays L.). Euphytica 91: 89–97. [Google Scholar]
- AgramaHAS, Zakaria AG, Said FB, Tuinstra M.1999. Identification of quantitative trait loci for nitrogen use efficiency in maize. Molecular Breeding 5: 187–195. [Google Scholar]
- AliML, Pathan MS, Zhang J, Bai G, Sarkarung S, Nguyen HT.2000. Mapping QTLs for root traits in a recombinant inbred population from two indica ecotypes in rice. Theoretical and Applied Genetics 101: 756–766. [Google Scholar]
- Alonso‐BlancoC, Koornneef M.2000. Naturally occurring variation in Arabidopsis: an underexploited resource for plant genetics. Trends in Plant Science 5: 22–29. [DOI] [PubMed] [Google Scholar]
- ArtlipTS, Madison JT, Setter TL.1995. Water deficit in developing endosperm of maize: cell division and nuclear DNA endoreduplication. Plant, Cell and Environment 18: 1034–1040. [Google Scholar]
- AustinDF, Lee M.1996. Comparative mapping in F2:3 and F6:7 generations of quantitative trait loci for grain yield and yield components in maize. Theoretical and Applied Genetics 92: 817–826. [DOI] [PubMed] [Google Scholar]
- BachemCWB, van der Hoeven RS, de Bruijin, SM, Vreugdenhil D, Zabeau M, Visser RGF.1996. Visualization of differential gene expression using a novel method of RNA fingerprinting based on AFLP: analysis of gene expression during potato tuber development. The Plant Journal 9: 745–753. [DOI] [PubMed] [Google Scholar]
- BajajS, Targolli J, Liu LF, Ho THD, Wu R.1999. Transgenic approaches to increase dehydration‐stress tolerance in plants. Molecular Breeding 5: 493–503. [Google Scholar]
- BartelsD.2001. Targeting detoxification pathways: an efficient approach to obtain plants with multiple stress tolerance? Trends in Plant Science 6: 284–286. [DOI] [PubMed] [Google Scholar]
- BeavisWD.1994. The power and deceit of QTL experiments: lessons from comparative QTL studies. Proceedings of the 49th Annual Corn and Sorghum Research Conference American Seed Trade Association, Washington, D.C., 250–266. [Google Scholar]
- BeavisWD.1998. QTL analysis: power, precision, and accuracy. In: Paterson AH, ed. Molecular dissection of complex traits Boca Raton, USA: CRC Press, 145–162. [Google Scholar]
- BeckDL, Darrah LL, Zuber MS.1987. An improved technique for measuring resistance to root pulling in maize. Crop Science 27: 356–358. [Google Scholar]
- BernacchiD, Beck‐Bunn T, Eshed Y, Lopez J, Petiard V, Uhlig J, Zamir D, Tanksley SD.1998. Advanced backcross QTL analysis in tomato. I. Identification of QTLs for traits of agronomic importance from Lycopersicon hirsutum Theoretical and Applied Genetics 97: 381–397. [Google Scholar]
- BernardoR.2001. What if we knew all the genes for a quantitative trait in hybrid crops? Crop Science 41: 1–4. [Google Scholar]
- BhattramakkiD, Dolan M, Hanafey M, Wineland R, Vaske D, Register JC III, Tingey SV, Rafalski A.2002. Insertion‐deletion polymorphisms in 3′ regions of maize genes occur frequently and can be used as highly informative genetic markers. Plant Molecular Biology 48: 539–547. [DOI] [PubMed] [Google Scholar]
- BlumA.1988. Plant breeding for stress environments. Boca Raton, USA: CRC Press. [Google Scholar]
- BohnertHJ, Bressan RA.2001. Abiotic stresses, plant reactions, and approaches towards improving stress tolerance. In: Nössberger J, ed. Crop Science: Progress and prospects Wallingford, UK: CABI International, 81–100. [Google Scholar]
- BolañosJ, Edmeades GO.1993. Eight cycles of selection for drought tolerance in lowland tropical maize. I. Responses in grain yield, biomass, and radiation utilization. Field Crops Research 31: 233–251. [Google Scholar]
- BolañosJ, Edmeades GO.1996. The importance of anthesis‐silking interval in breeding for drought tolerance in tropical maize. Field Crops Research 48: 65–80. [Google Scholar]
- BolañosJ, Edmeades GO.1997. The importance of the anthesis‐silking interval in breeding for drought tolerance in tropical maize. In: Edmeades GO, Bänziger M, Mickelson HR, Peña‐Valdivia CB, eds. Developing drought‐ and low N‐tolerant maize Proceedings of a symposium, March 25–29, 1996, CIMMYT, El Batán, Mexico, 355–368. [Google Scholar]
- BolañosJ, Edmeades GO, Martinez L.1993. Eight cycles of selection for drought tolerance in lowland tropical maize. III. Responses in drought adaptive physiological and morphological traits. Field Crops Research 31: 269–286. [Google Scholar]
- BorelC, Audran C, Frey A, Marion‐Poll A, Tardieu F, Simonneau T.2001. N. plumbaginifolia zeaxanthin epoxidase transgenic lines have unaltered baseline ABA accumulations in roots and xylem sap, but contrasting sensitivities of ABA accumulation to water deficit. Journal of Experimental Botany 52: 427–434. [DOI] [PubMed] [Google Scholar]
- BostB, Dillmann C, de Vienne D.1999. Fluxes and metabolic pools as model traits for quantitative genetics. I. The L‐shaped distribution of gene effects. Genetics 153: 2001–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BotsteinD, White RL, Skolnick M, Davis RW.1980. Construction of a genetic linkage map in man using restriction fragment length polymorphism. American Journal of Human Genetics 32: 314–331. [PMC free article] [PubMed] [Google Scholar]
- BowenB, Luedtke R.1997. Understanding maize through genomics. Proceedings of the 52nd American Seed Trade Association Corn Sorghum Research Conference, December 10–11, 1997, Chicago, Illinois Washington, D.C.: American Seed Trade Association, 29–43. [Google Scholar]
- BrayE.1997. Plant responses to water deficit. Trends in Plant Science 2: 48–54. [Google Scholar]
- BrennerS,Williams SR, Vermaas EH, Storck T, Moon K, McCollum C, Mao J, Luo S, Kirchner JJ, Eletr S, Robert B, DuBridge RB, Burcham T, Albrecht G.2000. In vitro cloning of complex mixtures of DNA on microbeads: physical separation of differentially expressed cDNAs. Proceedings of the National Academy of Sciences of the United States of America 97: 1665–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BruceW, Desbons P, Crasta O, Folkerts O.2001. Gene expression profiling of two related maize inbred lines with contrasting root‐lodging traits. Journal of Experimental Botany 52: 459–468. [DOI] [PubMed] [Google Scholar]
- BruceW, Folkerts O, Garnaat C, Crasta O, Roth B, Bowen B.2000. Expression profiling of the maize flavonoid pathway genes controlled by estradiol‐inducible transcription factors CRC and P. Plant Cell 12: 65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BruceWB, Edmeades GO, Barker TC.2002. Molecular and physiological approaches to maize improvement for drought tolerance. Journal of Experimental Botany 53: 13–25. [PubMed] [Google Scholar]
- BusV, Ranatunga C, Gardiner S, Bassett H, Rikkerink E, Geibel M, Fischer C.2000. Marker assisted selection for pest and disease resistance in the New Zealand apple breeding programme. Acta Horticulturae 538: 541–547. [Google Scholar]
- CardonLR, Bell JI.2001. Association study designs for complex diseases. Nature Review Genetics 2: 91–99. [DOI] [PubMed] [Google Scholar]
- CaronH, van Schaik B, van der Mee M, Baas F, Riggins G, van Sluis P, Hermus MC, van Asperen R, Boon K, Voute PA, Heisterkamp S, van Kampen A, Versteeg R.2001. The human transcriptome map: clustering of highly expressed genes in chromosomal domains. Science 291: 1289–1292. [DOI] [PubMed] [Google Scholar]
- CausseM, Rocher JP, Henry AM, Charcosset A, Prioul JL, de Vienne D.1995. Genetic detection of the relationship between carbon metabolism and early growth in maize with emphasis on key‐enzyme loci. Molecular Breeding 1: 259–272. [Google Scholar]
- CausseM, Santoni S, Damerval C, Maurice A, Charcosset A, Deatrick J, de Vienne D.1996. A composite map of expressed sequences in maize. Genome39: 418–432. [DOI] [PubMed] [Google Scholar]
- CeccarelliS, Grando S.1996. Drought as a challenge for the plant breeder. Plant Growth Regulation 20: 149–155. [Google Scholar]
- ChampouxMC, Wang G, Sarkarung S, Mackill DJ, O’Toole JC, Huang N, McCouch SR.1995. Locating genes associated with root morphology and drought avoidance in rice via linkage to molecular markers. Theoretical and Applied Genetics 90: 969–981. [DOI] [PubMed] [Google Scholar]
- ChapmanSC, Edmeades GO.1999. Selection improves drought tolerance in tropical maize populations: II. Direct and correlated responses among secondary traits. Crop Science 39: 1315–1324. [Google Scholar]
- CharcossetA, Gallais A.1996. Estimation of the contribution of quantitative trait loci (QTL) to the variance of a quantitative trait by means of genetic markers. Theoretical and Applied Genetics 93: 1193–1201. [DOI] [PubMed] [Google Scholar]
- ChoRJ, Mindrinos M, Richards DR, Sapolsky RJ, Anderson M, Drenkard E, Dewdney J, Reuber TL, Stammers M, Federspiel N, Theologis A, Yang WH, Hubbell E, Au M, Chung EY, Lashkari D, Lemieux B, Dean C, Lipshutz RJ, Ausubel FM, Davis RW, Oefner PJ.1999. Genome‐wide mapping with biallelic markers in Arabidopsis thaliana Nature Genetics 23: 203–207. [DOI] [PubMed] [Google Scholar]
- ChoiDW, Zhu B, Close TJ.1999. The barley (Hordeum vulgare L.) dehydrin multigene family: sequences, allele types, chromosome assignments, and expression characteristics of 11 Dhn genes of cv Dicktoo. Theoretical and Applied Genetics 98: 1234–1247. [Google Scholar]
- ChurchillGA, Doerge RW.1994. Empirical threshold values for quantitative trait mapping. Genetics 138: 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ClarksonDT, Touraine B.1994. Morphological responses of plants to nitrate‐deprivation: a role for ABA? In: Roy J, Garnia E, eds. A whole plant perspective on carbon‐nitrogen interactions. The Hague: SPB Academic, 187–196. [Google Scholar]
- CloseTJ.1996. Dehydrins: emergence of a biochemical role of a family of plant dehydration proteins. Physiologia Plantarum 97: 795–803. [Google Scholar]
- CloseTJ.1997. Dehydrins: a commonality in the response of plants to dehydration and low temperature. Physiologia Plantarum 100: 291–296. [Google Scholar]
- CloseTJ, Kortt AA, Chandler PM.1989. A cDNA‐based comparison of dehydration‐induced proteins (dehydrins) in barley and corn Plant Molecular Biology 13: 95–108. [DOI] [PubMed] [Google Scholar]
- CohenBA, Mitra RD, Hughes JD, Church GM.2000. A computational analysis of whole‐genome expression data reveals chromosomal domains of gene expression. Nature Genetics 26: 183–186. [DOI] [PubMed] [Google Scholar]
- ConsoliL, Lefévre CL, Zivy M, de Vienne D, Damerval C.2002. QTL analysis of proteome and transcriptome variations for dissecting the genetic architecture of complex traits in maize. Plant Molecular Biology 48: 575–581. [DOI] [PubMed] [Google Scholar]
- ContiS, Frascaroli E, Gherardi F, Landi P, Sanguineti MC, Stefanelli S, Tuberosa R.1994. Accumulation of and response to abscisic acid in maize. In: Bianchi A et al, eds. Proceedings of the XVI Conference of the Eucarpia Maize and Sorghum Section, Bergamo, Italy, June6–9, 1993, 212–223. [Google Scholar]
- CrosbieTM.1982. Changes in physiological traits associated with long‐term breeding efforts to improve grain yield of maize. Proceedings of the 37th Annual Corn and Sorghum Research Conference American Seed Trade Association, Washington, D.C., 206–223. [Google Scholar]
- CushmanJC, Bohnert HJ.2000. Genomic approaches to plant stress tolerance. Current Opinions in Plant Biology 3: 117–124. [DOI] [PubMed] [Google Scholar]
- DamervalC, Maurice A, Josse JM, de Vienne D.1994. Quantitative trait loci underlying gene product variation: a novel perspective for analyzing regulation of genome expression. Genetics 137: 289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DarvasiA, Soller M.1992. Selective genotyping for determination of linkage between a marker locus and a quantitative trait locus. Theoretical and Applied Genetics 85: 353–359. [DOI] [PubMed] [Google Scholar]
- DarvasiA, Weintreb A, Minke V, Weller JI, Soller M.1993. Detection of marker‐QTL linkage and estimating QTL gene effect and map location using a saturated genetic map. Genetics 134: 943–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DaviesWJ, Zhang J.1991. Root signals and the regulation of growth and development of plants in drying soil. Annual Review of Plant Physiology and Plant Molecular Biology 42: 55–76. [Google Scholar]
- DavisGL, McMullen MD, Polacco M, Grant D, Musket T, Baysdorfer C, Staebell M, Xu G, Koster L, Houchins K, Melia‐Hancock S, Coe EH.1998. UMC 1998 molecular marker map of maize ESTs, sequenced core markers, and nonmaize probes. Maize Genetics Cooperation Newsletter 72: 118–128. [Google Scholar]
- DavisGL, McMullen MD, Baysdorfer C, Musket T, Grant D, Staebell M, Xu G, Polacco M, Koster L, Melia‐Hancock S, Houchins K, Chao S, Coe EH Jr.1999. A maize map standard with sequenced core markers, grass genome reference points and 932 expressed sequence tagged sites (ESTs) in a 1736‐locus map. Genetics 152: 1137–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de VienneD, Leonardi A, Damerval C, Zivy M.1999. Genetics of proteome variation for QTL characterization: application to drought‐stress responses in maize. Journal of Experimental Botany 50: 303–309. [Google Scholar]
- DevosKM, Beales J, Nagamura Y, Sasaki T.1999. Arabidopsis‐rice: will colinearity allow gene prediction across the eudicot‐monocot divide? Genome Research 9: 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeyholosM, Galbraith DW.2001. High‐density microarrays for gene expression analysis. Cytometry 43: 229–238. [PubMed] [Google Scholar]
- DoebleyJ, Stec A, Gustus C.1995. Teosinte branched1 and the origin of maize: evidence for epistasis and the evolution of dominance. Genetics 141: 333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DoyleGG.1978. An ageotropic primary root mutant. Maize Genetics Cooperation Newsletter 52: 77. [Google Scholar]
- DuvickDN.1997. What is yield? In: Edmeades GO, Bänziger M, Mickelson HR, Peña‐Valdivia CB, eds. Developing drought‐ and low N‐tolerant maize Proceedings of a symposium, March 25–29, 1996. El Batán, Mexico: CIMMYT, 332–335. [Google Scholar]
- FalconerDS.1981. Introduction to quantitative genetics 2nd edn. London: Longman Inc. [Google Scholar]
- FincherRR, Darrah LL, Zuber MS.1985. Root development in maize as measured by vertical pulling resistance. Maydica 30: 383–394. [Google Scholar]
- FlintJ, Mott R.2001. Finding the molecular basis of quantitative traits: successes and pitfalls. Nature Review Genetics 2: 437–445. [DOI] [PubMed] [Google Scholar]
- ForsterBP, Ellis RP, Thomas WTB, Newton AC, Tuberosa R, This D, El Enein RA, Bahri MH, Ben Salem M.2000. The development and application of molecular markers for abiotic stress tolerance in barley. Journal of Experimental Botany 51: 19–27. [PubMed] [Google Scholar]
- FoyerCH, Noctor G.2000. Tansley Review No. 112. Oxygen processing in photosynthesis: regulation and signalling. New Phytologist 146: 359–388. [Google Scholar]
- FraryA, Nesbitt TC, Frary A, Grandillo S, van der Knaap E, Cong B, Liu JP, Meller J, Elber R, Alpert KB, Tanksley SD.2000. A quantitative trait locus key to the evolution of tomato fruit size. Science 289: 85–88. [DOI] [PubMed] [Google Scholar]
- FrascaroliE, Tuberosa R.1993. Effect of abscisic acid on pollen germination and tube growth of maize genotypes. Plant Breeding 110: 250–254. [Google Scholar]
- FridmanE, Pleban T, Zamir D.2000. A recombination hot spot delimits a wild‐species quantitative trait locus for tomato sugar content to 484 bp within an invertase gene. Proceedings of the National Academy of Sciences of the United States of America 97: 4718–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FrovaC, Krajewski P, Di Fonzo N, Villa M, Sari‐Gorla M.1999. Genetic analysis of drought tolerance in maize by molecular markers. I. Yield components. Theoretical and Applied Genetics 99: 280–288. [Google Scholar]
- GiulianiMM, Darrah LL, Salvi S, Sanguineti MC, Landi P, Conti S, Tuberosa R.2000. Comparative QTL analysis in maize for vertical root pulling resistance in the field and root traits in hydroponics. International Conference on Plant & Animal Genome VIII, January 9–12, San Diego, California, 114. [Google Scholar]
- GrahamGI, Wolff DW, Stuber CW.1997. Characterization of a yield quantitative trait locus on chromosome five of maize by fine mapping. Crop Science 37: 1601–1610. [Google Scholar]
- GuingoE, Hébert Y, Charcosset A.1998. Genetic analysis of root traits in maize. Agronomie 18: 225–235. [Google Scholar]
- HabbenJ, Helentjaris T, Sun Y, Zinselmeier C.2000. Utilizing new technologies to investigate drought tolerance in maize: a perspective from industry. In: Ribaut JM, Poland D, eds. Molecular approaches for the genetic improvement of cereals for stable production in water‐limited environments A Strategic Planning Workshop held at CIMMYT, El Batán, Mexico, June21–25, 1999, 154–155. [Google Scholar]
- HackettCA.2002. Statistical methods for QTL mapping in cereals. Plant Molecular Biology 48: 585–599. [DOI] [PubMed] [Google Scholar]
- HallauerAR, Miranda Fo JB.1988. Quantitative genetics in maize breeding 2nd edn Ames: Iowa State University Press. [Google Scholar]
- HarjesCE, Smith ME, McCouch SR, Tanksley SD.1999. Advanced backcross QTL analysis and introgression of perennial teosinte (Zea diploperennis) alleles to maize. Plant & Animal Genome VII Conference, January17–21, San Diego, California, 260. [Google Scholar]
- HelentjarisT, Wright S, Weber D.1986. Construction of a genetic linkage map in maize using restriction fragment polymorphisms. Maize Genetics Cooperation Newsletter 60: 118–120. [Google Scholar]
- HirelB, Bertin P, Quilleré I, Bourdoncle W, Attagnant C, Dellay C, Gouy A, Cadiou S, Retailliau C, Falque M, Gallais A.2001. Towards a better understanding of the genetic and physiological basis for nitrogen use efficiency in maize. Plant Physiology 125: 1258–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HochholdingerF, Feix G.1998. Early post‐embryonic root formation is specifically affected in the maize mutant Irtl The Plant Journal 16: 247–255. [DOI] [PubMed] [Google Scholar]
- HochholdingerF, Park WJ, Feix G.1998. Isolation of the new root mutant slrl affecting lateral root formation. Maize Genetics Cooperation Newsletter 72: 29–30. [Google Scholar]
- HoseE, Steudle E, Hartung W.2000. Abscisic acid and hydraulic conductivity of maize roots: a study using cell‐ and root‐pressure probes. Planta 211: 874–882. [DOI] [PubMed] [Google Scholar]
- HuX, Ribaut J‐M, Gonzales‐de‐Leon D.1997. Development of PCR‐based markers to facilitate large‐scale screening in molecular maize breeding. Maize Genetics Cooperation Newsletter 71: 61–62. [Google Scholar]
- HubankM, Schatz DG.1994. Identifying differences in mRNA expression by representational difference analysis of cDNA. Nucleic Acids Research 22: 5640–5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IngramJ, Bartels D.1996. The molecular basis of dehydration tolerance in plants. Annual Review of Plant Physiology and Plant Molecular Biology 47: 377–403. [DOI] [PubMed] [Google Scholar]
- InnesP, Blackwell RD, Quarrie SA.1984. Some effects of genetic variation in drought‐induced abscisic acid accumulation on the yield and water use of spring wheat. Journal of Agricultural Science 102: 341–351. [Google Scholar]
- IvanovicM, Quarrie SA, Djordjevic J, Pekic S.1992. Inheritance of abscisic acid production in maize (Zea mays L.) leaves in response to rapid drought stress and in the field. Maydica 37: 313–318. [Google Scholar]
- JaccoudD, Peng K, Feinstein D, Kilian A.2001. Diversity arrays: a solid state technology for sequence information independent genotyping. Nucleic Acids Research 29: e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaglo‐OttosenKR, Gilmour SJ, Zarka DG, Schabenberger O, Thomashow MF.1998. Arabidopsis CBF1 overexpression induces COR genes and enhances freezing tolerance. Science 280: 104–106. [DOI] [PubMed] [Google Scholar]
- JansenRC.1993. Interval mapping of multiple quantitative trait loci. Genetics 135: 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JansenRC.1996. Complex plant traits: time for polygenic analysis. Trends in Plant Science 1: 89–94. [Google Scholar]
- JansenRC, Stam P.1994. High resolution of quantitative traits into multiple loci via interval mapping. Genetics 136: 1447–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JansenRC, Van Ooijen JW, Stam P, Lister C, Dean C.1995. Genotype by environment interaction in genetic mapping of multiple quantitative trait loci. Theoretical and Applied Genetics 91: 33–37. [DOI] [PubMed] [Google Scholar]
- JensenAB, Busk PK, Figueras M, Albà MM, Peracchia G, Messeguer R, Goday A, Pagès M.1996. Drought signal transduction in plants. Plant Growth Regulation 20: 105–110. [Google Scholar]
- JonesRJ, Setter TL.2000. Hormonal regulation of early kernel development. In: Westgate ME, Boote KJ, eds. Physiology and modelling kernel set in maize Madison, Wisconsin: Crop Science Society of America, 25–42. [Google Scholar]
- JordeLB.2000. Linkage disequilibrium and the search for complex disease genes. Genome Research 10: 1435–1444. [DOI] [PubMed] [Google Scholar]
- KaoCH, Zeng ZB, Teasdale RD.1999. Multiple interval mapping for quantitative trait loci. Genetics 152: 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KasugaM, Liu Q, Miura S, Yamaguchi‐Shinozaki K, Shinozaki K.1999. Improving plant drought, salt and freezing tolerance by gene transfer of a single stress‐inducible transcription factor. Nature Biotechnology 17: 287–291. [DOI] [PubMed] [Google Scholar]
- KawasakiS, Deyholos M, Borchert C, Brazille S, Kawai K, Galbraith DW, Bohnert HJ.2001. Temporal succession of salt stress responses in rice by microarray analysis. Plant Cell 12: 889–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KearseyMJ, Farquhar AGL.1998. QTL analysis; where are we now? Heredity 80: 137–142. [DOI] [PubMed] [Google Scholar]
- KevernTC, Hallauer AR.1983. Relation of vertical root‐pull resistance and flowering in maize. Crop Science 23: 357–363. [Google Scholar]
- KhavkinE, Coe E.1997. Mapped genomic locations for developmental functions and QTLs reflect concerted groups in maize (Zea mays L.). Theoretical and Applied Genetics 95: 343–352. [Google Scholar]
- KiesselbachTA.1949. The structure and reproduction of corn. Agricultural Experiment Station, Lincoln, Nebraska. Research Bulletin 161. [Google Scholar]
- KnightH, Knight MR.2001. Abiotic stress signalling pathways specificity and cross‐talk. Trends in Plant Science 6: 262–267. [DOI] [PubMed] [Google Scholar]
- KorolAB, Ronin YI, Itskovich AM, Peng J, Nevo E.2001. Enhanced efficiency of quantitative trait loci mapping analysis based on multivariate complexes of quantitative traits. Genetics 157: 1789–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KrebsO, Feix G, Beaumont V, Schwall M.1999. Mapping of the root specific rtcs locus with the help of microsatellites. Maize Genetics Cooperation Newsletter 73: 33. [Google Scholar]
- KurataN, Umehara Y, Tanoue H, Sasaki T.1997. Physical mapping of the rice genome with YAC clones. Plant Molecular Biology 35: 101–113. [PubMed] [Google Scholar]
- LanderES, Botstein D.1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LandiP, Albrecht B, Giuliani MM, Sanguineti MC.1998. Seedling characteristics in hydroponic culture and field performance of maize genotypes with different resistance to root lodging. Maydica 43: 111–116. [Google Scholar]
- LandiP, Sanguineti MC, Conti S, Tuberosa R.2001a Direct and correlated responses to divergent selection for leaf abscisic acid concentration in two maize populations. Crop Science 41: 335–344. [Google Scholar]
- LandiP, Conti S, Gherardi F, Sanguineti MC, Tuberosa R.1995. Genetic analysis of leaf ABA concentration and of agronomic traits in maize hybrids grown under different water regimes. Maydica 40: 179–186. [Google Scholar]
- LandiP, Salvi S, Sanguineti MC, Stefanelli S, Tuberosa R.2002. Development and preliminary evaluation of near‐isogenic maize lines differing for a QTL which affects leaf ABA concentration. Maize Genetics Cooperative Newsletter http://www.agron.missouri. edu/mnl/76/71lendi.html [Google Scholar]
- LandiP, Giuliani MM, Darrah LL, Tuberosa R, Conti S, Sanguineti MC.2001b Variability for root and shoot traits in a maize population grown in hydroponics and in the field and their relationships with vertical root pulling resistance. Maydica (in press). [Google Scholar]
- Larqué‐SaavedraA, Wain RL.1976. Studies on plant‐growth regulating substance. XLII. Abscisic acid as a genetic character related to drought tolerance. Annals of Applied Biology 83: 291–297. [Google Scholar]
- LebretonC, Lazic‐Jancic V, Steed A, Pekic S, Quarrie SA.1995. Identification of QTL for drought responses in maize and their use in testing causal relationships between traits. Journal of Experimental Botany 46: 853–865. [Google Scholar]
- LeeM.1995. DNA markers and plant breeding programs. Advances in Agronomy 55: 265–344. [Google Scholar]
- LegareME, Bartlett FS, Frankel WN.2000. A major effect QTL determined by multiple genes in epileptic EL mice. Genome Research 10: 42–48. [PMC free article] [PubMed] [Google Scholar]
- LiJX, Wang XQ, Watson MB, Assmann SM.2000. Regulation of abscisic acid‐induced stomatal closure and anion channels by guard cell AAPK kinase. Science 287: 300–303. [DOI] [PubMed] [Google Scholar]
- LiangP, Pardee AB.1992. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science 257: 967–970. [DOI] [PubMed] [Google Scholar]
- LinYR, Schertz KF, Paterson AH.1995. Comparative analysis of QTLs affecting plant height and maturity across the Poaceae, in reference to an interspecific sorghum population. Genetics 141: 391–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LiuBH.1998. Statistical genomics: linkage, mapping and QTL analysis. Boca Raton, USA: CRC Press. [Google Scholar]
- LockhartDJ, Winzeler EA.2000. Genomics, gene expression and DNA arrays. Nature 405: 827–836. [DOI] [PubMed] [Google Scholar]
- LudlowMM, Muchow RC.1990. A critical evaluation of traits for improving crop yields in water‐limited environments. Advances in Agronomy 43: 107–153. [Google Scholar]
- LynchM, Walsh B.1998. Genetics and analysis of quantitative traits. Sunderland, Massachusetts, USA: Sinauer Associates. [Google Scholar]
- McDonaldAJS, Davies WJ.1996. Keeping in touch: responses of the whole plant to deficits in water and nitrogen supply. Advances in Botanical Research 22: 229–300. [Google Scholar]
- McMullenMD, Byrne PF, Snooks ME, Wiseman BR, Lee EA, Widstrom NW, Coe EH.1998. Quantitative trait loci and metabolic pathways. Proceedings of the National Academy of Sciences of the United States of America 95: 1996–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MartinezO, Curnow RN.1992. Estimating the locations and the sizes of the effects of quantitative trait loci using flanking markers. Theoretical and Applied Genetics 85: 480–488. [DOI] [PubMed] [Google Scholar]
- MatherDE, Tinker NA, La Berge DE, Edney M, Jones BL, Rossnagel BG, Legge WG, Briggs KG, Irvine RB, Falk DE, Kasha KJ.1997. Regions of the genome that affect grain and malt quality in a North American two‐row barley cross. Crop Science 37: 544–554. [Google Scholar]
- MatsumuraH, Nirasawa S, Terauchi R.1999. Transcript profiling in rice (Oryza sativa L.) seedling using serial analysis of gene expression (SAGE). The Plant Journal 20: 719–726. [DOI] [PubMed] [Google Scholar]
- MauricioR.2001. Mapping quantitative trait loci in plants: uses and caveats for evolutionary biology. Nature Review of Genetics 2: 370–381. [DOI] [PubMed] [Google Scholar]
- MelchingerAE, Utz HF, Schon CC.1998. Quantitative trait locus (QTL) mapping using different testers and independent population samples in maize reveals low power of QTL detection and large bias in estimates of QTL effects. Genetics 149: 383–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MeuwissenTHE, Goddard ME.2000. Fine mapping of quantitative trait loci using linkage disequilibria with closely linked marker loci. Genetics155: 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MichelmoreRW, Paran I, Kesseli RV.1991. Identification of markers linked to disease‐resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating population. Proceedings of the National Academy of Sciences of the United States of America 88: 9828–9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MiflinB.2000. Crop improvement in the 21st century. Journal of Experimental Botany 51: 1–8. [PubMed] [Google Scholar]
- MilborrowBW.2001. The pathway of biosynthesis of abscisic acid in vascular plants: a review of the present state of knowledge of ABA biosynthesis. Journal of Experimental Botany 52: 1145–1164. [PubMed] [Google Scholar]
- MugoSN, Bänziger M, Edmeades GO.2000. Prospects of using ABA in selection for drought tolerance in cereal crops. In: Ribaut JM, Poland D, eds. Molecular approaches for the genetic improvement of cereals for stable production in water‐limited environments. A Strategic Planning Workshop held at CIMMYT, El Batán, Mexico, June 21–25, 1999, 73–78. [Google Scholar]
- MullerB, Reymond M, Tardieu F.2001. The elongation rate at the base of a maize leaf shows an invariant pattern during both the steady‐state elongation and the establishment of the elongation zone. Journal of Experimental Botany 52: 1259–1268. [PubMed] [Google Scholar]
- NagyZ, Tuba Z, Zsoldos F, Erdei L.1995. CO2‐exchange and water relation responses of sorghum and maize during water and salt stress. Journal of Plant Physiology 145: 539–544. [Google Scholar]
- NassHG, Zuber MS.1971. Correlation of corn (Zea mays L.) roots early in development to mature root development. Crop Science 11: 655–658. [Google Scholar]
- NettingAG.2000. pH, abscisic acid and the integration of metabolism in plants under stressed and non‐stressed conditions: cellular responses to stress and their implication for plant water relations. Journal of Experimental Botany 51: 147–158. [DOI] [PubMed] [Google Scholar]
- NeufferMG, Coe EH, Wessler SR.1997. Mutants of maize. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press. [Google Scholar]
- OberES, Sharp RE.1994. Proline accumulation in maize (Zea mays L.) primary roots at low water potentials. I. Requirement for increased levels of abscisic acid. Plant Physiology 105: 981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OpenshawS, Frascaroli E.1997. QTL detection and marker‐assisted selection for complex traits in maize. Proceedings of the 52nd Annual Corn and Sorghum Research Conference American Seed Trade Association. Washington, D.C., 44–53. [Google Scholar]
- O’SullivanDM, Edwards D, Edwards KJ.1999. Maize genomics. AgBiotechNet 2: 1–6. [Google Scholar]
- O’TooleJC, Bland WL.1987. Genotypic variation in crop plant root systems. Advances in Agronomy 41: 91–145. [Google Scholar]
- OttoSP, Jones CD.2000. Detecting the undetected: estimating the total number of loci underlying a quantitative trait. Genetics 156: 2043–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OzturkZN, Talamé V, Deyholoska M, Michalowski CB, Galbraith DW, Gozukirmizi N, Tuberosa R, Bohnert HJ.2002. Monitoring large‐scale changes in transcript abundance in drought‐ and salt‐stressed barley. Plant Molecular Biology 48: 551–573. [DOI] [PubMed] [Google Scholar]
- PassiouraJB.1996. Drought and drought tolerance. Plant Growth Regulation 20: 79–83. [Google Scholar]
- PatersonAH, Damon S, Hewitt JK, Zamir D, Rabinowitch HD, Lincoln SE, Lander ES, Tanksley SD.1991. Mendelian factors underlying quantitative traits in tomato: comparison across species, generations, and environments. Genetics 127: 181–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PatersonAH, Lan TH, Reischmann KP, Chang C, Lin YR, Liu SC, Burow MD, Kowalski SP, Katsar CS, Del Monte TA, Feldmann KA, Schertz KF, Wendel JF. 1996. Toward a unified genetic map of higher plants, transcending the monocot‐dicot divergence. Nature Genetics 14: 380–382. [DOI] [PubMed] [Google Scholar]
- PelleschiS, Guy S, Kim J‐Y, Pointe C, Mahé A, Barthes L, Leonardi A, Prioul JL.1999. Ivr2, a candidate gene for a QTL of vacuolar invertase activity in maize leaves. Gene‐specific expression under water stress. Plant Molecular Biology 39: 373–380. [DOI] [PubMed] [Google Scholar]
- PhillipsRL, Kim TS, Kaeppler SM, Parentoni SN, Shaver DL, Stucker RI, Openshaw SJ.1992. Genetic dissection of maturity using RFLPs. Proceedings of the 47th Annual Corn and Sorghum Research Conference American Seed Trade Association, Washington, D.C., 135–150. [Google Scholar]
- PriceAH, Steele KA, Moore BJ, Barraclough PB, Clark LJ.2000. A combined RFLP and AFLP linkage map of upland rice (Oryza sativa L.) used to identify QTLs for root‐penetration ability. Theoretical and Applied Genetics 100: 49–56. [Google Scholar]
- PrioulJL, Quarrie S, Causse M, de Vienne D.1997. Dissecting complex physiological functions through the use of molecular quantitative genetics. Journal of Experimental Botany 48: 1151–1163. [Google Scholar]
- PrioulJL, Pelleschi S, Sene M, Thévenot C, Causse M, de Vienne D, Leonardi A.1999. From QTLs for enzyme activity to candidate genes in maize. Journal of Experimental Botany 50: 1281–1288. [Google Scholar]
- QuarrieSA.1991. Implications of genetic differences in ABA accumulation for crop production. In: Davies WJ, Jones HG, eds. Abscisic acid: physiology and biochemistry Oxford, UK: Bios Scientific Publishers, 227–243. [Google Scholar]
- QuarrieSA.1993. Understanding plant responses to stress and breeding for improved stress resistance – The generation gap. In: Close TJ, Bray EA, eds. Plant responses to cellular dehydration during environmental stress Proceedings of the 16th Annual Riverside Symposium in Plant Physiology, Riverside, California, 224–245. [Google Scholar]
- QuarrieSA.1996. New molecular tools to improve the efficiency of breeding for increased drought resistance. Plant Growth Regulation 20: 167–178. [Google Scholar]
- QuarrieSA, Jones HG.1977. Effects of abscisic acid and water stress on development and morphology of wheat. Journal of Experimental Botany 28: 192–203. [Google Scholar]
- QuarrieSA, Lazic‐Jancic V, Kovacevic, Steed A, Pekic S.1999. Bulk segregant analysis with molecular markers and its use for improving drought resistance in maize. Journal of Experimental Botany 50: 1299–1306. [Google Scholar]
- RagotM, Gay G, Muller JP, Durovray J.2000. Efficient selection for adaptation to the environment through QTL mapping and manipulation in maize. In: Ribaut JM, Poland D, eds. Molecular approaches for the genetic improvement of cereals for stable production in water‐limited environments. A Strategic Planning Workshop held at CIMMYT, El Batán, Mexico, June21–25, 1999, 128–130. [Google Scholar]
- RibautJM, Hoisington D.1998. Marker‐assisted selection: new tools and strategies. Trends in Plant Science 31: 236–239. [Google Scholar]
- RibautJM, Pilet PE.1991. Effects of water stress on growth, osmotic potential and abscisic acid content of maize roots. Physiologia Plantarum 81: 156–162. [Google Scholar]
- RibautJM, Edmeades G, Perotti E, Hoisington D.2000. QTL analyses, MAS results, and perspectives for drought‐tolerance improvement in tropical maize. In: Ribaut JM, Poland D, eds. Molecular approaches for the genetic improvement of cereals for stable production in water‐limited environments A Strategic Planning Workshop held at CIMMYT, El Batán, Mexico, 21–25 June 1999, 131–136. [Google Scholar]
- RibautJM, Hu XY, Hoisington D, Gonzales‐de‐Leon D.1997a Use of STSs and SSRs as rapid and reliable preselection tools in a marker assisted selection backcross scheme. Plant Molecular Biology Reporter 15: 156–164. [Google Scholar]
- RibautJM, Gonzalez‐de‐Leon D, Jiang C, Edmeades GO, Hoisington D.1997b Identification and transfer of ASI quantitative trait loci (QTL): a strategy to improve drought tolerance in maize lines and populations. In: Edmeades GO, Bänziger M, Mickelson HR, Peña‐Valdivia CB, eds. Developing drought‐ and low N‐tolerant maize Proceedings of a Symposium, March 25–29, 1996, CIMMYT, El Batán, Mexico, 396–400. [Google Scholar]
- RibautJM, Hoisington DA, Deutsch JA, Jiang C, Gonzales‐de‐Leon D.1996. Identification of quantitative trait loci under drought conditions in tropical maize. I. Flowering parameters and the anthesis‐silking interval. Theoretical and Applied Genetics 92: 905–914. [DOI] [PubMed] [Google Scholar]
- RibautJM, Jiang C, Gonzales‐de‐Leon D, Edmeades GO, Hoisington DA.1997c Identification of quantitative trait loci under drought conditions in tropical maize. II. Yield components and marker‐assisted selection strategies. Theoretical and Applied Genetics 94: 887–896. [DOI] [PubMed] [Google Scholar]
- RichardsRA.1988. Physiology and the breeding of winter‐grown cereals for dry areas. In: Srivastrava J, Porceddu E, Acevedo E, Barma S, eds. Drought tolerance in winter cereals Chicester, UK: Wiley, 133–149. [Google Scholar]
- RichardsRA.1996. Defining selection criteria to improve yield under drought. Plant Growth Regulation 20: 157–166. [Google Scholar]
- RiechmannJL, Ratcliffe OJ.2000. A genomic perspective on plant transcription factors. Current Opinion in Plant Biology 3: 423–434. [DOI] [PubMed] [Google Scholar]
- RobertsonDS.1985. A possible technique for isolating genic DNA for quantitative traits in plants. Journal of Theoretical Biology 117: 1–10. [Google Scholar]
- RuanY, Gilmore J, Conner T.1998. Towards Arabidopsis genome analysis: monitoring expression profiles of 1400 genes using cDNA microarrays. The Plant Journal 15: 821–833. [DOI] [PubMed] [Google Scholar]
- SaabIN, Ho THD, Sharp RE.1995. Translatable RNA populations with maintenance of primary root elongation and inhibition of mesocotyl elongation by abscisic acid in maize seedlings at low water potentials. Plant Physiology 109: 593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SainiHS, Westgate ME.2000. Reproductive development in grain crops during drought. Advances in Agronomy 68: 59–96. [Google Scholar]
- SalaminiF.1999. Plant breeding and sustainable agriculture – introduction to the problem. Proceedings of the 1999 World Seed Conference, September 6–8, Cambridge, UK, 59–62. [Google Scholar]
- SalviS, Tuberosa R, Phillips RL.2001. Development of PCR‐based assays for allelic discrimination in maize by using the 5‐nuclease procedure. Molecular Breeding 8: 169–176. [Google Scholar]
- SalviS, Tuberosa R, Sanguineti MC, Landi P, Conti S.1997. Molecular marker analysis of maize populations divergently selected for abscisic acid concentration in the leaf. Maize Genetics Cooperation Newsletter 71: 15–16. [Google Scholar]
- SalviS, Tuberosa R, Chiapparino E, Maccaferri M, Veillet S, van Beuningen L, Isaac P, Edwards K, Phillips RL.2002. Toward positional cloning of Vgt1, a QTL controlling the transition from the vegetative to the reproductive phase in maize. Plant Molecular Biology 48: 601–613. [DOI] [PubMed] [Google Scholar]
- SanguinetiMC, Conti S, Landi P, Tuberosa R.1996. Abscisic acid concentration in maize leaves: genetic control and response to divergent selection in two populations. Maydica 41: 193–203. [Google Scholar]
- SanguinetiMC, Giuliani MM, Govi G, Tuberosa R, Landi P.1998. Root and shoot traits of maize inbred lines grown in the field and in hydroponic culture and their relationships with root lodging. Maydica 43: 211–216. [Google Scholar]
- SanguinetiMC, Tuberosa R, Landi P, Salvi S, Maccaferri M, Casarini E, Conti S.1999. QTL analysis of drought‐related traits and grain yield in relation to genetic variation for leaf abscisic acid concentration in field‐grown maize. Journal of Experimental Botany 50: 1289–1297. [Google Scholar]
- Sari‐GorlaM, Krajewski P, Di Fonzo N, Villa M, Frova C.1999. Genetic analysis of drought tolerance in maize by molecular markers. II. Plant height and flowering. Theoretical and Applied Genetics 99: 289–295. [Google Scholar]
- SaxK.1923. The association of size differences with seed‐coat pattern and pigmentation in Phaseolus vulgaris Genetics 8: 552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SchafferR, Landgraf J, Pérez‐Amador M, Wisman E.2000. Monitoring genome‐wide expression in plants. Biotechnology 11: 162–167. [DOI] [PubMed] [Google Scholar]
- SchneiderK, Borchardt DC, Schafer‐Pregl R, Nagl N, Glass C, Jeppsson A, Gebhardt C, Salamini F.1999. PCR‐based cloning and segregation analysis of functional gene homologues in Beta vulgaris Molecular and General Genetics 262: 515–524. [DOI] [PubMed] [Google Scholar]
- SchwartzSH, Tan BC, Gage DA, Zeevaart JAD, McCarty DR.1997. Specific oxidative cleavage of carotenoids by VP14 of maize. Science 276: 1872–1874. [DOI] [PubMed] [Google Scholar]
- SenS, Churchill GA.2001. A statistical framework for quantitative trait mapping. Genetics 159, 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SetterTL.1997. Role of the phytohormone ABA in drought tolerance: potential utility as a selection tool. In: Edmeades GO, Bänziger M, Mickelson HR, Peña‐Valdivia CB, eds. Developing drought‐ and low N‐tolerant maize. Proceedings of a Symposium, March 25–29, 1996, CIMMYT, El Batán, Mexico, 142–150. [Google Scholar]
- SharopovaN, McMullen MD, Schultz L, Schroeder S, Sanchez‐Villeda H, Gardiner J, Bergstrom D, Houchins K, Melia‐Hancock S, Musket T, Duru N, Polacco M, Edwards K, Ruff T, Register JC, Brouwer C, Thompson R, Velasco R, Chin E, Lee M, Woodman‐Clikeman W, Long MJ, Liscum E, Cone K, Davis G, Coe EH.2002. Development and mapping of SSR markers for maize. Plant Molecular Biology 48: 463–481. [DOI] [PubMed] [Google Scholar]
- SharpRE.1996. Regulation of plant growth responses to low soil water potentials. Horticultural Science 31: 36–39. [Google Scholar]
- SharpRE, Wu Y, Voetberg GS, Saab IN, LeNoble ME, Wang TL Davies WJ, Pollock CJ.1994. Confirmation that abscisic acid accumulation is required for maize primary root elongation at low water potentials. Journal of Experimental Botany 45: 1743–1751. [Google Scholar]
- ShimketsRA, Lowe DG, Tai JT, Sehl P, Jin H, Yang R, Predki PF, Rothberg BE, Murtha MT, Roth ME, Shenoy SG, Windemuth A, Simpson JW, Simons JF, Daley MP, Gold SA, McKenna MP, Hillan K, Went GT, Rothberg JM.1999. Gene expression analysis by transcript profiling coupled to a gene database query. Nature Biotechnology 17: 798–803. [DOI] [PubMed] [Google Scholar]
- SignoraL, De Smet I, Foyer CH, Zhang H.2001. ABA plays a central role in mediating the regulatory effects of nitrate on root branching in Arabidopsis The Plant Journal 28: 655–662. [DOI] [PubMed] [Google Scholar]
- SimkoI, McMurry S, Yang HM, Manschot A, Davies PJ, Ewing EE.1997. Evidence from polygene mapping for a causal relationship between potato tuber dormancy and abscisic acid content. Plant Physiology 115: 1453–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SomervilleC, Somerville S.1999. Plant functional genomics. Science 285: 380–383. [DOI] [PubMed] [Google Scholar]
- SpollenWG, LeNoble ME, Samuels TD, Bernstein N, Sharp RE.2000. Abscisic acid accumulation maintains maize primary root elongation at low water potentials by restricting ethylene production. Plant Physiology 122: 967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- StuberCW, Edwards MD, Wendel JF.1987. Molecular marker‐facilitated investigations of quantitative trait loci in maize. II. Factors influencing yield and its component traits. Crop Science 27: 639–648. [Google Scholar]
- StuberCW, Polacco M, Senior ML.1999. Synergy of empirical breeding, marker‐assisted selection, and genomics to increase crop yield potential. Crop Science 39: 1571–1583. [Google Scholar]
- SullivanCY.1983. Genetic variability in physiological mechanisms of drought resistance. Iowa State Journal of Research 57: 423–439. [Google Scholar]
- TAGI (The Arabidopsis Genome Initiative). 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana Nature 408: 796–815. [DOI] [PubMed] [Google Scholar]
- TakahashiY,Shomura A, Sasaki T, Yano M.2001Hd6, a rice quantitative trait locus involved in photoperiod sensitivity, encodes the α subunit of protein kinase CK2. Proceedings of the National Academy of Sciences of the United States of America 98: 7922–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TanBC, Schwartz SH, Zeevaart JAD, McCarty DR.1997. Genetic control of abscisic acid biosynthesis in maize. Proceedings of the National Academy of Sciences of the United States of America 94: 12235–12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TanksleySD.1993. Mapping polygenes. Annual Review of Genetics 27: 205–233. [DOI] [PubMed] [Google Scholar]
- TanksleySD, Nelson JC.1996. Advanced backcross QTL analysis: a method for the simultaneous discovery and transfer of valuable QTLs from unadapted germplasm into elite breeding lines. Theoretical and Applied Genetics 92: 191–203. [DOI] [PubMed] [Google Scholar]
- TanksleySD, Ganal MW, Martin GB.1995. Chromosome landing: a paradigm for map‐based gene cloning in plants with large genomes. Trends in Genetics 11: 63–68. [DOI] [PubMed] [Google Scholar]
- TanksleySD, Grandillo S, Fulton TM, Zamir D, Eshed T, Petiard V, Lopez J, Beck‐Bunn T.1996. Advanced backcross QTL analysis in a cross between an elite processing line of tomato and its wild relative L. pimpinellifolium Theoretical and Applied Genetics 92: 213–224. [DOI] [PubMed] [Google Scholar]
- TaraminoG, Tingey S.1996. Simple sequence repeats for germplasm analysis and mapping in maize. Genome 39: 277–287. [DOI] [PubMed] [Google Scholar]
- TardieuF, Gowing D, Davies WJ.1993. A model of stomatal control by both ABA concentration in the xylem sap and leaf water status: test of the model and of alternative mechanisms for droughted and ABA‐fed field‐grown maize. Plant Cell and Environment 16: 413–420. [Google Scholar]
- TardieuF, Katerji N, Bethenod O, Zhang J, Davies WJ.1991. Maize stomatal conductance in the field: its relationship with soil and plant water potentials, mechanical constraints and ABA concentration in the xylem sap. Plant Cell and Environment 14: 121–126. [Google Scholar]
- TardieuF, Zhang J, Katerji N, Bethenod O, Palmer S, Davies WJ.1992. Xylem ABA controls the stomatal conductance of field‐grown maize subjected to soil compaction or soil drying. Plant Cell and Environment 15: 193–197. [Google Scholar]
- ThiellementH, Bahrman N, Damerval C, Plomion C, Rossignol M, Santoni V, de Vienne D, Zivy M.1999. Proteomics for genetic and physiological studies in plants. Electrophoresis 20: 2013–2026. [DOI] [PubMed] [Google Scholar]
- ThornsberryJM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ES.2001. Dwarf8 polymorphisms associated with variation in flowering time. Nature Genetics 28: 286–289. [DOI] [PubMed] [Google Scholar]
- TouzetP, Winkler RG, Helentjaris T.1995. Combined genetic and physiological analysis of a locus contributing to quantitative variation. Theoretical and Applied Genetics 91: 200–205. [DOI] [PubMed] [Google Scholar]
- TrejoCL, Clephan AL, Davies WJ.1995. How do stomata read abscisic acid signals? Plant Physiology 109: 803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TuberosaR, Salvi S, Phillips RL.1998a Bulked segregant analysis confirms the importance of the region near umc89a for days to pollen shed in maize. Maize Genetics Cooperative Newsletter 72: 71–72. [Google Scholar]
- TuberosaR, Sanguineti MC, Conti S.1986. Divergent selection for heading date in barley. Plant Breeding 97: 345–351. [Google Scholar]
- TuberosaR, Sanguineti MC, Landi P.1994. Abscisic acid concentration in the leaf and xylem sap, leaf water potential, and stomatal conductance in drought‐stressed maize. Crop Science 34: 1557–1563. [Google Scholar]
- TuberosaR, Parentoni S, Kim TS, Sanguineti MC, Phillips RL.1998b Mapping QTLs for ABA concentration in leaves of a maize cross segregating for anthesis date. Maize Genetics Cooperation Newsletter 72: 72–73. [Google Scholar]
- TuberosaR, Sanguineti MC, Landi P, Salvi S, Casarini E, Conti S.1998c RFLP mapping of quantitative trait loci controlling abscisic acid concentration in leaves of drought‐stressed maize (Zea mays L.). Theoretical and Applied Genetics 97: 744–755. [Google Scholar]
- TuberosaR, Sanguineti MC, Ribaut JM, Landi P, Giuliani M, Salvi S, Conti S.2000. QTL analysis of root characteristics in maize grown in hydroponics as related to field performance under drought conditions. International Conference on Plant & Animal Genome VIII, January 9–12, 2000, San Diego, California, 5. [Google Scholar]
- TuberosaR, Sanguineti MC, Landi P, Giuliani MM, Salvi S, Conti S.2002. Identification of QTLs for root characteristics in maize grown in hydroponics and analysis of their overlap with QTLs for grain yield in the field at two water regimes. Plant Molecular Biology 48: 697–712. [DOI] [PubMed] [Google Scholar]
- UtzHF, Melchinger AE.1996. PLABQTL. A program for composite interval mapping of QTL. Journal of Quantitative Trait Loci (http://probe.nalusda.gov:800/otherdocs/jqtl). [Google Scholar]
- VanBuurenM, Salvi S, Morgante M, Serhani B, Tuberosa R.2002. Comparative genomic mapping between a 754 kb region flanking DREB1A in Arabidopsis thaliana and maize. Plant Molecular Biology 48: 741–750. [DOI] [PubMed] [Google Scholar]
- VelculescuVE, Zhang L, Vogelstein B, Kinzler KW.1995. Serial analysis of gene expression. Science 270: 484–487. [DOI] [PubMed] [Google Scholar]
- VosP, Hogers R, Bleeker M, Reijans M, Van de Lee T, Hornes M, Freijters A, Pot J, Peleman J, Kuiper M, Zabeau M.1995. AFLP: a new technique for DNA fingerprinting. Nucleic Acid Research 23: 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VuylstekeM, Kuiper M, Stam P.2000a Chromosomal regions involved in hybrid performance and heterosis: their AFLP®‐based identification and practical use in prediction models. Heredity 85: 208–218. [DOI] [PubMed] [Google Scholar]
- VuylstekeM, Mank R, Brugmans B, Stam P, Kuiper M.2000b Further characterization of AFLP® data as a tool in genetic diversity assessments among maize (Zea mays L.) inbred lines. Molecular Breeding 6: 265–276. [Google Scholar]
- VuylstekeM, Mank R, Antonise R, Bastiaans E, Senior ML, Stuber CW, Melchinger AE, Lübberstedt T, Xia XC, Stam P, Zabeau M, Kuiper M.1999. Two high‐density AFLP® linkage maps of Zea mays L.: analysis of distribution of AFLP markers. Theoretical and Applied Genetics 99: 921–935. [Google Scholar]
- WangDG, Fan JB, Siao CJ, Berno A, Young P, Sapolsky R, Ghandour G, Perkins N, Winchester E, Spencer J, Kruglyak L, Stein L, Hsie L, Topaloglou T, Hubbell E, Robinson E, Mittmann M, Morris MS, Shen N, Kilburn D, Rioux J, Nusbaum C, Rozen S, Hudson TJ, Lipshutz R, Chee M, Lander ES.1998. Large‐scale identification, mapping, and genotyping of single‐nucleotide polymorphisms in the human genome. Science 280: 1077–1082. [DOI] [PubMed] [Google Scholar]
- WangGL, Paterson AH.1994. Assessment of DNA pooling strategies for mapping of QTLs. Theoretical and Applied Genetics 88: 355–361. [DOI] [PubMed] [Google Scholar]
- WangGL, Zhu J, Li ZK, Paterson AH.1999. Mapping QTLs with epistatic effects and QTL × environment interactions by mixed linear model approaches. Theoretical and Applied Genetics 99: 1255–1264. [Google Scholar]
- WenTJ, Schnable PS.1994. Analyses of mutants of three genes that influence root hair development in Zea mays (Gramineae) suggest that root hairs are dispensable. American Journal of Botany 81: 833–842. [Google Scholar]
- WestgateME, Boyer JS.1985. Osmotic adjustment and the inhibition of leaf, root, stem and silk growth at low water potentials in maize. Planta 164: 540–549. [DOI] [PubMed] [Google Scholar]
- WickingC, Williamson B.1991. From linked marker to gene. Trends in Genetics 7: 288–293. [DOI] [PubMed] [Google Scholar]
- WilkinsMR, Pasquali C, Appel RD, Ou K, Golaz O, Sanchez JC, Yan JX, Gooley AA, Huges G, Humphrey‐Smith I, Williams KL, Hochstrasser DF.1996. From proteins to proteomes: large‐scale protein identification by two‐dimensional electrophoresis and amino acid analysis. Biotechnology 14: 61–65. [DOI] [PubMed] [Google Scholar]
- WilsonWA, Harrington SE, Woodman WL, Lee M, Sorrels ME, McCouch SR.1999. Inferences on the genome structure of progenitor maize through comparative analysis of rice, maize and the domesticated Panicoids. Genetics 153: 453–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WinklerR, Helentjaris T.1995. The maize dwarf3 gene encodes a cytochrome P450‐mediated early step in gibberellin biosynthesis. Plant Cell 7: 1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WitcombeJR, Hash CT.2000. Resistance gene deployment strategies in cereal hybrids using marker‐assisted selection: gene pyramiding, three‐way hybrids, and synthetic parent populations. Euphytica 112: 175–186. [Google Scholar]
- WuYJ, Sharp RE, Durachko DM, Cosgrove DJ.1996. Growth maintenance of the maize primary root at low water potentials involves increases in cell‐wall extension properties, expansin activity, and wall susceptibility to expansins. Plant Physiology 111: 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XiaoJ,Li J, Grandillo S, Ahn SN, Yuan L, Tanksley SD, McCouch SR.1998. Identification of trait‐improving quantitative trait loci alleles from a wild rice relative, Oryza rufipogon Genetics 150: 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YamamotoT, Kuboki Y, Lin SY, Sasaki T, Yano M.1998. Fine mapping of quantitative trait loci Hd‐1, Hd‐2, and Hd‐3, controlling heading date of rice, as single Mendelian factors. Theoretical and Applied Genetics 97: 37–44. [Google Scholar]
- YanoM, Katayose Y, Ashikari M, Yamanouchi U, Monna L, Fuse T, Baba T, Yamamoto K, Umehara Y, Nagamura Y, Sasaki T.2000. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 12: 2473–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YatesJRI.1998. Mass spectrometry and the age of the proteome. Journal of Mass Spectrometry 33: 1–19. [DOI] [PubMed] [Google Scholar]
- YoungND.1999. A cautiously optimistic vision for marker‐assisted breeding. Molecular Breeding 5: 505–510. [Google Scholar]
- ZeevaartJAD, Creelman RA.1988. Metabolism and physiology of abscisic acid. Annual Review of Plant Physiology and Plant Molecular Biology 39: 439–473. [Google Scholar]
- ZengZB.1994. Precision mapping of quantitative trait loci. Genetics 136: 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZhangJ, Davies WJ.1990. Does ABA in the xylem control the rate of leaf growth in soil‐dried maize and sunflower plants? Journal of Experimental Botany 41: 1125–1132. [Google Scholar]
- ZhangJX, Klueva NY, Wang Z, Wu R, Ho TH, Nguyen HT.2000. Genetic engineering for abiotic stress resistance in crop plants. In Vitro Cellular and Developmental Biology 36: 108–114. [Google Scholar]
- ZhuT, Budworth P, Han B, Brown D, Chang HS, Zou GZ, Wang X.2001. Toward elucidating the global gene expression patterns of developing Arabidopsis: Parallel analysis of 8300 genes by a high‐density oligonucleotide probe array. Plant Physiology and Biochemistry 39: 221–242. [Google Scholar]
- ZinselmeierC, Jeong B‐R, Boyer JS.1999. Starch and the control of kernel number in maize at low water potentials. Plant Physiology 121: 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZinselmeierC, Westgate ME, Schussler JR, Jones RJ.1995. Low water potential disrupts carbohydrate metabolism in maize (Zea mays L.) ovaries. Plant Physiology 107: 385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZivyM, de Vienne D.2000. Proteomics: a link between genomics, genetics and physiology. Plant Molecular Biology 44: 575–580. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}