

Summary
Aims
The 24‐week, prospective, randomized, double‐blind ACTION study investigated the efficacy, safety, and tolerability of 13.3 versus 4.6 mg/24 h rivastigmine patch in patients with severe Alzheimer's disease (AD).
Methods
Patients had probable AD and Mini–Mental State Examination scores ≥3–≤12. Primary outcome measures were as follows: Severe Impairment Battery (SIB) and AD Cooperative Study–Activities of Daily Living scale–Severe Impairment Version (ADCS‐ADL‐SIV). Secondary outcomes were as follows: ADCS‐Clinical Global Impression of Change (ADCS‐CGIC), 12‐item Neuropsychiatric Inventory (NPI‐12), and safety/tolerability.
Results
Of 1014 patients screened, 716 were randomized to 13.3 mg/24 h (N = 356) or 4.6 mg/24 h (N = 360) patch. Baseline characteristics/demographics were comparable. Completion rates were as follows: 64.3% (N = 229) with 13.3 mg/24 h and 65.0% (N = 234) with 4.6 mg/24 h patch. The 13.3 mg/24 h patch was significantly superior to 4.6 mg/24 h patch on cognition (SIB) and function (ADCS‐ADL‐SIV) at Week 16 (P < 0.0001 and P = 0.049, respectively) and 24 (primary endpoint; P < 0.0001 and P = 0.025). Significant between‐group differences (Week 24) were observed on the ADCS‐CGIC (P = 0.0023), not NPI‐12 (P = 0.1437). A similar proportion of the 13.3 mg/24 h and 4.6 mg/24 h patch groups reported adverse events (AEs; 74.6% and 73.3%, respectively) and serious AEs (14.9% and 13.6%).
Conclusions
The 13.3 mg/24 h patch demonstrated superior efficacy to 4.6 mg/24 h patch on SIB and ADCS‐ADL‐SIV, without marked increase in AEs, suggesting higher‐dose patch has a favorable benefit‐to‐risk profile in severe AD.
Keywords: Clinical trial, Rivastigmine, Severe Alzheimer's disease, Transdermal patch
Introduction
As patients with Alzheimer's disease (AD) progress to severe stages, there is further degeneration of cortically projecting cholinergic neurons and changes in brain cholinesterase levels 1 associated with progressive impairments in memory, cognition, behavior, and performance of activities of daily living (ADL).
Cholinesterase inhibitors partially compensate for cholinergic deficits, providing symptomatic relief. Three cholinesterase inhibitors are widely approved for mild‐to‐moderate AD; rivastigmine (Exelon®, Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA), donepezil (ARICEPT®, Eisai Inc., Woodcliff Lake, NJ, USA), and galantamine (Razadyne®, Janssen Pharmaceutical N.V., Beerse, Belgium) 2, 3, 4, 5. Until recently, treatment options for severe AD were limited; donepezil is indicated for moderate‐to‐severe AD in the USA 4 and memantine (N‐methyl‐D‐aspartate receptor antagonist) for moderate‐to‐severe AD in the USA and several other countries worldwide 6.
Rivastigmine shows dose‐dependent efficacy on cognition, ADL, and global functioning 7, 8. The OPTIMA (OPtimising Transdermal Exelon In Mild‐to‐moderate AD) study demonstrated significantly greater efficacy on ADL with 13.3 mg/24 h (15 cm2) versus 9.5 mg/24 h (10 cm2) rivastigmine patch in patients with mild‐to‐moderate AD who showed functional and cognitive decline during preceding open‐label treatment with 9.5 mg/24 h patch 9.
Pooled analysis of clinical trial data suggests rivastigmine may continue to provide benefits at more advanced stages of disease 10, 11. A randomized, double‐blind, study demonstrated that oral rivastigmine was efficacious compared with placebo in moderately severe AD 12.
The objective of the ACTION (ACTivities of daily living and cognitION) study was to compare the efficacy, safety, and tolerability of 13.3 mg/24 h (15 cm2) versus 4.6 mg/24 h (5 cm2) rivastigmine patch in patients with severe AD.
Materials and Methods
Patients
Patients were male/female, aged ≥50 years, with probable AD (original 1984 National Institute of Neurological and Communicative Disorders and Stroke and AD and Related Disorders Association criteria) 13, and Mini–Mental State Examination (MMSE) 14 scores ≥3–≤12. Magnetic resonance imaging/computed tomography, used in the diagnosis of probable AD, was required within the prior 2 years. Patients were living with someone in the community or were in regular contact with their primary caregiver. Patients in assisted living facilities were eligible provided assessment could take place at the study site, and a caregiver was identified 15.
Exclusion criteria included any advanced/severe/progressive/unstable disease that could interfere with response to study treatment; patients living in/permanently placed during the study/likely (physicians' opinion) to be placed in a nursing home within the next 7 months; current medical/neurological condition other than AD that could be the primary cause of dementia; current diagnosis of probable/possible vascular dementia, uncontrolled seizure disorder, severe/unstable cardiovascular disease, bradycardia, sick‐sinus syndrome or conduction defects; current diagnosis of acute/severe/unstable asthmatic conditions; current diagnosis of uncontrolled peptic ulceration or gastrointestinal bleeding within the previous 3 months; and/or a history (past year) or current diagnosis of cerebrovascular disease. Patients were also excluded if they had a Diagnostic and Statistical Manual of Mental Disorders diagnosis of major depression 16, unless successfully treated (antidepressant without anticholinergic properties) in a stable regimen for ≥4 weeks; clinically significant urinary obstruction; allergy to vitamin E‐containing products, sensitivity to cholinergic drugs, or skin lesion/disorder that would prevent patch use; history of malignancy (≤5 years); use of cholinesterase inhibitors/other approved AD treatments 2 weeks prior (except stable memantine if taken for ≥3 months); use of centrally acting cholinergic drugs/any investigational drug for 4 weeks prior; use of peripheral anticholinergic drugs/selegiline, or new psychotropic/dopaminergic drugs if not taken at stable dose, for 4 weeks prior 15.
Standard Protocol Approvals, Registrations, and Patient Consents
The protocol and amendments were reviewed by Independent Ethics Committees or Institutional Review Boards. The study was conducted in accordance with Good Clinical Practice and the ethical principles of the Declaration of Helsinki. All patients, or if they lacked capacity, their legally authorized representative, provided written informed consent prior to participating. This study is registered (clinicaltrials.gov NCT00948766).
Protocol amendments after study‐start included clarifying enrollment eligibility requirements and revising instructions for patch application to prevent administration errors.
Study Design
ACTION was a 24‐week, prospective, randomized, double‐blind, double‐dummy, multicenter trial conducted at 82 centers across the USA between July 22, 2009 and January 10, 2012 (last‐patient‐last‐visit). Patients were randomized (1:1) at Week 0 to 13.3 mg/24 h or 4.6 mg/24 h rivastigmine patch. All patients initiated treatment on 4.6 mg/24 h patch. Patients randomized to 13.3 mg/24 h patch were up‐titrated (start of Week 4) to 9.5 mg/24 h patch and at the start of Week 8 to 13.3 mg/24 h patch. Patients randomized to 4.6 mg/24 h patch remained at that dose for the 8‐week titration. Patients were maintained at the target dose for the 16‐week maintenance period 15. The 4.6 mg/24 h patch group also received 10 cm2 (from start of Week 4) and 15 cm2 placebo patches (from start of Week 8–24). The 13.3 mg/24 h patch group received 5 cm2 placebo patches throughout.
For patients missing >3 consecutive days of treatment due to tolerability problems, treatment could be restarted (4.6 mg/24 h) and the dose increased after 2 weeks minimum. If tolerability was improved and the patient had missed treatment for ≤3 consecutive days, treatment could be restarted at the same dose level, and titration resumed. Further doses could be skipped if subsequent titration led to tolerability problems. In the maintenance phase, patients were required to be able to tolerate the maximum dose and were not permitted to down‐titrate, so as not to compromise blinding.
Primary Outcomes
Primary outcomes were the change from baseline–Week 24 on the Severe Impairment Battery (SIB) 17 and AD Cooperative Study–ADL scale–Severe Impairment Version (ADCS‐ADL‐SIV) 18.
Secondary Outcomes
Secondary outcomes were as follows: ADCS–Clinical Global Impression of Change (ADCS‐CGIC) 19 score at Week 24, and the change from baseline–Week 24 on the 12‐item Neuropsychiatric Inventory (NPI‐12) 20.
In addition to Week 24 (primary endpoint), all efficacy measures were assessed at Weeks 8 and 16.
Evaluations to maintain safety included the following: incidence of adverse events (AEs) and serious AEs (SAEs); laboratory tests; electrocardiogram analysis; assessments of skin irritation and vital signs; and the discontinuation rate due to AEs.
Sample Size, Randomization, and Blinding
It was estimated that 338 patients were required/group to achieve an effect size of 0.25 on the primary efficacy variables and overall power between 82% and 85%, assuming a correlation coefficient between the co‐primary efficacy variables of 0.3–0.6. To adjust for the 5% of patients estimated to be lost to follow‐up, a total sample size of 712 was planned.
Centralized block randomization was performed by an interactive voice response system. The investigator/his/her delegate was required to contact the interactive voice response system and confirm patient eligibility. The interactive voice response system assigned a randomization number, linking the patient to a treatment arm, and specified a unique medication number to dispense the first package of study medication. The randomization scheme was reviewed and approved by the Novartis Biostatistics Quality Assurance Group.
Patients, study investigators, and data analysts remained blinded from randomization until database lock. Unblinding occurred only in case of patient emergencies and at study end.
Statistical Analyses
The null hypotheses were 13.3 mg/24 h would not differ from 4.6 mg/24 h patch in the change from baseline–Week 24 on ADCS‐ADL‐SIV/SIB total score. The alternative was 13.3 mg/24 h differs from 4.6 mg/24 h patch in change from baseline–Week 24 in ADCS‐ADL‐SIV and SIB total score. Significant efficacy on both primary outcomes was required to demonstrate superiority of 13.3 mg/24 h over 4.6 mg/24 h patch.
Analyses of primary outcomes were based on the modified full analysis set (all randomized patients who received ≥1 dose of study medication and had ≥1 postbaseline measurement). Imputation of missing values was performed following the last‐observation‐carried‐forward approach. Treatment differences in the change from baseline on the ADCS‐ADL‐SIV, SIB, and NPI‐12 were compared using least‐squares means derived using analysis of covariance (ANCOVA) with treatment and pooled center as factors and corresponding baseline score as a covariate. ADCS‐CGIC scores were analyzed using the Cochran–Mantel–Haenszel test, with modification relative to an identified distribution integral transformation scores adjusting for pooled center.
Longitudinal analysis of the change from baseline for the co‐primary efficacy variables was performed for the modified full analysis set using observed cases. An unstructured covariance matrix for the repeated measures within each patient was applied. Explanatory variables included treatment, pooled center, week, treatment‐by‐week, and corresponding baseline. Treatment groups were compared based on least‐squares means. The SAS procedure PROC MIXED was used.
Sensitivity analyses were conducted using a pattern mixture model considering missing data (completers and noncompleters) for each co‐primary efficacy variable and were based on a repeated‐measures ANCOVA model with treatment, pooled center, week, dropout, treatment‐by‐week, treatment‐by‐dropout as factors, and baseline as a covariate, assuming an unstructured within‐subject covariance matrix.
Safety analyses were based on the safety set (all patients who received ≥1 dose of study medication and had ≥1 safety assessment postbaseline) and were summarized according to treatment received.
For continuous variables, number of patients with observed values (n), mean, standard deviation (SD), 95% confidence intervals (95% CI), minimum, and maximum were calculated. Categorical variables were summarized by frequency counts and percentages. Unless otherwise specified, all statistical tests were conducted against a two‐sided alternative hypothesis; P‐values below 0.05 were considered significant.
Results
Study Participants
Of 1014 patients screened, 716 were enrolled and randomized to 13.3 mg/24 h (N = 356) or 4.6 mg/24 h rivastigmine patch (N = 360). Similar proportions of each group completed the study (Figure 1). Baseline demographics and characteristics were comparable (Table 1).
Figure 1.
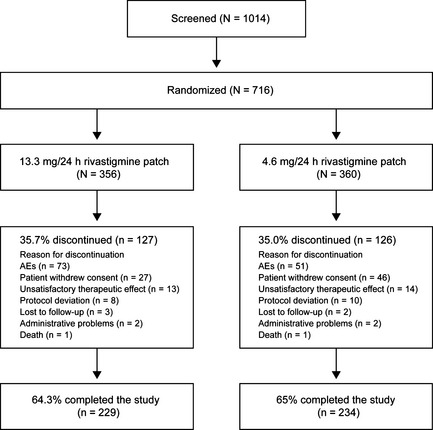
Patient disposition throughout the study (randomized population). AEs, adverse events; N, number of patients in the population; n, number of patients with an assessment. One patient in each treatment group was randomized, but was not exposed to study medication.
Table 1.
Patient demographics and background characteristics by treatment group (randomized set)
| 13.3 mg/24 h rivastigmine patch | 4.6 mg/24 h rivastigmine patch | Total | |
|---|---|---|---|
| N = 356 | N = 360 | N = 716 | |
| Age, years | |||
| Mean (SD) | 77.6 (8.7) | 76.5 (9.4) | 77.0 (9.0) |
| Range | 52–96 | 51–96 | 51–96 |
| Gender, % | |||
| Female | 63.8 | 65.0 | 64.4 |
| Predominant race, % | |||
| Caucasian | 86.0 | 88.6 | 87.3 |
| Black | 7.9 | 5.3 | 6.6 |
| Other | 6.2 | 6.1 | 6.2 |
| MMSE score | |||
| Mean (SD) | 8.8 (2.9) | 8.8 (3.0) | 8.8 (2.9) |
| Range | 3.0–13.0 | 3.0–19.0 | 3.0–19.0 |
| Years since diagnosis of AD | |||
| Mean (SD) | 4.3 (2.7) | 4.0 (2.7) | 4.1 (2.7) |
| Range | 0.0–19.1 | 0.0–18.3 | 0.0–19.1 |
| Years since diagnosis of severe dementia | |||
| Mean (SD) | 1.2 (1.9) | 1.2 (1.6) | 1.2 (1.7) |
| Range | 0.0–12.2 | 0.0–9.8 | 0.0–12.2 |
| Patients living situation, % | |||
| Home | 90.4 | 88.1 | 89.2 |
| Assisted living facility | 7.6 | 9.7 | 8.7 |
| Other | 2.0 | 2.2 | 2.1 |
AD, Alzheimer's disease; MMSE, Mini–Mental State Examination; N, number of patients in the randomized population; SD, standard deviation.
Dosing
All patients randomized to 4.6 mg/24 h patch received this dose at Week 24. Of those patients randomized to 13.3 mg/24 h patch, 85.1% received the target dose at Week 24; 6.5% and 8.5% received 9.5 mg/24 h or 4.6 mg/24 h patch, respectively. Mean (SD) duration of exposure was 19.6 (7.9) weeks in the 13.3 mg/24 h patch group and 20.1 (7.6) weeks in the 4.6 mg/24 h patch group.
Concomitant Medications
Overall, 96.1% and 95.8% of the 13.3 mg/24 h and 4.6 mg/24 h patch groups, respectively, were taking concomitant medication and/or using nondrug therapies. There were no notable differences in concomitant medication use between groups; the most commonly used were platelet aggregation inhibitors (43.9%, 13.3 mg/24 h; 40.4%, 4.6 mg/24 h patch group) and 3‐hydroxy‐3‐methyl‐glutaryl‐CoA reductase inhibitors (40.3% and 41.2%, respectively).
Psychotropic medications were taken by 83.9% and 82.5% of the 13.3 mg/24 h and 4.6 mg/24 h patch groups, respectively, and were most commonly antidementia drugs (primarily memantine hydrochloride; 60.6%), and selective serotonin reuptake inhibitors (38.7%).
Primary Outcome Analyses
SIB scores decreased from baseline in both groups throughout the study. Significantly less deterioration was observed at Weeks 16 (P < 0.0001; difference 4.9 points; 95% CI 2.8, 6.9) and 24 (primary endpoint; P < 0.0001; difference 4.9 points; 95% CI 2.8, 7.0) with 13.3 mg/24 h compared with 4.6 mg/24 h patch (Figure 2A; Table 2). Similar findings were observed in longitudinal (P < 0.0001; difference 5.3 points; 95% CI 3.1, 7.5 at Week 16 and P < 0.0001; difference 5.3 points; 95% CI 3.0, 7.7 at Week 24) and sensitivity analyses (P < 0.0001; difference 6.0 points; 95% CI 3.6, 8.3 at Week 16 and P < 0.0001; difference 6.1 points, 95% CI 3.6, 8.6 at Week 24).
Figure 2.
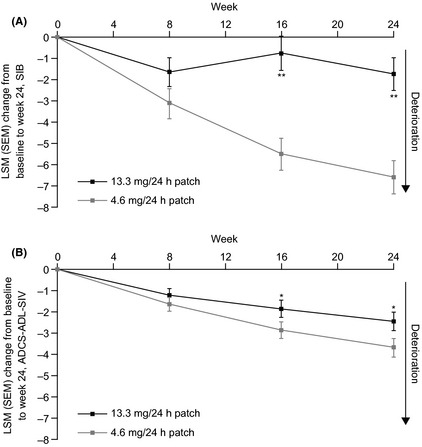
Least‐squares means change from baseline to Week 24 on (A) SIB and (B) ADCS‐ADL‐SIV (modified full analysis set). ADCS‐ADL‐SIV, Alzheimer's Disease Cooperative Study–Activities of Daily Living scale–Severe Impairment Version; SEM, standard error of the least‐squares means; SIB, Severe Impairment Battery. Error bars represent the SEM. *P < 0.05; **P < 0.0001 versus 4.6 mg/24 h patch.
Table 2.
Primary (SIB and ADCS‐ADL‐SIV) and secondary (ADCS‐CGIC and NPI‐12) efficacy outcomes (modified full analysis set)
| 13.3 mg/24 h rivastigmine patch | 4.6 mg/24 h rivastigmine patch | P‐value | |
|---|---|---|---|
| N = 338 | N = 335 | ||
| SIB | |||
| N (baseline) | 336 | 334 | |
| Mean (SD) score at baseline | 69.3 (21.5) | 68.3 (22.8) | |
| N (Week 24) | 313 | 316 | |
| Mean (SD) change from baseline at Week 24 | −1.6 (13.5) | −6.4 (14.0) | |
| Least‐squares means (SE) change from baseline at Week 24 | −1.7 (0.8) | −6.6 (0.8) | |
| Least‐squares means difference (95% CI) | 4.9 (2.8, 7.0) | <0.0001 | |
| ADCS‐ADL‐SIV | |||
| N (baseline) | 333 | 319 | |
| Mean (SD) score at baseline | 29.7 (11.3) | 29.1 (11.9) | |
| N (Week 24) | 310 | 303 | |
| Mean (SD) change from baseline at Week 24 | −2.6 (6.8) | −3.6 (7.7) | |
| Least‐squares means (SE) change from baseline at Week 24 | −2.4 (0.4) | −3.6 (0.4) | |
| Least‐squares means difference (95% CI) | 1.2 (0.2, 2.3) | 0.0247 | |
| ADCS‐CGIC | |||
| Week 24, n (%) | 0.0023 | ||
| Marked improvement | 3 (1.0) | 4 (1.3) | |
| Moderate improvement | 11 (3.5) | 11 (3.5) | |
| Minimal improvement | 63 (20.1) | 36 (11.4) | |
| No change | 107 (34.2) | 92 (29.2) | |
| Minimal worsening | 76 (24.3) | 99 (31.4) | |
| Moderate worsening | 44 (14.1) | 60 (19.0) | |
| Marked worsening | 9 (2.9) | 13 (4.1) | |
| NPI‐12 | |||
| N (baseline) | 335 | 331 | |
| Mean (SD) score at baseline | 17.3 (15.4) | 16.8 (16.7) | |
| N (Week 24) | 313 | 313 | |
| Mean (SD) change from baseline at Week 24 | −0.4 (14.0) | 1.2 (16.8) | |
| Least‐squares means (SE) change from baseline at Week 24 | −0.1 (0.8) | 1.5 (0.8) | |
| Least‐squares means difference (95% CI) | −1.6 (−3.8, 0.6) | 0.1437 | |
ADCS‐ADL‐SIV, Alzheimer's Disease Cooperative Study–Activities of Daily Living scale–Severe Impairment Version; ADCS‐CGIC, Alzheimer's Disease Cooperative Study–Clinical Global Impression of Change; CI, confidence interval; N, number of patients with an assessment at the given time point; n, number of patients in a given category; NPI‐12, 12‐item Neuropsychiatric Inventory; SD, standard deviation; SE, standard error; SIB, Severe Impairment Battery.
In both groups, the ADCS‐ADL‐SIV score decreased from baseline throughout the study. Significantly less deterioration was observed on the ADCS‐ADL‐SIV with 13.3 mg/24 h versus 4.6 mg/24 h patch at Weeks 16 (P = 0.049; difference 1.0 point; 95% CI 0.0, 2.0) and 24 (primary endpoint; P = 0.025; difference 1.2 points; 95% CI 0.2, 2.3; Figure 2B; Table 2). These findings were supported by the longitudinal (P = 0.057; difference 1.0 point; 95% CI −0.0, 2.1 at Week 16 and P = 0.031; difference 1.3 points; 95% CI 0.1, 2.6 at Week 24) and sensitivity analyses (P = 0.032; difference 1.3 points; 95% CI 0.1, 2.5 at Week 16 and P = 0.016; difference 1.6 points; 95% CI 0.3, 3.0 at Week 24).
Secondary Outcome Analyses
The between‐group difference in the distribution of ADCS‐CGIC ratings was significant (P = 0.0023; Table 2). A significantly higher percentage of patients receiving 13.3 mg/24 h compared with 4.6 mg/24 h patch displayed improvement in clinical status from baseline–Week 24 (P = 0.0094). There were no significant between‐group differences at Week 24 on the NPI‐12 (Table 2; P = 0.1437; difference −1.6 points; 95% CI −3.8, 0.6).
Safety and Tolerability
Overall, the incidence of AEs was similar between the 13.3 mg/24 h and 4.6 mg/24 h patch groups (74.6% [n = 265/355] vs. 73.3% [n = 263/359], respectively; Table 3). By preferred term, most AEs were more frequent with 13.3 mg/24 h than 4.6 mg/24 h patch (Table 3), with the exception of agitation, urinary tract infection, application site dermatitis, anxiety, confusional state, constipation, hallucination, and peripheral edema.
Table 3.
Most frequent AEsa by treatment and preferred term in the 24‐week treatment phase
| 13.3 mg/24 h rivastigmine patch N = 355 n (%) | 4.6 mg/24 h rivastigmine patch N = 359 n (%) | |
|---|---|---|
| Any AE | 265 (74.6) | 263 (73.3) |
| Application site erythema | 47 (13.2) | 42 (11.7) |
| Agitation | 41 (11.5) | 51 (14.2) |
| Urinary tract infection | 29 (8.2) | 34 (9.5) |
| Application site dermatitis | 27 (7.6) | 33 (9.2) |
| Fall | 27 (7.6) | 21 (5.8) |
| Insomnia | 25 (7.0) | 15 (4.2) |
| Vomiting | 25 (7.0) | 9 (2.5) |
| Diarrhea | 23 (6.5) | 19 (5.3) |
| Weight decreased | 23 (6.5) | 11 (3.1) |
| Nausea | 22 (6.2) | 10 (2.8) |
| Depression | 17 (4.8) | 15 (4.2) |
| Decreased appetite | 17 (4.8) | 5 (1.4) |
| Anxiety | 16 (4.5) | 16 (4.5) |
| Hypertension | 13 (3.7) | 9 (2.5) |
| Application site pruritus | 13 (3.7) | 8 (2.2) |
| Confusional state | 12 (3.4) | 13 (3.6) |
| Somnolence | 12 (3.4) | 9 (2.5) |
| Constipation | 11 (3.1) | 12 (3.3) |
| Urinary incontinence | 11 (3.1) | 10 (2.8) |
| Application site irritation | 11 (3.1) | 9 (2.5) |
| Dehydration | 11 (3.1) | 8 (2.2) |
| Dizziness | 11 (3.1) | 5 (1.4) |
AE, adverse event; N, number of patients in the population; n, number of patients reporting AE.
Only AEs with a ≥3% incidence in the 13.3 mg/24 h patch group are shown. A patient with multiple occurrences of an AE was counted only once in the AE category. AEs are presented by descending frequency in the 13.3 mg/24 h patch group.
Gastrointestinal AEs (nausea, vomiting, and diarrhea) were more frequent with 13.3 mg/24 h than 4.6 mg/24 h patch (nausea: 6.2% vs. 2.8%; vomiting 7.0% vs. 2.5%; diarrhea: 6.5% vs. 5.3%, respectively). Approximately a quarter of all patients experienced a skin irritation AE (26.5%, 13.3 mg/24 h patch; 24.0%, 4.6 mg/24 h patch).
The incidence of deaths during the study period and SAEs was comparable between the 13.3 mg/24 h and 4.6 mg/24 h patch groups (deaths: 0.3% in both groups; SAEs: 14.9% vs. 13.6%, respectively; Table 4). The deaths were not considered study‐drug‐related. SAEs were most commonly psychiatric disorders (3.1% and 4.2%, 13.3 mg/24 h and 4.6 mg/24 h patch group, respectively). Overall, 8.2% of the 13.3 mg/24 h and 4.5% of the 4.6 mg/24 h patch group discontinued due to SAEs. Discontinuations due to nonserious AEs (most commonly psychiatric disorders) were numerically higher with 13.3 mg/24 h (13.5%) than 4.6 mg/24 h patch (10.9%). Interestingly, discontinuations due to skin irritations at the application site were lower with 13.3 mg/24 h (1.7%) than 4.6 mg/24 h patch (2.5%; Table 4).
Table 4.
The incidence of deaths, SAEs, and discontinuations due to AEs and SAEs (safety set)
| 13.3 mg/24 h rivastigmine patch N = 355 n (%) | 4.6 mg/24 h rivastigmine patch N = 359 n (%) | |
|---|---|---|
| Deathsa | 1 (0.3) | 1 (0.3) |
| SAE(s) | 53 (14.9) | 49 (13.6) |
| Discontinuations due to SAE(s) | 29 (8.2) | 16 (4.5) |
| Discontinuations due to non‐serious AE(s) | 48 (13.5) | 39 (10.9) |
| Discontinuations due to nausea or vomiting | 9 (2.5) | 4 (1.1) |
| Discontinuations due to skin irritations at the application site | 6 (1.7) | 9 (2.5) |
AE, adverse event; N, number of patients in the population; n, number of patients reporting AE; SAE, serious adverse event.
Deaths that occurred during the study period.
Three clinically notable vital sign abnormalities were reported as AEs. Two patients experienced weight gain, classed as nonserious, mild in severity, and not suspected to be study‐drug‐related. One patient had an increase in systolic blood pressure, which was nonserious, mild, and suspected to be study‐drug‐related.
Conclusions
This was the first study to assess the efficacy, safety, and tolerability of 13.3 mg/24 h rivastigmine patch in patients with severe AD. As expected given the progressive nature of disease in this population, both treatment groups showed deterioration (SIB and ADCS‐ADL‐SIV) over the course of this 24‐week study. However, 13.3 mg/24 h patch was associated with superior efficacy on the SIB and ADCS‐ADL‐SIV, compared with 4.6 mg/24 h patch at Weeks 16 and 24 (co‐primary endpoint) in the primary last‐observation‐carried‐forward analyses. Supporting the primary findings, 13.3 mg/24 h patch demonstrated efficacy on global function (ADCS‐CGIC), providing evidence for clinical relevance of the high‐dose treatment effects. Longitudinal and sensitivity analyses were also supportive of the primary findings. No significant differences were observed on behavior (NPI‐12) or based on the similarity in incidence of psychiatric disorders as AEs between groups.
There tended to be a slight dose‐related increase in incidence of specific AEs, including gastrointestinal‐related (i.e., nausea, vomiting, decreased appetite, and weight loss), and application site erythema with 13.3 mg/24 h versus 4.6 mg/24 h patch. Yet, overall incidences of AEs were similar between groups suggesting that, generally, patients were able to tolerate higher doses without negatively impacting tolerability. Preliminary safety review of the 13.3 mg/24 h rivastigmine patch suggests a profile consistent with previous studies 9.
The efficacy findings are supported by previous clinical trials, pooled, and retrospective analyses, which have suggested rivastigmine may benefit patients with moderately severe or severe AD 10, 11, 12, 21, 22, 23. A 26‐week, randomized, controlled proof‐of‐concept trial of 3–12 mg/day oral rivastigmine in moderately severe to severe AD demonstrated significant improvements versus placebo on the SIB (co‐primary outcome measure) and ADCS‐CGIC 12, but did not reach significance on the NPI‐10 (co‐primary outcome measure), NPI‐4 or ADCS‐ADL 12. In a study of 24 mg oral galantamine in severe AD, cognitive function (SIB) was significantly improved, but no significant treatment effects were observed on the Minimum Data Set‐ADL scale (co‐primary efficacy measures) 24. Similarly, a study of 10 mg oral donepezil in severe AD demonstrated greater efficacy versus placebo on the SIB and Clinician's Interview‐Based Impression of Change‐Plus caregiver input (CIBIC‐Plus; co‐primary outcome measures), but not on ADCS‐ADL‐SIV or NPI 25. A randomized, double‐blind, 24‐week study of 23 mg/day versus 10 mg/day oral donepezil in moderate‐to‐severe AD demonstrated significantly greater efficacy of the higher dose at Week 24 on the SIB 26. No significant between‐group differences were observed on the co‐primary outcome measure, the CIBIC‐Plus, or secondary efficacy measures (ADCS‐ADL or MMSE) 26.
This is the first study to demonstrate efficacy of higher‐dose 13.3 mg/24 h rivastigmine patch on maintaining the ability to perform ADL, assessed as a co‐primary outcome. In addition to cognition, benefits on ADL in severe disease stages are important, not only for patients, but also for caregivers because patients with severe AD are more dependent and require greater care than patients in earlier disease stages 27, 28. Dependency on others to perform ADL impacts patient quality of life 29; minimizing functional decline could enhance quality of life and may decrease caregiver burden.
As the first study of rivastigmine patch in patients with severe AD, 4.6 mg/24 h patch was selected as a low‐dose active comparator to fully evaluate high‐dose patch in this patient population. Efficacy of 4.6 mg/24 h patch versus placebo has not been evaluated in a clinical trial setting; however, it was used as a titration dose in patients with mild‐to‐moderate AD during both the OPTIMA and Investigation of transDermal Exelon in ALzheimer's disease (IDEAL) studies 9, 30. The 4.6 mg/24 h patch provides comparable exposure to 3 mg twice daily 31, an oral titration dose associated with proven efficacy 32. Based on this comparison, it is conceivable that 4.6 mg/24 h patch could have masked the full extent of the 13.3 mg/24 h patch treatment effect in this trial. The 9.5 mg/24 h patch is currently the minimum effective dose and 13.3 mg/24 h patch the maximum effective dose for patients with mild‐to‐moderate AD, according to the US prescribing information 3. Efficacy of 9.5 mg/24 h patch and the benefit:risk ratio of 9.5 mg/24 h versus 13.3 mg/24 h patch in patients with severe AD remains to be investigated in a clinical trial setting.
In summary, higher‐dose 13.3 mg/24 h rivastigmine patch conferred benefits on cognition, ADL, and global functioning in patients with severe AD, without marked reduction in tolerability. These findings support previous data and suggest rivastigmine patch can benefit patients across the disease spectrum 9, 30. Based on the therapeutic benefit observed in this study population, the higher‐dose rivastigmine patch is now approved in the USA for the symptomatic treatment of severe AD.
Conflict of Interest
This article has not been previously published, nor is it under consideration for publication elsewhere.
Martin Farlow has served as a paid consultant for Accera, Alltech, Astellas, Avanir, Bayer, Biogen, Bristol‐Myers Squibb, Eisai Medical Research, GE Healthcare, Grifols, Helicon, INC Research, Medavante, Medivation Inc., Merck and Co. Inc., Novartis Pharma, Pfizer, Prana Biotech, QR Pharma, Sanofi Aventis Groupe, Schering‐Plough, Eli Lilly, Shire Pharmaceuticals, and Toyama; is a paid speaker for Eisai, Forest, Novartis, Eli Lilly and Pfizer; and receives research support from Accera, Biogen, Eisai, Eli Lilly and Co., Genentech, Navidea, Novartis Pharma, and Roche. George Grossberg has served as a consultant for Accera, Baxter Bioscience, Forest Labs, Lundbeck, Novartis, Otsuka, and Takeda; has received research support from Accera, Baxter, Elan, Forest, Janssen, NIH, Novartis, Noven, and Pfizer; and serves on a safety monitoring board for Merck. Carl Sadowsky has served on advisory boards for Novartis, Accera and Lilly, and speakers' bureau for Novartis, Accera, Lilly and Pamlab. Xiangyi Meng and Monique Somogyi are full‐time employees and stock holders of Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA.
Acknowledgments
The ACTION principal investigators are acknowledged for their contributions to the study.
This study was funded by Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA. Medical writing and editorial assistance in the development of this manuscript were provided by Katy Cooke at Fishawack Communications Ltd, Abingdon, Oxon, UK, and this service was supported by Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA.
References
- 1. Perry EK, Perry RH, Blessed G, et al. Changes in brain cholinesterases in senile dementia of Alzheimer type. Neuropathol Appl Neurobiol 1978;4:273–277. [DOI] [PubMed] [Google Scholar]
- 2. Exelon® . US prescribing information. 2006. [online]. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2006/020823s016,021025s008lbl.pdf. (Accessed on 24 January 2013).
- 3. Exelon Patch® . US Prescribing Information. 2009. Revised 2013. [online]. Available at: http://www.pharma.us.novartis.com/product/pi/pdf/exelonpatch.pdf. (Accessed on 24 January 2013).
- 4. ARICEPT . Prescribing Information. 2010. [online]. Available at: http://www.aricept.com/assets/pdf/AriceptComboFullPINovember2010.pdf. (Accessed on 24 January 2013).
- 5. Galantamine (Razadyne ER®) . Prescribing information. [online]. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2004/021615lbl.pdf. (Accessed on 24 January 2013).
- 6. Namenda US . Prescribing Information. 2011. [online]. Available at: http://www.frx.com/pi/namenda_pi.pdf. (Accessed on 24 January 2013).
- 7. Grossberg GT, Olin JT, Somogyi M, et al. Dose effects associated with rivastigmine transdermal patch in patients with mild‐to‐moderate Alzheimer's disease. Int J Clin Pract 2011;65:465–471. [DOI] [PubMed] [Google Scholar]
- 8. Anand R, Messina J, Hartman R. Dose‐response effect of rivastigmine in the treatment of Alzheimer's disease. Int J Geriatr Psychopharmacol 2000;2:68–72. [Google Scholar]
- 9. Cummings J, Froelich L, Black SE, et al. Randomized, double‐blind, parallel‐group, 48‐week study for efficacy and safety of a higher‐dose rivastigmine patch (15 vs. 10 cm2) in Alzheimer's disease. Dement Geriatr Cogn Disord 2012;33:341–353. [DOI] [PubMed] [Google Scholar]
- 10. Burns A, Spiegel R, Quarg P. Efficacy of rivastigmine in subjects with moderately severe Alzheimer's disease. Int J Geriatr Psychiatry 2004;19:243–249. [DOI] [PubMed] [Google Scholar]
- 11. Kurz A, Farlow M, Quarg P, et al. Disease stage in Alzheimer disease and treatment effects of rivastigmine. Alzheimer Dis Assoc Disord 2004;18:123–128. [DOI] [PubMed] [Google Scholar]
- 12. Lopez‐Pousa S, Vilalta‐Franch J, Hermandez B, et al. Efficacy of rivastigmine in patients with severe Alzheimer's Disease: A double‐blind, randomized pilot study. Brain Aging 2004;4:26–34. [Google Scholar]
- 13. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer disease: Report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 14. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 15. Farlow MR, Grossberg G, Gauthier S, et al. The ACTION study: Methodology of a trial to evaluate safety and efficacy of a higher dose rivastigmine transdermal patch in severe Alzheimer's disease. Curr Med Res Opin 2010;26:2441–2447. [DOI] [PubMed] [Google Scholar]
- 16. American Psychiatric Association . Diagnostic and statistical manual of mental disorders (DSM‐IV), 4th edn Washington, DC: American Psychiatric Association, 1994. [Google Scholar]
- 17. Saxton J, McGonigle‐Gibson K, Swihart A, et al. Assessment of the severely impaired patient: Description and validation of a new neuropsychological test battery. Psychol Assess 1990;2:298–303. [Google Scholar]
- 18. Galasko D, Bennett D, Sano M, et al. An inventory to assess activities of daily living for clinical trials in Alzheimer's disease. The Alzheimer's Disease Cooperative Study. Alzheimer Dis Assoc Disord 1997;11(Suppl 2):S33–S39. [PubMed] [Google Scholar]
- 19. Schneider LS, Olin JT, Doody RS, et al. Validity and reliability of the Alzheimer's Disease Cooperative Study‐Clinical Global Impression of Change. The Alzheimer's Disease Cooperative Study. Alzheimer Dis Assoc Disord 1997;11(Suppl 2):S22–S32. [DOI] [PubMed] [Google Scholar]
- 20. Cummings JL, Mega M, Gray K, et al. The Neuropsychiatric Inventory: Comprehensive assessment of psychopathology in dementia. Neurology 1994;44:2308–2314. [DOI] [PubMed] [Google Scholar]
- 21. Farlow MR, Grossberg GT, Meng X, et al. Rivastigmine transdermal patch and capsule in Alzheimer's disease: Influence of disease stage on response to therapy. Int J Geriatr Psychiatry 2011;26:1236–1243. [DOI] [PubMed] [Google Scholar]
- 22. Doraiswamy PM, Krishnan KR, Anand R, et al. Long‐term effects of rivastigmine in moderately severe Alzheimer's disease: Does early initiation of therapy offer sustained benefits? Prog Neuropsychopharmacol Biol Psychiatry 2002;26:705–712. [DOI] [PubMed] [Google Scholar]
- 23. Karaman Y, Erdogan F, Koseoglu E, et al. A 12‐month study of the efficacy of rivastigmine in patients with advanced moderate Alzheimer's disease. Dement Geriatr Cogn Disord 2005;19:51–56. [DOI] [PubMed] [Google Scholar]
- 24. Burns A, Bernabei R, Bullock R, et al. Safety and efficacy of galantamine (Reminyl) in severe Alzheimer's disease (the SERAD study): A randomised, placebo‐controlled, double‐blind trial. Lancet Neurol 2009;8:39–47. [DOI] [PubMed] [Google Scholar]
- 25. Black SE, Doody R, Li H, et al. Donepezil preserves cognition and global function in patients with severe Alzheimer disease. Neurology 2007;69:459–469. [DOI] [PubMed] [Google Scholar]
- 26. Farlow MR, Salloway S, Tariot PN, et al. Effectiveness and tolerability of high‐dose (23 mg/d) versus standard‐dose (10 mg/d) donepezil in moderate to severe Alzheimer's disease: A 24‐week, randomized, double‐blind study. Clin Ther 2010;32:1234–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Herrmann N, Gauthier S. Diagnosis and treatment of dementia: 6. Management of severe Alzheimer disease. CMAJ 2008;179:1279–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mohamed S, Rosenheck R, Lyketsos CG, et al. Caregiver burden in Alzheimer disease: Cross‐sectional and longitudinal patient correlates. Am J Geriatr Psychiatry 2010;18:917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Andersen CK, Wittrup‐Jensen KU, Lolk A, et al. Ability to perform activities of daily living is the main factor affecting quality of life in patients with dementia. Health and Qual Life Outcomes 2004;2:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Winblad B, Cummings J, Andreasen N, et al. A six‐month double‐blind, randomized, placebo‐controlled study of a transdermal patch in Alzheimer's disease – rivastigmine patch versus capsule. Int J Geriatr Psychiatry 2007;22:456–467. [DOI] [PubMed] [Google Scholar]
- 31. Mercier F, Lefèvre G, Huang HL, et al. Rivastigmine exposure provided by a transdermal patch versus capsules. Curr Med Res Opin 2007;23:3199–3204. [DOI] [PubMed] [Google Scholar]
- 32. Agid Y, Dubois B, Investigators IR, et al. Efficacy and tolerability of rivastigmine in patients with dementia of the Alzheimer type. Curr Ther Res 1998;59:837–845. [Google Scholar]