

Abstract
Blood metabolites can be detected as low-mass ions (LMIs) by mass spectrometry (MS). These LMIs may reflect the pathological changes in metabolism that occur as part of a disease state, such as cancer. We constructed a LMI discriminant equation (LOME) to investigate whether systematic LMI profiling might be applied to cancer screening. LMI information including m/z and mass peak intensity was obtained by five independent MALDI-MS analyses, using 1,127 sera collected from healthy individuals and cancer patients with colorectal cancer (CRC), breast cancer (BRC), gastric cancer (GC) and other types of cancer. Using a two-stage principal component analysis to determine weighting factors for individual LMIs and a two-stage LMI selection procedure, we selected a total of 104 and 23 major LMIs by the LOME algorithms for separating CRC from control and rest of cancer samples, respectively. CRC LOME demonstrated excellent discriminating power in a validation set (sensitivity/specificity: 93.21%/96.47%). Furthermore, in a fecal occult blood test (FOBT) of available validation samples, the discriminating power of CRC LOME was much stronger (sensitivity/specificity: 94.79%/97.96%) than that of the FOBT (sensitivity/specificity: 50.00%/100.0%), which is the standard CRC screening tool. The robust discriminating power of the LOME scheme was reconfirmed in screens for BRC (sensitivity/specificity: 92.45%/96.57%) and GC (sensitivity/specificity: 93.18%/98.85%). Our study demonstrates that LOMEs might be powerful noninvasive diagnostic tools with high sensitivity/specificity in cancer screening. The use of LOMEs could potentially enable screening for multiple diseases (including different types of cancer) from a single sampling of LMI information.
Keywords: serum profiling, MALDI-TOF mass spectrometry, pattern recognition
Many cancer-screening methods have been proposed and are currently under investigation, but numerous difficulties prevent them from becoming clinically useful. The heterogeneity of cell types and gene expression detected within each individual cancer patient and at different stages of disease progression hinders the development of cancer-screening methods.1 The effectiveness of certain screening methods remains controversial and can vary with different types of cancer. For instance, colonoscopy, sigmoidoscopy, barium enema, computed tomographic colonography, stool DNA and fecal occult blood tests (FOBT) are currently recommended as colorectal cancer (CRC) screening methods.2–4 However, these methods involve invasive, specialized procedures (e.g., colonoscopy) or have low sensitivity (e.g., FOBT). Extensive efforts to identify easy-to-measure, noninvasive biomarkers have led to the clinical implementation of serum carcinoembryonic antigen (CEA) levels as a CRC tumor biomarker5; however, CEA cannot be used as a screening marker for CRC. Moreover, although advances in genomics, proteomics and molecular pathology have suggested many candidate biomarkers with potential clinical value, such as blood-derived methylated SEPT9 DNA, N-Myc downstream-regulated gene 4 and fecal tumor M2 pyruvate kinase (PK),6–8 attempts to translate these research advances from bench to bedside have been disappointing.9 Currently, optimal blood markers for cancer screening are therefore lacking.
What’s new? —
It’s challenging to screen for cancer, not least because the prevalence of specific cell types and gene expression patterns vary among patients and at different stages of disease. In this paper, the authors explore the idea of screening by profiling metabolites in the blood. Metabolites can be detected as low-mass ions by mass spectrometry, and the authors created an algorithm to analyze these low-mass ions as a cancer screening tool. The test showed high sensitivity and specificity when applied to colorectal cancer, breast cancer and gastric cancer, and thus it could be developed into an effective non invasive screening tool.
Understanding the metabolic changes caused by cancer has become important not only for patient care but also for early detection. Indeed, metabolic profiling approaches have shown potential for cancer screening.10–12 Metabolic profiling using gas chromatography–mass spectrometry (GC-MS) and liquid chromatography (LC)-MS is very powerful in identifying metabolites, but usually no more than 100 distinct molecules are identified.13,14 Such a low number of metabolites may not reflect total metabolic changes. Furthermore, neither GC-MS nor LC-MS is acceptable as a high-throughput screening tool because of the relatively long time needed for the analysis of each sample. Alternative metabolic profiling using high-resolution magic angle spinning nuclear magnetic resonance and GC-MS robustly discriminated between normal and malignant mucosa in patients with CRC15; however, it is not possible to use this technique in the clinic because such an invasive tool is not appropriate for cancer screening.
We postulated that valuable information reflecting cancer-related metabolic changes may exist in the low-mass range. Systematic approaches examining metabolic changes, particularly those monitored in blood, have already received attention but, to the best of our knowledge, such information has never been systematically exploited by matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) MS. MALDI-TOF has several advantages compared to GC-MS and electrospray-ionization-based LC-MS (e.g., ion-trap MS and quadrupole-TOF): it requires less than a minute to analyze one sample, and more than 100 samples can be analyzed in a single target plate. Furthermore, even if the low-mass range contains numerous matrix peaks, they can be ruled out by their negligible weighting factors in a statistical process, as with other low-mass metabolites affected by diet, and used for mass calibration for low-mass ions (LMIs), which are mostly less than 800 mass-to-charge ratio (m/z) according to MALDI-TOF MS. Finally, in theory each targeted sample on a MALDI-TOF plate can be reanalyzed.
On the basis of the profiling of metabolites present in the serum that are detected as LMIs, we have developed specialized algorithms and the LOw-Mass-ion discriminant Equation (LOME) (for workflow, see Fig. 1) as a new concept for cancer screening. Herein, we introduce and discuss the clinical applications of LOME for discriminating not only CRC but also breast cancer (BRC) and gastric cancer (GC).
Figure 1.
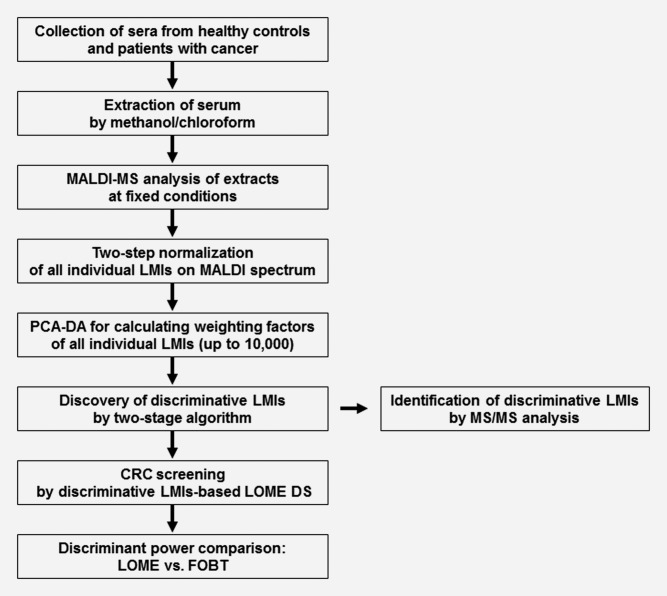
Overview of LOME construction for CRC screening and its clinical validation. Methanol/chloroform extraction of sera from healthy controls and patients with cancer was performed. Extracts were analyzed by MALDI-MS at fixed conditions allowing higher LMI resolution on the MALDI mass spectrum. Two-step normalization of the intensities of all individual LMIs was performed using the “total area sums” and “Pareto scaling” options, and weighting factors for all individual LMIs were calculated by PCA-DA. Two stratified algorithms were applied to select LMIs with strong discriminative power in CRC screening, and LOME was constructed by the selected LMIs. Serum samples were screened for CRC by discriminative LMIs-based LOME DS, and the discriminative powers in CRC LOME and FOBT (a noninvasive CRC screening tool) were compared. In addition, discriminative LMIs were identified by MS/MS analysis. Mass information from a nonmonoisotopic LMI was used to select a candidate metabolite from the Human Metabolome Database. The selected metabolite and the LMI were fragmented using the same method, and the resulting MS/MS spectra were compared to determine whether the compounds were identical. LMIs showing a monoisotopic peak pattern, a common feature of peptides in mass spectra, were identified by searching the MS/MS spectra against the Swiss-Prot Database.
Material and Methods
Patient samples
The study population consisted of patients with CRC, BRC, non-Hodgkin lymphoma (NHL), GC, ovarian cancer (OVC), carcinoma in situ or advanced adenoma of the colon, newly diagnosed by biopsy or radiologic imaging, and healthy individuals who were enrolled in the Health Screening Program including colonoscopy. The patients who were admitted for treatment and aged older than 18 years were eligible. Patients with previous or synchronous second primary malignancy, a known history of familial adenomatous polyposis, or hereditary nonpolyposis CRC and screenees with any detected neoplasms were excluded. Study participants were recruited consecutively between March 2009 and November 2011. The control sera were obtained from healthy individuals at the National Cancer Center (NCC), Korea (Supporting Information Table 1). The sera from patients with CRC, BRC, NHL, GC, OVC, carcinoma in situ or advanced adenoma of the colon were collected at NCC Hospital, Korea University Anam Hospital, Ewha Womans University Mokdong Hospital and Dong-A University Medical Center, Korea (Supporting Information Tables 2–7). Informed consent was obtained from all patients, and the research protocol was approved by the Institutional Review Boards of each participating institution. Obtained sera were processed as described in Supporting Information Methods.
Sample sets for LOME construction
Serum samples for CRC LOMEs were divided into two sets, A and B, representing the training and validation sets, respectively. Sets A and B were mutually exclusive (A ∩ B = ∅, where ∅ denotes the empty set). Set A was subdivided into Set A1 and Set A2 (A = A1 ∪ A2) for a two-stage training scheme. Sets A1 and A2 were mutually exclusive (A1 ∩ A2 = ∅). The weighting factors for individual LMIs were determined from Set A1 only. The discriminative biomarker LMIs were found using Sets A1 and A2. Set A0 was a subset of Set A1 (A0 ⊆ A1) and a principal component analysis-based discriminant analysis (PCA-DA) on Set A0 yielded a perfect classification of 100% sensitivity and specificity. The researchers were blinded to the clinical information of the serum samples.
Construction of LOMEs for cancer screening
Two-stage training scheme
The training set was involved in determining weighting factors and discriminative biomarker LMIs. The weighting factors were derived from Set A1 only. The training set was extended from Set A1 to Sets A1 and A2 when the discriminative biomarker LMIs were sought. This strategy was undertaken to alleviate overfitting by including Set A2, which was not used in determining the weighting factors.
Selection of preliminary LMI candidates
The preliminary LMI candidates were selected based on Set A0 through Algorithm 1 (Supporting Information Fig. 1). The loop in Algorithm 1 identifies statistically significant LMIs that make large contributions to the discriminant score (DS). The loop contains two steps: the first step selects LMIs whose weighted terms have a magnitude of more than a certain value (0.1 in this study). The second step selects LMIs that appear in more than a certain percentage of cases (50% in this study). The LOME with preliminary LMI candidates can be written as follows:
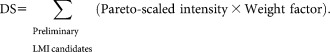 |
Discovery of discriminative biomarker LMIs
Discriminative biomarker LMIs were found by using the concepts of LMI-wise sensitivity and specificity. Algorithm 2 (Supporting Information Fig. 2) involved the following steps. (i) LMIs were divided into two sets, a high sensitivity set and a high specificity set. An LMI in the high sensitivity set has a higher sensitivity than specificity, and vice versa. Next, the LMIs in each set were arranged in descending order of the sum of their sensitivity and specificity: {Sns1, Sns2, Sns3 … SnsI} and {Spc1, Spc2, Spc3 … SpcJ}. (ii) The top two LMIs in each set, {Sns1, Sns2, Spc1, Spc2}, were taken to find the combination of LMIs with the best performance and a biomarker group was formed from them. (iii) The next top LMI in each set was taken and the performances of the four branches, {biomarker group}, {biomarker group, Sns3}, {biomarker group, Spc3} and {biomarker group, Sns3, Spc3}, were compared. The branch with the best performance was assigned to the biomarker group. This substep was iterated until one of the two sets bottomed out. Next, the next top LMI in the remaining set was taken, and the performances of the two branches, {biomarker group} and {biomarker group, Snsi or Spcj}, were compared, and the branch with the best performance was assigned to the biomarker group. This substep was iterated until no LMIs remained. (iv) The final biomarker group was extracted from the LMI candidates. (v) If at least two LMIs remained in each set, step i was performed again for the remaining LMI candidates. (vi) The performances of the final biomarker groups were evaluated and the biomarker groups with the highest performances were assembled.
Figure 2.

LOME construction procedures. (a) Classification results for Set A0 and the excluded cases with CRC LOMEs comprising LMIs selected by Algorithm 1. Set A0 and the excluded cases coincide with Set A1. Left: CRC vs. control (CRC LOME 1-278). Right: CRC vs. BRC/NHL/GC (CRC LOME 2-383). (b) Classification parameters of biomarker groups found by iteratively applying Algorithm 2. “Total accuracy” is the proportion of TPs and TNs in Set A. Left: CRC LOME 1. Right: CRC LOME 2. (c) Discriminative biomarker LMIs (104 and 23 for CRC LOMEs 1 and 2) and their associated weighting factors. The weighting factors for the selected LMIs in each LOME were rescaled for comparison so that their mean square value was unity. (d) Classification results for Set A with CRC LOMEs 1-104 and 2-23. Every sample was run in quintuplicate. All classification decisions were made using the mean DS.
When comparing the performances of the branches, the branch with the best performance was selected according to the following evaluation scheme: Priority 1) the branch maximizing the sum of sensitivity and specificity; Priority 2) the branch with the fewest LMIs and Priority 3) the branch maximizing the difference between the minimum mean DS of true positive (TP) cases and the maximum mean DS of true negative (TN) cases.
Statistical analysis
Categorical data were analyzed by the chi-squared, Fisher’s exact or Mann–Whitney U-tests. Quantitative data were analyzed using one-way analysis of variance with post hoc comparisons (Scheffe’s test). A two-tailed p value < 0.05 was considered statistically significant. All statistical analyses were performed using STATA 10.0 software (StataCorp LP, College Station, TX).
Results
Harvest of LMI information in sera as a first step for LOME construction
A total of 1,127 sera (Table 1) were obtained from healthy control individuals and from patients with CRC, BRC, GC, NHL, OVC, carcinoma in situ or advanced adenoma of the colon. We applied the Bligh and Dyer method to rule out most proteins and efficiently extract metabolic compounds from the sera.16 To avoid also removing metabolic lipid compounds,17 we did not change the organic solvent ratio in the extraction method.18 Different extraction processes led to different mass spectrum patterns and affected the resulting LOME. All results described in this article were based on our defined extraction protocol. All LMI information (i.e., m/z and mass peak intensity) was harvested from the serum extract by MALDI-TOF analysis and used for LOME construction.
Table 1.
Demographics of healthy control individuals and patients with CRC, BRC, NHL, GC, OVC, and TA [carcinoma in situ (Tis) or advanced adenoma of the colon]
| Number | Age (years) | ||||
|---|---|---|---|---|---|
| Total | Male | Female | Mean ± SD | Range | |
| Control | 295 | 137 | 158 | 53.9 ± 9.6 | 30–81 |
| CRC | 420 | 269 | 151 | 61.5 ± 11.0 | 33–88 |
| BRC | 161 | 0 | 161 | 50.0 ± 9.4 | 29–74 |
| NHL | 66 | 42 | 24 | 54.9 ± 15.4 | 24–80 |
| GC | 141 | 99 | 42 | 59.6 ± 12.5 | 31–82 |
| OVC | 25 | 0 | 25 | 56.3 ± 10.2 | 40–74 |
| TA | 19 | 11 | 8 | 60.3 ± 8.9 | 46–77 |
LOME construction and validation
We obtained a clear difference between CRC patients and the healthy control group by using mass peak data in PCA-DA for Set A1 (sensitivity, 98.50%; specificity, 98.69%). A single LOME for discriminating CRC from non-CRC (healthy control, BRC, NHL and GC) groups failed because of low specificity (69.28%) for control samples; thus, we constructed a second LOME that could discriminate CRC from other cancers (BRC, NHL and GC) (sensitivity, 99.25%; specificity, 95.45%). The PCA-DA results indicate that at least two LOMEs are required to distinguish CRC from healthy controls (CRC LOME 1) and CRC from BRC/NHL/GC (CRC LOME 2). Serum samples with two positive DSs by the two LOMEs were considered screen-positive (CRC), whereas samples with at least one negative DS were considered screen-negative (non-CRC).
The sensitivities and specificities for CRC LOMEs 1 and 2 did not all reach 100%. The workflow presented in Supporting Information Figure 3 yields subsets A01 (⊆A1, CRC LOME 1) and A02 (⊆A1, CRC LOME 2), which have 100% sensitivity and 100% specificity. Although there were only two false-positive (FP) and two false-negative (FN) cases for CRC LOME 1, applying PCA-DA again to the set, while excluding these four samples, did not directly lead to a complete discriminating feature. We generated Sets A01 and A02 by repeatedly excluding FP and FN cases. The weighting factors that produce a perfect classification of the subset can be determined through the workflow. Calculation of the DSs of the excluded cases for CRC LOME 1 (Supporting Information Fig. 4) revealed that they were FN or FP cases, as expected. Similar calculations were made for CRC LOME 2, and similar results were obtained.
Figure 3.
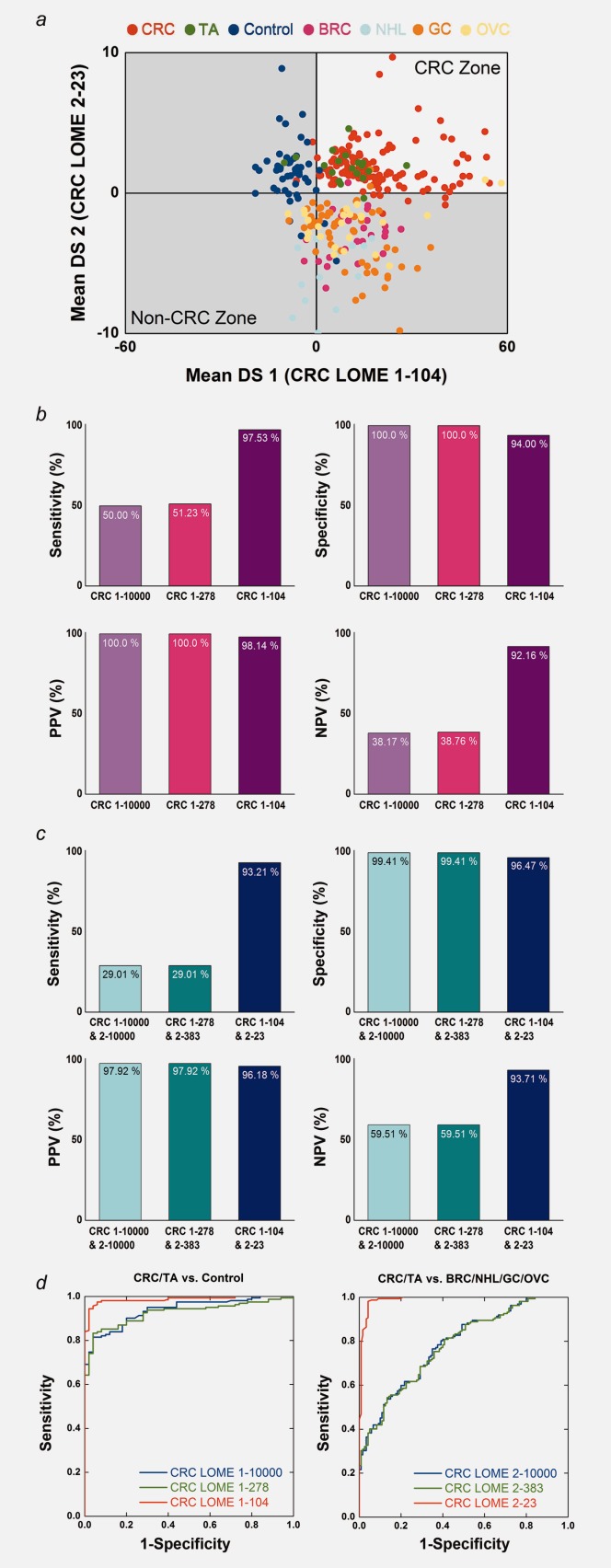
LOME validation results. (a) Classification results for Set B with CRC LOMEs 1-104 and 2-23. The validation set (Set B) also included OVC and TA samples. (b) Comparison of the discriminative power values of CRC LOME 1-10000, CRC LOME 1-278 and CRC LOME 1-104 for CRC/TA vs. control. (c) Comparison of the discriminative power values of CRC LOMEs 1-10000 and 2-10000, CRC LOMEs 1-278 and 2-383 and CRC LOMEs 1-104 and 2-23 for CRC/TA vs. non-CRC. Non-CRC includes control individuals and cases of BRC, NHL, GC and OVC. (d) Receiver operating characteristic (ROC) curves. Left: Comparison of CRC LOME 1-10000, CRC LOME 1-278 and CRC LOME 1-104 for CRC/TA vs. control. Right: Comparison of CRC LOME 2-10000, CRC LOME 2-383 and CRC LOME 2-23 for CRC/TA vs. BRC/NHL/GC/OVC.
Figure 4.

Identification and quantification of monoisotopic LMIs with 1465.6184 and 2450.9701 m/z and nonmonoisotopic LMI with 169.0653 m/z in the methanol/chloroform serum extracts. (a) Mass spectra of 1465.6184, 1466.6096 and 1467.5969 m/z. Left: Intensities of CRC and control samples. Middle: Pareto-scaled intensities of all samples. Right: Identification of the fibrinogen α chain. “% Intensity” of the ion with highest signal intensity in the given MS/MS spectrum was defined as 100. The positive ion with 1465.6184 m/z was unambiguously identified as fibrinogen α chain based on MS/MS (see online Supporting Information Methods for details). (b) Mass spectra of 2450.9701, 2451.9662 and 2452.9546 m/z. Left: Intensities of CRC and control samples. Middle: Pareto-scaled intensities of all samples. Right: Identification of transthyretin. MS/MS analysis identified the positive ion with 2450.9701 m/z as a transthyretin. (c) Mass spectra of 169.0653 m/z. Left: Intensities of CRC and control samples. Middle: Pareto-scaled intensities of all samples. Right: Identification of LMI with 169.0653 m/z as a PEP by MS/MS. (d) Plasma fibrinogen levels in healthy individuals and patients with CRC or colorectal adenoma. Error bar represents the mean ± standard deviation. The p values were given for the comparison of each with healthy individuals by Scheffe’s post hoc test. (e) Transthyretin levels in CRC patients and healthy individuals. (f) PEP levels in CRC patients and healthy individuals. The p value was given by Mann–Whitney U-test.
The CRC LOMEs yielding perfect classification results in Sets A01 and A02 involved 10,000 LMIs. We applied Algorithm 1 to select LMIs in view of their numerical significance, and Algorithm 1 suggested that the feature candidates consisted of 278 LMIs for Set A01 and 383 LMIs for Set A02. Notations such as “CRC LOME 1-278” or “CRC LOME 2-383” will be used hereafter, with the last number denoting the number of the LMIs involved in the LOME. Figure 2a shows the classification results by Algorithm 1 for Set A0 and the excluded cases. Although only 278 or 383 LMIs were involved in calculating the DSs, the classification performance remained excellent. Furthermore, the sharp decrease in the number of LMIs did not cause dramatic changes to the DS ranges; most of the LMIs are therefore unnecessary for DS calculation and for classification.
The core workflow (Supporting Information Fig. 2) was designed to overcome the lack of robustness. Every sample was run in quintuplicate. All classification decisions were made using the mean DS. PCA-DA and Algorithm 1 yielded nearly identical but poor classification results, in that the sensitivities for CRC LOMEs were low (Supporting Information Table 8). These data indicated that potentially problematic LMIs remained; thus, we carried out an additional discovery process. By iteratively applying Algorithm 2 while using the concepts of LMI-wise sensitivity and specificity and maximizing the sum of sensitivity and specificity for the biomarker group, we were able to identify multiple biomarker groups (Fig. 2b). Discriminative biomarker LMIs (Fig. 2c and Supporting Information Table 9) were determined by assembling the biomarker groups with the best performance. Figure 2d displays the distribution of the mean DSs of Set A by CRC LOMEs 1-104 and 2-23. Finally, we applied CRC LOMEs 1-104 and 2-23 to Set B to evaluate their predictive performance on the unseen data. CRC LOMEs 1-104 and 2-23 accomplished all performance parameters of more than 90% in Set B (Supporting Information Table 8). Figure 3a displays the distribution of the mean DSs of Set B. Substantial improvements in screening power were achieved by the two-stage LMI selection procedure (Algorithms 1 and 2) when compared to PCA-DA or Algorithm 1 only (Figs. 3b–3d).
Identification of biomarker LMIs and their clinical relevance to CRC
Among the LMIs used in CRC LOME 1, we found monoisotopic masses (1465.6184, 1466.6096, 1467.5969 and 2450.9701, 2451.9662, 2452.9546 m/z in Figs. 4a and 4b) that represent common peptide patterns observed in mass analysis. The 1465.6184 m/z intensity was higher among patients with CRC than in healthy control individuals (Fig. 4a, left and middle panels), whereas the reverse was true for 2450.9701 m/z (Fig. 4b, left and middle panels). Two main LMIs, 1465.6184 and 2450.9701 m/z, were further analyzed by MS/MS and unambiguously identified as fibrinogen α chain and transthyretin, respectively (Figs. 4a and 4b, right panels). We measured the fibrinogen levels in plasma and found them to be markedly increased in CRC Stages II, III and IV compared to healthy control individuals and patients with colorectal adenoma or CRC Stage I (Fig. 4d and Supporting Information Table 10). We also determined the transthyretin levels in sera from 52 CRC and 26 control samples. In contrast to fibrinogen, the serum transthyretin levels were slightly lower in the CRC samples (160.39 ± 62.41 ng/mL) than in the control samples (171.19 ± 30.86 ng/mL) (Fig. 4e). Nonmonoisotopic LMI with 169.0653 m/z showed higher intensity in the extracts of sera from CRC patients compared to healthy control individuals (Fig. 4c, left and middle panels). It was identified as a phosphoenolpyruvate (PEP) by comparing the pattern of MS/MS (Fig. 4c, right panel), and PEP level in sera from CRC patients (n = 30, 37.71 ± 13.03 μM) was significantly higher than that of controls (n = 30, 25.39 ± 9.88 μM) (p < 0.001) (Fig. 4f).
LOME reproducibility and the superior discriminating power of LOME compared to FOBT
Repeating the above experiments yielded 86.42% reproducibility. Predictions of four out of five cases with carcinoma in situ or advanced adenoma of the colon changed from “CRC” to “non-CRC.” When the five cases showing ambiguous screening results were excluded, the reproducibility was significantly increased to 90.79% (Supporting Information Table 11). Overall, the mean DS did not vary with the CRC stage, although Figure 5a shows a significant difference between Stage II and Stage III for CRC LOME 1-104. The LOME detection rate was not dependent on CRC stage (Fig. 5b and Supporting Information Table 12).
Figure 5.

Clinical perspectives on LOME. (a) The relevance of the LOME DS to clinical stage. Left: CRC LOME 1-104. The stage is based on the pathologic stage among patients with no preoperative treatment or on the clinical stage among patients who underwent preoperative chemoradiotherapy. Error bar represents the mean ± standard deviation. The p value was given for the comparison between Stage II and Stage III by Scheffe’s post hoc test. Right: CRC LOME 2-23. (b) The detection rates for CRC LOMEs 1-104 and 2-23, irrespective of CRC stage. (c) Comparison of the discriminative power values of CRC LOME 1-104, and of CRC LOMEs 1-104 and 2-23, versus FOBT.
To assess the discriminating power of LOME for CRC screening relative to that of FOBT, 96 patients with CRC and 49 healthy control individuals from Set B underwent FOBT. We found excellent CRC discriminating power for LOME 1-104 and LOME 2-23 (sensitivity, 94.79%; specificity, 97.96%), whereas FOBT-positive cases were detected in only half of the patients in Set B (sensitivity, 50.00%; specificity, 100.0%) (Fig. 5c and Supporting Information Table 13). However, it has to be noted that the FOBT sensitivity (50.00%) in our test set was slightly lower than previously reported (60–85%).2,19 Similar differences between LOME and FOBT were found for Set A.
Discussion
Many factors are expected to contribute to the pattern of LMIs in serum. For example, diet and chemotherapy may directly affect the pattern of LMIs in serum. However, it is not possible that all individuals consume the same meals or receive the same chemotherapy before collecting their blood. Therefore, their effect on the separating pattern seems trivial owing to the sample collection from multiple hospitals with no specific requirements for diet or treatment. The status of disease at the time of blood collection may be a primary factor in making the pattern of LMIs distinct. Given the body’s complex response to cancer, combinations of multiple biomarkers can better differentiate between cancer and healthy controls than a single biomarker alone. Our LOME method uses multiple LMIs to achieve an excellent and robust screening power for CRC, with much higher sensitivity than FOBT.
Based on our previous work,12 in which we developed a new LMI approach for NHL screening, we hypothesized that a new, noninvasive cancer-screening protocol could be established if LMI data were properly collected, analyzed by MALDI-TOF MS and statistically translated. We suspected that translation of the information from LMIs (mostly those less than 800 m/z) could be used as a tool to distinguish individuals with certain diseases from healthy individuals.
Applying the LOME derived from PCA-DA directly to test cases did not provide outstanding discrimination because it included numerous potentially interfering or unnecessary features. Although the effects of these features were not prominent in the training set, our analysis confirmed that they could adversely influence the classification performance on a validation set. Accordingly, we devised a two-stage LMI selection procedure to establish a robust classification framework. In the first stage (Algorithm 1), a set of LMI candidates was selected by considering the contribution of LMIs to DS. In the second stage (Algorithm 2), discriminative biomarker LMIs were discovered by maximizing classification performance (i.e., the sum of the sensitivity and specificity) with the training set, which was enlarged to reduce overfitting.
Although Algorithm 2, which used LMI-wise sensitivity and specificity concepts, was successful in this study, a couple of points should be considered. First, the LMI-wise sensitivity and specificity are determined only by the sign of the weighted intensity values and do not consider magnitude. In contrast, the sensitivity and specificity for the combination of LMIs depend not only on the signs but also on the magnitudes of the weighted intensities to be added. The ranges of sensitivity and specificity after combining two LMIs can be estimated using their LMI-wise information, but these ranges are usually too broad to be useful. Second, the inclusion or exclusion of a new LMI depends on the biomarker group of the former step or the other LMI to be compared. Therefore, the methods used to determine the starting biomarker group and the LMI arrangement should be carefully examined. Our scheme did not result in monotonically decreasing total accuracy in successive iterations (Fig. 2b, right panel).
One of the strengths of LOME construction is that it takes full advantage of all of the useful LMI information from various types of cancers. For instance, as a negative control for CRC LOME construction, we used LMI data from healthy control individuals and from patients with GC, NHL or BRC. LMI data from GC, NHL and BRC samples, respectively, represent other type of gastrointestinal cancer, nonsolid tumor and woman cancer, and so provide critical information regarding which LMIs are associated with CRC and which introduce false positives during CRC screening. Information from other types of cancer was also used for the construction of BRC and GC LOMEs (see online Supporting Information Material for details).
The screening strategy depended on the type of cancer. For example, the two-LOME scheme for CRC or BRC screening is different from the scheme used for GC screening. Whether this difference is owing to a limitation of the current LOME approach (it is not possible to proceed with a poor PCA-DA result) or it simply depends on the sample size (fewer samples were used for BRC or GC screening than for CRC screening) remains to be investigated. Accordingly, we may need to examine other well-known methods, such as partial least squares and support vector machines, in future work. Separation by the four-LOME scheme was superior to that of the two-LOME scheme for GC screening; however, the number of LOMEs should ideally be minimized. The use of multiple LOMEs will necessarily reduce the net sensitivity because at least one negative DS makes the sample screen-negative. Our GC example is one type of exception. GC LOMEs 3-50 and 4-46 gave a sensitivity of 100% for Set F, a validation set. Therefore, the effect of increasing the number of LOMEs was not evident.
The two major monoisotopic LMIs in the discriminative biomarker group were identified as fibrinogen α chain and transthyretin (Figs. 4a, 4b, 4d and 4e, and Supporting Information Table 10). Interestingly, fibrinogen α chain was a significant factor in the construction of LOMEs not only for CRC screening but also for BRC and GC screening. Fibrinogen α chain has been proposed to be a biomarker for prostate, bladder and breast cancers,20 and high fibrinogen levels have been linked to increased risk of CRC.21 However, the role of fibrinogen has also been reported in inflammatory disease such as colitis and bacterial infection.22 Fibrinogen α chain was identified as one of 104 LMIs in CRC LOME 1, which mainly functions in discriminating healthy controls from CRC. Therefore, discriminating power of fibrinogen α chain may be restricted to any of diseases accompanying inflammation, rather than only to CRC, and additional identification of LMIs in CRC LOME 2 becomes a pressing issue. Transthyretin, meanwhile, is a supportive marker for colorectal adenoma and cancer that represents nutritional status.23,24 Including these two proteins, the combination of unidentified metabolic compounds may serve to characterize CRC. Among 104 LMIs, the nonmonoisotopic LMI with 169.0653 m/z was identified as PEP (Figs. 4c and 4f). In tumor cells, the M2-PK isoform dominantly expresses and contributes to aerobic glycolysis and proliferation.25,26 Interaction of M2-PK with different oncoproteins can lead to the dimerization, which decreases the affinity of M2-PK for PEP and results in accumulation of phosphometabolites above M2-PK.27 The accumulated phosphometabolites can be used for synthesis of crucial molecules for proliferating cells such as nucleic acids, phospholipids and amino acids; however, it is remained to be clarified whether differential regulation of M2-PK in tumor cells affects the level of PEP in blood or overall pathophysiological change is a cause of aberrant PEP level. Fibrinogen, transthyretin and PEP alone cannot clearly separate CRC from control samples, but they showed substantial power as part of a set of 104 discriminators (Supporting Information Table 9). Moreover, CRC screening by LOME was quite reproducible. Although the predictive ability of LOMEs fluctuated in cases of carcinoma in situ or advanced adenoma of the colon, we have demonstrated their potential for identifying patients with Stage I CRC.
An ideal screening tool should be sensitive, specific and robust against inter-operator and inter-medical-center variability; furthermore, it should be cost-effective and quick to perform.9 Although the LOME protocol described here requires further refinement and confirmation in a large, independent screening population, it has proven its potential for cancer screening. New LOMEs could be constructed for a wide range of diseases, suggesting a new paradigm for disease screening.
Acknowledgments
The authors thank Drs. Sun Young Kong, Hyeon Seok Eom, Hyuk-Chan Kwon, Seung Cheol Kim and Woong Ju for their kind support in reviewing medical charts and collecting qualified sera from patients with cancer.
Glossary
- BRC
breast cancer
- CEA
carcinoembryonic antigen
- CRC
colorectal cancer
- DS
discriminant score
- FN
false negative
- FOBT
fecal occult blood test
- FP
false positive
- GC
gastric cancer
- GC-MS
gas chromatography—mass spectrometry
- LC
liquid chromatography
- LMI
low-mass ion
- LOME
LMI discriminant equation
- MALDI-TOF
matrix-assisted laser desorption/ionization-time of flight
- MS
mass spectrometry
- m/z
mass-to-charge ratio
- NCC
National Cancer Center
- NHL
non-Hodgkin lymphoma
- OVC
ovarian cancer
- PCA-DA
principal component analysis-based discriminant analysis
- PEP
phosphoenolpyruvate
- PK
pyruvate kinase
- ROC
receiver operating characteristic
- TA
carcinoma in situ (Tis) or advanced adenoma of the colon
- TN
true negative
- TP
true positive
Supporting Information
Additional Supporting Information may be found in the online version of this article
References
- Pollak MN, Foulkes WD. Challenges to cancer control by screening. Nat Rev Cancer. 2003;3:297–303. doi: 10.1038/nrc1042. [DOI] [PubMed] [Google Scholar]
- Levin B, Lieberman DA, McFarland B, et al. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. CA Cancer J Clin. 2008;58:130–60. doi: 10.3322/CA.2007.0018. [DOI] [PubMed] [Google Scholar]
- Davies RJ, Miller R, Coleman N. Colorectal cancer screening: prospects for molecular stool analysis. Nat Rev Cancer. 2005;5:199–209. doi: 10.1038/nrc1569. [DOI] [PubMed] [Google Scholar]
- Graser A, Stieber P, Nagel D, et al. Comparison of CT colonography, colonoscopy, sigmoidoscopy and faecal occult blood tests for the detection of advanced adenoma in an average risk population. Gut. 2009;58:241–8. doi: 10.1136/gut.2008.156448. [DOI] [PubMed] [Google Scholar]
- Duffy MJ. Carcinoembryonic antigen as a marker for colorectal cancer: is it clinically useful? Clin Chem. 2001;47:624–30. [PubMed] [Google Scholar]
- deVos T, Tetzner R, Model F, et al. Circulating methylated SEPT9 DNA in plasma is a biomarker for colorectal cancer. Clin Chem. 2009;55:1337–46. doi: 10.1373/clinchem.2008.115808. [DOI] [PubMed] [Google Scholar]
- Melotte V, Lentjes MH, van den Bosch SM, et al. N-Myc downstream-regulated gene 4 (NDRG4): a candidate tumor suppressor gene and potential biomarker for colorectal cancer. J Natl Cancer Inst. 2009;101:916–27. doi: 10.1093/jnci/djp131. [DOI] [PubMed] [Google Scholar]
- Haug U, Hundt S, Brenner H. Sensitivity and specificity of faecal tumour M2 pyruvate kinase for detection of colorectal adenomas in a large screening study. Br J Cancer. 2008;99:133–5. doi: 10.1038/sj.bjc.6604427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyers CL. The cancer biomarker problem. Nature. 2008;452:548–52. doi: 10.1038/nature06913. [DOI] [PubMed] [Google Scholar]
- Chen T, Xie G, Wang X, et al. Serum and urine metabolite profiling reveals potential biomarkers of human hepatocellular carcinoma. Mol Cell Proteomics. 2001;10:M110.004945. doi: 10.1074/mcp.M110.004945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asiago VM, Alvarado LZ, Shanaiah N, et al. Early detection of recurrent breast cancer using metabolite profiling. Cancer Res. 2010;70:8309–18. doi: 10.1158/0008-5472.CAN-10-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BC, Kong SY, Jang SG, et al. Identification of hypoxanthine as a urine marker for non-Hodgkin lymphoma by low-mass-ion profiling. BMC Cancer. 2010;10:55. doi: 10.1186/1471-2407-10-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly AD, Breitkopf SB, Yuan M, et al. Metabolomic profiling from formalin-fixed, paraffin-embedded tumor tissue using targeted LC/MS/MS: application in sarcoma. PLoS One. 2011;6:e25357. doi: 10.1371/journal.pone.0025357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiumi S, Kobayashi T, Ikeda A, et al. A novel serum metabolomics-based diagnostic approach for colorectal cancer. PLoS One. 2012;7:e40459. doi: 10.1371/journal.pone.0040459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EC, Koh PK, Mal M, et al. Metabolic profiling of human colorectal cancer using high-resolution magic angle spinning nuclear magnetic resonance (HR-MAS NMR) spectroscopy and gas chromatography mass spectrometry (GC/MS) J Proteome Res. 2009;8:352–61. doi: 10.1021/pr8006232. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–7. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Iverson SJ, Lang SLC, Cooper MH. Comparison of the Bligh and Dyer and Folch methods for total lipid determination in a broad range of marine tissue. Lipids. 2001;36:1283–7. doi: 10.1007/s11745-001-0843-0. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Stanley GHS. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Whitlock EP, Lin JS, Liles E, et al. Screening for colorectal cancer: a targeted, updated systematic review for the U.S. Preventive Services Task Force. Ann Intern Med. 2008;149:638–58. doi: 10.7326/0003-4819-149-9-200811040-00245. [DOI] [PubMed] [Google Scholar]
- Villanueva J, Shaffer DR, Philip J, et al. Differential exoprotease activities confer tumor-specific serum peptidome patterns. J Clin Invest. 2006;116:271–84. doi: 10.1172/JCI26022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okholm M, Iversen LH, Thorlacius-Ussing O, et al. Fibrin and fibrinogen degradation products in plasma of patients with colorectal adenocarcinoma. Dis Colon Rectum. 1996;39:1102–6. doi: 10.1007/BF02081408. [DOI] [PubMed] [Google Scholar]
- Davalos D, Akassoglou K. Fibrinogen as a key regulator of inflammation in disease. Semin Immunopathol. 2012;34:43–62. doi: 10.1007/s00281-011-0290-8. [DOI] [PubMed] [Google Scholar]
- Ingenbleek Y, Young V. Transthyretin (prealbumin) in health and disease: nutritional implications. Annu Rev Nutr. 1994;14:495–533. doi: 10.1146/annurev.nu.14.070194.002431. [DOI] [PubMed] [Google Scholar]
- Fentz AK, Spörl M, Spangenberg J, et al. Detection of colorectal adenoma and cancer based on transthyretin and C3a-desArg serum levels. Proteomics Clin Appl. 2007;1:536–44. doi: 10.1002/prca.200600664. [DOI] [PubMed] [Google Scholar]
- Mazurek S, Boschek CB, Hugo F, et al. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15:300–8. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–3. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- Mazurek S, Grimm H, Boschek CB, et al. Pyruvate kinase type M2: a crossroad in the tumor metabolome. Br J Nutr. 2002;87:S23–S29. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.