

Abstract
Breast cancer is the leading cause of new cancer diagnoses among women. Using peroxisome proliferator-activated receptor (PPAR)γ(+/−) mice, we showed normal expression of PPARγ was critical to stop 7,12-dimethylbenz[a]anthracene (DMBA)-induced breast tumorigenesis. PPARγ is expressed in many breast cell types including mammary secretory epithelial (MSE) cells. MSEs proliferate as required during pregnancy, and undergo apoptosis or reversible transdifferentiation during involution once lactation is complete. Thus, MSE-specific loss of PPARγ was hypothesized to enhance DMBA-mediated breast tumorigenesis. To test this, MSE cell-specific PPARγ knockout (PPARγ-MSE KO) and control (PPARγ-WT) mice were generated, mated and allowed to nurse for three days. One week after involution, dams were treated with DMBA to initiate breast tumors, and randomized on week 7 to continue receiving a normal chow diet (DMBA Only: PPARγ-WT, n = 15; PPARγ-MSE KO, n = 25) or one supplemented with a PPARγ activating drug (DMBA + ROSI: PPARγ-WT, n = 17; PPARγ-MSE KO, n = 24), and monitored for changes in breast tumor outcomes. PPARγ-MSE KOs had significantly lower overall survival and decreased mammary tumor latency as compared to PPARγ-WT controls. PPARγ activation significantly reduced DMBA-mediated malignant mammary tumor volumes irrespective of genotype. MSE-specific PPARγ loss resulted in decreased mammary gland expression of PTEN and Bax, increased superoxide anion production, and elevated serum eotaxin and RANTES, creating a protumorigenic environment. Moreover, PPARγ activation in MSEs delayed mammary tumor growth in part by down-regulating Cox-1, Cox-2 and cyclin D1. Collectively, these studies highlight a protective role of MSE-specific PPARγ during breast tumorigenesis, and support a novel chemotherapeutic role of PPARγ activation in breast cancer.
Keywords: breast cancer, PPARγ, mammary secretory epithelial cells, knockout mouse model, chemical carcinogenesis, chemotherapy
In 2012, over 250,000 North American women were diagnosed with breast cancer, and over 44,000 died from metastatic complications, with an equally poor prognosis among susceptible men.1,2 Advances in early detection and treatment for some types of breast tumors have steadily reduced associated deaths over the last decade, but predicting, which patients will suffer from aggressive forms of disease or respond poorly to current therapy remains a challenge. Interestingly, child-bearing and breast feeding are well known to reduce breast cancer risk, but a transient increased risk of breast cancer following childbirth is also established,3 which may be due to deregulated interactions between genetic and lifestyle risk factors during mammary gland involution. Improved understanding of these interactions may aid in reducing deaths among susceptible patients.
PPARγ is a ligand-activated transcription factor that plays a role in cancer, is essential for adipocyte differentiation, and regulates genes involved in sugar and fat metabolism.4 PPARγ forms a heterodimer with retinoid X receptor (RXR)α and interacts with specific DNA sequences known as peroxisome proliferator-response elements (PPREs) in the promoter regions of a broad range of target genes.5 In the absence of ligand, the PPARγ:RXRα complex associates with cell-specific corepressor molecules that silence target gene transcription. Upon ligand binding, a conformational change leads to the release of corepressors and recruitment of coactivators that promote target gene transcription. Alternatively, ligand activated PPARγ may also transrepress gene signaling through direct interaction with other transcription factors or competition for available coregulators.6 Examples of PPARγ ligands include naturally occurring lipids, such as fatty acids and eicosanoids, and a synthetic class of drugs known as the thiazolidinediones (TZDs).7 Rosiglitazone (ROSI), a TZD and gold standard PPARγ activator, improves insulin sensitivity and lowers plasma glucose levels in Type II diabetic patients.8
What’s new? —
PPARγ is a transcription factor that has been implicated in several types of cancer, including breast cancer. In this study, the authors examined the role of PPARγ during breast tumorigenesis in mice, and found that it has a significant protective effect. Drugs that activate PPARγ may thus provide a new chemotherapeutic option for breast cancer patients, especially during the postpregnancy period when risk is transiently increased.
PPARγ is expressed primarily in adipocytes,9 as well as many other cell types including mammary epithelial cells,10 a majority of human breast tumors11 and breast cancer cell lines.12 Several in vitro studies reported PPARγ ligands promote differentiation and reduce growth in MCF-7 and MDA-MB-231 cells.12,13 Others successfully induced in vivo regression of chemically-induced breast tumors in rodents using PPARγ ligands.12,14,15 Previously, DMBA-treated PPARγ+/− mice were shown to be more susceptible to the increased growth and spread of breast, and other tumors as compared to wild-type controls.16 DMBA-induced breast tumorigenesis was also enhanced by mammary epithelial-directed knock-down of PPARγ in vivo.17 More recently, stromal adipocyte-specific PPARγ expression and signaling was also protective in slowing the progression of DMBA-mediated breast tumors.18 These studies provide direct evidence that normal PPARγ expression is critical to suppressing chemical-induced breast carcinogenesis; however, the role of PPARγ in other mammary gland-associated cell types remains to be characterized.
The mammary gland undergoes many dynamic changes throughout a woman’s lifetime.19 During pregnancy, the alveolar compartment expands through extensive proliferation of epithelial cells that differentiate to become specialized mammary secretory epithelial (MSE) cells that occupy >90% of the breast and produce milk maximally at lactation.20 After lactation is complete, the mammary gland undergoes remodeling to a prepregnant-like resting structure wherein these milk-producing cells are cleared by apoptosis. Stromal adipocytes are also capable of transforming into MSE cells as required for pregnancy, and may revert back to their original form after nursing.20 Since PPARγ is essential for fat cell differentiation,21 it may also be required for this transdifferentiation process. Therefore, it was hypothesized that loss of MSE-specific PPARγ postpregnancy may alter normal breast cell function, and increase susceptibility to breast tumorigenesis. Here, it is shown for the first time that loss of PPARγ expression in a unique subset of transient-appearing mammary epithelial cells significantly enhances DMBA-mediated breast tumorigenesis, in part by maintaining a protumorigenic environment.
Material and Methods
Animals
All mice were housed and treated in accordance with Canadian Council for Animal Care (CCAC) guidelines under Queen’s University Animal Care Committee (UACC)-approved protocols. Animals were housed in microisolator cages throughout a 12 hr light/dark cycle with food and water provided ad libitum. Previously generated PPARγfl/fl (PPARγ-WT) mice16 were crossed with breeders, generously provided by Dr. Hennighausen (NIH, Bethesda), expressing the whey acidic protein (WAP) promoter fused to the Cre Recombinase (Cre) transgene22 to produce the PPARγfl/fl; WAP-Cre+ (referred to here as PPARγ-MSE KO) mice. Colonies were maintained by interbreeding for >20 generations. All mice were of mixed C57/6N;SV/129;FVB/N background. Mouse genotypes were confirmed by PCR analysis as described previously.16
In vivo carcinogenesis
Eight-week-old PPARγ-MSE KO and PPARγ-WT female virgin mice were mated with males from respective strains to achieve time-matched pregnancies. Three days following parturition, dams had their offspring removed, and were entered into tumorigenic studies. Prestudy nonfasted submandibular blood was separated to obtain serum samples that were frozen in liquid N2 for future analysis. One week after the start of involution, breast tumors were initiated in mice from each genotype with 6 individual doses of DMBA by oral gavage weekly, followed by randomization into groups continuing on a normal chow diet or one supplemented with the gold standard PPARγ activator ROSI (4 mg/kg/day). Mice were monitored for tumorigenic changes for 25 weeks, and tumor samples were harvested and assessed as previously described.18
Immunoblotting
Whole-cell extracts were prepared from normal and tumor tissue samples from PPARγ-WT and PPARγ-MSE KO mice. Briefly, tissues were homogenized in solubilization buffer, consisting of ddH2O, 100 μM sodium orthovanadate, 1M Tris-HCl (pH 7.5), 1M MgCl2, 100 mM PMSF, 7× protease inhibitor, and 10% SDS and then incubated for 1 hr at 4°C. Samples were spun at 15,000g at 4°C for 10 min and the supernatant was collected, flash frozen and stored at −80°C. Protein concentrations were quantified using the DC protein assay (BioRad). Proteins were separated by running 25 μg of protein/sample on an SDS–PAGE gel, transferred to a PVDF membrane and detected with primary antibodies for PPARγ (sc-7273; 1:500; Santa Cruz), cyclin D1 (sc-753; 1:500; Santa Cruz), Bax (sc-526; 1:500; Santa Cruz), β-actin (sc-47778; 1:1,000; Santa Cruz), α-actinin (sc-15335; 1:1,000; Santa Cruz), PTEN (#9559; 1:1,000; Cell Signaling), Cox-1 (#160109; 1:500; Cayman Chemical), Cox-2 (#160126; 1:500; Cayman Chemical) and 5-LPO (#160402; 1:500; Cayman Chemical) followed by appropriate HRP-conjugated secondary goat α-mouse (sc-2005; Santa Cruz) or goat α-rabbit (sc-2004; Santa Cruz) antibodies (1:10,000). Protein expression was assessed using ImageJ analysis software (rsbweb.NIH.gov).
Immunofluorescent (IF) staining
Sections (5 µm) of formalin-fixed paraffin-embedded involuting mammary glands isolated from untreated PPARγ-WT and PPARγ-MSE KO females were mounted on slides and incubated at 55°C overnight. Samples were deparaffinized and rehydrated by washing in consecutive dilutions of xylene, ethanol and ddH2O. Slides were placed in 1:10 sodium citrate buffer solution (Sigma) at 95°C for 20 min and then trypsinized for 20 min at 37°C. After washing, slides were placed in 0.025% Triton X/TBS buffer solution, followed by a 30 min incubation in 5% BSA/TBS. After washing, primary antibodies (Santa Cruz) for PPARγ (sc-7196, 1:500) and β-casein (sc-166520, 1:500) were applied in 5% BSA/TBS for 60 min at room temperature. Slides were rinsed with TBS and then incubated in secondary antibodies for FITC (Santa Cruz, 1:500) and Alexa Fluor 594 (Invitrogen, 1:500) in 5% BSA/TBS for 15 min at room temperature. After a final rinsing regimen with TBS, slides were coverslipped with mounting media containing DAPI stain (Vectashield). IF staining was visualized with a BX51 System Microscope (Olympus) and images were acquired with QCapture Pro 5.1 software (QImaging).
Serum assays
Whole blood was collected from all study mice at one week prior to the start of the DMBA dosing regimen (Week 0), at midstudy (Week 13) and at necropsy. Samples were then centrifuged at 9.8g force for 4.5 min, and serum was collected, flash frozen and stored at −80°C for future study. A Bio-Plex Pro Mouse Cytokine 23-plex serum assay kit (BioRad Laboratories) was used to assess sample cytokine expressions according to manufacturer’s protocols. Briefly, beads were washed twice with wash buffer, and serum was diluted 4-fold with the provided dilution solution and incubated with the beads for 1 hr at room temperature. After three washes, beads were incubated with detection antibody for 30 min. The beads were washed again and incubated with streptavidin–phycoerythrin for 10 min before a final wash and resuspension in assay buffer. Samples were read on the Luminex 100 system, and subsequent data analysis was carried out using Bio-Plex Manager 6.0 software. Clustering and heat map analyses were performed with NetWalker 1.0 software.23 Cytokine concentrations are reported as the mean ± standard deviation (SD) pg/ml for the following targets: IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12(p40), IL-12(p70), IL-13, IL-17A, eotaxin, G-CSF, GM-CSF, IFN-γ, KC, MCP-1, MIP-1α, MIP-1β, RANTES and TNF-α.
Prostaglandin E ELISA
Serum prostaglandin E (PGE) metabolites were analyzed using the PGE Metabolite EIA kit (Cayman Chemical) as per manufacturer’s protocol. Briefly, samples were first purified by acetone precipitation, and then derivatized. Using 96-well plates, standards and samples were incubated with PGEM AChE Tracer and EIA Antiserum for 18 hr at room temperature, followed by the addition of Ellman’s Reagent. Plates were developed in the dark for an additional 60 min and read at a wavelength of 405 nm. Data are reported as the mean ± standard error (SE) pg/ml.
Dihydroethidium (DHE) assay
The oxidative fluorescent dye, dihydroethidium (DHE), was used to evaluate in situ production of reactive oxygen species. DHE freely permeabolizes the cell membrane and in the presence of superoxide anion (O2−) converts to ethidium bromide (EtBr), which remains in the cell by intercalating DNA. Cryofrozen sections (8 μm) of lactating mammary glands from untreated PPARγ-WT and PPARγ-MSE KO females were coverslipped with a 1:1 mixture of DAPI mounting media and 0.5 mg/ml DHE (Cayman Chemical) and incubated for 30 min at 37°C. Fluorescent staining was imaged by a Quorum WaveFX-X1 spinning disk confocal system (Quorum Technologies) with a Borealis Synapse laser merge module (Spectral Applied Research). Images were collected through a Yokogawa CSU-X1 spinning disk head (field illumination corrected <4%) and two Ludl six position filter wheels with 50 ms adjacent speeds fitted with various emission filters. DAPI was excited with 405 nm and fluorescent emission collected through a 460/50 m filter. Orange EtBr was excited with 568 nm and fluorescent emission collected through a 620/60 m filter. Microscope control and data analysis were performed with Metamorph Offline 7.7 software (Molecular Devices).
Statistical analyses
Data were evaluated for statistical differences using Prism 6.0 software (GraphPad). Comparisons of multiple groups were performed using a Two-Way Analyses of Variance (ANOVA) followed by Tukey’s post hoc tests. Incidences were analyzed by Fisher’s exact tests. Survival proportions were analyzed by log-rank tests. A p-value of ≤0.05 was accepted as statistically significant during analysis.
Results
IF analysis of lactating mammary glands shows that PPARγ expression is maintained in the basal compartment of β-casein-positive MSE cells from PPARγ-WT mice, but is deleted specifically in MSE cells from PPARγ-MSE KOs (Fig. 1). Immunoblotting confirmed that PPARγ expression is maintained in virgin mammary glands from both PPARγ-WT and PPARγ-MSE KO strains (Fig. 1); whereas, this expression is significantly reduced by three days after initiation of involution in PPARγ-MSE KO mammary tissue, a timing that coincides with commencement of our in vivo tumor studies. In addition, as compared to their wild-type controls, untreated PPARγ-MSE KO mice are similar in mammary development and nursing ability, and do not have any increased spontaneous breast tumor formation when followed for over 1 year (data not shown), consistent with a report on a similar previously generated strain.24 This strain is no longer maintained (Dr. Hennighausen, personal communication).
Figure 1.
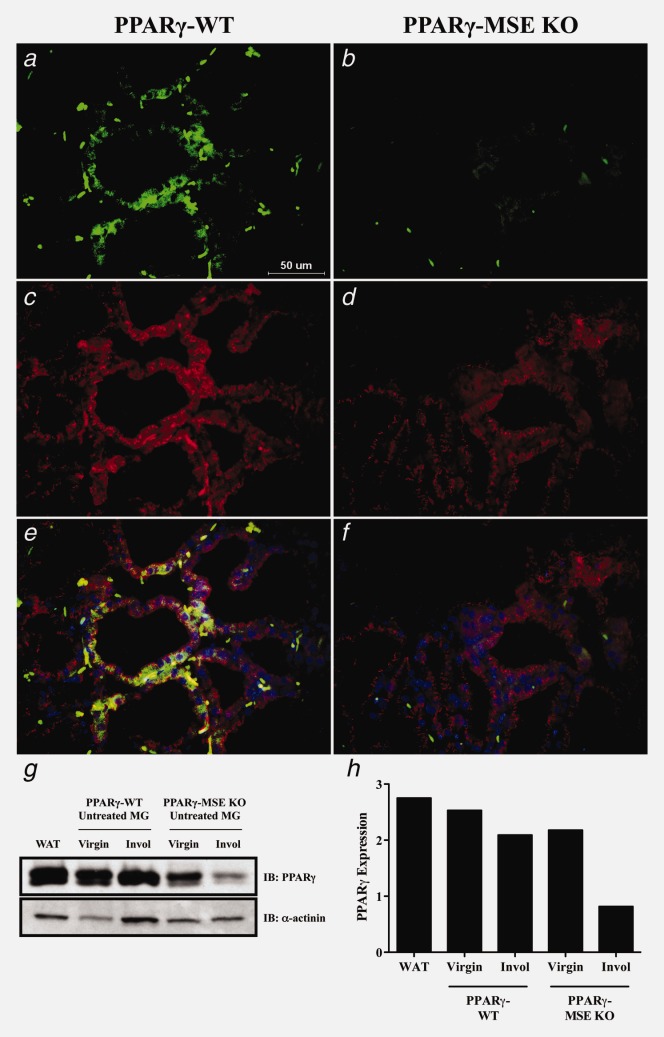
PPARγ protein expression in untreated mammary glands from PPARγ-WT and PPARγ-MSE KO strains. Representative immunofluorescence images illustrates (a and b) PPARγ (FITC; green) and (c and d) β-casein (Alexa Fluor 594; red) expression in lactating glands from both PPARγ-WT and PPARγ-MSE KO mice. An accompanying composite image (e and f) shows PPARγ and β-casein expression together with DAPI-stained nuclei. All photos were taken at ×600. (g) PPARγ expression was analyzed by Western blot in untreated mammary glands (MG) from both strains of virgin mice and mice three days after initiation of involution (Invol). White adipose tissue (WAT) from untreated PPARγ-WT mice was included as a positive control for PPARγ. α-actinin served as a loading control. (h) Densitometry was performed using ImageJ software.
Tumorigenic studies were initiated in postpregnant female mice from each genotype. At week 7, mice were randomized to continue receiving a normal chow diet (DMBA Only: PPARγ-WT, n = 15 and PPARγ-MSE KO, n = 25) or one supplemented with ROSI (DMBA+ROSI: PPARγ-WT, n = 17 and PPARγ-MSE KO, n = 24), and followed for 25 weeks for tumor outcomes. Two PPARγ-MSE KO mice treated with DMBA alone and four PPARγ-MSE KO mice treated with DMBA+ROSI were found dead in their cage due to undetermined circumstances. These mice were included in survival analyses but their tissues and tumors were excluded from further study due to possible sample degradation.
Throughout the study period, no significant differences were observed among average mouse body weights (Supporting Information Figs. 2a and 2b) or weekly food consumption (Supporting Information Fig. 2c) between genotypes irrespective of treatment. Analysis of survival proportions among DMBA only-treated mice (Fig. 2a) showed decreased overall survival among PPARγ-MSE KOs, wherein only 50% of animals were still alive by week 17, as compared to similarly treated PPARγ-WT mice, which did not reach this plateau even by the end of the observation period (p < 0.05). Similarly, DMBA + ROSI-treated PPARγ-MSE KO mice showed a strong trend toward decreased median overall survival (Fig. 2b), reaching a 50% plateau at week 18.5, as compared to similarly treated control mice that did not reach median overall survival by 25 weeks (p = 0.06). By study end, 53% and 24% of respective DMBA only-treated PPARγ-WT and PPARγ-MSE KO mice were still alive. Cotreatment with ROSI improved overall survival proportions by ∼10% in both strains, although this effect was not statistically significant.
Figure 2.
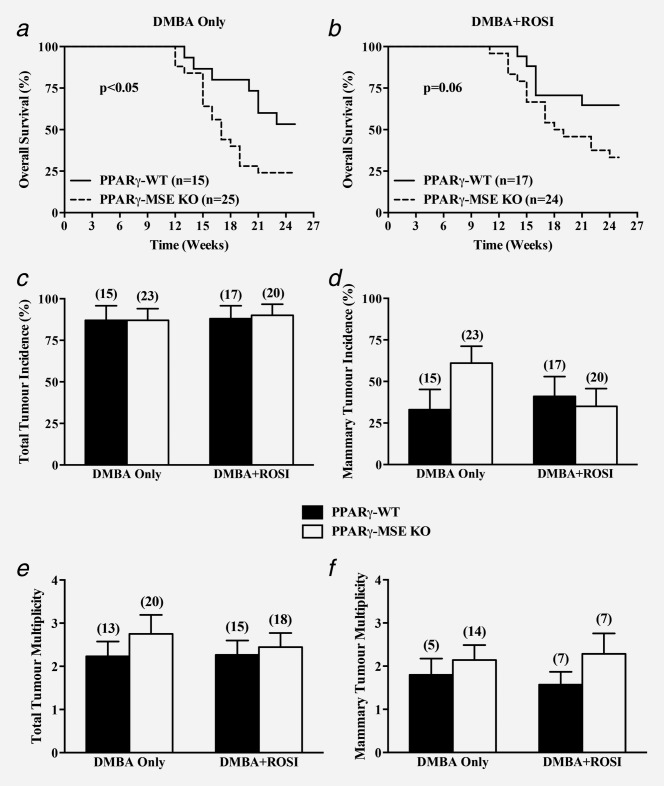
In vivo effects of MSE-specific PPARγ deletion on overall survival, tumor incidence and tumor multiplicity. Female PPARγ-WT and PPARγ-MSE KO mice were treated with DMBA only or DMBA + ROSI, and assessed as described in the Methods section. Overall survival was expressed as the percentage of mice per genotype per treatment group surviving in a given week. Solid lines, PPARγ-WT mice; dashed lines, PPARγ-MSE KO mice; n, number of mice. (a) DMBA only-treated groups; significantly different as compared to respective PPARγ-WT controls, p < 0.05. (b) DMBA + ROSI-treated groups; biologically different as compared to respective PPARγ-WT controls, p = 0.06. (c) Total tumor incidences were calculated as the number of mice with any tumor divided by the total number of mice within a given genotype and treatment group, and expressed as percent + standard error (SE). Black bars, PPARγ-WT mice; White bars, PPARγ-MSE KO mice; number in parentheses, number of mice. (d) Mammary tumor incidences were similarly calculated based on the number of mice with mammary tumors within a given genotype and treatment group, and expressed as percent + SE. (e) Total tumor multiplicity, expressed as a mean ± SE, was defined as the total number of lesions per tumor-bearing mice for a given genotype and treatment group. (f) Mammary tumor multiplicity was calculated as the number of mammary tumors per mouse afflicted with a breast lesion for a given genotype and treatment group, and expressed as a mean + SE.
Pathological examination of tumors yielded similar incidences of total tumors in DMBA Only-treated PPARγ-WTs and PPARγ-MSE KOs (percent ± SE: 87 ± 9% vs. 87 ± 7%, respectively) and DMBA + ROSI-treated strains (88 ± 8% vs. 90 ± 7%, respectively) (Fig. 2c). The pattern of DMBA-mediated tumor types were consistent with those previously associated with administration of this carcinogen, and comprised mainly mammary tumors, as well as skin, ovarian/uterine, thymic, and liver tumors, and lymphomas (Table 1). When compared to DMBA only-treated PPARγ-WT controls, mammary tumor incidences were 2-fold higher among similarly treated PPARγ-MSE KO mice, although this was not found to be significant (percent ± SE: 33 ± 12% vs. 61 ± 10%, respectively) (Fig. 2d). In contrast, mammary tumor incidences among DMBA + ROSI-treated PPARγ-WT and PPARγ-MSE KO mice were not significantly different (41 ± 12% vs. 35 ± 11%, respectively). Total tumor multiplicities between PPARγ-WT and PPARγ-MSE KO mice treated with DMBA only (mean ± SE: 2.2 ± 0.3 vs. 2.8 ± 0.4, respectively) or DMBA + ROSI (2.3 ± 0.3 vs. 2.4 ± 0.3, respectively) were not significantly different when compared by genotype or treatment (Fig. 2e). In addition, mammary tumor multiplicities between genotypes and across treatment groups showed a consistently higher trend among mice lacking PPARγ expression (DMBA only, PPARγ-WT vs. PPARγ-MSE KO: 1.8 ± 0.4 vs. 2.2 ± 0.3, respectively; DMBA + ROSI: 1.5 ± 0.3 vs. 2.3 ± 0.5, respectively) but were not significantly different (Fig. 2f). Interestingly, as compared to similarly treated PPARγ-WT mice, DMBA only-treated PPARγ-MSE KOs showed a 2-fold increase in the genotypic number of mammary tumors per mouse (mean ± SE: 0.60 ± 0.22 vs. 1.30 ± 0.27, respectively); however, these proportions were not significantly altered in either genotype following DMBA+ROSI-treatment (PPARγ-WT vs. PPARγ-MSE KO: 0.65 ± 0.19 vs. 0.80 ± 0.28, respectively) (Table 1). In addition, as compared with DMBA only-treated PPARγ-WT littermates, PPARγ-MSE KO mice had modestly larger median mammary tumor volumes (PPARγ-WT vs. PPARγ-MSE KO: 501.0 vs. 567.0 mm3, respectively) but these differences were not statistically significant (Fig. 3a). Notably, cotreatment with ROSI reduced median mammary tumor volumes by 3-fold for both PPARγ-WT (165.5 mm3) and PPARγ-MSE KO (126.0 mm3) mice as compared to their respective DMBA only-treated controls. Perhaps more importantly, when pathologically defined malignant mammary tumors were segregated irrespective of genotype, the median volumes from DMBA+ROSI-treated mice were significantly decreased over ∼6-fold as compared to those from DMBA only-treated controls (DMBA only vs. DMBA + ROSI: 1268.0 vs. 196.0 mm3, respectively; p < 0.05).
Table 1.
DMBA-induced tumors in PPARγ-WT and PPARγ-MSE KO mice
| DMBA only-treated mice | DMBA + ROSI-treated mice | |||
|---|---|---|---|---|
| PPARγ-WT (n = 15) | PPARγ-MSE KO (n = 23) | PPARγ-WT (n = 17) | PPARγ-MSE KO (n = 20) | |
| Mammary tumor type | Tumors/Mouse (# Tumors) | |||
| Benign tumor | 0.13 (2) | 0.30 (7) | 0.12 (2) | 0.10 (2) |
| Squamous cyst | 0.07 (1) | − | − | − |
| Fibrosis | − | 0.04 (1) | − | − |
| Other | 0.07 (1) | 0.26 (6) | 0.12 (2) | 0.10 (2) |
| Adenocarcinoma | 0.07 (1) | 0.22 (5) | 0.24 (4) | 0.15 (3) |
| Squamous cell carcinoma | 0.33 (5) | 0.39 (9) | 0.18 (3) | 0.35 (7) |
| Spindle cell carcinoma | 0.07 (1) | 0.04 (1) | − | − |
| Ductal carcinoma in situ | − | − | 0.06 (1) | − |
| Angiosarcoma | − | 0.04 (1) | − | − |
| Other carcinoma | − | 0.30 (7) | 0.06 (1) | 0.20 (4) |
| Total mammary tumors | 0.60 (9) | 1.30 (30) | 0.65 (11) | 0.80 (16) |
| Benign mammary | 0.13 (2) | 0.30 (7) | 0.12 (2) | 0.10 (2) |
| Malignant mammary | 0.47 (7) | 0.87 (20) | 0.53 (9) | 0.65 (13) |
| Metastatic mammary | − | 0.13 (3) | − | 0.05 (1) |
| Nonmammary tumor/tissue affected | Tumors/Mouse (# Tumors) | |||
| Skin | 0.53 (8) | 0.61 (14) | 0.76 (13) | 0.75 (15) |
| Squamous cell carcinoma | 0.07 (1) | 0.17 (4) | 0.41 (7) | 0.25 (5) |
| Spindle-cell carcinoma | − | 0.04 (1) | − | − |
| Other carcinoma | 0.07 (1) | 0.04 (1) | − | 0.15 (3) |
| Carcinoma in situ | − | − | 0.06 (1) | − |
| Epidermal inclusion cyst | − | 0.04 (1) | 0.06 (1) | − |
| Hyperkeratosis | 0.20 (3) | 0.13 (2) | − | 0.05 (1) |
| Hyperplasia | − | − | 0.06 (1) | 0.05 (1) |
| Sebaceous adenoma | − | 0.04 (1) | − | 0.05 (1) |
| Other | 0.20 (3) | 0.17 (4) | 0.18 (3) | 0.20 (4) |
| Ovarian/Uterine | 0.27 (4) | 0.09 (2) | 0.24 (4) | 0.20 (4) |
| Metastasis | 0.07 (1) | − | − | − |
| Adenocarcinoma | − | − | 0.06 (1) | − |
| Other carcinoma | − | − | 0.06 (1) | 0.10 (2) |
| Angiosarcoma | 0.07 (1) | − | − | − |
| Hyperplasia | − | 0.04 (1) | − | − |
| Other | 0.13 (2) | 0.04 (1) | 0.12 (2) | 0.10 (2) |
| Thymus | 0.13 (2) | 0.13 (3) | − | 0.15 (3) |
| Spleen | 0.13 (2) | − | − | − |
| Liver | 0.07 (1) | − | 0.06 (1) | 0.05 (1) |
| Adenoma | − | − | 0.06 (1) | − |
| Steatosis | − | − | − | 0.05 (1) |
| Other | 0.07 (1) | − | − | − |
| Lung | − | 0.04 (1) | − | − |
| Hyperplasia | − | 0.04 (1) | − | − |
| Gastrointestinal | − | 0.09 (2) | − | − |
| Squamous cell carcinoma | − | 0.09 (2) | − | − |
| Lymphoma | 0.20 (3) | 0.13 (3) | 0.29 (5) | 0.25 (5) |
| Localized | 0.13 (2) | 0.09 (2) | 0.06 (1) | 0.15 (3) |
| Infiltrating | 0.07 (1) | 0.04 (1) | 0.24 (4) | 0.10 (2) |
| Total tumors | 1.93 (29) | 2.39 (55) | 2.00 (34) | 2.20 (44) |
| Benign total | 0.6 (9) | 0.74 (17) | 0.47 (8) | 0.65 (13) |
| Malignant total | 1.27 (19) | 1.52 (35) | 1.53 (26) | 1.50 (30) |
| Metastatic total | 0.07 (1) | 0.13 (3) | − | 0.05 (1) |
The number of mammary tumors per mouse (multiplicity) is indicated with the total number of these tumors shown in parenthesis. Mammary tumors were also substratified and expressed as multiplicity of benign, malignant and metastatic tumors per genotype and treatment. Examples of specific benign tumor subtypes are also indicated. For nonmammary tissue, the numbers of each tumor per mouse is also indicated with the total number of each shown in parenthesis. Examples of specific tumor subtypes are specified under the title of the tissue type. Finally, total tumors were substratified and expressed as the multiplicity of benign, malignant and metastatic tumors per genotype and treatment.
Figure 3.
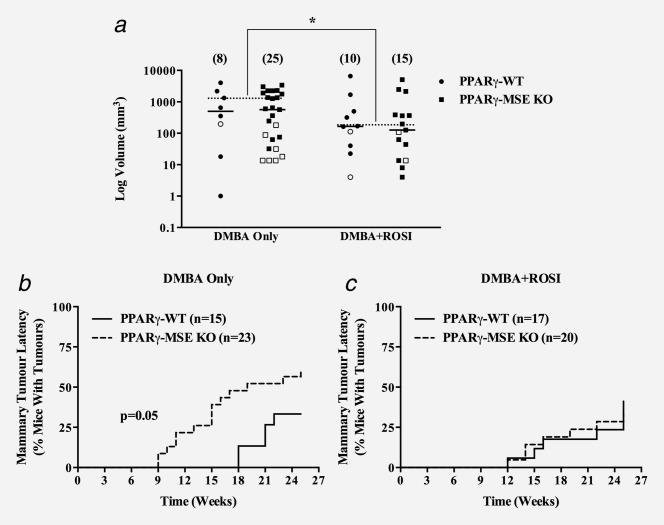
In vivo effects of MSE-specific PPARγ deletion on mammary tumor volume and latency. Mammary tumors were measured at necropsy, and volumes calculated using the standard formula (L × W2/2) and expressed as mean mm3 for each treatment group (a). Open and closed circles, benign and malignant mammary tumors, respectively from PPARγ-WT mice; Open and closed boxes, benign and malignant mammary tumors, respectively from PPARγ-MSE KO mice; Solid bars represent median values for all mammary tumors; dashed bars represent median values for pooled malignant mammary tumors for a given treatment; number in parentheses, number of tumors; *, significantly different from DMBA only-treated groups, p < 0.05. Latency of mammary tumors is expressed as the percentage of mice with palpable mammary tumors within a given genotype and treatment group for a given week. Solid lines, PPARγ-WT mice; dashed lines, PPARγ-MSE KO mice; n, number of mice. (b) DMBA only-treated groups; significantly different compared to respective PPARγ-WT controls, p < 0.05. (c) DMBA + ROSI-treated groups. n, number of mice.
When the time to mammary tumor onset was examined, latency was significantly reduced among DMBA only-treated PPARγ-MSE KOs, with a median onset by week 19, as compared to PPARγ-WT controls that never reached a 50% plateau (p = 0.05) (Fig. 3b). DMBA + ROSI treatment improved median mammary tumor latency such that by 25 weeks <50% of both mouse strains developed mammary tumors (Fig. 3c). Given that the majority of treated mouse groups did not reach median survival, quartile (25%) survival time was examined and showed DMBA + ROSI-treated mice (PPARγ-WT vs. PPARγ-MSE KO: week 25 vs. 22, respectively) had an improvement in mammary tumor free survival as compared to DMBA only-treated mice (PPARγ-WT vs. PPARγ-MSE KO: week 21 vs. 13, respectively). When compared to PPARγ-WTs treated with DMBA alone, mammary tumor onset was significantly earlier in DMBA only-treated PPARγ-MSE KOs (PPARγ-WT vs. PPARγ-MSE KO: week 20 vs. 15, respectively; p < 0.01) (Supporting Information Fig. 3). In contrast, mammary tumor onset was similar between both genotypes treated with DMBA + ROSI (PPARγ-WT vs. PPARγ-MSE KO: week 20 vs. 18, respectively).
Pathological analysis showed a similar histological pattern of untreated, involuting mammary glands and DMBA-mediated mammary tumor subtypes irrespective of mouse strain or treatment group (Fig. 4 and Table 1). Adenocarcinomas and squamous cell carcinomas were the most common mammary tumor subtypes identified. When subclassified by stage and compared to similarly treated controls, the number of tumors per mouse among DMBA only-treated PPARγ-MSE KO mice were increased 2-fold for both benign (PPARγ-WT vs. PPARγ-MSE KO, mean ± SE: 0.13 ± 0.09 vs. 0.30 ± 0.12, respectively) and malignant (0.47 ± 0.24 vs. 0.87 ± 0.20, respectively) mammary tumors. No difference was observed within DMBA + ROSI-treated groups (PPARγ-WT vs. PPARγ-MSE KO: benign, 0.12 ± 0.08 vs. 0.10 ± 0.10; malignant, 0.53 ± 0.21 vs. 0.65 ± 0.25, respectively) (Table 1). In addition, PPARγ-WTs had zero metastatic breast tumors per mouse, irrespective of treatment, as compared to the modest increased trend for PPARγ-MSE KO mice in both DMBA only-treated (0.13 ± 0.10) and DMBA + ROSI-treated (0.05 ± 0.05) groups.
Figure 4.
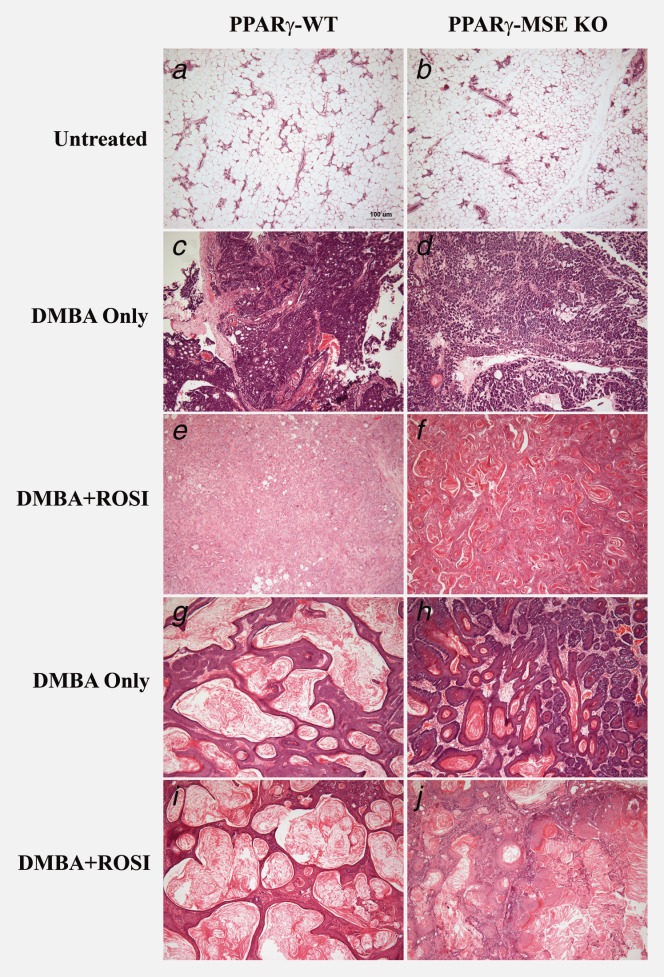
Mammary tumor subtypes among treated PPARγ-WT and PPARγ-MSE KO mice. Representative H&E images of: untreated, involuted mammary glands from (a) PPARγ-WT and (b) PPARγ-MSE KO strains; adenocarcinomas from (c) DMBA only-treated PPARγ-WT and (d) PPARγ-MSE KO mice and (e) DMBA + ROSI-treated PPARγ-WT and (f) PPARγ-MSE KO mice; and squamous cell carcinomas from (g) DMBA-only-treated PPARγ-WT and (h) PPARγ-MSE KO strains and (i) DMBA + ROSI-treated PPARγ-WT and (j) PPARγ-MSE KO strains. All photos were taken at ×100.
To further explore changes in cytokine/chemokine expressions, serum samples were evaluated using a serum cytokine array, with results compared by genotype and treatment group (Table 2 and Fig. 5d). Untreated PPARγ-MSE KO mice showed a significant increase in RANTES concentration as compared to PPARγ-WT mice (mean ± SD: 51.8 ± 6.2 vs. 24.7 ± 16.4, p < 0.05). For the DMBA + ROSI-treated group, PPARγ-MSE KO mice showed nonsignificant trends towards higher levels of IL-6 (60.4 ± 54.6 vs. 13.4 ± 9.7; p < 0.10) and lower levels of IL-12(p40) (728 ± 216 vs. 1804 ± 958; p < 0.10) as compared to PPARγ-WT controls. When analyzed by genotype alone (Supporting Information Table 1), PPARγ-MSE KOs collectively showed significantly lower levels of IL-5 (5.0 ± 10.8 vs. 17.7 ± 19.3, p < 0.05) and higher levels of eotaxin (798.9 ± 682.9 vs. 302.3 ± 492.6, p < 0.05) and RANTES (42.1 ± 14.1 vs. 21.8 ± 13.5, p < 0.001) as compared to PPARγ-WT mice. When compared by treatment alone (Supporting Information Table 2), DMBA + ROSI increased IL-6 (36.9 ± 44.2 vs. 5.4 ± 1.8, p < 0.05) and decreased IL-1α (28.4 ± 22.5 vs. 168.0 ± 172.8, p < 0.05) serum concentrations as compared to no treatment.
Table 2.
Serum concentrations of cytokines from untreated, DMBA only and DMBA + ROSI-treated strains
| PPARγ-WT | PPARγ-MSE KO | |||||
|---|---|---|---|---|---|---|
| Untreated (n = 6) | DMBA only (n = 4) | DMBA + ROSI (n = 4) | Untreated (n = 6) | DMBA only (n = 4) | DMBA + ROSI (n = 4) | |
| Cytokine [signif] | mean ± SD; all values expressed as pg/ml | |||||
| IL-1α [t] | 237.6 ± 218.8 | 50.9 ± 39.5 | 31.3 ± 21.3 | 98.4 ± 78.5 | 56.5 ± 79.1 | 25.5 ± 26.5 |
| IL-1β | 185.2 ± 45.4 | 349.1 ± 213.0 | 258.3 ± 44.1 | 230.5 ± 27.6 | 221.5 ± 37.1 | 208.3 ± 99.0 |
| IL-2 | 65.4 ± 74.1 | 33.0 ± 39.4 | 14.9 ± 17.4 | 20.9 ± 11.0 | 14.7 ± 15.0 | 32.1 ± 12.4 |
| IL-3 | 2.7 ± 6.6 | ND | 9.0 ± 17.9 | 5.4 ± 1.8 | 3.5 ± 5.0 | 29.9 ± 49.6 |
| IL-4 [t] | 10.4 ± 1.1 | 13.0 ± 2.9 | 10.7 ± 1.5 | 10.7 ± 1.9 | 12.1 ± 1.5 | 11.3 ± 0.6 |
| IL-5 [g] | 13.6 ± 12.1 | 9.3 ± 13.5 | 32.2 ± 27.9 | 3.4 ± 6.0 | ND | 12.3 ± 18.4 |
| IL-6 [t] | 4.9 ± 2.3 | 25.5 ± 26.0 | 13.4 ± 9.7 | 5.9 ± 1.1 | 22.0 ± 11.2 | 60.4 ± 54.6 # |
| IL-9 | ND | ND | ND | ND | ND | ND |
| IL-10 | 29.8 ± 23.7 | 109.1 ± 131.1 | 83.0 ± 54.2 | 9.3 ± 18.8 | 35.3 ± 20.2 | 66.4 ± 7.3 |
| IL-12(p40) [i,t,g] | 502.7 ± 97.6 | 1482.0 ± 910.8 | 1804.0 ± 958.1* | 694.6 ± 402.5 | 1001.0 ± 284.3 | 728.2 ± 215.8 |
| IL-12(p70) | ND | ND | ND | ND | ND | ND |
| IL-13 | 95.8 ± 88.0 | 157.5 ± 115.4 | 125.4 ± 99.0 | 80.2 ± 52.2 | 75.5 ± 29.4 | 86.5 ± 54.4 |
| IL-17A | 3.5 ± 8.6 | 12.2 ± 14.4 | 11.89 ± 13.84 | 4.7 ± 6.8 | 6.6 ± 2.9 | 12.9 ± 12.5 |
| Eotaxin [g] | 174.0 ± 271.3 | 379.3 ± 414.7 | 417.8 ± 835.7 | 406.8 ± 116.1 | 1207.0 ± 872.2 | 979.2 ± 815.2 |
| G-CSF [t] | 153.5 ± 105.6 | 2901.0 ± 4540.0 | 2269.0 ± 1963.0 | 106.0 ± 51.1 | 3071.0 ± 2484.0 | 1706.0 ± 1310.0 |
| GM-CSF | 94.4 ± 135.1 | 241.7 ± 153.2 | 126.3 ± 51.0 | 187.9 ± 73.8 | 174.8 ± 40.0 | 164.6 ± 32.4 |
| IFN-γ | 11.2 ± 9.4 | 15.3 ± 12.2 | 6.3 ± 1.9 | 10.7 ± 5.1 | 16.5 ± 5.4 | 17.2 ± 5.7 |
| KC [t] | 10.7 ± 4.3 | 27.5 ± 27.6 | 10.0 ± 8.9 | 7.0 ± 2.4 | 26.5 ± 13.5 | 17.1 ± 7.5 |
| MCP-1 | 382.2 ± 822.0 | 541.0 ± 847.1 | 138.5 ± 69.4 | 80.0 ± 40.0 | 173.7 ± 103.1 | 698.3 ± 1192.0 |
| MIP-1α [t] | 32.8 ± 20.1 | 65.7 ± 50.1 | 36.3 ± 15.1 | 34.0 ± 2.8 | 53.4 ± 9.3 | 66.0 ± 8.7 |
| MIP-1β [t] | 12.9 ± 9.3 | 33.0 ± 25.9 | 19.9 ± 6.0 | 19.4 ± 8.0 | 47.9 ± 40.7 | 17.5 ± 6.3 |
| RANTES [ggg] | 24.7 ± 16.4 | 23.6 ± 13.2 | 15.8 ± 10.0 | 51.8 ± 6.2* | 29.8 ± 14.4 | 39.7 ± 14.0 |
| TNF-α | 191.3 ± 149.4 | 448.5 ± 247.3 | 237.9 ± 80.0 | 203.7 ± 67.2 | 243.3 ± 81.3 | 379.4 ± 206.3 |
| PGE metabolites | 425.6 ± 235.7 (n = 5) | 1953.3 ± 3544.6 (n = 4) | 278.3 ± 102.0 (n = 4) | 713.0 ± 889.1 (n = 4) | 1350.7 ± 1865.2 (n = 4) | 484.3 ± 447.6 (n = 4) |
Concentrations reported as mean ± standard deviation (SD) and expressed as pg/ml. Except for PGE metabolites, which were analyzed with a separate ELISA kit, all cytokine concentrations were obtained by a multiplex array. *, Significantly different as compared to untreated PPARγ-WT, p < 0.05; #, significantly different compared to Untreated PPARγ-MSE KO, p < 0.05; i, interaction different, p < 0.05; t, treatment different, p < 0.05; g, genotype different, p < 0.05; ggg, genotype different, p < 0.001.
Figure 5.
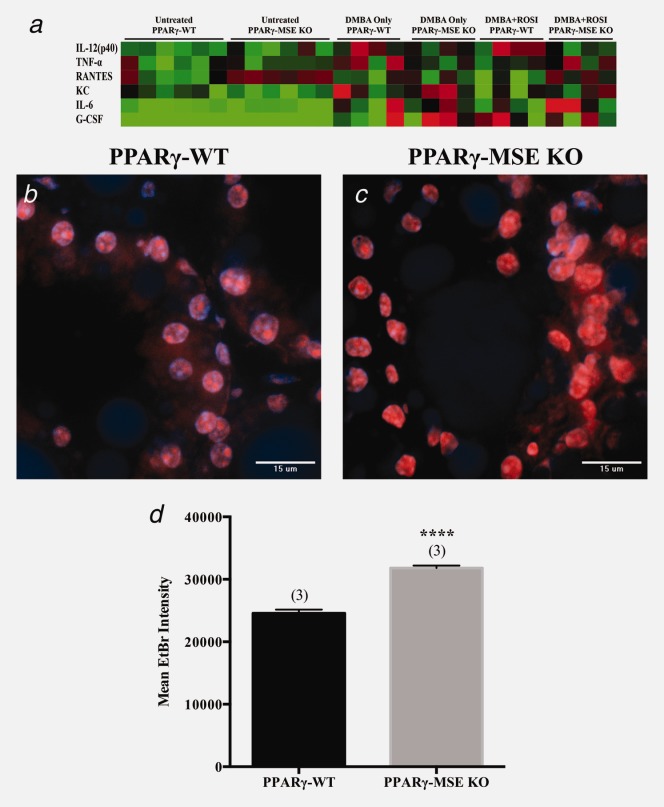
Serum cytokine and in situ superoxide production resulting from MSE-specific PPARγ loss. (a) Heat map illustrating serum cytokine expression profiles for genotypes and treatment groups. Values are Log2 (mean cytokine concentration, pg/ml) with red, black and green indicated high, median and low, respectively. DAPI- and EtBr-stained nuclei (orange) are shown in representative confocal images of lactating mammary glands from (b) untreated PPARγ-WT and (c) PPARγ-MSE KO mice. Both photos were taken at ×600. (d) EtBr fluorescence intensity, expressed as mean + SE, was measured using Metamorph imaging software. ****, significantly different from PPARγ-WT controls, p ≤ 0.0001.
To further extend our serum findings, the extent of reactive oxygen species production in lactating mammary glands from untreated mice was also assessed using the DHE staining assay. Confocal microscopy (Figs. 5a and 5b) and quantification of EtBr fluorescence (Fig. 5c), a consequence of O2−-dependent DHE oxidation, revealed that PPARγ-MSE KO mice produced significantly more O2− as compared to PPARγ-WT controls (Fig. 5c) (mean ± SE: 31815 ± 381 vs. 24577 ± 573, respectively; p < 0.0001).
To assess protein expression changes in proposed PPARγ anticancer targets resulting from MSE-cell specific deletion of PPARγ, Western blot analyses were performed on untreated involuting mammary glands from each genotype, and malignant mammary tumors from both strains of mice across treatment groups (Figs. 6a and 6b and Supporting Information Fig. 4). In untreated mammary glands from PPARγ-MSE KOs as compared to PPARγ-WTs, Bax and PTEN protein expressions were decreased 2-fold, and Cox-1 was decreased 3-fold (Fig. 6a). When target protein expression changes in mammary tumors from DMBA + ROSI-treated PPARγ-WTs as compared to DMBA only controls were examined by densitometry, significant 2-fold and 3.5-fold decreases in protein expression of Cox-1 and Cox-2 were observed, respectively (p < 0.05) (Fig. 5b). In contrast, protein expression of both Cox-1 and Cox-2 among mammary tumors from DMBA + ROSI-treated PPARγ-MSE KO mice increased ∼1.5-fold as compared to their respective DMBA Only-treated controls. Analysis of 5-lipoxygenase (5-LPO) showed no genotypic or treatment differences. Furthermore, within mammary tumors, DMBA + ROSI treatment significantly decreased cyclin D1 expression by 4-fold among PPARγ-WTs (p = 0.05), and nonsignificantly by 1.5-fold among PPARγ-MSE KOs as compared to their respective DMBA only-treated groups (Fig. 6b).
Figure 6.
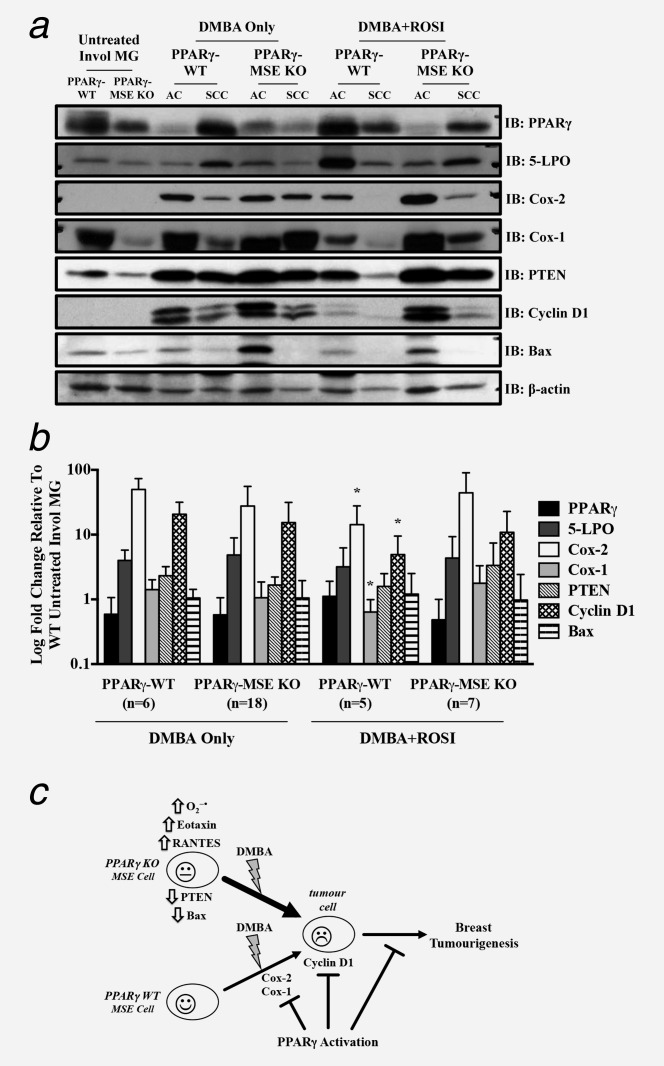
Molecular analysis from untreated mammary glands and mammary tumors from treated PPARγ-WT and PPARγ-MSE KO mice. Representative expression changes within untreated mammary glands (MG) and in vivo generated mammary tumors were analyzed by (a) Western blot as described in the Methods section. PPARγ, 5-LPO, Cox-2, Cox-1, PTEN, cyclin D1 and Bax protein levels were analyzed in untreated, involuted (Invol) MG from PPARγ-WT and PPARγ-MSE KO mice, as well as representative breast tumor subtypes from both strains of mice across both treatment groups. Mammary tumor subtypes include adenocarcinomas (AC), and squamous cell carcinomas (SCC). β-actin served as the loading control. (b) Densitometry was performed on malignant tumors using ImageJ software, and expressed as mean ± SD. Fold changes are relative to involuted mammary tissue from untreated WT. *, Significantly different from DMBA Only-treated groups, p ≤ 0.05. (c) Summary of proposed antibreast tumor MSE-specific PPARγ signaling. Deletion of PPARγ in MSE cells promotes a pro-tumorigenic environment, characterized by increased superoxide production, elevated eotaxin and RANTES circulation, as well as reduced PTEN and Bax protein expression in involuted mammary glands of PPARγ-MSE KO mice. Thus, upon exposure to DMBA, PPARγ-MSE KOs are more susceptible to breast tumorigenesis as compared to similarly treated PPARγ-WT mice. PPARγ activation protects against these effects, albeit more so in PPARγ-WT mice, at least in part by suppressing Cox-1, Cox-2 and Cyclin D1.
Discussion
The MSE cell-specific role of PPARγ in breast tumorigenesis was evaluated. PPARγ-MSE KO mice were generated, and had significantly decreased overall survival and shorter mammary tumor latency as compared to PPARγ-WT controls. Immunoblotting studies revealed a protumorigenic environment among mammary glands from PPARγ-MSE KOs that may contribute to enhanced breast cancer susceptibility. Cotreatment with a PPARγ activator provided a greater phenotypic rescue among PPARγ-WTs than PPARγ-MSE KO mice. The ROSI dose and regimen used in our study is effective in activating PPARγ signaling in mice25–27 and achieving murine serum profiles in the human therapeutic range.28,29 These studies implicate activation of PPARγ signaling in subpopulations of mammary gland-specific cells in suppressing carcinogen-induced breast tumors, consistent with our and other studies.12,13,16,18
Normal PPARγ expression was confirmed in virgin mammary glands of both mouse strains, but abolished in MSE cells of PPARγ-MSE KO mice from the time of lactation as compared to controls. Accordingly, this knockout model was useful for assessing our hypotheses. The pattern of PPARγ expression in a lactating mammary gland was previously implicated in the milk production process,30 and suggests PPARγ is normally expressed at specific times during lactation to suppress inflammatory lipid levels in milk.
Throughout the in vivo tumor studies, DMBA only-treated PPARγ-MSE KOs had significantly lower survival rates as compared to PPARγ-WT controls. Similar observations were found among DMBA + ROSI-treated strains, with modest improvements in overall survival. It cannot be discounted that the rapid growth of breast tumors in these mice and the ethical limitations on tumor study endpoints may have precluded the ability to observe additional carcinogen-mediated micro-metastases impacting on vital organs. Further, the influence of tumors arising in nonmammary tissue may have confounded the overall survival results; however, parallel tumor patterns observed in all genotypes and treatment groups suggests that any such contribution is minimal. Together, this data suggests that selective loss of PPARγ expression in MSE cells postpregnancy results in reduced overall survival.
That spontaneous tumor formation does not occur in PPARγ-MSE KO mice, necessitating DMBA administration to initiate breast tumorigenesis in our animal studies, implies that PPARγ acts as a suppressor of tumor progression. Therefore, it was not surprising that parameters of tumor initiation (i.e., mammary tumor incidence and multiplicity) were not different between DMBA only- and DMBA + ROSI-treated PPARγ-WTs and -MSE KOs. While the vast majority of mammary tumors are epithelial in nature, stromal influence on transformed cells may also influence carcinogenesis through signals that prime the tumor microenvironment and fuel disease progression.31 DMBA-mediated tumorigenesis, and PPARγ signaling activation, would be anticipated to exert similar responses in these mammary tumor parameters in both genotypes, which is consistent with the notion that MSE cells do not represent the mammary tumor cell of origin. Pathological sub-classification of mammary tumors also suggested no change in the incidence of mammary tumor malignancy between PPARγ-MSE KOs versus PPARγ-WT controls. These findings suggest MSE cells lacking PPARγ expression affect mammary tumor outcomes in a paracrine fashion rather than serving as primary tumor initiating cells themselves.
Upon examining specific parameters of tumor progression, DMBA only-treated PPARγ-MSE KO mice had a significantly earlier mean week of mammary tumor onset as compared to similarly treated PPARγ-WT mice. This suggests that loss of PPARγ expression in MSE cells decreases mammary tumor latency, presumably through enhanced pro-tumorigenic signaling. This further supports an MSE-specific paracrine influence during early mammary tumorigenesis in PPARγ-MSE KO mice. For this tumorigenic measure, it is notable that ROSI cotreatment increased mammary tumor latency among both strains. This suggests activation of signaling within other PPARγ-expressing cells associated with, or near, the mammary gland may contribute to this protective effect, and that activation of normal PPARγ signaling in other PPARγ-expressing cells common to both strains is chemotherapeutic. This is consistent with reports suggesting that, for some parameters, combination antitumor therapies including PPARγ activators may be beneficial.32
Although no genotypic difference in median mammary tumor volumes was observed for either treatment, cotreatment with ROSI significantly decreased pooled malignant tumor volumes versus those from DMBA only-treated mice. This is consistent with studies showing PPARγ activation suppresses carcinogen-induced mammary tumor growth in vivo.14,15 This was also apparent for the increased benign and malignant mammary tumor multiplicities among DMBA only-treated PPARγ-MSE KO mice, which were reduced to levels similar to those observed among PPARγ-WTs by ROSI cotreatment. The absence of a genotype-specific effect following ROSI cotreatment suggests that signaling activation within other PPARγ-expressing mammary epithelial or stromal cell types in both strains are involved in this protective antitumor effect, consistent with previous findings.18 Although no genotypic effect was observed with respect to these parameters, this study is the first to examine whether MSE-specific PPARγ activation would play a role in this process. Given the number of mammary-associated cell types that express PPARγ, such a determination is critical to understand how TZDs may in fact protect against tumorigenesis.
To further characterize the probreast tumorigenic environment promoted by MSE-specific PPARγ loss, increased serum eotaxin and RANTES was also observed from PPARγ-MSE KO mice. Both of these are chemotactic cytokines that selectively recruit leukocytes to sites of inflammation, and have been linked to the development of human cancer.33,34 Deletion of PPARγ in MSE cells also increases the pro-breast tumorigenic state of the mammary gland as exemplified by elevated endogenous levels of O2− species in PPARγ-MSE KO mice. Reactive oxygen species, like O2−, can alter macromolecular structures and signaling pathways, contributing to oxidative stress, inflammation and carcinogenic progression.35 Taken together, these findings may be responsible for the increased breast cancer risk observed in PPARγ-MSE KO mice.
Involuting mammary tissue from untreated PPARγ-MSE KOs exhibit lower levels of Bax and PTEN proteins as compared to PPARγ-WTs. This is consistent with previous findings that Bax degradation is mediated by the PI3K pathway,36 which is opposed by PTEN signaling. Since Bax promotes apoptosis and PTEN is a tumor suppressor, the postpregnant loss of MSE-specific PPARγ expression may further contribute to the suggested pro-tumorigenic environment in PPARγ-MSE KO mammary glands. This is also consistent with PPARγ activation upregulating both PTEN, via a PPRE, and Bax in human MCF-7 breast cancer cell lines.37,38 Furthermore, conditional deletion of PTEN in mouse mammary epithelium impairs apoptosis and delays involution.39 The loss of Bax also impairs cell death early in involution40 and facilitates tumor progression in mice.41 Given that the normal inflammatory and proteolytic processes that occur during involution have the capability of transforming the mammary gland microenvironment into one that favors tumorigenesis,42 disrupting and prolonging the involution program via impaired PTEN and Bax signaling may be responsible for the enhanced susceptibility to tumorigenesis observed among PPARγ-MSE KO mice. Schorr et al.40 also noted partial loss of Bax was most potent in impairing cell death during involution when combined with bcl-2 gain. Coupled with PTEN loss, decreased Bax expression in PPARγ-MSE KO postpregnant breast tissue may further delay involution. This effect may have contributed, at least in part, to the enhanced DMBA-mediated breast tumorigenesis among PPARγ-MSE KO mice.
The significant decrease in Cox-1 expression observed in untreated mammary glands of PPARγ-MSE KO mice was surprising considering its known involvement in tumorigenesis.43 The contributions of expression and signaling during tumorigenesis may depend on timing of expression, similar to the variable early versus late effects of TGF-β.44 Accordingly, it is possible pretumorigenic expression of Cox-1 protein protects target cells, whereas post-tumorigenic expression enhances progression in DMBA-treated PPARγ-MSE KOs. Though beyond the scope of this report, current work is examining the possibility that early decreases in Cox-1 in PPARγ-MSE KO mammary glands increases exposure of target cells to carcinogenic DMBA metabolites, and decreases mammary tumor initiation time. Perhaps more interestingly, PPARγ activation preferentially attenuated Cox-1 and Cox-2 expression in mammary tumors from DMBA + ROSI-treated PPARγ-WT, but not PPARγ-MSE KO mice. Inhibition of these enzymatic proteins mediates a reduction in metastatic capacity,43 and overexpression of Cox-2 is associated with poor breast cancer prognosis.45 Thus, mammary tumors in DMBA + ROSI-treated PPARγ-WTs may be associated with improved outcomes, which may be more apparent in extended studies. Moreover, activation of PPARγ signaling attenuated cyclin D1 expression in mammary tumors from both strains, likely contributing to the decreased tumor volumes in the DMBA + ROSI group. Suppression of this proliferative marker was greater in mammary tumors from DMBA + ROSI-treated PPARγ-WTs, further strengthening the argument that these mice might have exhibited more improved outcomes in extended studies.
Although not significant, it was interesting to note that cotreatment with ROSI upregulated expression of the p40 subunit of IL-12 in PPARγ-WT mice, despite the fact that the biologically active IL-12(p70) cytokine was undetectable by the array. IL-12(p40) is also known to heterodimerize with p19, another functionally related subunit, to form IL-23, which is known to have late antitumor and antimetastatic properties.46 This suggests that activation of PPARγ in MSE cells leads to the production of IL-23, which may contribute to the suppression of breast tumorigenesis, but remains to be confirmed.
A summary of proposed mechanisms contributing to the enhanced mammary tumorigenesis among postpregnant PPARγ-MSE KO mice is provided (Fig. 6c). Briefly, loss of MSE-specific PPARγ expression in the mammary gland results in early loss of PTEN and Bax expression, increased production of O2−, and increased serum eotaxin and RANTES, enhancing susceptibility to DMBA-mediated breast tumorigenesis. Activation of MSE-specific PPARγ among PPARγ-WT mice further suppresses DMBA-mediated breast tumorigenesis by decreasing Cox-1 and Cox-2. Among cells expressing PPARγ, activation of PPARγ-dependent signaling suppresses mammary tumor growth, in part by attenuating cyclin D1 levels. Previously, PPARγ activation was shown to inhibit cyclin D1 expression in vitro.47 These studies extend the former results, and provide the first in vivo evidence showing PPARγ activation suppresses cyclin D1, Cox-1 and Cox-2 in DMBA-mediated breast tumors. Attenuation of these targets is more pronounced in PPARγ-WT mice suggesting that MSE-specific PPARγ-dependent signaling contributes in large part to this effect.
It remains unclear whether loss of PPARγ alters MSE cell fate during the involution process, or how this may contribute improper paracrine signals to other normal and transformed cells in the mammary gland. Future studies examining this question, will aid in understanding the contributions of MSE cells during breast tumorigenesis. Studies evaluating the contributions of other specific PPARγ-expressing mammary gland-associated cell types are ongoing (Nicol et al., unpublished). These may aid in dissecting the complexity of epithelial-stromal crosstalk, and the global network and timing of protective signaling pathways involved in slowing the growth and spread of breast tumors, but is beyond the scope of this study.
These studies are the first to highlight the protective role of MSE-specific PPARγ expression and signaling in breast tumorigenesis, and suggest loss of MSE-specific PPARγ may contribute to the increased breast tumor risk following childbirth. These studies also add further support for a chemotherapeutic role of PPARγ activation in breast cancer treatment, and highlight a population of at risk women who may benefit from the use of PPARγ activators, in either mono- or combination therapy.
Acknowledgments
Special thanks to Dr. Wilma Hopman for assistance with statistical methodology, and Jeff Mewburn for help with confocal microscopy.
Author’s Contributions
AA performed animal handling and necropsies; Western blot, ELISA, and DHE assays; data collection and analysis; and drafted the manuscript. GSG assisted in animal handling, necropsies and sample collection. MD carried out immunofluorescence assays. RR and NP performed breeding of study animals, dosing and sample collection. MS and SS provided pathological expertise for staging of tumors. CN conceived the study and its design; coordinated and aided with sample collection; and drafted the manuscript. All authors read and approved the final manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
References
- American_Cancer_Society. Cancer Facts & Figures. Atlanta: American Cancer Society; 2012. [Google Scholar]
- Canadian_Cancer_Society. Canadian Cancer Statistics. Toronto, ON: Canadian Cancer Society; 2012. [Google Scholar]
- Lambe M, Hsieh C, Trichopoulos D, et al. Transient increase in the risk of breast cancer after giving birth. N Engl J Med. 1994;331:5–9. doi: 10.1056/NEJM199407073310102. [DOI] [PubMed] [Google Scholar]
- Lehrke M, Lazar MA. The many faces of PPARγamma. Cell. 2005;123:993–9. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- Campbell MJ, Carlberg C, Koeffler HP. A role for the PPARγamma in cancer therapy. PPAR Res. 2008;2008:314974. doi: 10.1155/2008/314974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M, Glass CK. PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta. 2007;1771:926–35. doi: 10.1016/j.bbalip.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman BM. PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;47:507–14. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- Werner AL, Travaglini MT. A review of rosiglitazone in type 2 diabetes mellitus. Pharmacotherapy. 2001;21:1082–99. doi: 10.1592/phco.21.13.1082.34615. [DOI] [PubMed] [Google Scholar]
- Braissant O, Foufelle F, Scotto C, et al. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology. 1996;137:354–66. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- Jain S, Pulikuri S, Zhu Y, et al. Differential expression of the peroxisome proliferator-activated receptor gamma (PPARγamma) and its coactivators steroid receptor coactivator-1 and PPAR-binding protein PBP in the brown fat, urinary bladder, colon, and breast of the mouse. Am J Pathol. 1998;153:349–54. doi: 10.1016/s0002-9440(10)65577-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller E, Sarraf P, Tontonoz P, et al. Terminal differentiation of human breast cancer through PPAR gamma. Mol Cell. 1998;1:465–70. doi: 10.1016/s1097-2765(00)80047-7. [DOI] [PubMed] [Google Scholar]
- Elstner E, Muller C, Koshizuka K, et al. Ligands for peroxisome proliferator-activated receptorgamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci U S A. 1998;95:8806–11. doi: 10.1073/pnas.95.15.8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HN, Lee YR, Noh EM, et al. Induction of G1 phase arrest and apoptosis in MDA-MB-231 breast cancer cells by troglitazone, a synthetic peroxisome proliferator-activated receptor gamma (PPARγamma) ligand. Cell Biol Int. 2008;32:906–12. doi: 10.1016/j.cellbi.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Suh N, Wang Y, Williams CR, et al. A new ligand for the peroxisome proliferator-activated receptor-gamma (PPAR-gamma), GW7845, inhibits rat mammary carcinogenesis. Cancer Res. 1999;59:5671–3. [PubMed] [Google Scholar]
- Pighetti GM, Novosad W, Nicholson C, et al. Therapeutic treatment of DMBA-induced mammary tumors with PPAR ligands. Anticancer Res. 2001;21:825–9. [PubMed] [Google Scholar]
- Nicol CJ, Yoon M, Ward JM, et al. PPARγamma influences susceptibility to DMBA-induced mammary, ovarian and skin carcinogenesis. Carcinogenesis. 2004;25:1747–55. doi: 10.1093/carcin/bgh160. [DOI] [PubMed] [Google Scholar]
- Yin Y, Yuan H, Zeng X, et al. Inhibition of peroxisome proliferator-activated receptor gamma increases estrogen receptor-dependent tumor specification. Cancer Res. 2009;69:687–94. doi: 10.1158/0008-5472.CAN-08-2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelhorne-Gross G, Reid AL, Apostoli AJ, et al. Stromal adipocyte PPARγamma protects against breast tumorigenesis. Carcinogenesis. 2012;33:1412–20. doi: 10.1093/carcin/bgs173. [DOI] [PubMed] [Google Scholar]
- Howard BA, Gusterson BA. Human breast development. J Mammary Gland Biol Neoplasia. 2000;5:119–37. doi: 10.1023/a:1026487120779. [DOI] [PubMed] [Google Scholar]
- Morroni M, Giordano A, Zingaretti MC, et al. Reversible transdifferentiation of secretory epithelial cells into adipocytes in the mammary gland. Proc Natl Acad Sci U S A. 2004;101:16801–6. doi: 10.1073/pnas.0407647101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM. PPARγamma: a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem. 2001;276:37731–4. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Wall RJ, St-Onge L, et al. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 1997;25:4323–30. doi: 10.1093/nar/25.21.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komurov K, Dursun S, Erdin S, et al. NetWalker: a contextual network analysis tool for functional genomics. BMC Genomics. 2012;13:282. doi: 10.1186/1471-2164-13-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Miyoshi K, Claudio E, et al. Loss of the peroxisome proliferation-activated receptor gamma (PPARγamma ) does not affect mammary development and propensity for tumor formation but leads to reduced fertility. J Biol Chem. 2002;277:17830–5. doi: 10.1074/jbc.M200186200. [DOI] [PubMed] [Google Scholar]
- Akiyama TE, Sakai S, Lambert G, Nicol CJ, Matsusue K, Pimprale S, Lee YH, Ricote M, Glass CK, Brewer HB,Jr, Gonzalez FJ. Conditional disruption of the peroxisome proliferator-activated receptor gamma gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Mol Cell Biol. 2002;22:2607–19. doi: 10.1128/MCB.22.8.2607-2619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilova O, Haluzik M, Matsusue K, et al. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem. 2003;278:34268–76. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- Nicol CJ, Adachi M, Akiyama TE, et al. PPARγamma in endothelial cells influences high fat diet-induced hypertension. Am J Hypertens. 2005;18:549–56. doi: 10.1016/j.amjhyper.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Rosak C, Standl E, Reblin T, et al. Rosiglitazone is effective and well-tolerated in a range of therapeutic regimens during daily practice in patients with type 2 diabetes. Int J Clin Pract. 2006;60:1040–7. doi: 10.1111/j.1742-1241.2006.01092.x. [DOI] [PubMed] [Google Scholar]
- Gerstein HC, Yusuf S, Bosch J, et al. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: a randomised controlled trial. Lancet. 2006;368:1096–105. doi: 10.1016/S0140-6736(06)69420-8. [DOI] [PubMed] [Google Scholar]
- Wan Y, Saghatelian A, Chong LW, et al. Maternal PPAR gamma protects nursing neonates by suppressing the production of inflammatory milk. Genes Dev. 2007;21:1895–908. doi: 10.1101/gad.1567207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Kalluri R. The role of the microenvironment in mammary gland development and cancer. Cold Spring Harb Perspect Biol. 2010;2:a003244. doi: 10.1101/cshperspect.a003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelhorne-Gross G, Nicol CJ. The key to unlocking the chemotherapeutic potential of PPARγamma ligands: having the right combination. PPAR Res. 2012;2012:946943. doi: 10.1155/2012/946943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen BM, Lokshin AE. Targeting CCL11 in the treatment of ovarian cancer. Exp Opin Ther Tar. 2010;14:157–67. doi: 10.1517/14728220903512983. [DOI] [PubMed] [Google Scholar]
- Lv D, Zhang Y, Kim HJ, Zhang L, Ma X. CCL5 as a potential immunotherapeutic target in triple-negative breast cancer. Cellular & molecular immunology. 2013;10:303–10. doi: 10.1038/cmi.2012.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012;2012:137289. doi: 10.5402/2012/137289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M, Deng X. Nicotine inactivation of the proapoptotic function of Bax through phosphorylation. J Biol Chem. 2005;280:10781–9. doi: 10.1074/jbc.M500084200. [DOI] [PubMed] [Google Scholar]
- Patel L, Pass I, Coxon P, et al. Tumor suppressor and anti-inflammatory actions of PPARγamma agonists are mediated via upregulation of PTEN. Curr Biol. 2001;11:764–8. doi: 10.1016/s0960-9822(01)00225-1. [DOI] [PubMed] [Google Scholar]
- Moon HS, Guo DD, Lee HG, et al. Alpha-eleostearic acid suppresses proliferation of MCF-7 breast cancer cells via activation of PPARγamma and inhibition of ERK 1/2. Cancer Sci. 2010;101:396–402. doi: 10.1111/j.1349-7006.2009.01389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Robinson GW, Lesche R, et al. Conditional loss of PTEN leads to precocious development and neoplasia in the mammary gland. Development. 2002;129:4159–70. doi: 10.1242/dev.129.17.4159. [DOI] [PubMed] [Google Scholar]
- Schorr K, Li M, Bar-Peled U, et al. Gain of Bcl-2 is more potent than bax loss in regulating mammary epithelial cell survival in vivo. Cancer Res. 1999;59:2541–5. [PubMed] [Google Scholar]
- Shibata MA, Liu ML, Knudson MC, et al. Haploid loss of bax leads to accelerated mammary tumor development in C3(1)/SV40-TAg transgenic mice: reduction in protective apoptotic response at the preneoplastic stage. EMBO J. 1999;18:2692–701. doi: 10.1093/emboj/18.10.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky DC, Hartmann LC. Mammary involution and breast cancer risk: transgenic models and clinical studies. J Mammary Gland Biol Neoplasia. 2009;14:181–91. doi: 10.1007/s10911-009-9123-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu N, Fulton AM. Selective cyclooxygenase (COX)-1 or COX-2 inhibitors control metastatic disease in a murine model of breast cancer. Cancer Res. 2002;62:2343–6. [PubMed] [Google Scholar]
- Tian M, Neil JR, Schiemann WP. Transforming growth factor-beta and the hallmarks of cancer. Cell Signal. 2011;23:951–62. doi: 10.1016/j.cellsig.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MD, Chen WY, Schnitt SJ, et al. COX-2 expression predicts worse breast cancer prognosis and does not modify the association with aspirin. Breast Cancer Res Treat. 2011;130:657–62. doi: 10.1007/s10549-011-1651-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao JS, Shan BE. Immune enhancement and anti-tumour activity of IL-23. Cancer Immunol Immunother CII. 2006;55:1426–31. doi: 10.1007/s00262-006-0171-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Fu M, D’Amico M, et al. Inhibition of cellular proliferation through IkappaB kinase-independent and peroxisome proliferator-activated receptor gamma-dependent repression of cyclin D1. Mol Cell Biol. 2001;21:3057–70. doi: 10.1128/MCB.21.9.3057-3070.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.