

Summary
Aim
Dysregulation of the activity of the disintegrin/metalloproteinase ADAM10 could contribute to the development of atherosclerosis. Although a number of genetic studies have focused on the association of ADAM10 gene polymorphisms with susceptibility to diseases, no genetic association studies of ADAM10 gene variability with atherosclerotic cerebral infarction (ACI) have been conducted. The aim of this study was to analyze the potential association between ADAM10 promoter polymorphisms and ACI.
Methods
The associations between rs653765 and rs514049 polymorphisms of the ADAM10 promoter and the possible risk of ACI were assessed among 347 patients with ACI and 299 matched healthy individuals in a case–control study.
Results
Overall, there was a significant difference in the genotypes frequencies of rs653765 (P = 0.04) between the ACI and control subjects. In addition, the rs653765 mutated allele of ADAM10 was significantly associated with increased ADAM10 expression in patients with ACI (P = 0.032). In contrast, the allele frequency of rs514049 was not statistically associated with ACI, and the rs514049 variant A > C did not affect the expression of ADAM10 either.
Conclusion
Our findings indicate a positive association between the rs653765 polymorphism of ADAM10 and ACI, as well as a negative result for rs514049. In addition, a significant increase in ADAM10 expression was observed in patients with ACI carrying the rs653765 C > T mutation. This new knowledge about ADAM10 might be clinically important and confirm a role for ADAM10 in the pathophysiology of ACI, with potentially important therapeutic implications.
Keywords: ADAM10, Atherosclerotic cerebral infarction, Polymorphism
Introduction
The ADAM (a disintegrin and metalloproteinase) family includes 34 transmembrane proteins, each with a disintegrin domain and a characteristic extracellular metalloproteinase domain 1, 2. By cleaving the extracellular domain of various cell surface molecules, for example, cytokines, adhesion molecules, and growth factor receptors, ADAMs play a role in various diseases, such as inflammation and cancer 2, 3, 4, 5. Recently, the role of ADAMs in the progression of atherosclerosis has come to light 6.
ADAM10 is a member of the ADAM family that is expressed in all vascular cell types, including vascular smooth cells, leukocytes, and endothelial cells 2. Various ADAM10 substrates have been considered to be relevant to the developmental, inflammatory, and regenerative processes of the vasculature, including TNF‐α, IL‐6R, L‐selectin, and CX3CL1 7, 8, which have been theorized to play important roles in atherosclerosis 9. In addition, ADAM10 expression is significantly increased during plaque progression from the early and advanced stages to the rupturing of atherosclerotic plaques 10. In addition, ADAM10‐deficient mice have exhibited multiple defects in both cardiovascular development and vasculogenesis, which were greatly attributable to the dysfunction of ADAM10 ectodomain shedding 1, 11. In summary, these lines of evidence have led us to formulate the hypothesis that ADAM10 could be of pathogenetic importance in atherosclerotic cerebral infarction (ACI).
The human ADAM10 gene is located on chromosome 15 at position 15q21.3‐q23 and contains 16 exons interrupted by 15 introns 12. Several single‐nucleotide polymorphisms located in the promoter of the ADAM10 gene have been shown to modify ADAM10 expression; for example, the ADAM10 rs514049–rs653765 C‐A promoter polymorphism has been reported to be associated with reduced ADAM10 expression and soluble amyloid precursor protein alpha (sAPPα) levels in cerebrospinal fluid 13. In addition, the associations between ADAM10 gene polymorphisms and possible disease susceptibility have been investigated in various diseases. Jian et al. 14 showed that the ADAM10 rs383902 polymorphism was associated with conduct disorder. Kavanagh et al. 15 found that ADAM10 was not strongly associated with diabetic nephropathy in white individuals with type 1 diabetes. Kim et al. 16 initially identified a genetic association in European populations between the ADAM10 rs2305421 polymorphism and Alzheimer's disease (AD). Song et al. 17 replicated the ADAM10 rs2305421 polymorphism in a Han Chinese population with AD, and no significant differences were found in the distributions of alleles or genotypes between the AD and control groups, although significant differences were observed in a subgroup stratified according to ApoE ε4 status. However, the association between human ADAM10 polymorphisms and ACI has not yet been determined. Therefore, we undertook a case–control study to ascertain whether ADAM10 promoter variations (rs653765 and rs514049) are associated with ACI susceptibility and whether these polymorphisms are associated with ADAM10 expression.
Methods
Study Population
Our study consecutively recruited 347 patients with atherosclerotic cerebral infarction (119 women and 228 men; mean age = 70.0 ± 10.1 years old) and 299 healthy controls (100 women and 199 men; mean age = 69.9 ± 8.92 years old) from the Department of Neurology of the Affiliated Hospital of Guangdong Medical College. Patient diagnoses were verified with either computed tomography (CT) or magnetic resonance imaging (MRI), and all of the patients with ACI were classified into subtypes according to the Trial of Org 10172 in Acute Stroke Treatment (TOAST) classification 18. Patients with histories of ischemic cerebrovascular diseases, cardiogenic cerebral infarctions, cerebral hemorrhage, coronary artery diseases, autoimmune diseases, systemic inflammatory diseases, blood diseases, and malignant tumors were excluded from the study. The control subjects were recruited from the Health Examination Center of the Affiliated Hospital of Guangdong Medical College and were comparable in age, sex, and race to the ACI subjects. Written informed consent was obtained from all of the enrolled participants, and this study was approved by the Ethics Committee of the Affiliated Hospital of Guangdong Medical College.
Ultrasound Imaging
Ultrasound measurements of left and right carotid arteries were performed at the Affiliated Hospital of Guangdong Medical College using a portable ultrasound machine (SonoSite MTurbo, SonoSite, Inc., USA) with a 13‐MHz linear‐array transducer. Optimized images of left and right carotid artery intima‐media thickness (CIMT) were selected and frozen at the end of diastole. The IMT of common carotid artery (CCA) was measured with Calc automated ultrasonic software at both the near and far walls of the left and right sides in an area free of atherosclerotic plaque. The value of the IMT of the CCA on the far wall of each side was used for analysis.
DNA Isolation and Genotyping
Genomic DNA was extracted from peripheral blood using the EZ‐10 Spin Column Whole Blood Genomic DNA Isolation Kit (Sangon Biotech®, Shanghai, China), according to manufacturer's instruction.
The polymorphisms of genes were analyzed by SNaPshot Multiplex Kit (Applied Biosystems Co., Ltd., Foster City, CA, USA). The primers used in the SNaPshot were as follows: rs653765F: AGCACCTCCCTCTCGCTCCAC, rs653765R:TGAGGCGGAGGTCTGAGTTTCGA; rs514049F:AGCACCTCCCTCTCGCTCCAC, rs514049R:TTTTTTTTTTTTTTTTTTTAAGAAGAAAAAAAACCTCTGTTACTTGTGAC. SNaPshot reactions were carried out in a 10 μL final volume containing 5 μL SNaPshot Multiplex Kit (ABI), 1 μL primer mix, 2 μL water, and 2 μL templates consisting of the multiplex PCR products from the different genes. SNaPshot response procedures: (1) initial denaturation at 96°C for 1 min, (2) denaturation at 96°C for 10 seconds, (3) annealing at 52°C for 5 seconds, (4) extension at 60°C for 30 seconds, and (5) for a total of 28 cycles. Amplified samples were stored at 4°C. Extension products were purified by 1‐h incubation with 1U of shrimp alkaline phosphatase (Takara:Otsu, shiga, Japan) at 37°C and 75°C for 15 min to inactivate the enzyme. The purified products (0.5 μL) were mixed with 9 μL of Hi‐Di and 0.5 μL Liz120 SIZE STANDARD. Samples were incubated at 95°C for 5 min and then loaded on an ABI 3130XL DNA sequence detector for capillary electrophoresis. The experimental results were analyzed with GeneMapper 4.0 (Applied Biosystems Co., Ltd.).
Mononuclear Cells Isolation
Peripheral blood mononuclear cells (PBMCs) were isolated using density gradient centrifugation method with Lymphoprep™ (Axis‐Shield PoCAS, Oslo, Norway). In brief, blood samples were mixed with equal volume of 0.9% NaCl. The diluted blood was then slowly added to tubes containing a Ficoll premium solution to make the blood layered upon the Ficoll. Samples were centrifuged at 800 × g for 30 min at room temperature. After centrifugation, the mononuclear cells form a distinct band at the medium interface. The cells were then shifted to other tubes using Pasteur pipette without removing the upper layer, and washed with 0.9% NaCl. Then, samples were centrifuged again at 250 × g for 10 min. The mononuclear cells were harvested and stored at −80°C.
RNA Extraction
Total cellular RNA was extracted from PBMCs using the RNAprep Pure Blood Kit (TianGen Biotech, Beijing, China), according to manufacturer's instructions. Briefly, PBMCs were pelleted and lysed using Trizol. The lysate was centrifuged at 13,400 × g for 2 min. The aqueous phase was transferred to a fresh microcentrifuge tube. Ethanol was added to the sample, and then, the sample was transferred to the silica surface of a spin column. All contaminating genomic DNA was eliminated by applying RNase‐free DNase I digestion to the silica membrane. The total RNA of all samples was further purified by performing wash steps and then eluted from the membrane into water. Then, the integrity of the RNA samples was verified by agarose gel electrophoresis and stored at −80°C.
Real‐Time PCR
Total RNA was converted to cDNA by First Strand cDNA Synthesis Kit (Thermo) according to the manufacturer's instructions. Resulting cDNAs were employed in quantitative real‐time PCR using the SYBR green method. Primers were designed with Primer Premier 5.0 according to the corresponding sequences in GenBank. The primers used in the assay were as follows: ADAM10 sense primer: CTGGCCAACCTATTTGTGGAA, ADAM10 antisense primer: GACCTTGACTTGGACTGCACTG; GAPDH sense primer: GAAGGGCTCATGACCACAGTCCAT, GAPDH antisense primer: TCATTGTCGTACCAGGAAATGAGCTT. For each cDNA sample, triplicates were analyzed, and the relative transcript level of each gene was obtained by the 2ΔDDCt method and normalized with respect to the housekeeping gene glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH).
PCR amplification was performed in a 20 μL final volume containing 10 μL 2 × SYBR Green PCR master mix (TaKaRa), 0.4 μL each of specific forward and reverse primers, 7.2 μL DNase‐free water, and 2 μL cDNA as template. Real‐time PCR was performed using a LightCycler®480 sequence detector system (Roche Applied Sciences, Indianapolis, IN, USA). The amplification program was as follows: 95°C for 30 seconds, 40 cycles of 95°C for 5 seconds, and 62°C for 20 seconds. The expression levels for each sample were calculated based on three technical replicates. The amplification products were validated by melting curve analysis.
Statistical Analyses
Statistical analyses were performed with SPSS software, version 19.0 (IBM, Armonk, NY, USA). These data are presented as percentage frequencies or means ± SDs. Allele frequencies were calculated from the genotypes of all of the subjects. The allele and genotype frequencies of ADAM10 between the patients and control subjects were compared using Fisher's exact test or the chi‐squared test. Hardy–Weinberg equilibrium (HWE) was assessed using HWE software. Risk factors were screened for using Student's t‐test or chi‐squared test. Haplotype analyses were conducted using Haploview, version 3.2.0. The relationships between different genotypes of ADAM10 and ACI were evaluated by an ANOVA. A P‐value < 0.05 was considered statistically significant. The associations of gene polymorphisms with CIMT were analyzed with a multiple linear regression model (SAS, version 6.12; SAS Institute Inc., Cary, NC, USA).
We performed multiple hypotheses testing using the Benjamini & Hochberg (BH) multiple testing correction to control for false discovery rate in the logistic regression analysis. Evaluating the false discovery rate is a way to address the problems associated with multiple comparisons, and false discovery rate provides a measure of the expected proportion of false positives among data. Analyses were performed using GraphPad Prism 4.0 (GraphPad Software, Inc., San Diego, CA, USA), StatsDirect Statistical Software Version 2.4.4 (StatsDirect Ltd., Altrincham, UK), and Medcalc (Version 7.4 for Windows; Mariakerke, Belgium).
Results
Baseline Characteristics
The baseline characteristics of all of the participants in the study are summarized in Table 1. Of the 646 participants, 347 were patients with ACI and 299 were healthy controls. There were no statistically significant differences between the patients and controls in terms of age or sex. The mean age was 70.0 years old (±10 years) for the ACI subjects and 69.9 years old (±8.9 years) for the control subjects. The sex (male‐to‐female) ratio was 1:1.09 in the case group and 1.05:1 in the control group. However, compared with the controls, the patients with ACI had higher total cholesterol (P < 0.0001), higher blood glucose (P = 0.0053), and higher high‐density lipoprotein (HDL; P = 0.0053) and were more likely to smoke (P = 0.0053) and have hypertension (P < 0.0001) than the control subjects. However, the differences in diabetes mellitus, triglycerides, and low‐density lipoprotein (LDL) between the two groups were not statistically significant (P > 0.05).
Table 1.
The characteristics of subjects between ACI and control group
| Variables | ACI (n = 347) | Control (n = 299) | P value | P value* |
|---|---|---|---|---|
| Mean age (years) | 70.0 ± 10.1 | 69.9 ± 8.92 | 0.745 | 0.953 |
| Male/female | 228/119 | 199/100 | 0.834 | 0.953 |
| Smokers, n (%) | 172 (49.6) | 93 (31.1) | 0.002 | 0.0053 |
| Hypertension, n (%) | 189 (54.5) | 74 (24.8) | <0.001 | <0.001 |
| Diabetes, n (%) | 70 (20.2) | 34 (11.4) | 0.214 | 0.428 |
| Blood glucose (mmol/L) | 5.93 ± 2.52 | 5.33 ± 1.67 | 0.002 | 0.0053 |
| Total cholesterol (mg/dL) | 5.23 ± 1.17 | 4.74 ± 1.96 | <0.001 | <0.001 |
| Triglycerides (mmol/L) | 1.59 ± 1.01 | 1.58 ± 1.27 | 0.996 | 0.996 |
| HDL (mmol/L) | 1.25 ± 0.51 | 1.57 ± 0.62 | 0.001 | 0.0053 |
| LDL (mmol/L) | 2.86 ± 0.98 | 2.87 ± 1.22 | 0.712 | 0.953 |
ACI, atherosclerotic cerebral infarction; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein.
Continuous data are expressed as the means ± SD.
Adjusted for age, sex, hypertension, diabetes mellitus, hyperlipidemia, and smoking.
*False discovery rate‐adjusted P value for multiple hypotheses testing using the Benjamini–Hochberg method.
Polymorphisms of ADAM10 Gene and the Risk of ACI
The genotype and allele frequencies of ADAM10 polymorphisms in the study are shown in Table 2. The deviation from Hardy–Weinberg equilibrium for the polymorphisms examined was not found in the distributions of genotypes in the patients and controls (data not shown). The comparison of genotype distributions between the ACI subjects and control subjects with the chi‐squared test revealed that there was a statistical association (P = 0.04) between the rs653765 polymorphism of ADAM10 and the risk of ACI. In a dominant model (CC + CT vs. TT), no significant difference was detected between the ACI group and controls (P = 0.89). However, in a recessive model (CC vs. CT + TT), a significant difference was observed in the ACI group compared with the controls (P = 0.04). However, the rs653765 C allele did not show a significant difference in the ACI group compared with the controls (P = 0.053) after Benjamini & Hochberg (BH) multiple testing correction. In contrast, there was no significant association between the rs514049 polymorphism of ADAM10 and the risk of ACI (P = 0.56). Accordingly, the frequencies of rs514049 of ADAM10 were not significantly different between the two groups, either in the dominant (P = 0.78) or recessive model (P = 0.38). There was also no statistically significant difference within the C allele of the ADAM10 rs514049 polymorphism in the ACI group compared with the controls (P = 0.38).
Table 2.
Frequencies of ADAM10 genotypes and alleles in patients with ACI and controls
| Genotypes | Patients with ACI n = 347 (%) | Controls n = 299 (%) | AOR (95% CI) | P value | P value |
|---|---|---|---|---|---|
| rs653765 | |||||
| CC | 230 (66.3) | 228 (76.3) | 0.02a | 0.04a | |
| CT | 106 (30.6) | 61 (20.4) | |||
| TT | 11 (3.17) | 10 (3.35) | |||
| Dominant model CC/CT versus TT | 336 (96.8) | 289 (96.7) | 1.06 (0.44–2.52) | 0.89 | 0.89 |
| Recessive model CC versus CT/TT | 117 (33.7) | 71 (23.8) | 0.61 (0.43–0.87) | 0.01a | 0.04a |
| C allele | 566 (81.6) | 517 (86.5) | 1.000 (reference) | ||
| T allele | 128 (18.5) | 81 (13.6) | 1.44 (1.07–1.95) | 0.04 | 0.053 |
| rs514049 | |||||
| AA | 298 (85.9) | 267 (89.3) | 0.42 | 0.56 | |
| AC | 46 (13.3) | 30 (10.0) | |||
| CC | 3 (0.86) | 2 (0.67) | |||
| Dominant model AA/AC versus CC | 343 (99.1) | 297 (99.3) | 0.77 (0.13–4.64) | 0.78 | 0.78 |
| Recessive model AA versus AC/CC | 49 (14.2) | 32 (10.7) | 0.73 (0.45–1.17) | 0.19 | 0.38 |
| A allele | 642 (92.5) | 564 (94.3) | 1.000 (reference) | ||
| C allele | 52 (7.49) | 34 (5.7) | 1.34 (0.86‐962.10) | 0.19 | 0.38 |
ACI, atherosclerotic cerebral infarction.
Adjusted for age, sex, hypertension, diabetes mellitus, hyperlipidemia, and smoking.
a P value of difference in genotypes between case group and control group.
*False discovery rate‐adjusted P value for multiple hypotheses testing using the Benjamini–Hochberg method.
We further performed a haplotype‐based association analysis of rs514049 and rs653765 using Haploview 19. However, no significant associations were observed between these haplotypes and ACI (Table 3).
Table 3.
The frequencies of haplotypes of ADAM10 gene in patients and controls
| rs514049–rs653765 | Case (n %) | Control (n %) | OR (95% CI) | P value | P value * |
|---|---|---|---|---|---|
| CA | 566 (81.6) | 495 (83.1) | 1 | – | |
| TA | 76 (11.0) | 67 (11.2) | 1.008 (0.710–1.43) | 0.964 | 0.964 |
| TC | 52 (7.49) | 34 (5.71) | 0.748 (0.477–1.17) | 0.203 | 0.405 |
Adjusted for age, sex, hypertension, diabetes mellitus, hyperlipidemia, and smoking.
*False discovery rate‐adjusted P value for multiple hypotheses testing using the Benjamini–Hochberg method.
The Association of ADAM10 Gene Polymorphisms with Demographic Characteristics
The associations of ADAM10 gene polymorphisms with demographic characteristics are shown in Table 4. In the analysis stratified by sex and age, increased risk associated with the variant genotypes (rs653765 CT and TT) and alleles was found in male patients (P = 0.008 and P = 0.004) and individuals greater than 70 years old (P = 0.03 and P = 0.031, respectively). However, in female subjects and those less than 70 years old, the associations between ADAM10 gene polymorphisms and ACI risk were not statistically significant. When the sample was stratified according to smoking status, the increased risk associated with the variant genotypes (rs653765 CT and TT) was found in the smokers (P = 0.016) compared with nonsmokers (P = 0.092) between the ACI cases and controls. In the analysis stratified according to hypertension and diabetes, the increased risk associated with the variant genotypes (rs653765 CT and TT) was observed in nonhypertensive (P = 0.026) and nondiabetic patients (P = 0.046; Table 4).
Table 4.
The relationship between baseline characteristics and ADAM10 rs653765 and rs514049 genotypes and alleles among subjects in case and control group
| Characteristics | rs653765 Case group | rs653765 Control group | P value | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype n (%) | Allele n (%) | Genotype n (%) | Allele n (%) | |||||||||||
| CC | CT | TT | C | T | CC | CT | TT | C | T | PG | PG * | PA | PA * | |
| Age | ||||||||||||||
| <70 years | 93 (62.4) | 50 (33.6) | 6 (4.02) | 236 (80.2) | 62 (19.8) | 105 (71.0) | 38 (25.7) | 5 (3.37) | 248 (83.8) | 48 (16.2) | 0.29 | 0.29 | 0.15 | 0.29 |
| ≥70 years | 137 (69.2) | 56 (28.3) | 5 (2.53) | 330 (83.3) | 66 (16.7) | 123 (81.5) | 23 (15.2) | 5 (3.31) | 269 (89.1) | 33 (10.9) | 0.015 | 0.03 | 0.031 | 0.031 |
| Gender | ||||||||||||||
| Male | 145 (63.6) | 73 (32.0) | 10 (4.39) | 363 (79.6) | 93 (20.4) | 154 (77.4) | 40 (20.1) | 5 (2.51) | 348 (87.4) | 50 (12.6) | 0.008 | 0.008 | 0.002 | 0.004 |
| Female | 86 (71.7) | 33 (27.5) | 1 (0.830) | 205 (85.4) | 35 (14.6) | 74 (74.0) | 21 (21.0) | 5 (5.00) | 169 (84.5) | 31 (15.5) | 0.096 | 0.192 | 0.789 | 0.789 |
| Hypertension | ||||||||||||||
| Yes | 130 (68.8) | 51 (27.0) | 8 (4.23) | 311 (82.3) | 67 (17.7) | 58 (78.4) | 13 (17.6) | 3 (4.05) | 129 (87.2) | 19 (12.8) | 0.253 | 0.253 | 0.173 | 0.346 |
| No | 100 (63.3) | 55 (34.8) | 3 (1.90) | 255 (80.7) | 61 (19.3) | 170 (75.6) | 48 (21.3) | 7 (3.11) | 388 (86.2) | 62 (13.8) | 0.013 | 0.026 | 0.04 | 0.04 |
| Smoking | ||||||||||||||
| Yes | 112 (65.1) | 52 (30.2) | 8 (4.65) | 276 (80.2) | 68 (19.8) | 75 (80.7) | 17 (18.3) | 1 (1.08) | 167 (89.8) | 19 (10.2) | 0.016 | 0.016 | 0.005 | 0.01 |
| No | 118 (67.4) | 54 (30.9) | 3 (1.71) | 290 (82.9) | 60 (17.1) | 153 (74.3) | 44 (21.4) | 9 (4.37) | 350 (85.0) | 62 (15.1) | 0.046 | 0.092 | 0.432 | 0.432 |
| Diabetes | ||||||||||||||
| Yes | 45 (64.3) | 20 (28.6) | 5 (7.14) | 110 (78.6) | 30 (21.4) | 26 (76.5) | 6 (17.7) | 2 (5.88) | 58 (85.3) | 10 (14.7) | 0.426 | 0.426 | 0.248 | 0.496 |
| No | 185 (66.8) | 86 (31.1) | 6 (2.17) | 456 (82.3) | 98 (17.7) | 202 (76.2) | 55 (20.8) | 8 (3.02) | 459 (86.6) | 71 (13.4) | 0.023 | 0.046 | 0.051 | 0.051 |
| Characteristics | rs514049 Case group | rs514049 Control group | P value | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype n (%) | Allele n (%) | Genotype n (%) | Allele n (%) | |||||||||||
| AA | AC | CC | A | C | AA | AC | CC | A | C | PG | PG * | PA | PA * | |
| Age | ||||||||||||||
| <70 years | 123 (82.6) | 25 (16.8) | 1 (0.670) | 271 (90.9) | 27 (9.06) | 128 (86.5) | 19 (12.8) | 1 (0.67) | 275 (92.9) | 21 (7.09) | 0.63 | 0.63 | 0.38 | 0.76 |
| ≥70 years | 175 (88.4) | 21 (10.6) | 2 (1.01) | 371 (93.7) | 25 (6.31) | 139 (92.1) | 11 (7.28) | 1 (0.67) | 289 (95.7) | 13 (4.30) | 0.53 | 0.53 | 0.25 | 0.5 |
| Gender | ||||||||||||||
| Male | 191 (83.8) | 34 (14.9) | 3 (1.32) | 416 (91.2) | 40 (8.77) | 179 (90.0) | 20 (10.05) | 0 (0) | 378 (95.0) | 20 (5.03) | 0.079 | 0.158 | 0.163 | 0.163 |
| Female | 107 (89.9) | 12 (10.1) | 0 (0) | 226 (95.0) | 12 (5.04) | 88 (88.0) | 10 (10.00) | 2 (2.00) | 186 (93.0) | 14 (7.00) | 0.206 | 0.412 | 0.388 | 0.388 |
| Hypertension | ||||||||||||||
| Yes | 163 (86.2) | 24 (12.7) | 2 (1.06) | 350 (92.6) | 28 (7.41) | 68 (91.9) | 5 (6.76) | 1 (1.35) | 141 (95.3) | 7 (4.73) | 0.348 | 0.348 | 0.268 | 0.536 |
| No | 135 (85.4) | 22 (13.9) | 1 (0.630) | 292 (92.41) | 24 (7.59) | 199 (88.4) | 25 (11.1) | 1 (0.44) | 423 (94.0) | 27 (6.00) | 0.687 | 0.687 | 0.383 | 0.766 |
| Smoking | ||||||||||||||
| Yes | 143 (83.1) | 26 (15.1) | 3 (1.74) | 312 (90.7) | 32 (9.30) | 88 (94.6) | 5 (5.38) | 0 (0) | 181 (97.31) | 5 (2.69) | 0.011 | 0.011 | 0.004 | 0.008 |
| No | 155 (88.6) | 20 (11.4) | 0 (0) | 330 (94.3) | 20 (5.71) | 179 (86.9) | 25 (12.1) | 2 (0.97) | 383 (92.96) | 29 (7.04) | 0.282 | 0.564 | 0.458 | 0.458 |
| Diabetes | ||||||||||||||
| Yes | 60 (85.7) | 10 (14.3) | 0 (0) | 130 (92.9) | 10 (7.14) | 30 (88.2) | 3 (8.82) | 1 (2.94) | 63 (92.65) | 5 (7.35) | 0.255 | 0.51 | 0.956 | 0.956 |
| No | 238 (85.9) | 36 (13.0) | 3 (1.08) | 512 (92.4) | 42 (7.58) | 237 (89.4) | 27 (10.2) | 1 (0.38) | 501 (94.53) | 29 (5.47) | 0.395 | 0.395 | 0.161 | 0.322 |
PG: P value of difference in genotypes between case group and control group.
PA: P value of difference in alleles between case group and control group.
Adjusted for age, sex, hypertension, diabetes mellitus, hyperlipidemia, and smoking.
*False discovery rate‐adjusted P value for multiple hypotheses testing using the Benjamini–Hochberg method.
Interestingly, when the sample was stratified according to smoking status, the increased risk associated with the variant genotypes (rs514049 AC and CC) and allele frequencies was more significant in patients with ACI compared with the controls (P = 0.011 and P = 0.008, respectively). However, when the sample was stratified by age, sex, hypertension, and diabetes, no significant differences in the genotype or allele frequencies between the ACI cases and the controls were detected in the ADAM10 rs514049 polymorphism carriers (Table 4).
We also analyzed the associations of the ADAM10 polymorphisms with clinical parameters, including blood glucose, total cholesterol, triglycerides, HDL, and LDL. The results are shown in Table 5. CC carriers of rs653765 were associated with lower total cholesterol (P < 0.0001), HDL (P < 0.0001), and higher blood glucose (P = 0.009) among the patients with ACI compared with the controls. AA carriers of rs514049 were nominally associated with lower total cholesterol (P < 0.0001), HDL (P < 0.0001), and higher blood glucose (P = 0.009) among the patients with ACI compared with the control group. Subjects carrying rs653765 CT and the rs514049 AC were coincidently associated with lower HDL levels (P < 0.0001 and P = 0.006, respectively) among patients with ACI compared with the controls. There were no statistically significant associations between ADAM10 polymorphisms and triglycerides or LDL in patients with ACI compared with the controls. Studies with greater sample sizes are needed to confirm these associations.
Table 5.
Quantitative traits stratified according to ADAM10 genotypes in case and control group
| Characteristics | Case group genotype | Control group genotype | P value | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs653765 | CC | CT | TT | CC | CT | TT | P CC | P CC * | P CT | P CT * | P TT | P TT * |
| Blood glucose (mmol/L) | 5.84 ± 2.39 | 5.79 ± 2.41 | 8.33 ± 4.07 | 5.30 ± 1.34 | 5.71 ± 2.19 | 5.75 ± 1.76 | 0.003 | 0.009 | 0.083 | 0.083 | 0.08 | 0.12 |
| Total cholesterol (mg/dL) | 5.12 ± 0.97 | 5.11 ± 1.62 | 5.03 ± 0.79 | 4.48 ± 1.07 | 4.82 ± 1.06 | 4.56 ± 1.57 | <0.001 | <0.001 | 0.098 | 0.147 | 0.408 | 0.408 |
| Triglycerides (mmol/L) | 1.60 ± 0.95 | 1.51 ± 1.01 | 1.56 ± 0.67 | 1.58 ± 1.51 | 1.56 ± 1.04 | 1.52 ± 1.01 | 0.853 | 0.919 | 0.751 | 0.919 | 0.919 | 0.919 |
| HDL (mmol/L) | 1.27 ± 0.54 | 1.28 ± 0.32 | 1.48 ± 0.38 | 1.60 ± 0.40 | 1.52 ± 0.37 | 1.60 ± 0.46 | <0.001 | <0.001 | <0.001 | <0.001 | 0.054 | 0.054 |
| LDL (mmol/L) | 2.79 ± 0.88 | 2.8 ± 0.89 | 2.65 ± 1.20 | 2.89 ± 0.87 | 2.87 ± 1.10 | 2.84 ± 0.70 | 0.193 | 0.579 | 0.63 | 0.945 | 0.665 | 0.665 |
| rs514049 | AA | CA | CC | AA | CA | CC | P AA | P AA * | P CA | P CA * | P CC | P CC * |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Blood glucose (mmol/L) | 5.90 ± 2.48 | 6.12 ± 2.66 | 4.95 ± 0.14 | 5.37 ± 1.52 | 5.62 ± 2.01 | 6.61 ± 1.46 | 0.003 | 0.009 | 0.381 | 0.381 | 0.123 | 0.185 |
| Total cholesterol (mg/dL) | 5.16 ± 1.02 | 5.07 ± 1.93 | 5.43 ± 0.34 | 4.79 ± 1.07 | 4.77 ± 1.18 | 4.00 ± 1.22 | <0.001 | <0.001 | 0.242 | 0.242 | 0.219 | 0.329 |
| Triglycerides (mmol/L) | 1.56 ± 0.93 | 1.64 ± 1.17 | 1.29 ± −0.80 | 1.56 ± 1.43 | 1.73 ± 1.28 | 1.66 ± 0.71 | 0.944 | 0.944 | 0.731 | 0.944 | 0.635 | 0.944 |
| HDL (mmol/L) | 1.28 ± 0.50 | 1.25 ± 0.32 | 1.27 ± 0.32 | 1.59 ± 0.41 | 1.50 ± 0.35 | 1.86 ± 0.48 | <0.001 | <0.001 | 0.004 | 0.006 | 0.185 | 0.185 |
| LDL (mmol/L) | 2.80 ± 0.88 | 2.75 ± 0.97 | 2.16 ± 0.87 | 2.87 ± 0.95 | 2.87 ± 0.90 | 2.82 ± 1.14 | 0.206 | 0.618 | 0.6 | 0.6 | 0.514 | 0.771 |
HDL, high‐density lipoprotein; LDL, low‐density lipoprotein.
Continuous data are expressed as the means ± SD.
PCC: P value of CC genotype between case group and control group; PCT: P value of CT genotype between case group and control group; PTT: P value of TT genotype between case group and control group; PAA: P value of AA genotype between case group and control group; PCA: P value of CA genotype between case group and control group.
Adjusted for age, sex, hypertension, diabetes mellitus, hyperlipidemia, and smoking.
*False discovery rate‐adjusted P value for multiple hypotheses testing using the Benjamini–Hochberg method.
Association of ADAM10 Gene Polymorphisms with ACI Subtypes
To explore whether the effects of ADAM10 polymorphisms are confined to a specific subtype or related to generalized risk, we further separated the ACI group into two subgroups according to the TOAST classification 18. As shown in Table 6, the rs653765 and rs514049 polymorphisms were not associated with ACI when stratified according to the two major ACI subtypes (large‐artery atherosclerosis and small‐artery atheroslcerosis) in the case cohorts.
Table 6.
The realationship between ADAM10 genotypes and ACI stratified by TOAST classification in patients with ACI
| Genotypes | TOAST classification | P value | |
|---|---|---|---|
| LA | SA | ||
| rs653765 | |||
| CC | 162 | 68 | 0.669 |
| CT | 73 | 33 | |
| TT | 9 | 2 | |
| CT/TT | 82 | 35 | 0.946 |
| rs514049 | |||
| AA | 217 | 81 | 0.568 |
| AC | 34 | 12 | |
| CC | 3 | 0 | |
| AC/CC | 37 | 12 | 0.693 |
ACI, atherosclerotic cerebral infarction; LA, Large‐artery atheroslcerosis; SA, Small‐artery atheroslcerosis; TOAST, Trial of Org 10172 in Acute Stroke Treatment.
Effect of ADAM10 Gene Polymorphisms on ADAM10 Expression
A comparative determination was performed on the ADAM10 mRNA expression levels in PBMCs of 50 patients with ACI and 50 healthy controls. According to the data obtained, the mean value of the ADAM10 mRNA expression was 1.3‐fold higher in the patients with ACI than the controls (Figure 1), and this difference was significant (P = 0.025).
Figure 1.
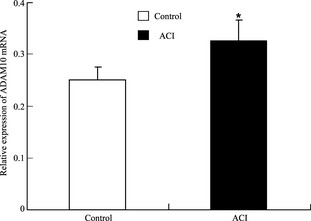
Mean values ± SD of relative ADAM10 mRNA in peripheral blood mononuclear cells (PBMCs) from patients with atherosclerotic cerebral infarction (ACI, n = 50) and healthy subjects (controls, n = 50). The blank box and the black box represent the relative expressions of ADAM10 in the controls and patients with ACI, respectively, and the median is indicated by a bar across the box. P = 0.025 when comparing relative ADAM10 mRNA levels between patients with ACI and controls.
In addition, we also assessed whether there was an association between the mean values of the ADAM10 mRNA levels in patients with ACI with the ADAM10 genotype. The results obtained are presented in Figure 2A. A significant increase in ADAM10 mRNA expression was observed in patients with ACI who carried the mutated rs653765 CT or TT genotype of ADAM10 (P = 0.032), while in healthy subjects who carried the mutated allele, a slight but not significant increase was detected (P = 0.062). For the rs514049 polymorphism, no statistically significant difference in ADAM10 mRNA expression was found between the population carrying the mutated allele and the population carrying the major AA in either the patients with ACI or control subjects (Figure 2A). When the ADAM10 mRNA expression was further stratified by age, an increased expression of ADAM10 was detected in patients with ACI over 70 years old (P = 0.041; Figure 2B). However, in the normal control group, the expression of ADAM10 was indistinctive in individuals aged ≥70 or < 70 years old (Figure 2B).
Figure 2.
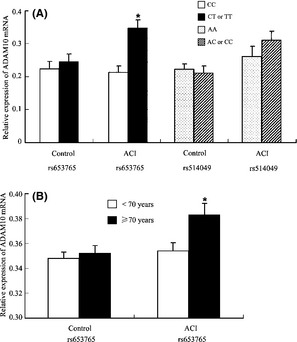
Mean values ± SD of ADAM10 mRNA in PBMCs from the healthy subjects (n = 50) and patients with atherosclerotic cerebral infarction (ACI; n = 50) stratified according to the presence of the mutated allele (A) (P = 0.032) or age (B) (P = 0.041).
Effect of ADAM10 Gene Polymorphisms on Carotid Atherosclerosis
Associations of the ADAM10 gene polymorphisms with CIMT and ACI were explored, and the results are shown in Figure 3. The mean of CIMT in the patients with ACI with the variant genotypes (rs653765 CT and TT) was not significantly different compared with the mean CIMT in those subjects with the CC genotype (P = 0.259). However, unexpectedly, the mean CIMT in the patients with ACI carrying the rs514049 AA genotype was significantly greater than in those carrying a mutated C allele (P = 0.046).
Figure 3.
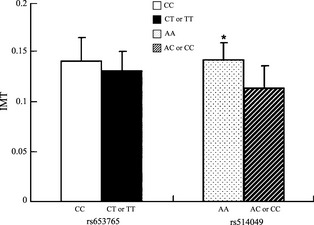
Intima‐media thickness mean values ± SD of patients with atherosclerotic cerebral infarction (ACI; n = 264) stratified according to the presence of the mutated allele. P = 0.046.
Discussion
There is increasing evidence of the potential impact of members of the ADAM family on the pathogenic mechanisms leading to ACI 3. ADAM10 has been implicated in the development and progression of atherosclerosis via various cellular processes, such as cytokine and growth factor shedding, proliferation, migration, and extracellular matrix degradation 6. ADAM10 might increase vascular permeability and leukocyte transmigration by cleaving JAM‐A and VE‐cadherin 20. By mediating the ectodomain shedding of VEGFR2, ADAM10 could increase vascular permeability and facilitate vascular endothelium migration 21, 22. ADAM10 also contributes to cell proliferation by shedding VE‐cadherin and modulating the β‐catenin signaling pathway 23. The observation that ADAM10 was present in carotid lesions and that its expression was significantly increased from early and advanced to ruptured atherosclerotic plaques confirmed its involvement in atherosclerosis pathogenesis 3. However, despite ample in vitro and in vivo evidence, no data are available on the role of common genetic variants in ADAM10 in ACI risk.
In 2005, Prinzen et al. reported that nucleotide −2179 to −1 represented a functional TATA‐less promoter of ADAM10 and also identified several polymorphisms in this promoter region of ADAM10. However, no significant differences were detected between patients with AD and control subjects. Bekris et al. 13, 24 reported that the rs653765 and rs514049 genetic variations within the ADAM10 promoter region are associated with ADAM10 expression levels and the levels of sAPPα in the cerebrospinal fluid of patients with AD, although no association was observed between AD risk and ADAM10 promoter polymorphisms. In this hospital‐based case–control study, we show for the first time that the rs653765 polymorphism of ADAM10 is associated with ACI, while the rs514049 polymorphism of ADAM10 is negatively associated with ACI. Interestingly, the prevalence of ADAM10 polymorphisms in our study was different from that previously reported in white individuals (26). In our population, the rs653765 allele frequency was 86.5% for the C allele, and the rs514049 allele frequency was 94.3% for the A allele, which were significantly different from the rs653765 C allele frequency (24.1%) and rs514049 A allele frequency (39.8%) in white individuals. This discrepancy might result from profound ethnic differences and the relatively small sample size of the earlier study 12.
As increased ADAM10 expression was observed in unstable plaques, primarily in macrophages and leukocytes 6, we employed real‐time PCR to detect ADAM10 mRNA expression in peripheral blood mononuclear cells. Consistent with a previous study 6, the expression of ADAM10 in mononuclear cells was significantly increased in patients with ACI, compared with the control subjects. We also found that the individuals carrying the ADAM10 rs653765 mutated T allele expressed higher ADAM10 mRNA levels, compared with populations that carry the C allele. The ADAM10 rs653765 C > T polymorphism in the promoter region could be a functional SNP. Regarding the rs653765 polymorphism in patients with AD, ADAM10 expression in the brain was lower for AA carriers than AG/GG carriers and control AA carriers 13. In addition, ADAM10 rs514049 promoter haplotype activity in SHSY5Y cells was significantly lower than that with the ADAM10 rs653765 promoter haplotype 13. This evidence supports the view that ADAM10 rs653765 polymorphisms could affect ADAM10 expression by altering its promoter activity. Additionally, bioinformatic analysis indicated that the rs653765 C > T polymorphism is located one base downstream from the potential binding site for the myc‐associated zinc finger protein (MAZ) transcription factor 12. Thus, the C > T polymorphism of ADAM10 could affect its binding with MAZ, thereby influencing the regulation of gene expression. In the two populations studied, the effects exerted by the mutation occurred mostly in patients with ACI, and the same tendencies were also observed in controls. A possible explanation for these stronger effects in patients with ACI compared with healthy subjects might be the risk factors that apply during ACI development, such as smoking. Cigarette smoke, which is a major risk factor for ACI, is known to activate matrix metalloproteinases (MMPs) 5. Because ADAMs share so many structural and functional similarities with MMPs, it is conceivable that smoking might induce increased expression of ADAM10. This presumption is supported by our observation that the smokers carrying a mutated T allele have a higher risk of ACI (Table 4). It is reported that positive expression of ADAM10 was correlated with age, size of tumor, location of tumor, depth of invasion, vessel invasion, lymph node, and distant metastasis and TNM stage in gastric cancer lesions 25. In the present study, we found that the expression of ADAM10 was correlated with age in patients with ACI, indicating the individuals over 70 years old carrying a rs653765 C > T polymorphism may run a higher risk of ACI.
A previous study showed an association between MMP9 R279Q and internal carotid artery bulb IMT 26. The progression of atherosclerosis in carotid artery lesions is associated with excessive degradation of extracellular matrix components of the vessel wall, especially elastic and collagen fibers 14, 27. ADAMs, proteins that are evolutionally closely related to MMPs, with similar functions in atherosclerotic carotid plaque progression, might be implicated in early vascular remodeling, thus affecting CIMT thickening. The use of IMT as a noninvasive tool to track changes in arterial walls has made it a worthwhile predictor of future coronary heart disease and stroke 28. We have therefore sought to examine the associations of ADAM10 polymorphisms with CIMT in patients with ACI. Unexpectedly, the rs653765 T allele group, which ran a higher risk of ACI, did not show a significant difference in CIMT compared with the C allele group. However, carriers of the rs514049 A allele had a higher CIMT than those carrying the mutated C allele among the patients with ACI. Our findings provide little support for polymorphisms of ADAM10 being direct risk factors for IMT, at least in the early stages of ACI development, which are characterized by vessel remodeling and thickening, although ADAM10 has been implicated in both the early and advanced stages of atherosclerosis. However, IMT provides a measurement of early atherosclerosis and remodeling, and IMT thickening appears before cerebral infarcts formation. An IMT greater than 0.9–1 mm is almost certainly are indicative of atherosclerosis and increased risk of cardiovascular disease 29. The prediction of stroke or death was significantly improved when ultrasound parameters were included 30. So, IMT progression was associated with the occurrence of future cardiovascular events. However, the influence of ADAM10 polymorphisms on IMT should be further verified in studies with considerably larger sample sizes.
In this study, we stratified the patients with ACI into large‐artery atherosclerosis (also called atherothrombotic stroke) and small‐artery atheroslcerosis (also referred to as lacunar stroke), according to the TOAST classification 18. Atherothrombosis is characterized by a sudden, unpredictable, atherosclerotic plaque disruption, leading to platelet activation and thrombus formation 31. Lacunar stroke results from occlusion of one of the penetrating arteries that provides blood to the brain's deep structures. Although the pathogenic mechanisms of atherothrombotic and lacunar stroke are considered to be different, there are several clues link atherosclerosis and ADAM10 to lacunar infarction. First, microatheroma now is regarded as the most common mechanism of arterial occlusion 32. Miller Fisher indicates that the diseases in the larger perforating arteries resulting from atherosclerosis cause one single or multiple larger lacunar infarcts 33. In this point of view, atherosclerosis is likely to play a role in lacunar stroke. Moreover, considerable evidence suggests that endothelial dysfunction contributes to lacunar stroke 34. ADAM10 was required for VEGF‐induced VE‐cadherin cleavage, vascular permeability, and endothelial cells migration 10. Thus, the significant role ADAM10 played in vasculature and endothelial cells makes it possible to be related to lacunar stroke. Finally, an inflammatory process with activated monocytes/macrophages may play a role in lacunar infarction 32. Thus, through cleavage of several cell surface molecules such as IL‐6R, TNF‐α, L‐selectin, CX3CL, and HB‐EGF, ADAM10 is involved in inflammatory processes in the vasculature and thereby may play a key role in lacunar stroke. Nevertheless, in future study, we will further clarify the specific role ADAM10 played in atherothrombotic and lacunar stroke, separately.
This case–control study had some limitations; the study sample was not sufficiently large, perhaps leading to nonrepresentative results. The other clinical characteristics of the study group, such as hypertension, diabetes, or hypercholesterolemia, might have complicated the associations between ADAM10 polymorphisms and ACI. Larger prospective studies are necessary to fully elucidate the role of these polymorphisms in ACI. Other functional polymorphisms might also influence the expression of ADAM10, and their combined effects must be studied to improve the prediction of the occurrence, severity, and outcomes of ACI.
In summary, our study shows for the first time a significant association between the ADAM10 rs653765 polymorphism and increased risk of ACI. Genetic variation in ADAM10 is therefore a promising new candidate for playing a role in regulating the development of atherosclerosis. The current study provides new information about the possible role of ADAM10 in the development of ACI; however, more work is still required to shed light on the role of ADAM10 in the pathogenesis of atherosclerosis and further clarify its prognostic and therapeutic potential.
Conflict of Interest
The authors have no actual or potential conflicts of interest related to this manuscript. Appropriate approval and procedures were used concerning human subjects.
Acknowledgments
This work was supported by funding from the National Nature Science Foundation of China (grant numbers 31171219, 81271213, 81070878, 81271214, and 81261120404), the Natural Science Foundation of Guangdong Province, China (No. S2012010008222), and the Science and Technology Innovation Fund of Guangdong Medical College (No. STIF 201101).
The first two authors contributed equally to this work.
References
- 1. Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med 2008;29:258–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dreymueller D, Pruessmeyer J, Groth E, Ludwig A. The role of ADAM‐mediated shedding in vascular biology. Eur J Cell Biol 2012;91:472–485. [DOI] [PubMed] [Google Scholar]
- 3. van der Vorst EP, Keijbeck AA, de Winther MP, Donners MM. A disintegrin and metalloproteases: Molecular scissors in angiogenesis, inflammation and atherosclerosis. Atherosclerosis 2012;224:302–308. [DOI] [PubMed] [Google Scholar]
- 4. Murphy G. The ADAMs: Signalling scissors in the tumour microenvironment. Nat Rev Cancer 2008;8:929–941. [DOI] [PubMed] [Google Scholar]
- 5. Duffy MJ, Mullooly M, O'Donovan N, et al. The ADAMs family of proteases: New biomarkers and therapeutic targets for cancer? Clin Proteomics 2011;8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pelisek J, Pongratz J, Deutsch L, Reeps C, Stadlbauer T, Eckstein HH. Expression and cellular localization of metalloproteases ADAMs in high graded carotid artery lesions. Scand J Clin Lab Invest 2012;72:648–656. [DOI] [PubMed] [Google Scholar]
- 7. Hundhausen C, Misztela D, Berkhout TA, et al. The disintegrin‐like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1‐mediated cell‐cell adhesion. Blood 2003;102:1186–1195. [DOI] [PubMed] [Google Scholar]
- 8. Hikita A, Tanaka N, Yamane S, et al. Involvement of a disintegrin and metalloproteinase 10 and 17 in shedding of tumor necrosis factor‐alpha. Biochem Cell Biol 2009;87:581–593. [DOI] [PubMed] [Google Scholar]
- 9. Pruessmeyer J, Ludwig A. The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Semin Cell Dev Biol 2009;20:164–174. [DOI] [PubMed] [Google Scholar]
- 10. Donners MM, Wolfs IM, Olieslagers S, et al. A disintegrin and metalloprotease 10 is a novel mediator of vascular endothelial growth factor‐induced endothelial cell function in angiogenesis and is associated with atherosclerosis. Arterioscler Thromb Vasc Biol 2010;30:2188–2195. [DOI] [PubMed] [Google Scholar]
- 11. Zhang C, Tian L, Chi C, et al. Adam10 is essential for early embryonic cardiovascular development. Dev Dyn 2010;239:2594–2602. [DOI] [PubMed] [Google Scholar]
- 12. Prinzen C, Muller U, Endres K, Fahrenholz F, Postina R. Genomic structure and functional characterization of the human ADAM10 promoter. FASEB J 2005;19:1522–1524. [DOI] [PubMed] [Google Scholar]
- 13. Bekris LM, Lutz F, Li G, et al. ADAM10 expression and promoter haplotype in Alzheimer's disease. Neurobiol Aging 2012;33:2229 e2221–2229 e2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jian XQ, Wang KS, Wu TJ, Hillhouse JJ, Mullersman JE. Association of ADAM10 and CAMK2A polymorphisms with conduct disorder: Evidence from family‐based studies. J Abnorm Child Psychol 2011;39:773–782. [DOI] [PubMed] [Google Scholar]
- 15. Kavanagh D, McKay GJ, Patterson CC, McKnight AJ, Maxwell AP, Savage DA. Association analysis of Notch pathway signalling genes in diabetic nephropathy. Diabetologia 2011;54:334–338. [DOI] [PubMed] [Google Scholar]
- 16. Kim M, Suh J, Romano D, et al. Potential late‐onset Alzheimer's disease‐associated mutations in the ADAM10 gene attenuate {alpha}‐secretase activity. Hum Mol Genet 2009;18:3987–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Song JH, Yu JT, Liu M, Yan CZ, Tan L. Genetic association between ADAM10 gene polymorphism and Alzheimer's disease in a Northern Han Chinese population. Brain Res 2011;1421:78–81. [DOI] [PubMed] [Google Scholar]
- 18. Adams HP Jr., Bendixen BH, Kappelle LJ, et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993;24:35–41. [DOI] [PubMed] [Google Scholar]
- 19. Barrett JC, Fry B, Maller J, Daly MJ. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005;21:263–265. [DOI] [PubMed] [Google Scholar]
- 20. Saftig P, Reiss K. The “A Disintegrin And Metalloproteases” ADAM10 and ADAM17: Novel drug targets with therapeutic potential? Eur J Cell Biol 2011;90:527–535. [DOI] [PubMed] [Google Scholar]
- 21. Schulz B, Pruessmeyer J, Maretzky T, et al. ADAM10 regulates endothelial permeability and T‐Cell transmigration by proteolysis of vascular endothelial cadherin. Circ Res 2008;102:1192–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Claesson‐Welsh L. ADAM‐mediated shedding, a new flavor in angiogenesis regulation. Arterioscler Thromb Vasc Biol 2010;30:2087–2088. [DOI] [PubMed] [Google Scholar]
- 23. Rocks N, Paulissen G, El Hour M, et al. Emerging roles of ADAM and ADAMTS metalloproteinases in cancer. Biochimie 2008;90:369–379. [DOI] [PubMed] [Google Scholar]
- 24. Bekris LM, Galloway NM, Millard S, et al. Amyloid precursor protein (APP) processing genes and cerebrospinal fluid APP cleavage product levels in Alzheimer's disease. Neurobiol Aging 2011;32:556 e513–556 e523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang YY, Ye ZY, Li L, Zhao ZS, Shao QS, Tao HQ. ADAM10 is associated with gastric cancer progression and prognosis of patients. J Surg Oncol 2011;103:116–123. [DOI] [PubMed] [Google Scholar]
- 26. Armstrong C, Abilleira S, Sitzer M, Markus HS, Bevan S. Polymorphisms in MMP family and TIMP genes and carotid artery intima‐media thickness. Stroke 2007;38:2895–2899. [DOI] [PubMed] [Google Scholar]
- 27. Wilton SB, Kavanagh KM, Aggarwal SG, et al. Association of rate‐controlled persistent atrial fibrillation with clinical outcome and ventricular remodelling in recipients of cardiac resynchronization therapy. Can J Cardiol 2011;27:787–793. [DOI] [PubMed] [Google Scholar]
- 28. Polak JF, Pencina MJ, O'Leary DH, D'Agostino RB. Common carotid artery intima‐media thickness progression as a predictor of stroke in multi‐ethnic study of atherosclerosis. Stroke 2011;42:3017–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Molinari F, Suri JS, Kathuria C. Atherosclerosis Disease Management. Germany: Springer, 2010. ISBN: 1‐4419‐7221‐8 [Google Scholar]
- 30. Ziegelbauer K, Schaefer C, Steinmetz H, Sitzer M, Lorenz MW. Clinical usefulness of carotid ultrasound to improve stroke risk assessment: Ten‐year results from the Carotid Atherosclerosis Progression Study (CAPS). Eur J Prev Cardiol 2012;22:22. [DOI] [PubMed] [Google Scholar]
- 31. Munger MA, Hawkins DW. Atherothrombosis: Epidemiology, pathophysiology, and prevention. J Am Pharm Assoc 2004;44:S5–S13. [DOI] [PubMed] [Google Scholar]
- 32. Del Bene A, Palumbo V, Lamassa M, Saia V, Piccardi B, Inzitari D. Progressive lacunar stroke: Review of mechanisms, prognostic features, and putative treatments. Int J Stroke 2012;7:321–329. [DOI] [PubMed] [Google Scholar]
- 33. Fisher C. The arterial lesions underlying lacunes. Acta Neuropathol 1969;12:1–15. [DOI] [PubMed] [Google Scholar]
- 34. Markus HS. Genes, endothelial function and cerebral small vessel disease in man. Exp Physiol 2008;93:121–127. [DOI] [PubMed] [Google Scholar]