

Abstract
Genetic screening in Alzheimer's disease (AD) has identified only a handful of genes that are mutated in the disorder. Thus, for a very large proportion of patients, the biology of their disease is poorly understood. Epigenetic alterations may provide an explanation in these cases. Using DNA methylation profiles of human hippocampus from controls and patients, we have identified the presence of promoter hypermethylation of the dual-specificity phosphatase 22 (DUSP22) gene in AD. DUSP22 is a likely candidate gene for involvement in the pathogenesis of the disorder since, as we demonstrate here, it inhibits PKA activity and thereby determines TAU phosphorylation status and CREB signaling. © 2014 The Authors. Hippocampus Published by Wiley Periodicals, Inc.
Keywords: epigenetics, DNA methylation, hippocampus, brain and neurodegeneration
Alzheimer's disease (AD) is the most frequent degenerative brain disorder in western society. It affects 11% of people older than 65 years and 50% of those older than 85 years. It is characterized by a progressive decline in mental abilities, neuronal loss and accumulation of amyloid plaques and neurofibrillary tangles of phosphorylated TAU (Querfurth et al., 2010). Epidemiologically, there are two main variants of AD, a rare familial early-onset variant, caused by mutations in amyloid precursor protein (APP), presenilin 1 (PSEN1) or presenilin 2 (PSEN2), and a sporadic late-onset variant, representing 95% of cases and for which only one gene, APOE, has been clearly associated as a risk factor (Querfurth et al., 2010). In recent decades, attempts to discover new risk factors for AD using genetic screening have not been as successful as expected (Bertram et al., 2008). A consideration of epigenetics offers additional opportunities to identify disease-associated genes. In this context, initial epigenetic screening has already identified genes that are deregulated by DNA methylation in case-control studies of AD (Seigmund et al., 2009; Bakulski et al., 2012; Rao et al., 2012). Herein, we focus our attention on the DNA methylation profile in one of the brain regions most commonly affected in AD, the hippocampus.
We compared the DNA methylation status of 27,578 individual CpG sites corresponding to 14,475 unique genes (Bibikova et al., 2009) in five DNA samples of control hippocampus grey matter, five Braak stage I–II, five Braak stage III–IV, and five Braak stage V–VI DNA samples from sporadic late-onset AD cases (Supporting Information). The available clinicopathological information (Braak staging, age, and gender) for the control samples in this discovery cohort is described in Supporting Information. To detect sites that are differentially methylated in AD, we selected those CpGs that yielded a more than 25% average beta-value difference between controls and Braak stage V–VI samples with increasing CpG methylation differences according to their Braak stage. The DNA methylation data obtained are freely available in the Gene Expression Omnibus database: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ttmzxyksmukiafk&acc=GSE45775.
Using this approach, four CpG methylation probes corresponding to three individual genes showed a significant difference (Mann–Whitney test, FDR adjusted P < 0.05): dual-specificity phosphatase 22 (DUSP22), claudin 15 (CLDN15), and quiescin Q6 sulfhydryl oxidase 1 (QSCN6). DUSP22 is represented by two CpG probes that are differentially methylated between control and AD samples (Fig. 1a). There was a linear relationship between DUSP22 methylation and the Braak stage (Pearson correlation coefficient R = 0.95, P < 0.05) (Supporting Information). In addition, because cognitively normal controls above age 80 might also have Braak I & II, we pooled these samples with the five AD Braak stage I–II samples as our reference population. Using this approach, the two DUSP22 CpG probes retained the highest differences between the reference population and the pooled AD Braak stage III-IV-V-VI group with a 13.5% and 12.7% DNA methylation difference. For all these reasons, we studied this candidate gene further for evidence of epigenetic dysregulation.
FIGURE 1.
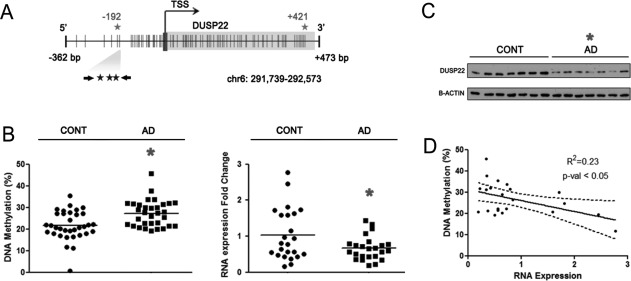
DUSP22 promoter hypermethylation and diminished expression in hippocampus of AD patients. (a) Schematic representation of DUSP22 promoter. CpGs interrogated by microarray and pyrosequencing are represented by grey and black stars, respectively. Vertical lines represent CpG dinucleotides. (b) Left, DNA methylation in control and AD samples measured by pyrosequencing. Mean values for three CpGs are shown. Right, fold change in RNA expression between control and AD samples. (c) DUSP22 protein levels in control and AD samples measured by western blot. (d) Correlation of DUSP22 DNA methylation and RNA expression. *P < 0.05 in two-tailed Student's t-test. Correlation p-value corresponds to the linear regression coefficient.
We next confirmed the presence of DUSP22 hypermethylation in the disease-associated DNAs using a different DNA methylation detection technology (pyrosequencing) in an independent set of 25 control hippocampus grey matter and 25 AD samples (Supporting Information and Fig. 1b). The available clinicopathological information (Braak staging, age, and gender) for the control samples in this validation cohort is described in Supporting Information. The lower methylation difference found in the validation set in comparison to the discovery set could be explained because the pyrosequencing data is an average across a larger region that contains three CpGs where only one is a critical CpG derived from the DNA methylation microarray. We observed downregulation of the DUSP22 RNA transcript (0.67 ± 0.07 fold change; t-test P < 0.05) (Fig. 1b) and protein (0.50 ± 0.07 fold change; t-test P < 0.05) (Fig. 1c) in the AD hippocampus samples compared with the control tissues. In keeping with the accepted epigenetic concept that links gene promoter hypermethylation and transcriptional inactivation (Portela et al., 2010), we observed a correlation between DUSP22 DNA methylation and diminished RNA expression (R2 = 0.23, P < 0.05) (Fig. 1d).
Once we had identified the presence of DUSP22 promoter hypermethylation and downregulation in the hippocampus of AD patients, we examined its potential functional consequences. Dual-specific phosphatases (DUSPs) can dephosphorylate both tyrosine and serine/threonine residues (Patterson et al., 2009). DUSP22, also known as JNK pathway-associated phosphatase (JKAP), is expressed in various types of tissues and organs (Chen et al., 2009), including the hippocampus (Hawrylycz et al., 2012), suggesting that it may participate in essential biological processes. The TAU protein could be an important target for DUSP22-mediated dephosphorylation in AD. TAU Thr231 phosphorylation is one of the first phosphorylation events in AD and has a major role in TAU regulation (Luna-Muñoz et al., 2007). Although phosphoseryl/phosphothreonyl protein phosphatase-2A (PP-2A) appears to be the most active enzyme in dephosphorylating the abnormal TAU to a near-normal state (Wang et al., 2013), other phosphatases might also play a role. Due to the lack of human cell culture models of AD, we screened a panel of seven cell lines from neuronal lineages (the neuroblastoma cell lines SH-SY5Y, SK-N-AS, SK-N-BE(2), SK-N-F1, SKN-JD, and SK-N-SH) for TAU Thr231 phosphorylation (Fig. 2a). In this setting, SK-N-BE(2) presented the highest levels of TAU Thr231 phosphorylation (Fig. 2a) and was selected for further experimentation. Stable short-hairpin RNA (shRNA) depletion of DUSP22 in SK-N-BE(2) cells caused a loss of TAU Thr231 phosphorylation in comparison with the shRNA scramble cells (Fig. 2b). In the reciprocal experiment we found the equivalent result, whereby stable transfection of DUSP22 in SK-N-BE(2) cells caused an increase in TAU Thr231 phosphorylation (Fig. 2c). These data suggest that DUSP22 affects TAU Thr231 phosphorylation status but in an inverse manner, contrary to what was expected. Thus, TAU is not itself a direct target of DUSP22, but its phosphorylation depends indirectly on the studied phosphatase.
FIGURE 2.

DUSP22 regulates TAU phosphorylation through PKA. (a) Levels of TAU Thr231 phosphorylation across different neuronal cell lines. (b) Stable clones interfering (top) and overexpressing (bottom) DUSP22. (c) Levels of TAU Thr231 phosphorylation across stable clones. (d) Levels of TAU Thr231 phosphorylation between interfered and overexpressing DUSP22 SK-N-BE(2) cells after 24 h of 20 mM LiCl, 30 min of 500 mM sorbitol and 30 min of 50 µM forskolin treatments. (e) Top, immunoprecipitation between DUSP22-myc tagged and catalytic subunits of PKA. Below, western blot to confirm cellular fractionation using membrane (AKAP79), cytosol (NSE) and nuclear (Histone H3) located proteins. (f) Basal levels of PKA Thr197 of wild-type, scramble, interfered and overexpressing DUSP22 SK-N-BE(2) cells.
Several candidates could mediate the effect of DUSP22 on TAU phosphorylation, the most likely targets being those kinases that phosphorylate TAU. In this regard, we studied the glycogen synthase kinase 3 (GSK3β), p38 mitogen-activated protein kinase (p38) and cAMP-dependent protein kinase (PKA) proteins which are all able to phosphorylate TAU at the Thr231 site (Wang et al., 2013). The use of a GSK3β inhibitor (lithium) caused a similar reduction of TAU Thr231 phosphorylation in both DUSP22 shRNA-depleted (0.7 ± 0.12) and transfected SK-N-BE(2) (0.5 ± 0.13) cells (Fig. 2d). In a similar way, the use of a p38 activator (sorbitol) induced a similar increase in TAU Thr231 phosphorylation in both DUSP22 experimental models, stable downregulation (2.7 ± 0.5) and stable overexpresssion (2.2 ± 0.6) (Fig. 2d). These findings suggest that the two TAU Thr231 phosphorylation pathways are unaffected by DUSP22 expression. A different pattern was observed for PKA, wherein the use of the PKA activator forskolin was able to induce TAU Thr231 phosphorylation in DUSP22 shRNA-depleted cells (2.2 ± 0.5), but not in DUSP22-overexpressing SK-N-BE(2) cells (0.8 ± 0.3) (Fig. 2d). This result suggests that DUSP22 inhibits PKA activity, and the link between the two pathways was confirmed by the observation that both proteins are located in the cellular membrane subfraction (Fig. 2e) and co-immunoprecipitate (Fig. 2e).
PKA has a complex regulation in which two catalytic and two regulatory subunits are differentially associated and phosphorylated (Taylor et al., 2012). In response to cAMP production, PKA regulatory subunits release the catalytic components which are concomitantly phosphorylated at Thr197 constituting the active form of the enzyme (Taylor et al., 2012). DUSP22 shRNA depletion increased PKA Thr197 phosphorylation, while DUSP22 transfection decreased phosphorylation. Wild-type and shRNA scramble cells showed intermediate phosphorylation (Fig. 2f). These data reinforce the idea of DUSP22 acting as a PKA inhibitor.
In addition to the DUSP22 role on PKA-mediated TAU phosphorylation, the epigenetic silencing of the studied phosphatase can have another physiopathological role in PKA and the disease: its impact on the cAMP response element-binding protein (CREB) signaling pathway. CREB activity is nowadays considered critical for neuronal function and regulates synaptic plasticity, long-term memory formation and neuronal survival (Lonze et al., 2002); all processes that are altered in AD (Querfurth et al., 2010). Given that PKA activates CREB by phosphorylation of Ser133 (Johannessen et al., 2004), we analyzed this pathway in our model. We found that in SK-N-BE(2) cells with stable overexpression of DUSP22 the phosphorylation of CREB Ser133 upon the use of the PKA activator was delayed relative to DUSP22-shRNA-depleted cells (Fig. 3a). Most significantly, we also observed that DUSP22 through PKA was able to regulate the expression of CREB target downstream genes, such as somatostatin (SST), nuclear receptor subfamily 4 group member 2 (NR4A2) and FBJ osteosarcoma oncogene (FOS). We found that in DUSP22-shRNA-depleted cells the use of the PKA activator induced the expression of the described CREB-response genes in comparison with the lack of effect in DUSP22 stable transfected cells (Fig. 3b). With respect to the CREB target genes, the effect on SST is of particular note because its expression is a protective factor against amyloid beta (Aβ) toxicity (Rubio et al., 2012). In this regard, we were able to show that DUSP22-shRNA-depleted cells have a significantly higher survival ability (measured by the MTT assay) than the cells with stable overexpression of DUSP22 (Fig. 3c). Most importantly, the external addition of STT in the DUSP22-overexpressing cells can partially rescue the survival ability of these cells upon Aβ use (Fig. 3c), providing evidence of the role of STT as a neuroprotective factor and the contribution of DUSP22 to these processes. Thus, these latter data imply that the inhibition of PKA by DUSP22 also causes a loss of CREB phosphorylation and of the activity that affects its downstream effector genes and functions, such as STT and cell survival.
FIGURE 3.
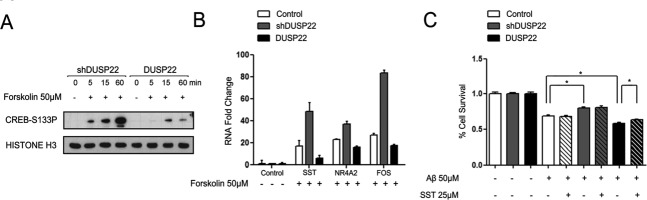
DUSP22 regulates CREB phosphorylation and activity through PKA. (a) Different kinetics of CREB activation between interfered and overexpressing DUSP22 SK-N-BE(2) cells measured by Ser133 phosphorylation. (b) RNA expression patterns of genes downstream CREB wild type, interfered and overexpressing DUSP22 SK-N-BE(2) cells after 2 h of 50 µM forskolin treatment. (c) Percentage cell survival after 24 h of 50 µM Aβ treatment in wild type, interfered and overexpressing DUSP22 SK-N-BE(2) cells with and without external addition of 25 µM SST peptide measured by MTT assay. *P < 0.05 in two-tailed Student's t-test.
DUSP22, VHR-related MKPX (VHX), JNK stimulatory phosphatase-1 (JSP-1), and LMW-DSP2 are members of the atypical dual specificity phosphatases family (DUSPs); these small proteins lack the typical N-terminal regulatory domain of DUSPs proteins. DUSPs are important signaling regulators that dephosphorylate tyrosine and serine/threonine residues and can act on many substrates (Patterson et al., 2009). Herein, we have demonstrated that DUSP22 is epigenetically deregulated in AD and that it affects PKA signaling in neuroblastoma cells. Our results suggest that DUSP22 expression could elicit a handicap in PKA/GSK3β regulation and TAU phosphorylation. Furthermore, this suggests that high-expressing DUSP22 cells would have a special tendency to accumulate hyperphosphorylated TAU, thereby reducing the viability of such cells. Therefore, the increase in DNA promoter methylation in DUSP22 observed in AD could be consequence of an imbalance of cell populations, by which high DUSP22-expressing cells would persistently dwindle in parallel with the progression of the disease, and only epigenetically-silenced DUSP22 cells would remain viable. We cannot reject the possibility that cells, in order to increase their survival, actively methylate DUSP22 promoter when exposed to Aβ-induced toxicity. Nevertheless, it highlights the role of PKA/GSK3β signaling in AD and supports the idea that treatments inducing DUSP22 inhibition could benefit AD patients.
Additionally, our results suggest that DUSP22 expression could also compromise Aβ-induced cell survival through PKA/CREB inhibition outstanding the pro-survival role of the downstream SST gene. Complete disruption of CREB is lethal (Rudolph et al., 1998), but its partial disturbance has been associated to several neurological disorders. Coffin-Lowry syndrome and Rusbenstein-Taybi Syndrome are caused by mutations in CREB pathway genes (Petrij et al., 1995; Trivier et al., 1996). CREB signaling deficits have also been reported in Huntington's disease (Steffan et al., 2000), Parkinson's disease (Andersson et al., 2001) and AD (Saura et al., 2011). Namely, much time has passed since CREB was initially associated with AD in seminal studies reporting a decrease of CREB phosphorylation in AD (Yamamoto-Sasaki et al., 1999). Similarly, the AD Tg2756 mice model also exhibits reduced CREB expression (Pugazhenthi et al., 2011) and phosphorylation (Dineley et al., 2001), suggesting a general disturbance of CREB signaling in AD. In fact, Aβ can impair PKA/CREB signaling at different levels. On one hand, Aβ decreases PKA activity and CREB phosphorylation in hippocampal neurons (Vitolo et al., 2002). On the other hand, Aβ also inhibits CREB regulated transcription coactivator 1 (CRTC1) with similar results in CREB signaling (España et al., 2010); in both cases, this resulted in reduced synaptic plasticity and memory formation. Our data is in line with these results and reinforce the idea that CREB signaling could be affected in AD.
The importance of epigenetic alterations to our understanding of human disease is increasingly well established and such changes are probably also involved in neurodegenerative disorders, such as AD. Herein, we have shown that in the hippocampus of AD patients a particular phosphatase (DUSP22) undergoes promoter hypermethylation and loss of expression. We further demonstrate that this enzyme could be relevant, as a PKA inhibitor, in TAU phosphorylation and CREB signaling, two pathways that are essential in AD pathobiology. However, a limitation of the studies doing whole tissue epigenetics, such as this one, derives from the recognition that the number of neurons in a hippocampus varies wildly between Braak V and Braak I samples, thus the distinct DNA methylation profiles could also be due to changing cell type populations with disease, rather than a mechanism of disease in neurons. These issues will be the focus of future research in this area.
Acknowledgments
M.E. is an ICREA Research Professor.
Supporting Information
Additional Supporting Information may be found in the online version of this article
Supporting Information
REFERENCES
- Andersson M, Konradi C, Cenci MA. cAMP response element-binding protein is required for dopamine-dependent gene expression in the intact but not the dopamine-denervated striatum. J Neurosci. 2001;21:9930–9943. doi: 10.1523/JNEUROSCI.21-24-09930.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakulski KM, Dolinoy DC, Sartor MA, Paulson HL, Konen JR, Lieberman AP, Albin RL, Hu H, Rozek LS. Genome-wide DNA methylation differences between late-onset Alzheimer's disease and cognitively normal controls in human frontal cortex. J Alzheimers Dis. 2012;29:571–588. doi: 10.3233/JAD-2012-111223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE. Thirty years of Alzheimer's disease genetics: The implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9:768–778. doi: 10.1038/nrn2494. [DOI] [PubMed] [Google Scholar]
- Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, Gunderson KL. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics. 2009;1:177–200. doi: 10.2217/epi.09.14. [DOI] [PubMed] [Google Scholar]
- Chen AJ, Zhou G, Juan T, Colicos SM, Cannon JP, Cabriera-Hansen M, Meyer CF, Jurecic R, Copeland NG, Gilbert DJ, Jenkins NA, Fletcher F, Tan TH, Belmont JW. The dual specificity JKAP specifically activates the c-Jun N-terminal kinase pathway. J Biol Chem. 2002;277:36592–36601. doi: 10.1074/jbc.M200453200. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- España J, Valero J, Miñano-Molina AJ, Masgrau R, Martín E, Guardia-Laguarta C, Lleó A, Giménez-Llort L, Rodríguez-Alvarez J, Saura CA. beta-Amyloid disrupts activity-dependent gene transcription required for memory through the CREB coactivator CRTC1. J Neurosci. 2010;30:9402–9410. doi: 10.1523/JNEUROSCI.2154-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, van de Lagemaat LN, Smith KA, Ebbert A, Riley ZL, Abajian C, Beckmann CF, Bernard A, Bertagnolli D, Boe AF, Cartagena PM, Chakravarty MM, Chapin M, Chong J, Dalley RA, Daly BD, Dang C, Datta S, Dee N, Dolbeare TA, Faber V, Feng D, Fowler DR, Goldy J, Gregor BW, Haradon Z, Haynor DR, Hohmann JG, Horvath S, Howard RE, Jeromin A, Jochim JM, Kinnunen M, Lau C, Lazarz ET, Lee C, Lemon TA, Li L, Li Y, Morris JA, Overly CC, Parker PD, Parry SE, Reding M, Royall JJ, Schulkin J, Sequeira PA, Slaughterbeck CR, Smith SC, Sodt AJ, Sunkin SM, Swanson BE, Vawter MP, Williams D, Wohnoutka P, Zielke HR, Geschwind DH, Hof PR, Smith SM, Koch C, Grant SG, Jones AR. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391–399. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen M, Delghandi MP, Moens U. What turns CREB on? Cell Signal. 2004;16:1211–1227. doi: 10.1016/j.cellsig.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Riccio A, Cohen S, Ginty DD. Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron. 2002;34:371–385. doi: 10.1016/s0896-6273(02)00686-4. [DOI] [PubMed] [Google Scholar]
- Luna-Muñoz J, Chávez-Macías L, García-Sierra F, Mena R. Earliest stages of tau conformational changes are related to the appearance of a sequence of specific phospho-dependent tau epitopes in Alzheimer's disease. J Alzheimers Dis. 2007;12:365–375. doi: 10.3233/jad-2007-12410. [DOI] [PubMed] [Google Scholar]
- Patterson KI, Brummer T, O'Brien PM, Daly RJ. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem J. 2009;418:475–489. doi: 10.1042/bj20082234. [DOI] [PubMed] [Google Scholar]
- Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ, Breuning MH. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Pugazhenthi S, Wang M, Pham S, Sze CI, Eckman CB. Downregulation of CREB expression in Alzheimer's brain and in Aβ-treated rat hippocampal neurons. Mol Neurodegener. 2011;6:60. doi: 10.1186/1750-1326-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Rao JS, Keleshian VL, Klein S, Rapoport SI. Epigenetic modifications in frontal cortex from Alzheimer's disease and bipolar disorder patients. Transl Psychiatry. 2012;2:e132. doi: 10.1038/tp.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio A, Sánchez-Mut JV, García E, Velasquez ZD, Oliver J, Esteller M, Avila J. Epigenetic control of somatostatin and cortistatin expression by β amyloid peptide. J Neurosci Res. 2012;90:13–20. doi: 10.1002/jnr.22731. [DOI] [PubMed] [Google Scholar]
- Rudolph D, Tafuri A, Gass P, Hämmerling GJ, Arnold B, Schütz G. Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc Natl Acad Sci USA. 1998;95:4481–4486. doi: 10.1073/pnas.95.8.4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura CA, Valero J. The role of CREB signaling in Alzheimer's disease and other cognitive disorders. Rev Neurosci. 2011;22:153–169. doi: 10.1515/RNS.2011.018. [DOI] [PubMed] [Google Scholar]
- Siegmund KD, Connor CM, Campan M, Long TI, Weisenberger DJ, Biniszkiewicz D, Jaenisch R, Laird PW, Akbarian S. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One. 2007;2:e895. doi: 10.1371/journal.pone.0000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Ilouz R, Zhang P, Kornev AP. Assembly of allosteric macromolecular switches: Lessons from PKA. Nat Rev Mol Cell Biol. 2012;13:646–658. doi: 10.1038/nrm3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivier E, De Cesare D, Jacquot S, Pannetier S, Zackai E, Young I, Mandel JL, Sassone-Corsi P, Hanauer A. Mutations in the kinase Rsk-2 associated with Coffin-Lowry syndrome. Nature. 1996;384:567–570. doi: 10.1038/384567a0. [DOI] [PubMed] [Google Scholar]
- Vitolo OV, Sant'Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M. Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: Reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci USA. 2002;99:13217–13221. doi: 10.1073/pnas.172504199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JZ, Xia YY, Grundke-Iqbal I, Iqbal K. Abnormal hyperphosphorylation of tau: Sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis. 2013;33:S123–S139. doi: 10.3233/JAD-2012-129031. [DOI] [PubMed] [Google Scholar]
- Yamamoto-Sasaki M, Ozawa H, Saito T, Rösler M, Riederer P. Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type. Brain Res. 1999;824:300–303. doi: 10.1016/s0006-8993(99)01220-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information