

Abstract
The fluorination of phenolphthalein and naphtholphthalein was performed with diluted fluorine gas under acidic conditions. For both compounds we observed an electrophilic fluorination into ortho position to the hydroxyl group. Through the use of this reaction we synthesized and characterized mono-and difluorinated derivatives of phenolphthalein and naphtholphthalein. The compounds were also prepared in the 18F labeled form, which are usable as a new type of probe for in vivo pH measurement in biological objects using Cerenkov imaging or combination of light absorption and PET.
Keywords: F-18, Electrophilic fluorination, Phenolphthalein, Naphtholphthalein
1. Introduction
Direct electrophilic fluorination is an infrequently used synthetic method for the labeling of complex organic molecules with the positron emitting isotope 18F. The electrophilic fluorination reaction produces compounds with relatively low specific activity and in most cases results in non-specific decomposition of the reagent. Despite this, we have recently achieved the specific electrophilic fluorination of phenolsulfonphthalein [1] and its derivatives [2], and used this reaction for the synthesis of 18F-labeled pH indicators [3].
In the current paper we report the synthesis of 18F-labeled derivatives of two other types of triarylmethane indicators, phenolphthalein and naphtholphthalein. These compounds represent a new class of dual-mode indicators with high potential as in vivo pH probes in biological objects.
2. Materials and methods
All reagents were purchased from Sigma–Aldrich, with the exception of the fluorine-neon mixture, which was obtained from Matheson TriGas Inc. (Twinsburg, OH). Labeling of the molecules with F2 and [18F]-F2 was performed at the Cyclotron facility of the University of Pennsylvania using a custom-made fluorination unit. The [18F]-F2 gas was prepared by the 20Ne(d,α)18F reaction using an IBA 18/9 Cyclone accelerator. The labeling reaction was performed by bubbling of 0.1% F2 or [18F]-F2 in Ne (total 100–200 mCi of activity in 35 micromoles of fluorine carrier) for 5 min through a solution of the correspondent indicator in trifluoroacetic acid (2 mg/mL). The reaction mixture was evaporated in a vacuum at 100 °C, redissolved in 2 mL of water/acetonitrile mixture, injected into a semi-preparative HPLC column (Phenomenex, Synergi 4 µm Hydro-RP 80 Å 10 mm × 250 mm) and eluted with 40% acetonitrile–water for phenolphthalein and 55% acetonitrile–water buffer for naphtholphthalein at a rate of 3 mL/min with simultaneous detection of radioactivity and absorption at 276 nm. Under these conditions the elution times for the products of interest were in the range of 20–35 min with good separation of initial precursor and fluorination products on both UV and radioactivity detectors.
In order to analyze the reaction products, corresponding fractions were collected and analyzed by analytical HPLC, mass-spectroscopy, UV–vis spectroscopy, and 1H and 19F NMR. The presence of individual compounds in the fractions was confirmed by analytical HPLC performed with a 4.6 mm × 250 mm column of the same type and using a gradient of acetonitrile–ammonium acetate buffer (0.1 M, pH 4.7). The incorporation of fluorine atoms into the indicator molecule was proven by mass-spectroscopy on a Thermo Fisher Scientific (Bremen, Germany) Orbitrap Exactive with Xcalibur software. Since the relative amount of fluorine-18 in radioactive [18F]-F2 gas was very small, and as thus would not give any distinct signal on mass-spectra, the molecular mass of the products was determined after complete decay of the radioactive label. UV–vis spectroscopic investigation of the absorption and indicator properties of the products was performed on a Beckman-Coulter DU 530 spectrophotometer using acetate, phosphate, TRIZMA, and borate buffers for different pH values. A combination of radioactivity and optical properties was used as a supplementary method for confirming the incorporation of the fluorine atoms into the indicator molecule. The position of the fluorination in the molecules was determined from 1H and 19F NMR spectra obtained for neutral (colorless) forms of the indicators on a Bruker DMX-360 spectrometer using deuterated methanol solvent. Decoupling experiments were carried out with set parameters O1 = 4.5 ppm, O2 = −138 ppm for 1H {19F} and O1 = −100 ppm, O2 = 6.95 ppm for 19F {1H}, respectively.
3. Experimental
The reactants and synthesized products were characterized by mass spectra, visible spectrophotometry, and proton and fluorine NMR. The results are given below.
3.1. Phenolphthalein
m/z 317.079 amu (negative), 319.097 amu (positive); pKa = 9.75, λbasic = 553 nm.
1H (360 MHz, CD3OD) δ 6.735 ppm (dd, J = 6.5, 2.1 Hz 4H); δ 7.089 ppm (dd, J = 6.5, 2.1 Hz 4H); δ 7.593 (t, J = 7.6 Hz, 1H); δ 7.603 (d, J = 7.2 Hz, 1H); δ 7.754 (td, J = 7.5, 1.3 Hz, 1H); δ 7.882 (dt, J = 7.2, 1.3 Hz, 1H).
3.2. (RS)-3-(3-fluoro-4-hydroxyphenyl)-3-(4-hydroxyphenyl)isobenzofuran-1(3H)-one (monofluorophenolphthalein, MFPP)
m/z 335.069 amu (negative), 337.087 amu, (positive); pKa = 9.33, λbasic = 558 nm.
1H (360 MHz, CD3OD) δ 6.750 (ddd, JHH = 8.9, 3.0, 2.2 Hz 2H); δ 6.891 (dd, JHF = 8.0 Hz, JHH = 8.3 Hz 1H); δ 6.914 (dd, JHH = 8.5, 2.1 Hz, 1H); δ 6.948 (dd, JHF = 12.1 Hz, JHH = 2.1 Hz 1H); δ 7.089 (ddd, JHH = 8.9, 3.0, 2.2 Hz 4H); δ 7.619 (td, JHH = 7.5, 0.9 Hz, 1H); δ 7.640 (d, JHH = 7.8 Hz, 1H); δ 7.779 (td, JHH = 7.6, 1.1 Hz, 1H); δ 7.897 (d, JHH = 7.6 Hz, 1H).
19F (338 MHz, CD3OD): δ −138.185 (dd, JFH = 8.1, 11.9 Hz, 1F).
3.3. 3,3-Bis(3-fluoro-4-hydroxyphenyl)isobenzofuran-1(3H)-one (difluorophenolphthalein, DFPP)
m/z 353.060 amu (negative), 355.079 amu (positive); pKa = 8.70, λbasic = 564 nm.
1H (360 MHz, CD3OD): δ 6.897 (ddd, JHH = 8.3, 0.7 Hz, JHF = 8.3 Hz, 2H); δ 6.921 (dd, JHH = 9.7, 1.8 Hz, 2H); δ 6.951 (dd, JHF = 11.7 Hz, JHH = 1.8 Hz 2H); δ 7.638 (td, JHH = 7.6, 0.7 Hz, 1H); δ 7.675 (d, JHH = 7.8 Hz, 1H); δ 7.799 (td, JHH = 7.6, 1.1 Hz, 1H); δ 7.909 (d, JHH = 7.6 Hz, 1H).
19F (338.8 MHz, CD3OD): δ −137.954 (dd, JFH = 11.8, 7.5 Hz, 2F).
3.4. Naphtholphthalein
m/z 417.110 amu (negative), 419.128 amu (positive); pKa = 7.83, λbasic = 653 nm.
1H (360 MHz, CD3OD) δ 6.712 (d, J = 7.9 Hz, 2H); δ 7.129 (td, J = 7.9, 1.4 Hz, 2H); δ 7.290 (td, J = 7.6, 1.1 Hz, 2H); δ 7.590 (d, J = 7.6 Hz, 1H); δ 7.600 (td, J = 7.6, 1.4 Hz, 1H); δ 7.657 (td, J = 7.6, 1.4 Hz, 1H); δ 7.885 (d, J = 9.0 Hz, 2H); δ 7.976 (dd, J = 7.6, 1.4 Hz 1H); δ 8.215 (d, J = 8.3 Hz 2H).
3.5. (RS)-3-(3-fluoro-4-hydroxynaphthalen-1-yl)-3-(4-hydroxynaphthalen-1-yl)isobenzofuran-1(3H)-one (monofluoronaphtholphthalein, MFNP)
m/z 435.100 amu (negative), 437.119 amu (positive); pKa = 7.66, λbasic = 659 nm.
1H (360 MHz, CD3OD) δ 6.725 (d, J = 8.3 Hz, 1H); δ 7.104 (t, J = 7.9 Hz, 1H); δ 7.165 (td, J = 7.9, 1.4 Hz, 1H); δ 7.306 (td, J = 7.6, 1.1 Hz, 1H); δ 7.330 (t, J = 7.6 Hz, 1H); δ 7.617 (d, J = 6.5 Hz, 1H); δ 7.624 (td, J = 7.6, 1.4 Hz, 1H); δ 7.683 (td, J = 7.6, 1.4 Hz, 1H); δ 7.861 (d, J = 8.6 Hz, 1H); δ 7.873 (d, J = 8.6 Hz, 1H); δ 7.997 (dd, J = 7.6, 1.1 Hz 1H); δ 8.220 (d, J = 7.9 Hz 1H); δ 8.229 (d, J = 7.9 Hz, 1H).
19F (338 MHz, CD3OD): δ -143.965 (bs, 1F).
3.6. 3,3-Bis(3-fluoro-4-hydroxynaphthalen-1-yl)isobenzofuran-1(3H)-one (difluoronaphtholphthalein, DFNP)
m/z 453.090 amu (negative), 455.110 amu (positive); pKa = 7.24, λbasic = 665 nm.
1H (360 MHz, CD3OD) δ 7.161 (td, J = 7.9, 1.4 Hz, 2H); δ 7.367 (t, J = 7.6 Hz, 2H); δ 7.667 (d, J = 6.8 Hz, 1H); δ 7.693 (td, J = 7.6, 1.4 Hz, 1H); δ 7.799 (td, J = 7.6, 1.4 Hz, 1H); δ 7.858 (d, J = 8.6 Hz, 2H); δ 8.043 (dd, J = 7.2, 1.4 Hz 1H); δ 8.247 (d, J = 7.9 Hz 2H).
19F (338 MHz, CD3OD): δ −143.955 (bs, 2F).
4. Results
The reaction between phenolphthalein and [18F]-F2 gas in trifluoroacetic acid demonstrates a certain degree of similarity with the previously observed fluorination of Phenol Red and its derivatives [1–3]. In both cases significant amounts of the product of fluorine-hydrogen substitution were obtained via specific fluorination of the precursor molecules. Separation of the reaction mixture by HPLC indicated the presence of the non-reacted precursor and the synthesis of two major radioactive components. These products had higher retention time on the reverse-phase column, which is typical for more hydrophobic fluorinated derivatives [1–3]. The ratio of the radioactivity to UV absorption was almost 1–2 for the obtained products, suggesting successive incorporation of two fluorine atoms into the phenolphthalein molecule.
Mass-spectroscopic investigation of the three HPLC fractions reveal main peaks at m/z 317.079, 335.069, and 353.060 amu for negative mode and 319.097, 337.087, and 355.079 amu for positive mode. These data indicate the successive substitution of one or two hydrogen atoms by fluorine in the phenolphthalein molecule.
Based on the mass-spectroscopic data and radioactivity, two reaction products were identified as mono- (MFPP) and difluorophenolphthalein (DFPP). The corrected radiochemical yield of these products varied for the two products and was in the ranges of 10–15% and 3–4%, respectively. The chemical yield of the products was calculated from the absorption of their basic forms using the extinction coefficient of phenolphthalein, 3 × 104 M−1 cm−1 [4]. The value of the chemical yield was the same as the radiochemical for MFPP and halved for DFPP.
Both compounds acted as pH indicators with properties similar to phenolphthalein. Colorless neutral forms of these molecules have weak absorption maxima in the UV range (270–280 nm), while the purple basic forms of indicators absorbed at 558 nm for the monofluorinated and 564 nm for the difluorinated derivative. The pKa values for the transfer between these two forms were found to be 9.33 for MFPP and 8.70 for DFPP. Thus the successive incorporation of fluorine atoms into the phenolphthalein molecule (pKa = 9.75, λbasic = 553 nm) causes a decrease in the pKa values and a bathochromic shift of λbasic, which is consistent with the behavior of fluorinated derivatives of Phenol Red [1–3].
The position of fluorine incorporation into the phenolphthalein molecule was determined by comparison of the 1H and 19F NMR spectra of the compounds. The 1H NMR spectrum of phenolphthalein (Fig. 1a) has four signals in the range of δ 7.6–7.9 ppm from the protons of the aromatic ring containing the carboxyl group. Incorporation of fluorine atoms into the molecule causes only minor (<0.1 ppm) shifts of these signals without any alteration of their shape. From these results we gather that substitution of hydrogen by fluorine does not occur in the carboxyl ring. Two other signals in the 1H NMR spectrum of phenolphthalein have δ 6.735 ppm (4H) and 7.089 ppm (4H) and correspond to protons located in the phenolic rings in ortho and meta positions to the hydroxyl groups, respectively. Introduction of one fluorine atom into the molecule (Fig. 1b) decreases their integral intensity by a half, while a new group of signals appears in the range of δ 6.8–7.0 ppm (3H). The second fluorine atom (Fig. 1d) causes complete disappearance of the signals from the non-substituted phenol rings of phenolphthalein, while the new group of peaks is doubled in integral intensity without significant change in their shape. Spectra with fluorine decoupling (Fig. 1c and e) shows that these new signals represent protons adjacent to 19F.
Fig. 1.

1H NMR spectra of phenolphthalein and its fluorinated derivatives. Successive introduction of fluorine atoms into the phenolphthalein molecule (a) causes only a minor shift of the signals from the carboxylic ring (δ 7.6–7.9 ppm) and gradual replacement of the signals from phenolic rings (δ 6.7 and 7.1 ppm) by a new group of peaks (δ 6.8–7.0 ppm) for mono- (b) and difluorophenolphthalein (d). Decoupling of the fluorine-hydrogen coupling (spectra (c) and (e) for mono- and difluorophenolphthalein, respectively) shows, that these new peaks are coupled with fluorine and allows an assignment of the proton signals.
The 1H NMR data indicate that the first fluorine atom substitutes a hydrogen in the phenol ring, and the second fluorine atom is located in the identical positions on the second phenol ring. The latter conclusion is also supported by 19F NMR data (Fig. 2), showing only one signal from two identical nuclei in the difluorophenolphthalein spectrum. The possibility of substitutions in the carboxyl ring, or introduction of both fluorine atoms into the same phenol ring, are thus both ruled out as well. The exact position of the substitution can be deduced from the chemical shift in 19F NMR spectra (Fig. 2). Location of the signals at −138 ppm is characteristic of fluorine atoms in ortho positions in fluorophenols and excludes the possibility of meta location (which has much lower value chemical shift of −113 ppm) [5]. From these data we can identify the monofluorophenolphthalein (MFPP) as (RS)-3-(3-fluoro-4-hydroxyphenyl)-3-(4-hydroxyphenyl)isobenzofuran-1(3H)-one and difluorophenolphthalein (DFPP) as 3,3-bis(3-fluoro-4-hydroxyphenyl)isobenzofuran-1(3H)-one and propose a mechanism of the reaction for fluorination of phenolphthalein, shown in Fig. 3.
Fig. 2.

19F NMR spectra of mono- (a) and di- (c) fluorophenolphthalein show that all the signals have a chemical shift close to δ −138 ppm, which is specific for fluorine atoms in ortho position to hydroxyl group of the phenolic rings. Decoupling of the hydrogen-fluorine coupling (b and d, respectively) indicates that for both compounds no isomers are present and both fluorine atoms in difluorophenolphthalein molecule are identical.
Fig. 3.
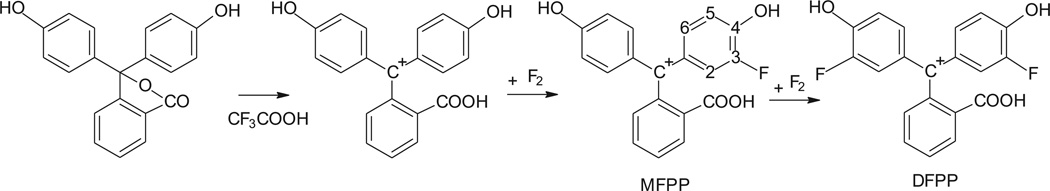
Mechanism of fluorination of phenolphthalein in trifluoroacetic acid. In trifluoroacetic acid solution the compound is turned into the cation form, which is fluorinated in the ortho-positions to the hydroxyl group.
By decoupling fluorine from the proton spectrum, we were also able to identify all the signals in the fluorophenol ring and assign them to three different protons, as presented in Section 3. The signal at δ 6.90 ppm corresponds to the proton at carbon 5 (the numbering is shown in Fig. 3 on the MFPP structure); δ 6.92 ppm is from proton 6, and δ 6.95 ppm from 2. The close values of chemical shift for these protons can be explained if the opposite effects of the hydroxyl group and fluorine atom are considered. In the spectrum of phenolphthalein, the hydroxyl group activates protons in the ortho position, shifting signals from protons 3 and 5 upfield to δ 6.74 ppm, and deactivates protons in the meta position, shifting signals from protons 2 and 6 downfield to δ 7.09. The introduction of the fluorine atom into position 3 has just the opposite effect: shifting protons at 2 and 6 upfield, and 5 downfield. The fluorine-hydrogen coupling on both protons 2 and 5 creates a very complicated multiplet structure, which we were able to resolve only after fluorine-hydrogen decoupling.
The direct fluorination of naphtholphthalein occurs in a similar manner. We were also able to detect only two major radioactive products, which were separable by HPLC due to their higher retention time on the reverse-phase column. The ratio of radioactivity to absorption was doubled for the second product, suggesting the presence of one and two fluorine atoms in the molecules of the fluorinated derivatives, respectively. The mass-spectroscopy of the initial compound and the fluorinated products showed m/z values of 417.110, 435.100, and 453.090 amu for negative mode, and 419.128, 437.119, and 455.110 amu for positive mode. Collectively these data indicate the synthesis of mono- (MFNP) and difluoronaphtholphthalein (DFNP). The corrected radiochemical yields of these products varied for different experiments in the range of 3–5% and 6–8%, respectively, while the chemical yields were virtually the same.
Both compounds act as pH indicators, having a pKa value of 7.66 and absorption maximum for the basic form at 659 nm for MFNP, and pKa = 7.24 and λbasic = 665 nm for DFNP. Introduction of the fluorine atom into the molecule of naphtholphthalein (pKa = 7.83, λbasic = 653 nm) also causes a decrease of the pKa values and a bathochromic shift of the absorption maximum of the basic form, which seems to be a common behavior for all the triarylmethane indicators.
Analysis of 1H and 19F NMR spectra of naphtholphthalein and its fluorinated derivatives reveals a mechanism for its fluorination reaction. Comparison of the 1H NMR spectra of naphtholphthalein and its derivatives (Fig. 4) shows that only one proton signal is drastically affected by successive introduction of the fluorine atoms. The signal from two protons at δ 6.7 ppm in the spectrum of naphtholphthalein loses half of its intensity for MFNP and completely disappears for DFNP, indicating that these two protons represent the position of the fluorine-hydrogen substitution. Disappearance of the signal in the spectrum of DFNP also confirms that the two fluorine atoms are introduced into identical positions, which suggests a hydrogen–fluorine exchange in the naphthol rather than benzoic rings. This signal is located in the spectrum of naphtholphthalein in the most shielded field and therefore is the most prone to electrophilic attack. By modeling the NMR spectra in various online programs, we determined that this signal belongs to the proton located in the ortho position to the hydroxyl group and marked as 3 and 3′ on Fig. 5.
Fig. 4.

1H NMR spectra of naphtholphthalein (top) and its mono- (middle) and di- (bottom) fluorinated derivatives. Disappearance of the signal at δ 6.7 indicates the position of the fluorine-hydrogen substitution; assignment of other signals is discussed in the text.
Fig. 5.

A proposed mechanism for the reaction of naphtholphthalein fluorination. Trifluoroacetic acid converts the reagent into the cation form, which can survive direct fluorination producing mono- and difluorinated products. The formula of monofluorinated derivative has a numbering scheme for the carbon atoms, which is used in the text.
The effect of the introduction of fluorine atoms into the molecule of naphtholphthalein on other proton NMR signals supports the presumption that fluorine substitution occurs in the naphthol systems in the ortho position. Introduction of the electron-withdrawing fluorine atom causes a significant shift of the signals from the naphthol aromatic system without alteration of their shape. As both naphthol systems in DFNP are substituted and therefore identical, for these naphthol protons we should observe only shifted peaks with the same intensity as compared with naphtholphthalein. In contrast, both substituted and non-substituted naphthol systems are present in the molecule of MFNP. As a result, the signals from the naphthol protons should be doubled and halved in the spectrum of MFNP, and its spectrum should look like overlapped spectra of naphtholphthalein and DFNP. We observe this effect for four signals: doublets at δ 8.2 ppm (from protons at 5 and 5′ in Fig. 5) and δ 7.9 ppm (8 and 8′) and triplets at δ 7.1 ppm (6 and 6′) and δ 7.3 ppm (7 and 7′), thus also supporting the successive introduction of fluorine into the naphthol rings.
Signals from the benzoic ring do not demonstrate any change in intensity in the spectra of MFNP and DFNP, showing only a slight shift of the peaks to lower field due to successive addition of fluorine atoms into the molecule. This is observed for the doublet at δ 8.0 ppm (proton at 3″) and a group of peaks at δ 7.6–7.7 ppm (overlapping doublet from 6″ and two triplets from 4″ and 5″), and provides further evidence for the absence of substitution in the benzoic ring.
19F NMR spectra of MFNP and DFNP have only one signal at δ −144 ppm, which is also attributable to the fluorine atom located in the ortho- position to the hydroxyl group in the aromatic system. Based on these data, we can identify monofluoronaphtholphthalein (MFNP) as (RS)-3-(3-fluoro-4-hydroxynaphthalen-1-yl)-3-(4-hydroxynaphthalen-1-yl)isobenzofuran-1(3H)-one and difluoronaphtholphthalein (DFNP) as 3,3-bis(3-fluoro-4-hydroxynaphthalen-1-yl)isobenzofuran-1(3H)-one and as such propose a mechanism of fluorination of naphtholphthalein (Fig. 5) identical to phenolphthalein and phenolsulfonphthaleins [1–3].
The 1H NMR spectra of naphtholphthalein and its derivatives have one quite unusual feature: in all the cases we were not able to detect any distinct signal from the protons at carbon atoms 2 and 2′. On the other hand, we can see a broad signal in the range of δ 7.1–7.4, which might be attributed to these protons. 19F NMR spectra of MFNP and DFNP also differ from the data we observed for phenolphthalein derivatives. They contain only one broad fluorine signal, which does not demonstrate any splitting on the adjacent proton. Both these observations can be attributed to steric hinderance in the molecule of naphtholphthalein. Bulk naphthol substitute groups are not able to face each other, placing 2 and 2′ protons in very close position, subsequently increasing their relaxation time and potentially causing a coupling with variable distance, causing broadening of the signal.
5. Discussion
We were recently able to perform specific electrophilic fluorination of phenolsulfonphthalein [1] and cresolsulfonphthalein [2] with dilute fluorine gas in acetic acid solution. This reaction was used to synthesize new types of pH indicators, 18F-labeled derivatives of phenolsulfonphthalein, which have a potential for in vivo pH measurement in biological objects using Cerenkov imaging or combination of light absorption and PET [3].
Our initial attempts to accomplish the same fluorination for phenolphthalein were unsuccessful and resulted in non-specific destruction of the reagent [1]. The failure of the reaction was attributed to different structures of the molecules of these indicators in acetic acid solution. The acidic form of phenolsulfonphthalein exists as a zwitterion [6] with a positive charge distributed through the conjugated system of two phenol rings and a central carbon atom [1]. This structural feature decreases the overall energy of the fluorination reaction, promoting a specific substitution in the phenol rings rather than destruction of the molecule [2]. In contrast, at neutral and mild acidic conditions phenolphthalein exists as an uncharged cyclic lactone structure and does not survive an attack by the aggressive fluorine molecule.
Analyzing the literature we found [7] that under strong acidic conditions, such as in sulfuric acid, phenolphthalein forms a yellow solution, which contains its molecule in the form of positively charged ion. This charge is distributed through the conjugated phenol rings of phenolphthalein, forming a structure similar to the zwitterion of the uncharged form of phenolsulfonphthalein. This similarity led us to the conclusion that in very strong acidic conditions we might be able to avoid the non-specific destruction of the molecule and perform a specific electrophilic fluorination of phenolphthalein.
Trifluoroacetic acid is a much stronger acid in comparison with the acetic acid (with pKa of 0.5 vs. 4.76, respectively [8]), which we initially used as a solvent for the fluorination reaction. We found that trifluoroacetic acid could act similarly to sulfuric acid, dissolving phenolphthalein and forming a yellow solution. Addition of dilute fluorine gas to this solution validated our initial assumptions about the reactivity of the positively charged form of phenolphthalein. Instead of the destruction of the target molecule, which we observed in acetic acid, the reaction between dilute fluorine gas and phenolphthalein in trifluoroacetic acid resulted in formation of two major fluorination products (Fig. 3). We characterized these as mono- and difluorinated derivatives of phenolphthalein, which were produced with quite significant chemical yields, 10–15% and 1.5–2%, respectively. We can thus expand the list of novel 18F-labeled indicators with the addition of various derivatives of phenolphthalein and other similar phthaleins. These results also support our initial presumption [1,2] that the presence of the positive charge in phenolsulfonphthalein derivatives is a necessary condition for their successful specific fluorination, making strong acidic conditions suitable for reactions of electrophilic fluorination [9].
In general, phenolphthalein demonstrates lower stability toward fluorination reaction in comparison with phenolsulfonphthalein derivatives. Although it has four available ortho-positions, it forms only two fluorination products (vs. up to four for phenolsulfonphthaleins [2,3]) with relatively lower yields. Nevertheless, the reaction leads to the synthesis of fluorinated products in amounts sufficient for their practical application.
Incorporation of the fluorine atoms into molecule of phenolphthalein alters the properties as indicators in a similar fashion to that observed for phenolsulfonphthalein derivatives. The value of pKa for phenolphthalein is equal to 9.75 [10] and the maximum wavelength of absorption of its basic form is 553 nm. Successive incorporation of fluorine atoms into molecule of phenolphthalein decreases its pKa value to 9.33 and 8.70 and causes a bathochromic shift of the basic form maximum absorption to 558 nm and 564 nm, respectively. Although these compounds are not ideal for pH measurements in biological objects, our ability to perform the reaction itself is quite a fascinating result. Through the use of the same reaction we can almost certainly synthesize other indicators of this class, namely fluorinated derivatives of cresolphthalein and thymolphthalein. This, however, is of only theoretical interest as the effects of methyl and fluorine substituting groups on pH and absorption are additive [2], and as such these indicators should have even higher values of pKa.
The reaction of α-naphtholphthalein fluorination is similar to that of phenolphthalein. Naptholphthalein was also destroyed by electrophilic fluorination in acetic acid, but in trifluoroacetic acid naphtholphthalein forms a blue-green solution, which produced only two fluorinated products in the reaction with dilute fluorine gas. Given the various different ways in which the fluorination could occur in the naphthol rings, this is quite an unexpected result. Nevertheless, our results indicate an incorporation of only one fluorine atom per naphthol group into the ortho position to the hydroxyl group. A lack of products with higher number of fluorine atoms is most likely associated with the lower stability to oxidation of trialylphthaleins as compared to triarylsulfonphthaleins.
Fluorinated derivatives of α-naphtholphthalein demonstrate a very high potential for their practical applications. As discussed previously [3], 18F-labeled pH indicators represent a new type of probe for in vivo investigation of hydrogen ion concentration. These compounds have three potential modes of detection: pH-dependent optical absorption, pH dependent Cerenkov emission [11], and concentration-dependent γ-radiation by PET technique. The combination of the 3-dimensional distribution of the basic form of the indicator with the PET image of its concentration will lead to in vivo 3D mapping of pH in biological objects. A functional probe for this kind of investigation should contain a radioactive PET label (18F) within the indicator molecule with an optimal value of pKa (between 6 and 8), and an appropriate absorption maximum of the basic form (above 600 nm.) The latter limitation considers the presence of porphyrin-based molecules in biological objects. For example, hemoglobin has strong absorption peaks for both Hb (555 nm) and HbO2 (542 nm and 577 nm) [12] and will interfere with the color change of indicator in the range of 500–600 nm. The parental compound α-naphtholphthalein has pKa = 7.95 and absorption maximum of the basic form at 653 nm, which makes it more suitable for biological objects in comparison with phenolphthalein and phenolsulfonphthalein derivatives. Introduction of fluorine atoms into the molecule of α-naphtholphthalein leads to additional improvement of its properties, since the absorption maxima of the basic form undergoing a bathochromic shift to 659 nm for MFNP and 665 nm DFNP, moving them even further away from the porphyrins. Moreover, the values of pKa are shifted to 7.66 for MFNP and 7.24 for DFNP, meaning that the strongest color change occurs in the range of physiological pH. This combination of the properties makes both compounds particularly useful for in vivo pH measurement in biological objects.
6. Conclusions
Direct fluorination of phenolphthalein and naphtholphthalein with [18F]-F2 in acidic conditions leads to synthesis of 18F-labeled mono- and difluorinated derivatives. The fluorinated products act like pH indicators with decreased values of pKa and a bathochromic shift of absorption maxima for their basic forms. Derivatives of phenolphthalein demonstrate a shift of pKa to 9.33 and 8.70 and λbasic to 558 nm and 564 nm for mono- and difluorinated compounds, respectively. Fluorination of α-naphtholphthalein results in the synthesis of two compounds with optimal values of pKa (7.66 and 7.24) and high wavelengths of absorption (659 nm and 665 nm). Both mono- and difluorinated derivatives of α-naphtholphthalein have very high potential for in vivo pH measurement in biological objects.
Acknowledgements
Supported by the Institute for Translational Medicine and Therapeutics (ITMAT) Transdisciplinary Awards Program in Translational Medicine and Therapeutics Translational Biomedical Imaging Core Grant (TAPITMAT-TBIC) UL1RR024134 from the National Center For Research Resources and by Grant # IRG -78-002-31 from the American Cancer Society. The authors would like to thank Diana Sardelis and Charles N. McEwen at the University of the Sciences for help with mass-spectroscopy.
References
- 1.Kachur AV, Popov AV, Karp JS, Delikatny EJ. Cell Biochem. Biophys. 2012 doi: 10.1007/s12013-012-9390-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kachur AV, Sardelis D, Bentzley C, Popov AV, Delikatny EJ, Karp J. J. Fluorine Chem. 2013;145:112–117. doi: 10.1016/j.jfluchem.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kachur AV, Delikatny EJ, Karp JS. J. Nucl. Med. 2012;53(S1):187. [Google Scholar]
- 4.Barnes MD, La-Mer VK. J. Am. Chem. Soc. 1942;64:2312–2316. [Google Scholar]
- 5.Dolbier WR. Guide to Fluorine NMR for Organic Chemists. Hoboken, NJ: John Wiley & Sons, Inc.; 2009. [Google Scholar]
- 6.Tamura Z, Maeda M. Yakugaku Zasshi. 1997;117:764–770. doi: 10.1248/yakushi1947.117.10-11_764. [DOI] [PubMed] [Google Scholar]
- 7.Hopkinson AC, Wyatt PAH. J. Chem. Soc. B. 1970:530–535. [Google Scholar]
- 8.Dean JA, editor. Lange’s Handbook of Chemistry. 13th ed. New York: McGraw-Hill Book Company; 1985. [Google Scholar]
- 9.Dolbier WR, Li AR, Koch CJ, Shiue CY, Kachur AV. Appl. Radiat. Isot. 2001;54:73–80. doi: 10.1016/s0969-8043(00)00102-0. [DOI] [PubMed] [Google Scholar]
- 10.Lalanne JR. J. Chem. Educ. 1971;48:266–268. [Google Scholar]
- 11.Czupryna J, Kachur A, Blankemeyer E, Popov A, Karp J, Delikatny E. J. Nucl. Med. 2012;53(S1):73. [Google Scholar]
- 12.Horecker BL. J. Biol. Chem. 1943;148:173–183. [Google Scholar]