

Abstract
Current models hold that serum antibody titers are maintained chiefly by long-lived bone marrow (BM) plasma cells (PCs). Here we characterize the role of subpopulations of BM PCs in long-term humoral responses to T-cell dependent antigen. Surprisingly, our results indicate that 40–50% of BM PCs are recently formed cells, defined in part by rapid steady state turnover kinetics and secretion of low affinity immunoglobulin-M (IgM) antibodies. Further, for months after immunization with a hapten-protein conjugate newly formed antigen-induced IgM-secreting BM PCs were detected in parallel with longer-lived IgG-secreting cells, suggesting ongoing and parallel input to the BM PC pool from two distinct pools of activated B cells. Consistent with this interpretation, IgM and IgG antibodies secreted by cells within distinct PC subsets exhibited distinct light chain usage. We conclude that long-term antibody responses are maintained by a dynamic BM PC pool comprised of both recently formed and long-lived PCs drawn from clonally disparate precursors.
Keywords: B cells, antibodies, cell differentiation, memory, plasma cells
INTRODUCTION
Conventional wisdom holds that lasting serum antibody titers reflect the activity of long-lived plasma cells (PCs) localized in specialized niches in the bone marrow (BM) (1). It is generally assumed that the vast majority of BM PCs are long-lived and derived from germinal centers (GCs), specialized microenvironments enriched for antigen-stimulated B cells undergoing class switch recombination (CSR), somatic hypermutation (SHM), and affinity-driven selection (2). However, BM PCs also arise via GC-independent pathways (3–6), and many memory B cells also arise without maturing in GCs (7, 8). However the extent to which the GC-driven and GC-independent pathways contribute to long-lived BM PC pools is unknown.
Newly formed PCs are thought to enter the BM as immature cells where they must compete for limited survival niches in the BM (1). This possibility is consistent with three reports describing immature plasmablast-like PCs in the BM (5, 9, 10). Furthermore, it has been proposed that newly formed PCs must supplant previously generated PCs to achieve longevity (11). However, to our knowledge a comprehensive study of the cellular dynamics of the BM PC pool together with an assessment of the kinetics and duration with which recently induced PCs enter and persist in the BM has not been performed.
Our work examines the cellular dynamics of distinct BM PC populations and their role in long-lived humoral immunity. Surprisingly, at steady state more than 40% of antibody-secreting BM PCs share multiple features with immature PCs in the spleen or lymph nodes, as these cells express the B cell surface antigens B220 and CD19, exhibit a rapid 50% renewal rate of 2–3 days, and possess relatively low concentrations of the PC-requisite transcription factor Blimp-1 (10). These results indicate that a considerable fraction of the BM PC pool relies on routine input from peripheral tissues. Equally surprisingly, upon immunization of C57BL/6 (B6) adults with the hapten-protein conjugate nitrophenyl-chicken γ-globulin (NP-CγG), some 50% of NP-specific PCs were detected within the immature BM PC subset for months post-immunization, suggesting that antigen depots drive the generation of new PCs long after immunization. Furthermore, although NP-specific responses in B6 mice are dominated by a single clone defined in part by utilization of λ1 Ig light chains, nearly all cells within the newly formed B220+ fraction of BM PCs secreted IgM+κ+ antibodies. These results suggest that IgM+ memory cells produce PCs continuously throughout much of the primary immune response. Altogether these results indicate that lasting antibody responses are mediated by the combined activity of immature and mature BM PCs derived from clonally distinct pools of activated B cells.
MATERIALS AND METHODS
Mice
C57BL/6 (B6) females (age 8–10 weeks) were obtained from Jackson Laboratories and housed in our colony for at least 2 weeks before analysis. B6.Blimp1+/GFP mice (10) were kindly provided by Dr. Mark Pescovitz (Indiana University) with permission from Dr. Stephen Nutt (WEHI). All animal procedures were approved by the University of Pennsylvania Office of Regulatory Affairs.
Flow cytometry and cell sorting
Spleen and BM cells were harvested and stained with the indicated antibodies as described (12). Unless noted otherwise all reagents were purchased from eBiosciences: FITC-anti-IgM (R26-46, Pharmingen), and PNA (Sigma); phycoerythrin (PE)-anti-CD138 (281-2, Pharmingen); PE- TexasRed-anti-B220 (RA3-6B2); PE-Cy7-anti-CD4 (RM4-5), anti-CD8& (53-6.7), anti-Gr-1 (RB6- 8C5), anti-F4/80 (BM8), and anti-TER119; allophycocyanin (APC)-Cy5.5-anti-CD19 (1D3); Alexa405 anti-IgD (11-26), and Biotin-anti-CD138 (281-2, Pharmingen). Biotinylated antibodies were revealed with Streptavidin-APC-Cy7 (Pharmingen). APC-NP was conjugated in our laboratory. Nonviable cells were eliminated from analyses with the UV-excited DNA dye DAPI (Molecular Probes), and doublets were excluded based on the width of the forward and side scatter signals. For intracellular stains, live cells were identified by pre-incubation with AquaLIVE/DEAD fixable live/dead stain (Invitrogen). Cells were fixed and permeabilized using solutions A and B (Caltag). For PC and B cell subset labeling in vivo, B6.Blimp1+/GFP adults were given a single intravenous injection of 0.4 μg PE-anti-CD138 or PE-anti-CD19, then sacrificed 2 minutes later. Resulting cell preps were then subjected to standard cell surface staining protocols. Flow cytometry was performed on a BD LSRII and cell sorting was performed on a four laser Aria (Becton Dickinson). Analysis was done using FlowJo8.8 (Tree Star, Inc.).
In vivo BrdU labeling
Adult B6 mice were fed drinking water containing 0.5mg/ml BrdU and 1mg/ml sucrose. Flow cytometric analysis of BrdU incorporation was accomplished as previously described (12) using FITC-anti-BrdU antibodies (Becton Dickinson).
Computational modeling
The maximum number of actively dividing PCs based on BrdU pulse-chase labeling data was estimated with the equations listed below.
For the increasing parts of the curve:
And for the decreasing parts of the curve:
Where U and L indicate unlabeled and labeled cell numbers, respectively, and p and d represent proliferation and death rates (1/time). These formulas can be converted into the following fractions:
Because there are only two components, Uf(t) + Lf(t) = 1. Results from these calculations for the increasing part of the curve are shown in supplemental Table 1.
Immunizations
8–12 week old mice were immunized intraperitoneally (i.p.) with 50μg NP16-CGG in alum. Multiscreen HTS plates (Millipore) were coated with 10μg/well of either Goat anti-Mouse Ig(H+L) (Southern Biotech), or NP33-BSA, or NP4-BSA (BioSearch) in sodium bicarbonate buffer.
ELISPOT and ELISA analyses
For ELISPOTs, cells were serially diluted across the plate, and then incubated for 4–8 hr at 37°C. Biotin-Goat anti-IgG, Goat-anti-IgM, or Goat-anti-IgG1 (Southern Biotech) diluted in block buffer was added, followed by three washes with 0.1% Tween-20 detergent, and a secondary incubation with ExtrAvidin-alkaline phosphatase (Sigma). Spots were detected using BCIP/NBT (Sigma) and scanned and counted with an ImmunoSpot Analyzer (Cellular Technology Ltd.). To assess frequencies of hapten-specific PCs within defined subsets, 2–20 × 103 B220+ CD138high or B220− CD138high BM cells were sorted into ELISPOT plates, and the fraction of these cells secreting NP-specific antibodies determined as described above. This number was multiplied by the overall frequency of cells within the relevant subpopulation as a function of all BM cells followed by the total number of BM cells as determined by Opstelten and Osmond (13). For ELISAs, 2-fold dilutions of sera from immunized or control animals were added to NP26-BSA coated 96-well plates and processed via standard procedures. Plates were developed with HRP-conjugated heavy or light-chain isotype-specific antibodies at optimal dilutions. After subtracting average background readings, titers were calculated as the greatest dilution to achieve an optical density of 0.1.
Cell morphology
104 sorted cells were centrifuged onto microscope slides (800rpm, 3min) with a Cytospin, fixed in ethanol (95% EtOH for 10 min, 70% EtOH for 30 sec) and stained with eosin for 3 min followed by hematoxylin for 1 min. Images were obtained on a Nikon Eclipse TE2000-U microscope and captured using a Nikon DA-Fi2 camera.
Statistical Analysis
Significances in differences in plasma cell frequencies between two experimental groups were evaluated with the unpaired two-tailed t-test using Excel software.
RESULTS
Resolution of three bone marrow plasma cell subsets
We noted recently that many NP-specific PCs in the BM of NP-LPS immunized mice retain expression of the naïve B cell surface protein B220 (3). Since B220 and CD19 expression are thought to be down-regulated during early phases of PC differentiation (14), this observation, together with other recent reports of BM PCs with properties of immature antibody-secreting cells (5), led us to evaluate B220 and CD19 surface expression on all BM PCs. To identify rare BM PCs effectively, we first focused on cells co-expressing the PC-associated surface protein CD138 (15) and Blimp-1 in adult B6.Blimp1+/GFP reporter knock-in mice that were not immunized intentionally (10). Because some T cells are Blimp1low (16, 17), we intentionally excluded TcRβ+ cells from these analyses. As shown, in B6.Blimp1+/GFP adults some 60% of BM PCs (TcRβ− Blimp1/GFP+ CD138high) co-expressed B220 and CD19 (Fig. 1A), and surface CD19 expression was also readily evident among approximately two-thirds of the remaining B220− Blimp1/GFP+ CD138high cells. We also resolved three populations of CD138high BM cells in conventional B6 adults defined by differential B220 and CD19 surface expression (Fig. 1B). These included B220+ CD138high cells (gate b) that were also CD19+ (corresponding to the B220+ CD19+ population illustrated in Figure 1A, and B220− CD138high cells that consisted of both CD19+ (gate c) and a minor CD19− population (gate d) corresponding to the B220− CD19+ and B220− CD19− subsets in Figure 1A. Note: although the levels of surface B220 expression on the BM PC populations defined here are somewhat lower than typically observed on mature B cells and encompass a broad range in surface protein expression, for simplicity we will refer to these PC subsets here as B220+ and B220−.
Figure 1. Resolution of three populations of BM PCs.

(A) BM cells from an 8-week-old female B6.Blimp1+/GFP mouse were stained with the indicated antibodies before analysis of 2 × 106 events. The left-most plot is pre-gated on DAPI− cells. Arrows indicate each successive gate. “Dump” antibodies were F4/80, Gr-1, Ter-119, CD4, and CD8α. Representative of four separate analyses. (B) BM cells from an 8-week-old female B6 mouse were stained and analyzed as in (A). Representative of ten separate analyses. (C) Cells from the indicated populations were sorted and added in triplicate to ELISPOT plates coated with anti-mouse Igκ antibodies. Representative of two separate experiments. (D) cDNA prepared from CD3+ T cells, splenic CD23+ follicular (FOL) B cells, FOL B cells stimulated with LPS + IL-4 for three days, or cells within the indicated BM PC subsets (B) were sorted from adult B6 mice and subjected to RT-PCR using Taqman probes for Blimp1 (Prdm1), Pax5, or GAPDH. Resulting signals were normalized to FOL B cells. (E) Cells were sorted from adult B6 mice using the gates shown in (B) and stained eosin and hematoxylin as described in Methods. (F) BM or spleen cells from an 8-week-old female B6.Blimp1+/GFP mouse were stained as in (B), and relative GFP expression determined using the gating strategies shown in (B) and summarized in the legend below.
Based on ELISPOT analyzes with cells sorted from B6 adults all three CD138high subpopulations were highly enriched for cells characterized by active antibody secretion (Fig. 1C). These cells are clearly distinct from a previously described B220low CD138low pre-B cell fraction found in gate a in Figure 1B (18), as this population lacked cells secreting antibody and Blimp-1 expression (Fig. 1C, F). We should also note that B220+ CD138high BM PCs do not appear to correspond to the pre-PCs described in Ig transgenic mice, because pre-PCs do not actively secrete antibodies (19). As expected, when derived from B6 adults, cells within these subpopulations exhibited substantial Prdm1 (Blimp1) transcript abundance, although Prdm1 transcripts were lower for B220+ CD138high cells. These populations also exhibited minimal transcript levels for the B-lineage master transcription factor Pax5, which is down regulated upon induced PC differentiation (Fig. 1D) (14). Cells within the CD138high B220+ and CD138high B220− fractions also exhibited cell morphology consistent with full PC differentiation (Fig. 1E). Finally, when we applied the gating strategy illustrated in Figure 1B to BM cells derived from a B6.Blimp1+/GFP adult, it was clear that cells in all three BM CD138high subpopulations exhibit substantial levels of Blimp1 expression (Fig. 1F), although it should be noted that cells within the CD138high B220+ BM fraction possessed significantly lower Blimp1/GFP levels compared to their B220− counterparts in the BM yet similar levels to immature splenic B220+ PCs. Together these data indicate that BM PCs can be subdivided into at least three subsets based on differential B220 and CD19 surface expression. Furthermore, data revealing relatively low Blimp1 expression for B220+ CD138high BM cells suggest that these cells are the least mature PCs within the BM PC pool (10).
The majority of B220+ BM PCs are recently formed
Past work has shown that immature splenic PCs label with rapid and linear kinetics, achieving near 100% labeling within 3 days (20). Accordingly we defined steady state cellular renewal rates for each BM PC subpopulation using continuous in vivo BrdU labeling. We gave cohorts of B6 adults BrdU for up to 60 days, and determined the proportion of BrdU+ cells for the total BM PC pool as well as for each BM PC subset at multiple time points. Small non-dividing pre-B cells (FSClow B220low CD43− IgM−), which exhibit near complete cellular turnover every 3 days (21), were used to control for the efficiency of BrdU labeling. As shown (Fig. 2A), some 30% of the total BM PC pool became BrdU+ within 5 days, and within 25 days just over 40% were BrdU+. As expected, within 3 days small pre-B cells were nearly 100% BrdU+. Most notably, when subdivided based on B220 surface expression, B220+ PCs in the BM exhibited markedly rapid labeling kinetics, achieving 80% labeling within 5–6 days with a 50% renewal rate of 2–2.5 days (Fig. 2B). These labeling kinetics are comparable to extrafollicular splenic PCs (22). In contrast, labeling rates for B220− BM PCs were relatively protracted, reaching 35% BrdU+ by day 25, then plateauing at later time points. Labeling kinetics for B220− CD19+ and B220− CD19− PCs were indistinguishable from one another.
Figure 2. Many BM plasma cells are recently formed.

(A) B6 mice were fed BrdU for the indicated days before determination of the % BrdU+ cells among all Dump− IgD− CD138high BM cells. Small pre-B cells were gated as FSClow B220low AA4+ IgM− cells. Best-trend lines were drawn across the mean % BrdU+ cells for each population using 3–4 mice per time point. (B) The flow cytometric data in (A) were gated as shown in Figure 1B to determine the fraction of BrdU+ cells in the indicated populations at each time point. (C) B6 adults were given BrdU for up to six days, and then given BrdU-free drinking water (chase) for another 12 days. The fraction BrdU+ cells in each BM population was determined as in (A) and (B). (D) BM cells from 12-week old B6 mice were stained to resolve large pre-B cells (FSChigh B220low AA4+ IgM−) or PC subsets based on differential B220 expression, fixed and permeabilized, and then stained with anti-Ki67 antibodies and DAPI. (E) Data summarized for 3 mice using gates shown in (D). Representative of two separate experiments. (F) 8-week old B6 mice were inoculated with BrdU. Sixty minutes later BrdU+ cells were identified among the indicated BM and spleen cell populations. GC B cells were identified as CD19+ IgD− CD38− PNAhigh cells.
To examine the turnover kinetics and potential precursor-successor relationships for these BM PC subsets more closely, we performed pulse-chase experiments in which mice were fed BrdU for 6 days, with per-cent labeling assessed at several time points before and after terminating BrdU labeling. As shown (Fig. 2C), during the chase period the fraction of BrdU+ cells within the B220+ BM PC fraction dropped from 80% to under 20% within 4 days, reaching background levels by day 6. Surprisingly, although B220+ PCs comprise some 50% of the BM PC pool and were 80% BrdU+ on day 6, we detected few if any BrdU+ cells within the B220− fraction during either the labeling or chase periods. These data suggest that, at steady state, relatively few B220+ BM PCs give rise to longer-lived B220− PCs.
Because early PC populations may include dividing cells (23), we used two separate strategies to assess proliferation among B220+ and B220− BM PCs. First, we measured the DNA content of cells within each subset with the DNA dye DAPI together with Ki67 antibodies. Recent work has shown that this combined approach allows resolution of cells in the G0 (Ki67−, 2N), G1 (Ki67+, 2N), and S + G2/M (Ki67+, >2N) phases of the cell cycle (24). We were unable to detect cells in the S or G2/M phase within any BM PC population (Fig. 2D, E), although cell division was readily apparent in large pre-B cells (FSChigh B220low CD43− IgM−) (25). Because exposure to BrdU can induce division of BM progenitors (24), we also performed these analyses with mice given BrdU for 6 days; however exposure to BrdU did not alter Ki67 or DNA content profiles for any BM PC subpopulation (not shown). Second, we employed a 60-minute in vivo BrdU pulse strategy shown to label dividing B-lineage cells selectively and efficiently (26). Whereas this approach led to the identification of proliferative GC B cells and a small fraction of BrdU+ PCs in the spleen, we were unable to detect BrdU+ B220+ PCs in the BM with this approach (Fig. 2F and supplemental Figure 1). Together these data suggest that B220+ BM PCs are largely non-proliferative cells derived from proliferative precursors.
To evaluate the role of cell division in the homeostasis of BM PC populations more closely, we employed mathematical modeling algorithms formulated to determine the minimal degree of cell division required to explain the BrdU labeling kinetics observed for B220+ BM PCs (see supplement) (27). When we fit the on or off labeling segments of the BrdU pulse-chase data in Figure 2C to a simple single compartment proliferation model (see methods), we find that for the observed BrdU labeling kinetics to reflect active cell division, 16–30% of B220+ BM PCs would need to undergo cell division every day. Because the observed fraction of actively dividing cells is far less, we conclude that most newly formed BM PCs are non-dividing cells derived from actively dividing precursors, as described previously for memory B cells (27). When considered together, the BrdU labeling and cell cycle analysis data allow two conclusions. First, we conclude that 40–50% of BM PCs are replaced by incoming cells every 5–6 days. Second, given the non-linear nature of the labeling kinetics obtained for B220− BM PCs (Fig. 2B), we suggest that this PC fraction contains at least 2 subpopulations characterized by intermediate versus lengthy lifespans.
Immature BM PCs are not enriched in marrow sinusoids
One possibility is that B220+ PCs harvested from BM cell preparations are localized in sinusoids in situ, allowing for ready recirculation throughout the vasculature. Alternatively, B220+ BM PCs may be positioned within bona fide PC niches in the BM parenchyma. To distinguish these possibilities, we adopted an in vivo antibody labeling approach used previously to identify B-lineage progenitors localized to sinusoids versus the BM parenchyma (28). With this strategy, cells localized within sinusoids become labeled only 2 minutes after intravenous injection of PE-labeled antibodies specific for relevant cell surface proteins, whereas within this time frame cells within the BM parenchyma remain unlabeled. We inoculated B6.Blimp1+/GFP adults with PE-anti-CD138 or PE-anti-CD19 antibodies, then 2 minutes later assessed PE labeling among B220+ and B220− Blimp1/GFP+ cells in the spleen and BM and pre-B and immature B cells in the BM. As shown (Fig. 3A, C), whereas a large fraction of splenic PCs became labeled with this approach, cells within the B220+ and B220− BM PC fractions remained unlabeled. By contrast, consistent with the notion that a considerable fraction of CD19+ immature B cells are found in BM sinusoids (28), immature BM B cells were readily labeled upon i.v. inoculation with anti-CD19 antibodies, but pro and pre-B cells, known to be localized within the BM parenchyma, were not (Fig. 3B, C). We conclude that B220+ BM PCs are not found within BM sinusoids.
Figure 3. B220+ marrow plasma cells are not located in bone marrow sinusoids.

(A, B) B6.Blimp1+/GFP adults were given a single i.v. inoculation of 0.4 μg of PE-CD138 or PE-CD19. Two minutes later mice were sacrificed and BM and spleen cells stained with the indicated antibodies (excluding CD138 and CD19) before flow cytometric analysis of 2.5 × 106 events. (A) Representative CD138 labeling of spleen (top) or BM (bottom) cells is shown. Left-most plots are pre-gated on viable Dump− IgD− cells as in Figure 1. Overlay histograms illustrate CD138 signal on the indicated PC subsets (black line), with the indicated B220high Blimp1/GFP− gates used to establish background signal (gray filled). (B) CD19 labeling for BM pro/pre-B cells (GFP− B220low IgM−) and immature (GFP− B220+ IgMhigh IgDlow) B cells. Gray filled curves are from mice that were not inoculated with PE-labeled antibodies. (C) Mean and SEM of % labeled cells within the indicated populations, with 3–5 mice per group summarized from two separate experiments.
Role of B220+ BM PCs in long-term responses
Next we probed the roles played by B220+ and B220− BM PCs in induced T cell dependent antibody responses. To establish the kinetics with which PCs enter and persist within each population, we quantified NP-specific PCs among sorted B220+ and B220− BM PCs at several time points between 6–130 days after a single inoculation with NP-CγG (Fig. 4A). Remarkably, we detected hapten-specific cells in both pools at all time points examined, even out to day 130, although after 100 days somewhat fewer NP-specific PCs localized to the B220+ BM fraction (Fig. 4A). When considered in tandem with the BrdU labeling data in Figure 2, these data suggest that recently formed antibody-secreting cells continue to seed the BM PC pool long after a single immunization. To test this idea further, we immunized additional mice with NP-CγG, and 30 days later added BrdU to their drinking water for an additional 7 days before assaying for BrdU+ NP-binding BM PCs. As shown (Fig. 4B), some 9% of all detectable NP-binding BM PCs were BrdU+, and as expected these cells were B220+. We conclude that newly formed PCs contribute to the BM PC pool well after initiation of T cell dependent humoral responses.
Figure 4. Antigen-induced PCs colonize the B220+ BM PC subset for months post-immunization.

(A) Adult B6 mice were immunized with NP-CγG, and at the indicated time points B220+ and B220− BM PCs were sorted into NP26-BSA coated ELISPOT plates that were subsequently probed with anti-Igκ and Igλ antibodies. Numbers of hapten-specific PCs in the BM at each time point were calculated as described in Methods. Error bars represent SEM of triplicate ELISPOT wells. Background has been subtracted out. *, p<0.05. (B) B6 mice were immunized with NP-CγG. At d30 post-immunization mice were fed BrdU (or not) for an additional 7 days, and BM CD138high B220+/− PCs evaluated for intracellular NP-binding and BrdU incorporation. 8 × 106 events were collected per file.
Past studies indicate that antigen-activated or antigen-experienced B cell populations can be identified in peripheral tissues long after exposure to antigen and without maturing in GCs. For instance, Hsu et al detected extrafollicular plasmablasts for extended periods in the spleens of mice immunized once with the type 2 TI antigen NP-Ficoll (29), and recent findings indicate that many memory B cells are GC-independent IgM+ cells (7, 8), and hence consist mainly of cells bearing relatively low-affinity antigen receptors. Therefore we considered the possibility that some BM PCs that form late in the NP-CγG response might not secrete high-affinity class-switched antibodies. Accordingly, we examined the heavy chain isotype of antibodies secreted by B220+ and B220− BM PCs at multiple time points ranging from 27–134 days post-immunization with NP-CγG. To measure PCs secreting high affinity class switched antibodies, we used ELISPOT plates coated with proteins with a low NP-carrier molar ratio (30). Strikingly, whereas the B220− fraction contained cells secreting either IgM or high affinity IgG antibodies, within the B220+ fraction hapten-specific PCs were comprised almost exclusively of IgM secreting cells (Fig. 5A–B). These results indicate that B220+ and B220− BM PCs play functionally distinct roles in maintaining antibody titers in response to NP-CγG, while further suggesting that antibody-secreting cells in each pool arise from clonally unrelated precursors. To probe the latter notion further, we performed additional experiments to characterize Ig light chain usage by NP-specific PCs in each population, again at several time points post-immunization. As shown in Figure 5C, whereas hapten-specific PCs within the B220− BM PC fraction secreting antibodies utilizing either Igκ or Igλ light chains, beginning 50 days post- immunization NP-specific antibodies secreted by B220+ BM PCs were almost exclusively Igκ+. The failure to detect Igλ+ PCs among the B220+ BM fraction at later time points may be due to the failure of GCs induced by NP-CγG immunization to persist for extended periods (31), but may also reflect an exceptionally long-lifespan for IgM+ memory B cells suggested by several recent studies (8, 31, 32). Finally, as expected early post-immunization NP-specific serum antibodies were mainly Igλ+. However, consistent with our ELISPOT data, by 50 days post-immunization a considerable fraction of NP-specific serum antibodies were Igκ+ (Figure 6).
Figure 5. Long-term colonization of the BM by IgM-secreting PCs.

(A) BM PC subsets were sorted and assayed at the indicated time points post-immunization with NP-CγG as described in Figure 3A using ELISPOT plates coated with either NP26-BSA or NP4-BSA. These plates were probed subsequently with anti-μ chain or anti-γ chain antibodies, respectively. *, p<0.01. (B) Separate experiment wherein sorted BM PC subsets were assayed by ELISPOT at the indicated time points post-immunization using NP4-BSA-coated plates and anti-γ chain antibodies. *, p<0.01. (C) Additional experiment wherein the indicated BM PC subsets were sorted and assayed for secretion of NP-specific antibodies at the indicated time points using NP26-BSA-coated plates and anti-κ or anti-λ antibodies. Numbers of hapten-specific PCs in the BM at each time point were calculated as described in Methods. *, p<0.01.
Figure 6. Kinetics of serum NP-specific antibody responses.
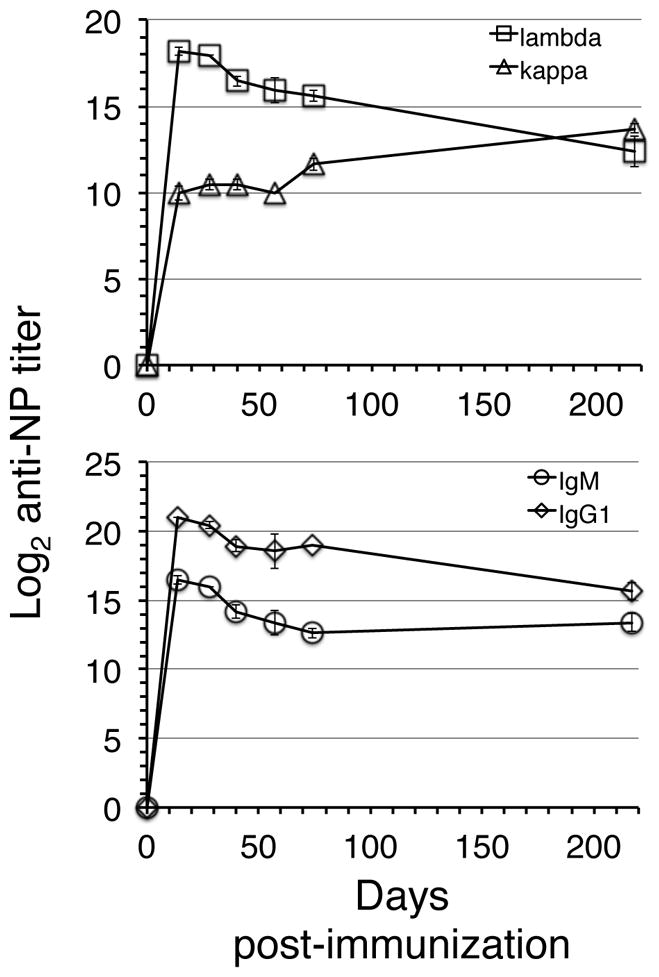
Adult B6 mice were immunized with NP-CγG. On the indicated days post-immunization serum samples were evaluated for relative titers of NP-specific Igλ (squares), Igκ (triangles), IgM (circles), or IgG1 (diamonds)-bearing antibodies by ELISA as described in Methods. Data are means and SEMs established from 3–4 individual mice at each time point.
Candidate memory B cell populations
The predominance of cells secreting IgMκ+ antibodies in the NP-specific B220+ BM fraction led us to examine light chain usage among peripheral NP-specific IgM+ memory cells using criteria for memory B cells established by Shlomchik and colleagues (33). Although rather infrequent, we did detected NP-binding IgM+ IgD− CD19+ CD38+ splenic B cells in mice immunized with NP-CγG 57 days previously. Notably, 40–50% of these cells were κ+ (Fig. 7A), and upon exposure to BrdU for 5 days 25% of NP-binding κ+ cells were BrdU+ (Fig. 7A). Consistent with their designation as memory cells, NP+ IgM+ CD38+ κ+ B cells also exhibited surface expression of CD73 and PDL-2, although CD73+ cells were less frequent among IgM+ cells compared to their IgM− counterparts (Fig. 7B). Surprisingly however, we were unable to detect BrdU+ NP-binding kappa+ cells in 60 minute pulse experiments using the approach described in Figure 2F, indicating that this population is also not enriched for actively dividing cells. Nonetheless, based on the data in Figures 4–7 together with the BrdU chase data in Figure 2B, we conclude that enduring antibody responses to NP-CγG are mediated by a combination of long-lived GC-derived and recently generated BM PCs, with the latter population derived chiefly from IgM+ memory B cells.
Figure 7. Resolution of NP-specific kappa+ IgM+ memory B cells.

(A) Adult B6 mice were immunized with NP-CγG. On day 52 mice were given BrdU for 5 days, and on day 57 splenocytes were stained with the indicated reagents to evaluate Igκ surface expression and BrdU incorporation. Numbers in bottom of kappa vs. BrdU plot indicate the mean and standard deviation for the % BrdU+ cells among NP-binding IgMκ+ B cells for 5 mice. All plots representative of 5 mice. (B) Separate experiment evaluating CD73 and PDL2 surface expression on IgM+ and IgM− NP-binding splenic B cells 60 days post-immunization with NP-CγG. Naïve IgD+ B cells were used as a comparison. Data in (A) and (B) each representative of two separate experiments. For all samples we collected 10 × 106 events/file.
DISCUSSION
In this work we characterized subpopulations of BM PCs and their roles in long-term humoral responses to a T-cell dependent antigen. Perhaps most notably, we find that approximately 40% of BM PCs are replaced by recently formed cells every 3–6 days. Thus, although past work introduced the notion that BM PC populations include immature cells (5, 10), we suggest that the BM PC pool is far more dynamic and heterogeneous than expected, with a substantial fraction consisting of recently formed cells. Indeed, based on our BrdU labeling and cell cycle data we propose the existence of three subpopulations of BM PCs: short-lived B220+ cells, B220− cells characterized by an intermediate half-life of 2–3 months, and a relatively rare population of long-lived B220− cells. We also find that hapten-specific BM PCs induced by NP-CγG immunization do not consist chiefly of cells secreting high-affinity IgG antibodies, rather our data reveal substantial diversity in Ig heavy chain usage by BM PCs. In fact, by summing frequencies of IgM and IgG secreting PCs across all BM PC subsets more than 70 days post-immunization, we estimate that 80% of all NP-specific BM PCs secrete IgM antibodies. Together these findings indicate that PCs derived from several source populations coexist in the BM where they are further characterized by a wide range of half-lives.
The complex nature of the BM PC compartment raises many questions about the factors regulating its size and the survival of individual cells within long-lived pools. While many factors may influence PC survival, the extensive heterogeneity among BM PCs raises the possibility that these factors target distinct PC subsets. For instance, PCs often locate adjacent to CXCL12-producing stromal cells in the BM (34). It would be informative to determine whether access to relevant stromal cells and the signals they deliver differs for rapidly versus slowly renewing and/or IgG- versus IgM-secreting BM PCs. Likewise, whereas depletion of cytokines such as BLyS and APRIL or major cell types that secrete these cytokines markedly reduces frequencies of IgG-producing BM PCs (35–37), it is not known whether APRIL also promotes the generation of immature IgM-secreting PCs or their entry into long-lived PC pools. It is tempting to speculate that many immature PCs fail to thrive due to a failure to gain access to APRIL and perhaps other factors. These general considerations apply to cell intrinsic regulators of PC differentiation and survival as well. One such example concerns the Blimp1 transcription factor encoded by Prdm1. Past studies showed that induced deletion of Prdm1 resulted in a roughly 80% drop in BM PC frequencies (38). Based on our data, we surmise that the decline in BM PC numbers in this study was likely due to the death of pre-existing PCs and an arrest in PC differentiation upon induction of Prdm1 deletion. It would be necessary to evaluate the impact of induced Blimp1 deletion on B220+ and B220− BM PCs to address this and related issues further.
Administration of BrdU in vivo has been an effective tool to identify proliferative cells and to define the steady-state dynamics of cell populations enriched for non-proliferative cells. With this approach, past work has defined steady-state renewal rates for a variety of non-proliferative B-lineage cell population such as immature IgM+ IgD−/low BM B cells and their immature/transitional counterparts in the adult spleen, where BrdU incorporation occurs mainly within highly mitotic large pre-B cells in the BM. Due to the rapid rate at which immature B cells in the BM and spleen either die or progress into more mature cells, cells in both pools achieve near 100% labeling within 3–4 days (39–41). By contrast, immature PCs in the spleen are thought to consist chiefly of actively dividing PCs or plasmablasts, and also exhibit rapid BrdU labeling rates (20, 22). Therefore rapid BrdU labeling may be due to active cell division, but may instead reflect rapid cellular turnover of cells within a population enriched for non-dividing cells. To address this potentially confusing issue we evaluated cell division among BM PC subpopulations directly (Fig. 2D–F). Our analyses indicate that, unlike in the spleen, relatively few PCs within the B220+ BM fraction are actively dividing. Therefore, we suggest that the bulk of B220+ BM PCs are immature but non-dividing PCs, derived from a proliferative precursor within B memory and/or early plasma cell pools in peripheral lymphoid tissues.
Our inability to detect BrdU+ B220− PCs in the BM after a 6-day pulse, despite labeling of the bulk of B220+ PCs within this time frame (Fig. 2C), suggests that most B220+ BM PCs fail to gain entry to long-lived niches. Why might so many of these cells fail to mature? As alluded to above, additional experiments are needed to establish the capacity of immature BM PCs to utilize survival factors needed to promote PC longevity and whether the signals they provide are indeed limiting. In this regard, an alternative but not widely considered view is that PC longevity is determined very early during differentiation, irrespective of the environment in which they settle. From this standpoint, PC lifespan would be determined by limiting signals available only to small numbers of antigen-responsive B cells or newly formed PCs at the inception of PC differentiation. In this scenario, only a small fraction of immature PCs would possess the capacity to receive and translate signals into the transcriptional and epigenetic changes needed for long-term survival. Alternatively, the BM may harbor distinct pools of PCs with unique roles in long-term protection. For instance, B220+ BM PCs may arise from specialized naïve B cells in the marginal zone and B1 compartments, as IgM-secreting PCs derived from these pools play unique and important functions in response to bacterial and viral pathogens (42, 43).
Whereas analyses of unimmunized animals revealed the steady-state cellular dynamics of BM PC pools, controlled immunization experiments allowed us to characterize the roles of the BM PC subsets in T-cell dependent humoral immunity. Two aspects of these results were rather unanticipated. First, we originally posited that B220+ BM PCs are a temporary reservoir for GC-derived PCs. However, NP-specific PCs in this pool lacked classic characteristics of GC-derived cells (Fig. 5), and we were able to capture such cells in this population after GC responses evoked by NP-CγG immunization wane (31). Indeed, our data suggest that IgM+ memory B cells contribute substantially to the BM PC pool long after initiation of antibody responses. These results are consistent with the notion that residual persisting antigen plays important roles in shaping the BM PC pool. However, our results may also reflect chronic stimulation of toll-like receptors on memory B cells as proposed by Lanzavecchia and co-workers (44). Second, despite the dominant role played by Igλ1+ cells in the NP-specific antibody response in B6 mice, at later time points nearly all hapten-specific IgM+ B220+ BM PCs employed Igκ light chains. Past work has shown that NP-specific hybridomas bearing Igκ light chains can be readily derived from B6 mice, provided that such mice are immunized multiple times (45). By contrast, limiting dilution experiments indicate that approximately 50% of the naïve NP-reactive B cell pool consists of Igκ+-bearing cells (46). Taken together these results suggest that, while both Igκ+ and Igλ1+ cells become engaged in responses to NP, the latter cells undergo a greater number of cell divisions during early phases of the primary response, and thus are more likely to become engaged with follicular helper T cells and form GCs where they eventually yield class switched cells. By contrast NP-responsive Igκ+ cells readily generate PCs via a GC-independent pathway (3), but are in general less likely to contribute substantially to classical GC responses.
We were surprised to find that cells secreting NP-specific IgMκ+ antibodies can be captured within the B220+ BM PC pool for more than 100 days post-immunization. We speculate that IgM memory cells possessing stem cell-like properties including limited self-renewal activity contribute to the BM PC pool over extended time frames post-immunization. Stimulation of CD8+ T cells by residual persistent minor alloantigens has been reported to drive the production of memory stem-like cells (47). These cells retain certain characteristics of naïve T cells, yet appear to divide slowly while also producing effector T cell populations. Memory IgM+ B cells may possess similar properties, provided that they are stimulated appropriately by residual antigen or other ligands with the capacity to evoke activation and PC differentiation. Clearly further work is needed to explore these issues.
Given the multi-layered nature of long-term antibody responses to NP-CγG, we are further tempted to speculate about potential advantages of functionally heterogeneous antibody responses. It has been shown that the BCRs expressed by memory B cells possess fewer SHM events compared to BM PCs (48). Moreover, many IgM+ memory cells arise independently of GC-associated maturation pathways (7). Consequently, as proposed by Purtha et al., memory B cells may be readily positioned to respond to alternative epitope structures such as those expressed by viral escape mutants (49). Therefore IgM+ memory cells may play a key role in defending against rapidly evolving viruses by quickly generating short-lived pools of B220+ BM PCs secreting anti-viral IgM antibodies in a manner analogous to the swiftness with which B cells in the splenic marginal zone generate antibodies to blood-borne bacteria (43, 50).
In sum, we found unexpectedly that the BM PC compartment is rather heterogeneous, both with respect to rates of turnover for discreet subpopulations of PCs, and the types of antibodies these cells secrete. We suggest that this heterogeneity reflects the multilayered nature of the antigen-responsive peripheral B cell pool, with input from GC B cells and memory B cells derived from this compartment, and B cells with minimal or no participation in GCs such as IgM+ memory, marginal zone, and perhaps B1 B cells. Understanding the combined roles played by discrete pools of activated and memory B cells and the PCs they produce in fending off pathogens and in autoimmunity hopefully leads to improved vaccines and strategies to constrain the activity of pathogenic PCs.
Supplementary Material
Acknowledgments
We thank Drs. Michael Cancro, Avinash Bhandoola and Stephen Emerson for helpful discussions.
Footnotes
This work was supported by the NIH grants RO1AI097590 (D.A.), F30HL112628 (I.C.), T32CA009140 (D.D.J.), and T32 AI070099 (A.B.).
The authors have no financial conflicts of interest.
References
- 1.Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dorner T, Hiepe F. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. 2006;6:741–750. doi: 10.1038/nri1886. [DOI] [PubMed] [Google Scholar]
- 2.Tarlinton DM. Evolution in miniature: selection, survival and distribution of antigen reactive cells in the germinal centre. Immunol Cell Biol. 2008;86:133–138. doi: 10.1038/sj.icb.7100148. [DOI] [PubMed] [Google Scholar]
- 3.Bortnick A, Chernova I, Quinn WJ, 3rd, Mugnier M, Cancro MP, Allman D. Long-lived bone marrow plasma cells are induced early in response to T cell-independent or T cell-dependent antigens. J Immunol. 2012;188:5389–5396. doi: 10.4049/jimmunol.1102808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foote JB, Mahmoud TI, Vale AM, Kearney JF. Long-term maintenance of polysaccharide-specific antibodies by IgM-secreting cells. J Immunol. 2012;188:57–67. doi: 10.4049/jimmunol.1100783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Racine R, McLaughlin M, Jones DD, Wittmer ST, MacNamara KC, Woodland DL, Winslow GM. IgM production by bone marrow plasmablasts contributes to long-term protection against intracellular bacterial infection. J Immunol. 2011;186:1011–1021. doi: 10.4049/jimmunol.1002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taillardet M, Haffar G, Mondiere P, Asensio MJ, Gheit H, Burdin N, Defrance T, Genestier L. The thymus-independent immunity conferred by a pneumococcal polysaccharide is mediated by long-lived plasma cells. Blood. 2009;114:4432–4440. doi: 10.1182/blood-2009-01-200014. [DOI] [PubMed] [Google Scholar]
- 7.Kaji T, Ishige A, Hikida M, Taka J, Hijikata A, Kubo M, Nagashima T, Takahashi Y, Kurosaki T, Okada M, Ohara O, Rajewsky K, Takemori T. Distinct cellular pathways select germline-encoded and somatically mutated antibodies into immunological memory. J Exp Med. 2012;209:2079–2097. doi: 10.1084/jem.20120127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor JJ, Pape KA, Jenkins MK. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med. 2012;209:597–606. doi: 10.1084/jem.20111696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hauser AE, Debes GF, Arce S, Cassese G, Hamann A, Radbruch A, Manz RA. Chemotactic responsiveness toward ligands for CXCR3 and CXCR4 is regulated on plasma blasts during the time course of a memory immune response. J Immunol. 2002;169:1277–1282. doi: 10.4049/jimmunol.169.3.1277. [DOI] [PubMed] [Google Scholar]
- 10.Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, Nutt SL. Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J Exp Med. 2004;200:967–977. doi: 10.1084/jem.20040973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Odendahl M, Mei H, Hoyer BF, Jacobi AM, Hansen A, Muehlinghaus G, Berek C, Hiepe F, Manz R, Radbruch A, Dorner T. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood. 2005;105:1614–1621. doi: 10.1182/blood-2004-07-2507. [DOI] [PubMed] [Google Scholar]
- 12.Lindsley RC, Thomas M, Srivastava B, Allman D. Generation of peripheral B cells occurs via two spatially and temporally distinct pathways. Blood. 2007;109:2521–2528. doi: 10.1182/blood-2006-04-018085. [DOI] [PubMed] [Google Scholar]
- 13.Opstelten D, Osmond DG. Pre-B cells in mouse bone marrow: immunofluorescence stathmokinetic studies of the proliferation of cytoplasmic mu-chain-bearing cells in normal mice. J Immunol. 1983;131:2635–2640. [PubMed] [Google Scholar]
- 14.Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, Giltnane JM, Yang L, Zhao H, Calame K, Staudt LM. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002;17:51–62. doi: 10.1016/s1074-7613(02)00335-7. [DOI] [PubMed] [Google Scholar]
- 15.Sanderson RD, Lalor P, Bernfield M. B lymphocytes express and lose syndecan at specific stages of differentiation. Cell regulation. 1989;1:27–35. doi: 10.1091/mbc.1.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kallies A, Hawkins ED, Belz GT, Metcalf D, Hommel M, Corcoran LM, Hodgkin PD, Nutt SL. Transcriptional repressor Blimp-1 is essential for T cell homeostasis and self-tolerance. Nat Immunol. 2006;7:466–474. doi: 10.1038/ni1321. [DOI] [PubMed] [Google Scholar]
- 17.Martins GA, Cimmino L, Shapiro-Shelef M, Szabolcs M, Herron A, Magnusdottir E, Calame K. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nat Immunol. 2006;7:457–465. doi: 10.1038/ni1320. [DOI] [PubMed] [Google Scholar]
- 18.Tung JW, Mrazek MD, Yang Y, Herzenberg LA. Phenotypically distinct B cell development pathways map to the three B cell lineages in the mouse. Proc Natl Acad Sci U S A. 2006;103:6293–6298. doi: 10.1073/pnas.0511305103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Connor BP, Cascalho M, Noelle RJ. Short-lived and long-lived bone marrow plasma cells are derived from a novel precursor population. J Exp Med. 2002;195:737–745. doi: 10.1084/jem.20011626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho F, Lortan JE, MacLennan IC, Khan M. Distinct short-lived and long-lived antibody-producing cell populations. Eur J Immunol. 1986;16:1297–1301. doi: 10.1002/eji.1830161018. [DOI] [PubMed] [Google Scholar]
- 21.Cancro MP, Sah AP, Levy SL, Allman DM, Schmidt MR, Woodland RT. xid mice reveal the interplay of homeostasis and Bruton’s tyrosine kinase-mediated selection at multiple stages of B cell development. Int Immunol. 2001;13:1501–1514. doi: 10.1093/intimm/13.12.1501. [DOI] [PubMed] [Google Scholar]
- 22.Sze DM, Toellner KM, Garcia de Vinuesa C, Taylor DR, MacLennan IC. Intrinsic constraint on plasmablast growth and extrinsic limits of plasma cell survival. J Exp Med. 2000;192:813–821. doi: 10.1084/jem.192.6.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Makela O, Nossal GJ. Autoradiographic studies on the immune response. II. DNA synthesis amongst single antibody-producing cells. J Exp Med. 1962;115:231–244. doi: 10.1084/jem.115.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, Lio P, Macdonald HR, Trumpp A. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 25.Srivastava B, Quinn WJ, 3rd, Hazard K, Erikson J, Allman D. Characterization of marginal zone B cell precursors. J Exp Med. 2005;202:1225–1234. doi: 10.1084/jem.20051038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schittek B, Rajewsky K, Forster I. Dividing cells in bone marrow and spleen incorporate bromodeoxyuridine with high efficiency. Eur J Immunol. 1991;21:235–238. doi: 10.1002/eji.1830210136. [DOI] [PubMed] [Google Scholar]
- 27.Anderson SM, Khalil A, Uduman M, Hershberg U, Louzoun Y, Haberman AM, Kleinstein SH, Shlomchik MJ. Taking advantage: high-affinity B cells in the germinal center have lower death rates, but similar rates of division, compared to low-affinity cells. J Immunol. 2009;183:7314–7325. doi: 10.4049/jimmunol.0902452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pereira JP, An J, Xu Y, Huang Y, Cyster JG. Cannabinoid receptor 2 mediates the retention of immature B cells in bone marrow sinusoids. Nat Immunol. 2009;10:403–411. doi: 10.1038/ni.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsu MC, Toellner KM, Vinuesa CG, Maclennan IC. B cell clones that sustain long-term plasmablast growth in T-independent extrafollicular antibody responses. Proc Natl Acad Sci U S A. 2006;103:5905–5910. doi: 10.1073/pnas.0601502103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takahashi Y, Dutta PR, Cerasoli DM, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V. Affinity maturation develops in two stages of clonal selection. J Exp Med. 1998;187:885–895. doi: 10.1084/jem.187.6.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dogan I, Bertocci B, Vilmont V, Delbos F, Megret J, Storck S, Reynaud CA, Weill JC. Multiple layers of B cell memory with different effector functions. Nat Immunol. 2009;10:1292–1299. doi: 10.1038/ni.1814. [DOI] [PubMed] [Google Scholar]
- 32.Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science. 2011;331:1203–1207. doi: 10.1126/science.1201730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomayko MM, Steinel NC, Anderson SM, Shlomchik MJ. Cutting edge: Hierarchy of maturity of murine memory B cell subsets. J Immunol. 2010;185:7146–7150. doi: 10.4049/jimmunol.1002163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tokoyoda K, Egawa T, Sugiyama T, Choi BI, Nagasawa T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity. 2004;20:707–718. doi: 10.1016/j.immuni.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 35.Belnoue E, Tougne C, Rochat AF, Lambert PH, Pinschewer DD, Siegrist CA. Homing and adhesion patterns determine the cellular composition of the bone marrow plasma cell niche. J Immunol. 2012;188:1283–1291. doi: 10.4049/jimmunol.1103169. [DOI] [PubMed] [Google Scholar]
- 36.Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, Noelle RJ. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol. 2008;180:3655–3659. doi: 10.4049/jimmunol.180.6.3655. [DOI] [PubMed] [Google Scholar]
- 37.Chu VT, Frohlich A, Steinhauser G, Scheel T, Roch T, Fillatreau S, Lee JJ, Lohning M, Berek C. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat Immunol. 2011;12:151–159. doi: 10.1038/ni.1981. [DOI] [PubMed] [Google Scholar]
- 38.Shapiro-Shelef M, Lin KI, Savitsky D, Liao J, Calame K. Blimp-1 is required for maintenance of long-lived plasma cells in the bone marrow. J Exp Med. 2005;202:1471–1476. doi: 10.1084/jem.20051611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allman D, Lindsley RC, DeMuth W, Rudd K, Shinton SA, Hardy RR. Resolution of three nonproliferative immature splenic B cell subsets reveals multiple selection points during peripheral B cell maturation. J Immunol. 2001;167:6834–6840. doi: 10.4049/jimmunol.167.12.6834. [DOI] [PubMed] [Google Scholar]
- 40.Allman DM, Ferguson SE, Lentz VM, Cancro MP. Peripheral B cell maturation. II. Heat-stable antigen(hi) splenic B cells are an immature developmental intermediate in the production of long-lived marrow-derived B cells. J Immunol. 1993;151:4431–4444. [PubMed] [Google Scholar]
- 41.Hartley SB, Cooke MP, Fulcher DA, Harris AW, Cory S, Basten A, Goodnow CC. Elimination of self-reactive B lymphocytes proceeds in two stages: arrested development and cell death. Cell. 1993;72:325–335. doi: 10.1016/0092-8674(93)90111-3. [DOI] [PubMed] [Google Scholar]
- 42.Baumgarth N, Herman OC, Jager GC, Brown LE, Herzenberg LA, Chen J. B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J Exp Med. 2000;192:271–280. doi: 10.1084/jem.192.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin F, Oliver AM, Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 2001;14:617–629. doi: 10.1016/s1074-7613(01)00129-7. [DOI] [PubMed] [Google Scholar]
- 44.Bernasconi NL, Traggiai E, Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science. 2002;298:2199–2202. doi: 10.1126/science.1076071. [DOI] [PubMed] [Google Scholar]
- 45.Reth M, Hammerling GJ, Rajewsky K. Analysis of the repertoire of anti-NP antibodies in C57BL/6 mice by cell fusion. I. Characterization of antibody families in the primary and hyperimmune response. Eur J Immunol. 1978;8:393–400. doi: 10.1002/eji.1830080605. [DOI] [PubMed] [Google Scholar]
- 46.Stashenko P, Klinman NR. Analysis of the primary anti-(4-hydroxy-3-nitrophenyl) acetyl (NP) responsive B cells in BALB/C and B10.D2 mice. J Immunol. 1980;125:531–537. [PubMed] [Google Scholar]
- 47.Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat Med. 2005;11:1299–1305. doi: 10.1038/nm1326. [DOI] [PubMed] [Google Scholar]
- 48.Smith KG, Light A, Nossal GJ, Tarlinton DM. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. 1997;16:2996–3006. doi: 10.1093/emboj/16.11.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Purtha WE, Tedder TF, Johnson S, Bhattacharya D, Diamond MS. Memory B cells, but not long-lived plasma cells, possess antigen specificities for viral escape mutants. J Exp Med. 2011;208:2599–2606. doi: 10.1084/jem.20110740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oliver AM, Martin F, Kearney JF. IgMhighCD21high lymphocytes enriched in the splenic marginal zone generate effector cells more rapidly than the bulk of follicular B cells. J Immunol. 1999;162:7198–7207. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.