

Abstract
p53 is a major suppressor of human malignancy. The protein levels and activity are tightly regulated in cells. Early experiments identified nuclear localization signal 1 (NLS1) as a regulator of p53 localization. We have generated mice bearing a mutation in p53NLS1, designated p53NLS1. Our experiments confirm a role for NLS1 in regulating p53 function. Murine embryonic fibroblasts generated from homozygous p53NLS1 animals are partially defective in cell cycle arrest and do not respond to inhibitory signals from oncogenic Ras. In addition, p53-dependent apoptosis is abrogated in thymocytes. Contrary to predicted results, fibroblasts from homozygous p53NLS animals have a greater rate of proliferation than p53-null cells. In addition, p53NLS cells are more resistant to UV-induced death. Surprisingly, the homozygous p53NLS1 animals exhibit embryonic and peri-natal lethality, with a significant portion of the animals developing exencephaly. Thus, p53NLS1/NLS1 embryos exhibit a reduced viability relative to p53-null mice. These studies indicate that the NLS1 is a major regulator of p53 activity in vivo.
Keywords: p53, Nuclear localization signal 1, Exencephaly, Apoptosis, Embryonic lethality
Introduction
p53 is a major suppressor of human malignancy (Hainaut and Hollstein 2000). p53 is activated in cells in response to various stimuli, including DNA damage, oncogenic transformation and environmental stress. The protein levels and activity are under tight regulation in the cells (Liang and Clarke 2001). In unstressed cells, p53 protein half-life is short and the protein is targeted for degradation in the cytoplasm. This process is regulated by the E3 ligase Mdm2, as well as additional newly identified E3 ligases (Brooks and Gu 2006). Mdm2 binds and ubiquitinates p53 whereby p53 is exported to the cytoplasm and degraded by the proteosome (O’Brate and Giannakakou 2003). In response to DNA damage, inhibition of the p53-Mdm2 complex formation and p53 degradation results in elevated p53 protein levels. p53 is imported to or retained in the nucleus to undertake one of its primary roles as a transcriptional activator. Thus, in response to DNA damage, there is both inhibition of p53 degradation and increased nuclear accumulation of p53.
Early experiments demarcated the sequences involved in nuclear import and nuclear export. Three nuclear import sequences (NLS1, NLS2, and NLS3) were identified in the carboxyl terminus based on mutagenesis studies (Dang and Lee 1989; Shaulsky et al. 1990). NLS1 forms a basic core region similar to SV40 Large T-Antigen NLS and is conserved across species (Addison et al. 1990; Dang and Lee 1989; Shaulsky et al. 1990). In addition, a basic and spacer region has been shown to cooperate in regulating p53 cytoplasmic location, demonstrating NLS1 functions as a bipartite signal (Liang and Clarke 1999a, b; Liang et al. 1998). p53 nuclear import is mediated by binding of the NLS to the importin-α complex (Liang and Clarke 1999a). Proteins have been identified that act to tether p53 in the cytoplasm (Gannon and Lane 1991; O’Brate and Giannakakou 2003; Wadhwa et al. 1998). Nuclear localization is important for p53 activity. In an E1A-Ras transformation assay, the suppressive properties of wildtype p53 were dependent on an intact NLS1 (Shaulsky et al. 1991). In addition, nuclear retention has been shown to be critical for p53-dependent apoptosis (Ryan et al. 1994) and growth inhibition (Knippschild et al. 1996; Moll et al. 1996). However, p53 NLS1 has been shown to be a weak monopartite nuclear localization signal (Liang and Clarke 1999a, b).
Although critical in developing a model of p53 regulation, these in vitro expression studies have the disadvantage of analyzing p53 function in culture cells. In order to examine the role of p53 cellular localization in vivo, we have generated a knock-in mouse expressing a p53 protein with a mutated NLS1, designated p53NLS1. Several p53 functions were defective in cells derived from homozygous p53NLS1 animals. However, additional characteristics were identified. For instance, p53NLS1/NLS1 cells grow more rapidly than p53-null cells. Interestingly, the homozygous p53NLS1 mice exhibit embryonic and peri-natal lethality with a high degree of penetrance. Therefore, p53NLS1 animals are not simply recapitulating p53-null mice phenotype. These studies demonstrate a definitive role of the NLS1 in regulating p53 function and an additional role of the p53 NLS1 in the regulation of embryonic development.
Results
Generation of p53 NLS1 mutant animals
The p53 gene consists of 11 exons and spans about 14 kb. The sequences of murine p53 nuclear localization 1 (NLS1) include amino acids 313–322 in exon 9 (Fig. S1A). We disrupted the core domain by substituting Alanines for Lysines 316, 317 and 318 (Fig. 1a) and introduced a NotI restriction site (Fig. S1B). This substitution has previously been made in human p53 cDNA and, when overexpressed in MCF-7 cells, lead to a cytoplasmic localization and diffuse nuclear staining (Liang and Clarke 1999a). This mutant was shown to bind weakly to importin-α. NLS1 resides outside of the tetramerization domain, and does not affect the tetramerization of the protein (Liang and Clarke 1999a). To introduce an Alanine substitution at Lys316–318, we built a targeting vector that contained LoxP sites flaking a neomycin selection marker, and introduced the mutation by site-directed mutagenesis (Fig. 1b). The targeting vector was sequenced to confirm that no other mutations were present in the p53 sequence. We electroporated PC3 ES cells (O’Gorman et al. 1997). We identified 10 positive clones for recombination by southern analysis. The targeted allele is depicted in Fig. 1c.
Fig. 1.
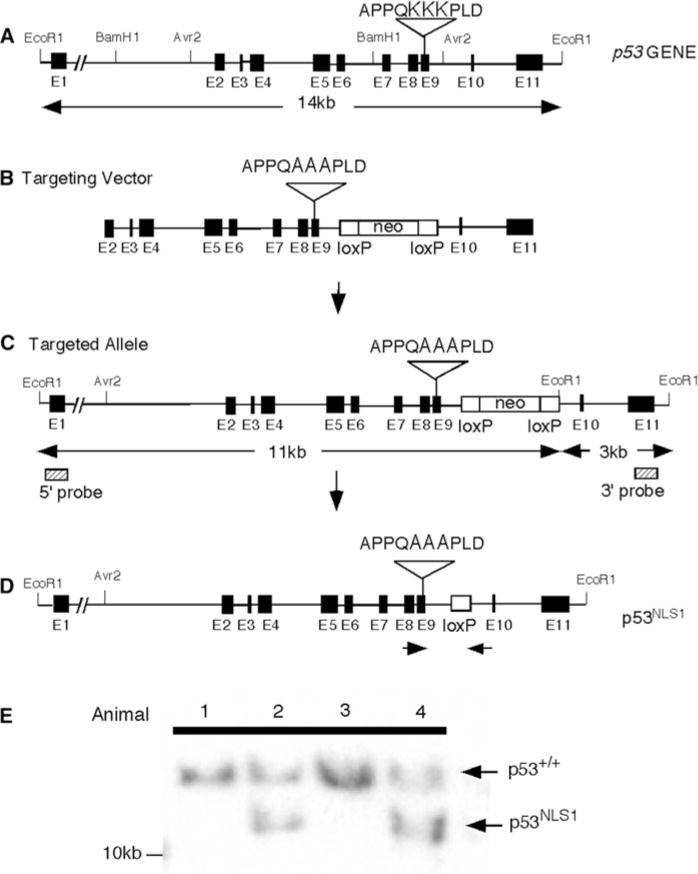
Mutation of Lysines 316–318 (NLS1) in the p53 locus. a Organization of the p53 locus. Filled boxes represent exons. Restriction enzymes sites are indicated. b The targeting vector encodes a triple Lysine to Alanine missense mutation in exon 9. A floxed NeoR gene was introduced in intron 9 to permit positive selection of ES cells. c Homologous recombination of the targeting vector in ES cells is illustrated. Probes used for Southern blot analysis are shown (hatched boxes). d The NeoR gene is excised in the chimera due to expression of a protamine-Cre transgene. e Southern blot analysis of targeted ES cell DNA using a 5′ external probe. EcoR1 digest of ES cell DNA indicates the presence of the mutant allele (unexcised NeoR gene, 11 kb) and the wildtype allele (14 kb)
One positive ES cell line was injected into blastocysts and chimeras were established. High degree chimeras were generated and mated with C57/B6 to produce progeny. The PC3 cells contain the Cre recombinase under the protamine reporter, which excises the neo in the male germline (O’Gorman et al. 1997). Therefore, after Cre excision, the resulting allele contained a loxP site and the exon containing the mutation of p53 (Fig. 1d). We identified animals that were heterozygous for the mutation. We confirmed excision of the floxed allele by southern analysis (Fig. 1e). The presence of the Alanine mutation in the mice tail DNA was confirmed by PCR and subsequent digestion of the PCR fragments with NotI (Fig. S1C). The mutation was also confirmed by sequencing the PCR product.
Growth characteristics of p53 NLS1- deficient embryonic fibroblasts
To explore the role of Lys316–318 (NLS1) in p53-mediated control of cell growth, we generated murine embryonic fibroblasts (MEFs) from p53+/+, p53 heterozygous (p53−/+), p53-null (p53−/−), p53NLS1/+ and p53NLS1/NLS1 embryos. Previous studies showed that p53−/− MEFs are distinguishable from wildtype cells in their growth rate and plating efficiency (Harvey et al. 1993). Therefore, the growth rates of p53NLS1/+ and p53NLS1/NLS1 fibroblasts were compared with wildtype and p53−/− cells (Fig. 2a). The p53NLS1/+ and p53NLS1/NLS1 MEFs had a faster proliferation rate than wildtype MEFs. There was no significant difference in the proliferation rates or saturation densities of p53+/− and p53NLS1/+ MEFs. p53−/− MEFs grew very rapidly and reached saturation on day 5. However, p53NLS1/NLS1 MEFs grew faster at low densities than p53−/− cells and achieved confluent densities at day 4. The increased growth rate was significant 2 and 3 days after plating. The cells were analyzed for bromodeoxyuridine (BrdU) uptake. p53NLS1/+ and p53NLS1/NLS1 cells demonstrated an increased S-phase population compared to wildtype (Fig. 2b). In addition, p53−/− cells exhibited an increase in S-phase compared to wildtype cells, however, it was not significantly greater than p53NLS1/NLS1 cells. These data suggest that growth is abnormal in p53NLS1/NLS1 MEFs and that the mutation in both alleles gives MEFs a growth advantage.
Fig. 2.
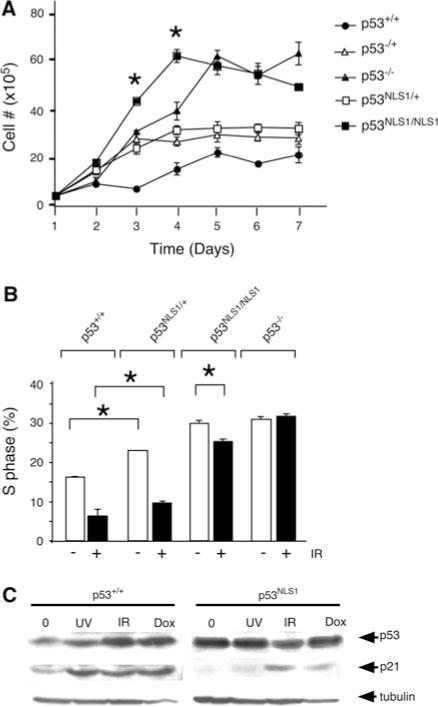
Growth characteristics of p53NLS1 cells. a Effects of p53 NLS1 mutation on cell growth. Cells were plated in triplicate and cell number was determined at the indicated times. b Loss of p53 NLS1 leads to defects in cell cycle arrest. Cell cycle arrest of heterozygous and homozygous p53NLS1 MEFs in response to gamma radiation, compared to wild type and p53−/− MEFs. Cells were harvested 18 h post-treatment. The decrease in S-phase in p53NLS1/NLS1 MEFs is significant (*, P<0.05) c Expression of p53NLS1 protein. Western analysis of wildtype (left panel) and p53NLS1/NLS1 cells (right panel) in response to various genotoxic stress [UV (50 J/m2), gamma irradiation (IR 20 Gy) and doxorubicin (Dox 50 μg/ml)] and harvested at 8 h post-treatment. α-tubulin is the loading control
To determine the role for Lys316–318 (NLS1) in p53-mediated cell cycling, we analyzed the DNA-damage induced growth arrest in response to gamma irradiation (IR). Wildtype MEFs displayed a 70% reduction in S-phase population of cells following IR treatment (Fig. 2b). The p53NLS1/+ cells arrested in response to IR, with a 60% reduction in S-phase. p53NLS1/NLS1 cells exhibited a 14% reduction in S-phase. In contrast, p53−/− cells exhibit no reduction in S-phase (Fig. 2b). Therefore, p53NLS1/NLS1 cells exhibited a partial response to DNA damage.
Western analysis was performed to examine the levels of p53 and p21, a downstream target of p53, in response to DNA damaging agents (Fig. 2c). In wildtype cells treated with IR, ultra-violet light (UV), and doxorubicin (dox), p53 protein levels were induced (Fig. 2c, lanes 2–4). Surprisingly, in untreated p53NLS1/NLS1 cells, p53NLS1 protein levels were elevated (Fig. 2c, lane 5). There was no further induction of p53 in p53NLS1/NLS1 cells in response to UV, IR, or doxorubicin treatment (Fig. 2c, lanes 6–8)).
Role of p53 NLS1 in Ras induced senescence
To further examine the effects of Lys316–318 (NLS1) in p53 regulated cell growth, we performed a senescence assay with oncogenic Ras, which causes cells to undergo p53-dependent senescence (Serrano et al. 1997). The effect of expressing oncogenic Ras was examined in wildtype, p53−/− and p53NLS1/NLS1 MEFs (Fig. 3). Wildtype cells infected with a pBabe control vector doubled in population over 6 days and expression of oncogenic Ras inhibited this growth (Fig. 3a). In addition, the number of cells positive for senescence marker was increased with oncogenic Ras expression in wildtype cells (Fig. 3b, c). Wildtype cells infected with Ras demonstrated the flat cells phenotype characteristic of cells that have exited the cell cycle (Fig. 3b). p53−/− cells did not have any growth change in the presence of Ras (Fig. 3a) and very few senescent cells were detected (Fig. 3b, c). The p53NLS1/NLS1 MEFs also did not respond to the growth-suppressive properties of oncogenic Ras (Fig. 3a), and very few senescent cells were present (Fig. 3b, c). Thus, p53NLS1/NLS1 cells were able to escape Ras induced senescence.
Fig. 3.
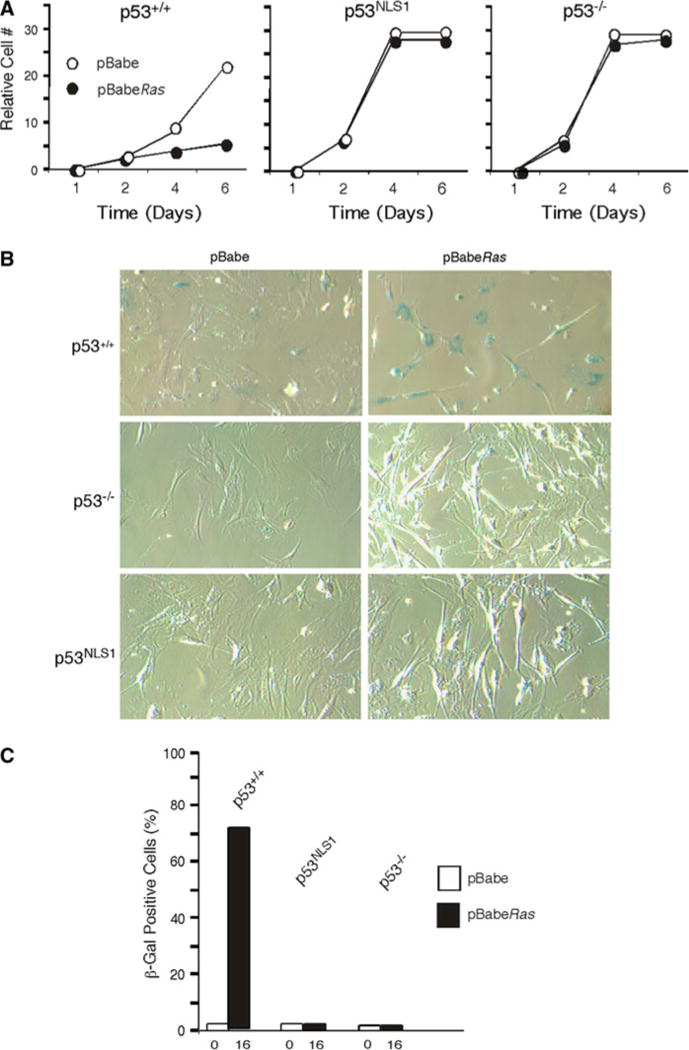
p53NLS1/NLS1 cells escape Ras induced cellular senescence. a Ras infection does not reduce viability of p53NLS1/NLS1 cells. Cells were infected with pBabe (vector) or pBabe-HaRas were plated and growth recorded at the indicated time points. b p53NLS1/NLS1 cells do not senescence in response to Ras. Phase-contrast micrograph of cells infected with oncogenic HaRas stained with senescence marker β-galactosidase. c Number of cells staining positive for β-galactosidase in cells in response to oncogenic Ras
p53NLS1 homozygous cells have defective expression of p53
The increased levels of p53 under non-stressed conditions (Fig. 2c) suggested that basal p53NLS1 protein levels would be increased. Therefore, we examined the cellular location of the p53NLS1 protein. We performed immunofluorescence for the p53 protein on wildtype (p53+/+) and p53NLS1 homozygous (p53NLS1/NLS1) cells (Fig. 4). The p53 protein in wildtype cells is barely discernable under basal conditions (data not shown). In contrast, p53NLS1 protein was readily detected in non-stressed p53NLS1/NLS1 cells (Fig. 4, top panel). The detection of the protein at basal levels supports the observation of increased levels of p53 in the Western analysis (Fig. 2a, c). The staining pattern of p53 in the non-stressed p53NLS1/NLS1 cells was not uniform. The cells either had a predominantly cytoplasmic staining, with very little nuclear staining, or the cells had staining with distribution of p53 in the nucleus and the cytoplasm. Examples of both cells are labeled in Fig. 4. We examined p53 cellular distribution in 30 p53NLS1/NLS1 cells. We determined that 53% of the cells had cytoplasmic p53 staining and 46% had distribution of p53 in the cytoplasm and nucleus. Thus, under basal conditions, the protein can be detected in the cytoplasm and/or nucleus. This observation supports previous studies that additional sequences (such as NLS2, NLS3 and the basic region, Fig. S1A) are also contributing to nuclear localization of the p53NLS1 protein (Liang and Clarke 1999a, b).
Fig. 4.
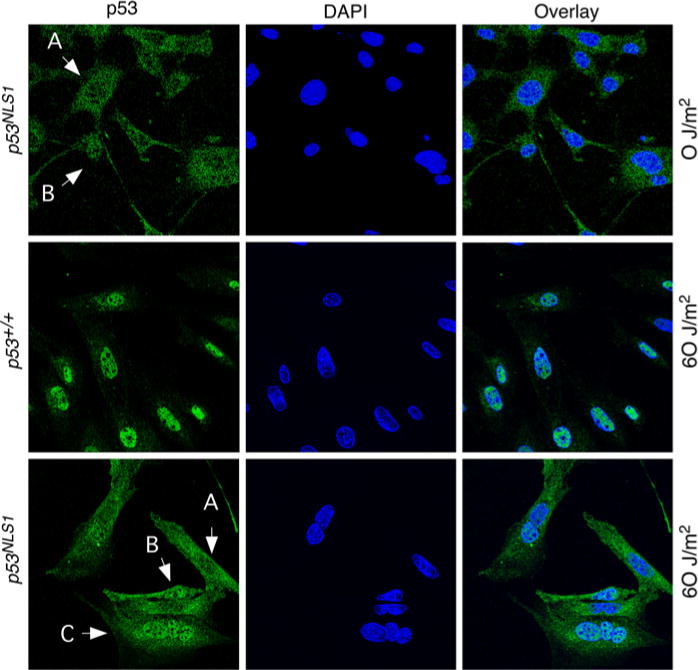
Analysis of p53NLS1 localization. Representative staining in cells with or without UV treatment. Staining of untreated p53NLS/NLS1 cells (top panels), wildtype cells treated with UV (middle panels), and UV-treated p53NLS1/NLS1 cells (bottom panels). Cells were stained 18 h post-treatment with the anti-p53 antibody Ab-7 and DAPI. Nuclear accumulation in response to UV is defective in p53NLS1 cells
In order to examine p53 localization in response to DNA damage, we examined p53 location after UV treatment in wildtype and p53NLS1 cells. There was a dramatic increase in p53 in wildtype cells treated with UV with greatly induced nuclear staining (Fig. 4, middle panels). The distribution of p53 in UV-treated p53NLS1/NLS1 cells varied from cell to cell (Fig. 4, bottom panels). We detected the same expression pattern that had either a diffuse staining (cytoplasmic and nuclear), or a predominantly cytoplasmic staining such as observed in basal conditions (Fig. 4, top panels). In addition, very few p53NLS1/NLS1 cells exhibited nuclear stabilization of p53. In response to UV, 16% of the cells showed the predominantly cytoplasmic staining, whereas 75% had a diffuse staining, and another 16% had nuclear stabilization of p53.
p53NLS1 homozygous thymocytes are resistant to p53-mediated death
In order to explore the effect of loss of p53 NLS1 on p53-dependent apoptosis in vivo, we examined thymocyte apoptosis in response to DNA-damage. CD4+/CD8+ thymocytes undergo p53-dependent apoptosis following IR (Clarke et al. 1993; Lowe et al. 1993b). We compared the relative population of CD4+/CD8+ cells in thymocytes from wildtype (p53+/+), p53−/−, p53NLS1/NLS1 and p53NLS1/+ mice, treated or untreated with IR (8 Gy). CD4+/CD8+ staining was performed 24 h post-IR. A significant depletion of CD4+/CD8+ thymocytes was observed in wildtype mice (Fig. 5a). p53NLS1/+ cells showed a decrease in depletion of CD4+/CD8+ population in response to IR compared to wildtype thymocytes. The partial depletion of CD4+/CD8+ in p53NLS1/+ cells was similar to that previously observed in p53−/− animals (Sluss et al. 2004). This result suggests the presence of the p53NLS1 allele in the heterozygous animal was not exerting a dominant negative effect on wildtype p53. p53NLS1/NLS1 CD4+/CD8+ thymocytes were resistant to death, similar to that observed in p53−/− cells (Fig. 5a).
Fig. 5.
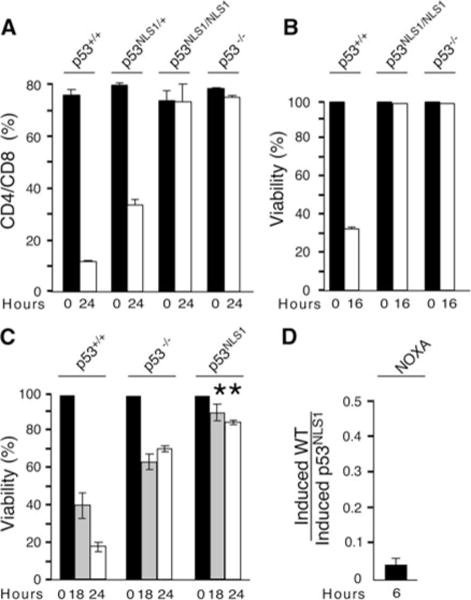
Loss of p53 Lys316–318 (NLS1) abrogates p53-dependent death. a Protection of CD4+/CD8+ thymocytes in p53NLS1/NLS1 animals. Mice were treated in vivo with IR (8 Gy) or untreated and the thymocytes harvested 24 h post treatment. Cells were stained for CD4 and CD8. b Loss of p53 NLS1 leads to increased survival in thymocytes. Thymocytes were removed from untreated and treated (8 Gy) animals 16 h post-treatment. The percent of viable cells is presented (cells negative for annexin-V and 7AAD). c Loss of p53 NLS1 leads to increased survival in E1A-MEFs. MEFs were exposed to UV radiation (5 J/m2) and survival was measured at 18 and 24 h post-treatment. All numbers are normalized to the number of cells in untreated samples. Increased survival in p53NLS1/NLS1 MEFs compared to p53−/− MEFs is significant (*, P < 0.05). d Defective NOXA induction in response to UV in p53NLS1 cells. The expression of NOXA mRNA was measured by quantitative real time PCR analysis of UV-treated MEFs (6 h post-treatment). The amount of Gapdh mRNA in each sample was used to calculate relative mRNA expression (mean ± -S.E.M.; n = 3). The induction of NOXA was presented as the ratio of induced in wildtype/induced in p53NLS1 MEFs
The fraction of non-apoptotic thymocytes was also measured at multiple time points post-irradiation by staining cells for annexin-V and 7AAD (Fig. 5b). annexin-V negative and 7AAD negative populations were determined, and compared to untreated thymocytes. Following DNA damage, a significant decrease in non-apoptotic cells and concomitant increase in Annexin V positive cells was observed in wild-type thymocytes (Fig. 5b). p53NLS1/NLS1 thymocytes displayed almost 100% survival following DNA damage, indicating no apoptosis was occurring (Fig. 5b). These results demonstrated that loss of p53 Lys316–318 abrogated p53-dependent death in thymocytes.
p53NLS1 exerts a gain of function phenotype
To further examine the effects of p53NLS1 on cell survival, we examined the survival of E1A-transformed MEFs in response to UV. MEFs are normally resistant to death by radiation and chemotherapeutic agents (Lowe et al. 1993a). However, MEFs co-expressing the adenovirus E1A protein undergo apoptosis when treated with DNA-damaging agents or chemotherapeutic agents (Attardi et al. 2004; Lowe et al. 1993a). E1A—transformed cells exhibit p53-dependent and independent death (a portion of EIA-p53−/− MEFs die). We examined viability of cells at 0, 18, and 24 h after treatment with 5 J/m2 of UV (Fig. 5c). Whereas wildtype cells were lost viability with treatment, p53NLS1/NLS1 cells were resistant to UV-induced death. In addition, we observed a significant increase in survival of p53NLS1/NLS1 cells compared to p53−/− cells. Thus, there was an increase in the portion of cells resistant to death at low doses of UV, suggesting a gain-of-function for the p53NLS1 allele.
In order to examine a possible mechanism of this defect, we found that p53NLS1 cells exhibited very little induction of NOXA, compared to wildtype cells. There was a 27-fold reduction in the amount of UV-induced NOXA in the mutant cells compared to wildtype (Fig. 5d). Therefore, although nuclear p53NLS1 was able to induce p21, it was severely defective in inducing NOXA.
p53NLS1 animals exhibit embryonic lethality and develop exencephaly
In addition to studies in cells, we examined the effects of p53 NLS1 on whole animals. Mice heterozygous for the p53NLS1 allele (p53NLS1/+) are viable (Table 1). These mice were intercrossed and we noted that mice homozygous for the mutant allele were recovered at low frequency (Table 1). There was a 90% reduction in the expected number of p53NLS1/NLS1 offspring at birth. This surprising result suggested the homozygous p53NLS1 allele could be causing embryonic lethality. We performed an analysis on timed embryos to determine how early we could detect homozygous p53NLS1 embryos. We detected almost the expected ratio of p53NLS/NLS1 embryos at embryonic day 14.5 and 12.5 (83 and 78%, respectively). We detected p53NLS1/NLS1 embryos as young as embryonic day 9.5, although there was no reduction in expected ratio of p53NLS1/NLS1 embryos. This suggested p53NLS1/NLS1 embryos were dying mid to late gestation. During the examination of embryos day 9.6–14.5, we detected embryos that exhibited hindbrain exencephaly (Fig. 6a, b). Examination of embryonic day 14.5 embryos revealed that 40% p53NLS1/NLS1 embryos exhibited hindbrain exencephaly (Table 1). This percentage was similar in other embryos. Most of the females exhibited exencephaly (75%), whereas one out of six males exhibited exencephaly. Exencephaly is observed in a low frequency of p53−/− animals (Armstrong et al. 1995; Sah et al. 1995). Unlike p53NLS1/NLS1 mice, p53−/− mice are mostly viable. Thus, the reduced viability of p53NLS1/NLS1 mice relative to p53−/− mice could be attributed to an increase in the rate of exencephaly. Exencephalic p53−/− embryos show defects in apoptosis (Sah et al. 1995). Studies are underway to examine apoptosis in the p53NLS1/NLS1 exencephalic embryos.
Table 1.
Genotype of embryos from p53NLS1 heterozygous intercross
| Genotype | p53+/+ | p53NLS1/+ | p53NLS1/NLS1 |
|---|---|---|---|
| Birth (n = 131) | |||
| Obtained | 61 | 67 | 3 |
| Expected | 32.75 | 65.5 | 32.75 |
| Genotype | p53+/+ | p53NLS1/+ | p53NLS1/NLS1 | Reabsorptions |
|---|---|---|---|---|
| Embryo Day 14.5 (n = 48) | ||||
| Obtained | 12 | 26 | 10 | 3 |
| Expected | 12 | 24 | 12 | |
| Genotype | p53+/+ | p53NLS1/+ | p53NLS1/NLS1 | Reabsorptions |
|---|---|---|---|---|
| Embryo day 12.5 (n = 46) | ||||
| Obtained | 12 | 25 | 9 | 6 |
| Expected | 11.5 | 23 | 11.5 | |
| Genotype | p53+/+ | p53NLS1/+ | p53NLS1/NLS1 | Reabsorptions |
|---|---|---|---|---|
| Embryo day 9.5 (n = 36) | ||||
| Obtained | 6 | 18 | 10 | 0 |
| Expected | 9 | 18 | 9 | |
| Ratio of p53NLS1/NLS1 embryos exhibiting exencephaly | |
|---|---|
| Embryo day 14.5 | |
| Males/exencephaly | 6/1 |
| Females/exencephaly | 4/3 |
Fig. 6.
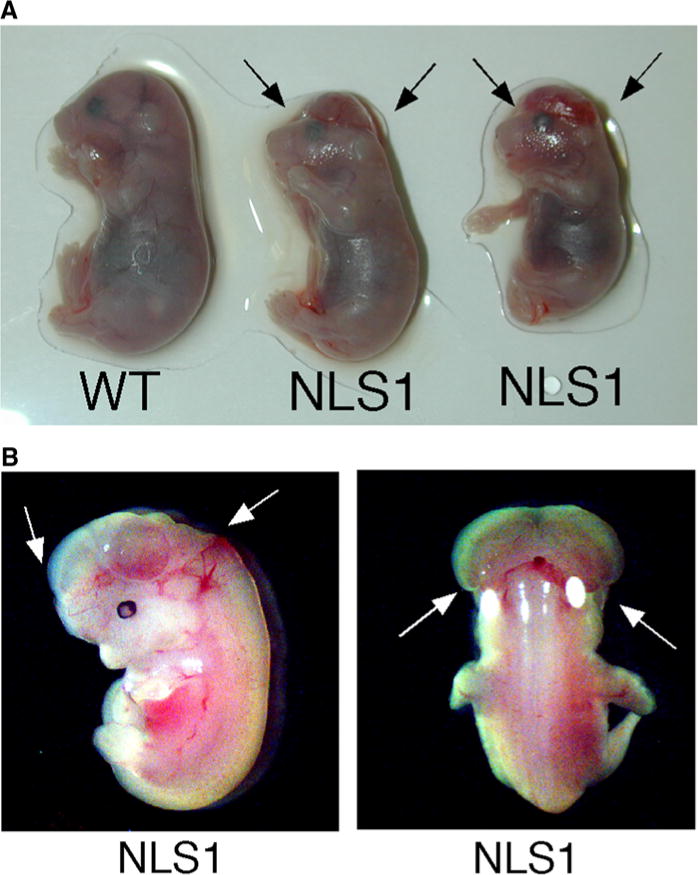
p53NLS1/NLS1 animals develop exencephaly. a Embryonic Day 15.5 embryos from a heterozygous p53NLS1 cross. Wildtype animal (on left), with complete closure of hindbrain. p53NLS1/NLS1 animals in the middle (female, NLS1-1) and right (male, NLS1-2), showing hindbrain exencephaly. b p53NLS1/NLS1 exencephalic embryo showing the eversion and exposure of neural tissue (arrows) pictured in frontal section (left panel) and back sagittal section (right panel)
Discussion
Early experiments showed the p53 protein translocates to the nucleus via a NLS1 (Dang and Lee 1989; Shaulsky et al. 1990), a secondary NLS2 and NLS3 (Shaulsky et al. 1990), and a basic and spacer sequences adjacent to NLS1 that are critical for nuclear import (Liang and Clarke 1999a, b; Liang et al. 1998) (s). The importance of nuclear import of p53 is underscored by the observation that p53 function can be inactivated in tumors by cytoplasmic sequestration (Bosari et al. 1995; Moll et al. 1996; Ryan et al. 1994). Although many studies have demonstrated nuclear localization is central to p53 function, recent studies have implicated a role for cytoplasmic p53 in p53 function (Green and Kroemer 2009). Therefore, to address the contribution of NLS1 to p53 function, we generated a mouse knock-in mutation in the p53 locus that abolished the primary Lysines in the NLS1 (Lys316–318). We have utilized gene targeting in mouse ES cells to generate mice bearing a NLS1 defective p53 allele, designated p53NLS1. Our results confirm a critical role for p53 NLS1 in p53 function.
Interestingly, the mutant protein was stabilized in basal conditions, and could be detected by Western and immunoflouresence, whereas wildtype protein can not be detected. We detected either a diffuse staining pattern or a predominantly cytoplasmic staining pattern in unstressed p53NLS1/NLS1 cells (Fig. 4). This observation has been shown in in vitro overexpression studies. Cells with expressing a p53cDNA with the three Lysines in the NLS1 mutated to Alanine exhibited a cytoplasmic and partial nuclear p53 staining (Liang and Clarke 1999a).
The elevated p53NLS1 protein in basal conditions was unexpected as wildtype p53 is under tight control in the non-stressed cells and maintained in low levels. It is interesting that the p53NLS1 protein is not degraded in the cytoplasm, as that is the major site of p53 degradation (Liang and Clarke 2001). Thus, p53NLS1 is likely not being targeted for degradation, despite its cytoplasmic localization (Yu et al. 2000). The site in the NLS1 is also a site for ubiquitination, therefore, it is possible the protein is not receiving the proper signal for degradation, such as mono-ubiquitination by Mdm2 (Li et al. 2003). Studies are underway to look at the ubiquitination status of the p53NLS1 protein.
In addition, we observed very little nuclear accumulation/stabilization of p53 in p53NLS1/NLS1 cells in response to UV (Fig. 4, bottom panels). Whereas almost 100% of wild type cells had strong induction of p53 in the nucleus, 16% of p53NLS1/NLS1 cells were observed to have nuclear induction. These observation suggests that the NLS1 is a predominant site for control of subcellular localization for p53 upon UV treatment.
Supporting a role for NLS1 in the regulation of p53 activity, cells derived from p53NLS1/NLS1 mice exhibit loss of p53 function. MEFs from p53NLS1/NLS1 animals had a faster proliferation rate than wildtype MEFs (Fig. 2a). p53NLS1/NLS1 cells grew faster than p53−/− cells, although they reached similar saturation densities. It is not clear why this occurred, however, basal p53 levels are greater in p53NLS1/NLS1 cells. This would argue that increased p53 should inhibit cell growth. It is possible the mutation in p53NLS1 is providing a gain-of-function, in either the cytoplasm or nucleus (where it can be detected in non-stressed cells). There is data that the p53NLS1 protein can have some normal function. For instance, p53NLS1 cells exhibited a partial defect in cell cycle arrest in response to gamma radiation and induced p21 (Fig. 2b, c). However, several p53 functions were disrupted. p53NLS1/NLS1 cells were able to escape Ras-induced senescence (Fig. 3). Thymocytes from p53NLS1/NLS1 animals exhibited complete survival in response to gamma radiation (Fig. 5a, b). In addition, p53NLS1/NLS1 E1A-MEFs were more resistant to death than p53−/− E1A-MEFs (Fig. 5c). The loss of apoptosis correlated with defective induction of NOXA (Fig. 5d). This observation, along with the cell growth advantage, suggests that the p53 NLS1 mutation imparted a gain of function phenotype to the cells.
p53 can mediate a cytoplasmic death program [reviewed in (Fuster et al. 2007)]. In one model, p53 exerts a BH3-like role. Studies have suggested this is transcription-independent or dependent on priming by induction of PUMA (Chipuk et al. 2005). Another model of cytoplasmic p53-dependent death involves translocation of p53 to the mitochondria. Recent work suggests that monoubiquitination of p53 allows for mitochondrial targeting (Marchenko et al. 2007). Studies have demonstrated Mdm2 can ubiquitinate proteins in the cytoplasm (Yu et al. 2000). Therefore, it is interesting the p53NLS1/NLS1 cells do not die even with increased levels of p53 protein in the cytoplasm. The mutation lies within ubiquitination sites that are important to mitochondrial targeting as well as nuclear targeting.
We observed a dramatic loss of induction of NOXA in response to UV (Fig. 5d). Therefore, this mutation may be contributing to the loss of function of the residual p53NLS1 protein in the nucleus. In support of this possibility, transgenic mice have been generated wherein the PCAF1/NLS1 site in p53 was mutated, and Lys317 was replaced with an Arg residue (Chao et al. 2006). Increased apoptosis and NOXA and PUMA expression was observed in these p53Lys317Arg cells, suggesting that acetylation negatively regulates p53-dependent transcription of NOXA. Thus, Alanine, which can mimic neutralization imparted by acetylation (Ren and Gorovsky 2001), would be predicted to have less expression NOXA. It is interesting to speculate that the protein may also exert some functions as a mimic of charge patch neutralization. Irrespective of the model, the p53NLS1 protein is not competent of undergoing p53-dependent cytoplasmic/mitochondrial death. Furthermore, it appears that p53NLS1 mutation interferes with induction of p53-mediated apoptotic genes.
Surprisingly, we found that the loss of the NLS1 lead to embryonic and peri-natal lethality. p53NLS1/NLS1 animals develop exencephaly as early as embryonic day 9.5. A subset of p53−/− mice exhibit exencephaly (Armstrong et al. 1995; Sah et al. 1995). It is possible loss of p53-mediated gene expression during embryonic development induces exencephaly, which would suggest the nuclear p53NSL1 protein is mimicking a p53-null allele (where there is no nuclear p53 expression). Mice deficient in the p53 transactivation domain (p53QS animals) also exhibit embryonic lethality (Johnson et al. 2005). However, these mice die earlier than p53NLS1/NLS1 mice, and mostly reabsorptions are detected. Thus, it is unlikely that the exencephaly observed in p53NLS1/NLS1 embryos was due solely to the loss if p53-transcriptional activity. The p53NLS1/NLS1 mice will be important for examining the layers of complex regulation that govern p53 functions during development and growth suppression.
Methods
Generation of targeting vector
The targeting vector was generated using a portion of the p53 gene (129SVBrd-strain) spanning an XhoI to HindIII fragment 5496 base pair from exon 2 to intron 11 of the p53 gene. Lysines 316–318 in exon 9 were mutated using oligonucleotide site directed mutagenesis (QuickChange Kit, Stratagene) with a sub-fragment of the targeting vector. The subfragment was sequenced to confirm the mutation and subcloned back into the targeting vector. Additional sequencing of the targeting vector revealed no mutations (data not shown). A NeoR cassette flanked by LoxP sites was introduced in an Avr2 restriction endonuclease site. An MC1-TK gene was added to the 3′ end of the targeting vector.
Generation of targeted ES cells and p53NLS1 mice
PC3 ES cells (O’Gorman et al. 1997) were electro-porated with the targeting vector and drug selected with G418 and FIAU. ES cells were screened and correctly targeted clones were identified by Southern blot and PCR analysis of EcoR1-digested genomic DNA using 5′ and 3′ probes external to the targeting vector. We identified 10 positive ES clones for recombination. The presence of the Alanine mutation in the ES clones was confirmed by PCR as described below. Chimeric mice were created by microinjection of the ES cells into E3.5 blastocysts (C57Bl/6 strain) and the embryos were surgically implanted into pseudopregnant foster mice using standard procedures. The male chimeric mice were bred with female C57BL6/J mice (The Jackson Laboratories). Excision of the NeoR cassette in the F1 generation was confirmed by digestion with EcoRI and NotI and Southern analysis using a p53 intron 1 probe (data not shown). All mice were on a mixed 129/Sv × C57BL/6 background. All research with mice complies with federal and institutional policies, as well as guidelines established by the Institutional Animal Care and Use Committee (IACUC) at the University of Massachusetts Medical School.
PCR genotyping and sex determination
Lysine 316, 317 and 318 were mutated to an Alanine. A NotI restriction site was introduced by mutagenesis. The presence of the Alanine mutation in the mice tail DNA was confirmed by PCR using oligonucleotide primers to exon 8 forward (5′-GAGAGACCGCCGTACAGAAG -3′) and intron 9 reverse (5′-CCATCCCCAGTGGAGGAGCACCTGTTTAT G-3′) and subsequent digestion of the PCR fragments with NotI. The wild-type PCR product was digested to yield one fragment, 477 bp. The mutated PCR product yielded 279 bp and 198 bp DNA fragments following NotI digestion (Fig. S2B).
The PCR to determine embryo sex was done with Forward primer: CCGCTGCCAAATTCTTTGG and Reverse primer: 5′-TGAAGCTTTTGGCTTTGAG-3′. PCR of a male will give 2 bands and female one band at 300 bp (Mroz et al. 1999).
Cell growth studies and immunoflouresence
Murine embryonic fibroblasts (MEFs) were generated and maintained as described (Sluss et al. 2004). Growth curves were performed with triplicate plating of wildtype, p53NLS1/+, p53NLS1/NLS1, p53−/+, and p53-null early passage MEFs (E13.5). Cells were plated (5 × 105 cells per 10 cm dish) in triplicate and cell number was determined each day 7 days post-plating using a hemocytometer. Proliferation assays were done with two independent lines of each genotype, and one representative experiment is presented.
Immunoflouresence was performed on low passage primary MEFs. Cells were either untreated or treated (60 J/m2 UV) and fixed 18 h post treatment. Cells were stained with sheep anti-p53 (Ab-7) and biotin anti- sheep (Calbiochem), and then p53 was visualized with Strepavidin-Alexa 488. DNA was stained with DAPI. Cells were analyzed on a Leica Confocal Microscope.
Ras transformation assay
Retroviral transduction studies were performed with recombinant viruses generated using Bosc cells and the vectors pBabe-Puro and pBabe-HaRas61L-Puro as described (Armata et al. 2007). Transduced MEFs were plated at 25,000 cells/well (12-well multi-dish). Relative viability was determined at days 2,4, and 6 post-plating by trypan blue staining. The number of senescent cells was determined by staining for the senescence marker acidic β-galactosidase (Dimri et al. 1995).
Protein analysis and westerns
For western analysis, MEFs were treated with DNA damaging agents [UVC light (50 J/m2), gamma irradiation (IR 20 Gy) and doxorubicin (Dox 50 μg/ml)] and harvested 8 h post treatment. Following treatment, cells were lysed in NP-40 lysis buffer and western performed for p53 as described (Armata et al. 2007). p53 antibody (Ab-7) was obtained from Calbiochem, anti-α-Tubulin from Sigma and anti-p21 from Santa Cruz. Bound Antibody was visualized by enhanced chemiluminescence (ECL, Amersham).
DNA damage response
For growth arrest, cells were treated with 8 Gy, labeled with bromodeoxyuridine (BrdU) for 3 h and harvested 18 h post-treatment. Replicative DNA synthesis was quantified using bivariate flow cytometry from propidium iodide-complexes as described (Sluss et al. 2004) on a FACStar flow cytometer. Cell cycle distribution was determined using FLOW Jo software. Similar numbers of cells were analyzed for each sample.
Thymocyte CD4/CD8 and viability analysis
For CD4/CD8 staining, animals were irradiated (8 Gy) with a Gammacell 40 irradiator and thymocytes harvested (24 h post-treatment). Thymocytes were stained with anti-CD4-PE and anti-CD8-FITC (Oncogene Research Products) as described (Sluss et al. 2004) and analyzed by FACS. After staining, the fraction of CD4/CD8-double positive thymocytes were quantified using FLOW Jo software. For viability analysis, thymocytes were removed from untreated and treated (8 Gy) animals 16 h post-treatment. Thymocytes were stained with fluorescein isothiocyanate-labeled annexin V antibody (Pharmingen) and PI (Sigma) as described (Sluss et al. 2004). Relative amounts of apoptotic cells were determined by binding of annexin V and subsequent FACS analysis. The fraction of non-apoptotic thymocytes post irradiation was measured by calculating annexin-V- and 7AAD- negative populations and compared to untreated thymocytes.
Generation and analysis of E1A-MEFs
Retroviral transduction studies were performed as described (Armata et al. 2007) with recombinant viruses generated from the vectors pBabe-Puro and pBabe-E1A. Cells were selected with puromyocin. For death assays, E1A-expressing cells were plated in triplicate 3–4 days prior to treatment, at 1.25 × 105 cells/6-well. Cells were treated with 5 J/m2 of UV light. Relative cell number was obtained by the crystal violet staining method.
RNA preparation and analysis
RNA was prepared from tissues collected in RNA-later (Ambion) and snap frozen in liquid nitrogen. Total RNA was prepared with RNeasy kits (Qiagen) following manufacturer’s instructions. The purified RNA was subjected to an additional DNAse treatment (Ambion) to ensure removal of contaminating genomic DNA prior to final column purification. The relative expression of mRNA was examined by quantitative PCR analysis. cDNA was prepared using Superscript III (Invitrogen) with random hexamers and 0.5–1 μg of RNA per tissue. Quantitative realtime PCR was performed on a Biorad iCycler using SyBr Green master mix (Biorad). The primer sequences for the murine genes were: Gapdh (5′ -CT TCACCACCATGGAGAAGGC-3′; 5′-GGCATGGA CTGTGGTCAT-3′); p53 (5′-TGAAACGCCGACC TATCCTTA-3′; 5′-GGCACAAACACGAACCTCA AA-3′); and NOXA (5′-AGGAAGGAAGTTCCGCC G-3′, 5′-AGCGTTTCTCTCATCACATCACA-3′). All samples were examined in triplicate and values were normalized for baseline expression and for expression of Gapdh. Calculations of values were made using the DDCt method. Statistical significance was calculated using CT values.
Data analysis
To calculate statistical changes, statistically significant differences (P < 0.05) between groups were examined using the two-tailed Student’s t-test. Microsoft Excel was used for statistical calculations.
Supplementary Material
Acknowledgments
We thank the members of the Davis lab and Dr. Irene Rainville for helpful discussions. We are grateful to Drs. Venkatachalam and Donehower for providing p53 genomic DNA, and the Rice lab for generous use of their equipment. This research was supported by, in part, by NIH CA-077735 (SNJ) and the Dutch Cancer Society (AR). Core resources supported by the Diabetes and Endocrinology Research Center grant DK32520 were also used. HKS and SNJ are members of the UMass DERC (DK32520).
Footnotes
Conflict of interest The authors declare no conflict of interest.
Electronic supplementary material The online version of this article (doi:10.1007/s11248-010-9468-4) contains supplementary material, which is available to authorized users.
Contributor Information
Anouk Regeling, Department of Medicine, University of Massachusetts Medical School, 55 Lake Ave North, Worcester, MA 01605, USA.
Heather L. Armata, Department of Medicine, University of Massachusetts Medical School, 55 Lake Ave North, Worcester, MA 01605, USA
Judy Gallant, Department of Cell Biology, University of Massachusetts, Medical School, 55 Lake Ave North, Worcester, MA 01605, USA.
Stephen N. Jones, Department of Cell Biology, University of Massachusetts, Medical School, 55 Lake Ave North, Worcester, MA 01605, USA
Hayla K. Sluss, Email: hayla.sluss@umassmed.edu, Department of Medicine, University of Massachusetts Medical School, 55 Lake Ave North, Worcester, MA 01605, USA
References
- Addison C, Jenkins JR, Sturzbecher HW. The p53 nuclear localisation signal is structurally linked to a p34cdc2 kinase motif. Oncogene. 1990;5:423–426. [PubMed] [Google Scholar]
- Armata HL, Garlick DS, Sluss HK. The ataxia telangiectasia-mutated target site Ser18 is required for p53-mediated tumor suppression. Cancer Res. 2007;67:11696–11703. doi: 10.1158/0008-5472.CAN-07-1610. [DOI] [PubMed] [Google Scholar]
- Armstrong JF, Kaufman MH, Harrison DJ, Clarke AR. High-frequency developmental abnormalities in p53-deficient mice. Curr Biol. 1995;5:931–936. doi: 10.1016/s0960-9822(95)00183-7. [DOI] [PubMed] [Google Scholar]
- Attardi LD, de Vries A, Jacks T. Activation of the p53-dependent G1 checkpoint response in mouse embryo fibroblasts depends on the specific DNA damage inducer. Oncogene. 2004;23:973–980. doi: 10.1038/sj.onc.1207026. [DOI] [PubMed] [Google Scholar]
- Bosari S, Viale G, Roncalli M, Graziani D, Borsani G, Lee AK, Coggi G. p53 gene mutations, p53 protein accumulation and compartmentalization in colorectal adenocarcinoma. Am J Pathol. 1995;147:790–798. [PMC free article] [PubMed] [Google Scholar]
- Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell. 2006;21:307–315. doi: 10.1016/j.molcel.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao C, Wu Z, Mazur SJ, Borges H, Rossi M, Lin T, Wang JY, Anderson CW, Appella E, Xu Y. Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Mol Cell Biol. 2006;26:6859–6869. doi: 10.1128/MCB.00062-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309:1732–1735. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- Dang CV, Lee WM. Nuclear and nucleolar targeting sequences of c-erb-A, c-myb, N-myc, p53, HSP70, and HIV tat proteins. J Biol Chem. 1989;264:18019–18023. [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JJ, Sanz-Gonzalez SM, Moll UM, Andres V. Classic and novel roles of p53: prospects for anticancer therapy. Trends Mol Med. 2007;13:192–199. doi: 10.1016/j.molmed.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Gannon JV, Lane DP. Protein synthesis required to anchor a mutant p53 protein which is temperature-sensitive for nuclear transport. Nature. 1991;349:802–806. doi: 10.1038/349802a0. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–1130. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. 2000;77:81–137. doi: 10.1016/s0065-230x(08)60785-x. [DOI] [PubMed] [Google Scholar]
- Harvey M, Sands AT, Weiss RS, Hegi ME, Wiseman RW, Pantazis P, Giovanella BC, Tainsky MA, Bradley A, Donehower LA. In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice. Oncogene. 1993;8:2457–2467. [PubMed] [Google Scholar]
- Johnson TM, Hammond EM, Giaccia A, Attardi LD. The p53QS transactivation-deficient mutant shows stress-specific apoptotic activity and induces embryonic lethality. Nat Genet. 2005;37:145–152. doi: 10.1038/ng1498. [DOI] [PubMed] [Google Scholar]
- Knippschild U, Oren M, Deppert W. Abrogation of wild-type p53 mediated growth-inhibition by nuclear exclusion. Oncogene. 1996;12:1755–1765. [PubMed] [Google Scholar]
- Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–1975. doi: 10.1126/science.1091362. [DOI] [PubMed] [Google Scholar]
- Liang SH, Clarke MF. A bipartite nuclear localization signal is required for p53 nuclear import regulated by a carboxyl-terminal domain. J Biol Chem. 1999a;274:32699–32703. doi: 10.1074/jbc.274.46.32699. [DOI] [PubMed] [Google Scholar]
- Liang SH, Clarke MF. The nuclear import of p53 is determined by the presence of a basic domain and its relative position to the nuclear localization signal. Oncogene. 1999b;18:2163–2166. doi: 10.1038/sj.onc.1202350. [DOI] [PubMed] [Google Scholar]
- Liang SH, Clarke MF. Regulation of p53 localization. Eur J Biochem. 2001;268:2779–2783. doi: 10.1046/j.1432-1327.2001.02227.x. [DOI] [PubMed] [Google Scholar]
- Liang SH, Hong D, Clarke MF. Cooperation of a single lysine mutation and a C-terminal domain in the cytoplasmic sequestration of the p53 protein. J Biol Chem. 1998;273:19817–19821. doi: 10.1074/jbc.273.31.19817. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993a;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993b;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 2007;26:923–934. doi: 10.1038/sj.emboj.7601560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll UM, Ostermeyer AG, Haladay R, Winkfield B, Frazier M, Zambetti G. Cytoplasmic sequestration of wildtype p53 protein impairs the G1 checkpoint after DNA damage. Mol Cell Biol. 1996;16:1126–1137. doi: 10.1128/mcb.16.3.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroz K, Carrel L, Hunt PA. Germ cell development in the XXY mouse: evidence that X chromosome reactivation is independent of sexual differentiation. Dev Biol. 1999;207:229–238. doi: 10.1006/dbio.1998.9160. [DOI] [PubMed] [Google Scholar]
- O’Brate A, Giannakakou P. The importance of p53 location: nuclear or cytoplasmic zip code? Drug Resist Updat. 2003;6:313–322. doi: 10.1016/j.drup.2003.10.004. [DOI] [PubMed] [Google Scholar]
- O’Gorman S, Dagenais NAMQ, Marchuk Y. Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. PNAS. 1997;26:14602–14607. doi: 10.1073/pnas.94.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Q, Gorovsky MA. Histone H2A.Z acetylation modulates an essential charge patch. Mol Cell. 2001;7:1329–1335. doi: 10.1016/s1097-2765(01)00269-6. [DOI] [PubMed] [Google Scholar]
- Ryan JJ, Prochownik E, Gottlieb CA, Apel IJ, Merino R, Nunez G, Clarke MF. c-myc and bcl-2 modulate p53 function by altering p53 subcellular trafficking during the cell cycle. Proc Natl Acad Sci USA. 1994;91:5878–5882. doi: 10.1073/pnas.91.13.5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T. A subset of p53-deficient embryos exhibit exencephaly. Nat Genet. 1995;10:175–180. doi: 10.1038/ng0695-175. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Shaulsky G, Goldfinger N, Ben-Ze’ev A, Rotter V. Nuclear accumulation of p53 protein is mediated by several nuclear localization signals and plays a role in tumorigenesis. Mol Cell Biol. 1990;10:6565–6577. doi: 10.1128/mcb.10.12.6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulsky G, Goldfinger N, Tosky MS, Levine AJ, Rotter V. Nuclear localization is essential for the activity of p53 protein. Oncogene. 1991;6:2055–2065. [PubMed] [Google Scholar]
- Sluss HK, Armata H, Gallant J, Jones SN. Phosphorylation of serine 18 regulates distinct p53 functions in mice. Mol Cell Biol. 2004;24:976–984. doi: 10.1128/MCB.24.3.976-984.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhwa R, Takano S, Robert M, Yoshida A, Nomura H, Reddel RR, Mitsui Y, Kaul SC. Inactivation of tumor suppressor p53 by mot-2, a hsp70 family member. J Biol Chem. 1998;273:29586–29591. doi: 10.1074/jbc.273.45.29586. [DOI] [PubMed] [Google Scholar]
- Yu ZK, Geyer RK, Maki CG. MDM2-dependent ubiquitination of nuclear and cytoplasmic P53. Oncogene. 2000;19:5892–5897. doi: 10.1038/sj.onc.1203980. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.