

Abstract
Diabetic cardiomyopathy as an important threat to health occurs with or without coexistence of vascular diseases. The exact mechanisms underlying the disease remain incompletely clear. Although several pathological mechanisms responsible for diabetic cardiomyopathy have been proposed, oxidative stress is widely considered as one of the major causes for the pathogenesis of the disease. Hyperglycemia-, hyperlipidemia-, hypertension- and inflammation-induced oxidative stress are major risk factors for the development of microvascular pathogenesis in the diabetic myocardium, which results in abnormal gene expression, altered signal transduction and the activation of pathways leading to programmed myocardial cell deaths. In the present article, we aim to provide an extensive review of the role of oxidative stress and anti-oxidants in diabetic cardiomyopathy based on our own works and literature information available.
Keywords: Anti-oxidants, Diabetic cardiomyopathy, Oxidative stress
Introduction
The occurrence of systemic hyperglycemia in diabetes can ultimately cause long-term damage to multiple organs, leading to severe complications. The microvasculature is a key target of hyperglycemic damage, and damage to small blood vessels can initiate and/or exacerbate systemic complications. Diabetic cardiovascular injury includes both cardiac and peripheral blood vessels. However, research has focused largely on peripheral vascular injury. Retinopathy, neuropathy and nephropathy are all caused, in part, by changes in blood flow or abnormal vessel growth1–3. A disorder of the heart muscle in patients with diabetes is called diabetic cardiomyopathy (DCM), which can lead to inability of the heart to circulate blood through the body effectively, a state known as heart failure4. DCM is only said to exist if there is no coronary artery disease to explain the heart muscle disorder, although most heart failure in patients with diabetes results from coronary artery disease. Although recent studies have extended our understanding of the pathogenesis of the DCM, the mechanism by which hyperglycemia causes DCM remains incompletely understood1–3.
Oxidative stress has been linked both to the onset of diabetes and its complications5–9. Recent studies have shown that oxidative damage induced by reactive oxygen or nitrogen species (ROS and/or RNS) derived from hyperglycemia plays a critical role in diabetic injury in multiple organs. Thus, the relationship between oxidative stress and DCM is a major focus of current research. Although these pathogenic factors probably cause DCM through different mechanisms10–15, the major contribution of these pathogenic factors to DCM is oxidative stress16. Therefore, anti-oxidant prevention and therapy is critically important to prevent or postpone the development of diabetic cardiovascular complications. Here, we review the features of DCM in term of its physiological and pathogenic bases, the critical role of oxidative stresses in the pathogenesis of DCM and potential anti-oxidant therapy for DCM, with a focus on targets of the nuclear factor, erythroid-2-related factor 2 (Nrf2), and metallothionein (MT) based on the literature and our own studies.
Pathophysiological Base of DCM
Emerging evidence from experimental, pathological, epidemiological and clinical studies has shown that diabetes mellitus causes cardiac functional and structural changes, independent of hypertension, coronary artery disease or any other known cardiac diseases, which supports the existence of DCM17–19. DCM is characterized by a disproportionate increase in left ventricular (LV) mass and myocardial fibrosis, and, as an early complication of diabetes, is manifested by diastolic dysfunction followed by abnormalities in systolic function20. One particularity of DCM is the long latent phase, during which the disease progresses, but is completely asymptomatic. A prominent ‘a’ wave can also be noted in the jugular venous pulse, and the cardiac apical impulse might be overactive or sustained throughout systole. After the development of systolic dysfunction, LV dilation and symptomatic heart failure, the jugular venous pressure might become elevated, and the apical impulse would be displaced downward and to the left. These changes are accompanied by a variety of electrocardiographic changes that could be associated with DCM in 60% of patients without structural heart disease, although usually not in the early asymptomatic phase. Later in the progression, a prolonged QT interval might be indicative of fibrosis.
In most cases, DCM is detected with concomitant hypertension or coronary artery disease. When presenting with other cardiovascular complications (i.e., ischemic heart disease or hypertension), diabetic patients have a much worse prognosis than non-diabetic patients, and are more prone to progress to congestive heart failure21,22. Diastolic dysfunction characterized by increased ventricular wall stiffness, and increased diastolic relaxation time is prevalent at early stages of the cardiomyopathy. Cardiomyocyte death is paralleled by fibroblast replacement, and leads to interstitial fibrosis mediated primarily by transforming growth factor-β (TGF-β), whereas cell death is considered to be a result of the induction of cardiac oxidative stress, although several other mechanisms have been also associated with DCM10,23,24.
General Mechanisms Underlying DCM
Extensive evidence from experimental models of type 125 and type 226 diabetes implicates hyperglycemia as one of the main pathogenic mechanisms in DCM. An important consequence of high glucose-induced cellular injury is the formation of advanced glycation end-products (AGEs) resulting from the non-enzymatic glycation and oxidation of proteins and lipids27. Recently, high levels of AGE products have been found in cardiac tissue of diabetic patients28. Activation of the protein kinase C/diacylglycerol (PKC/DAG) signaling pathway is one of the mechanisms by which hyperglycemia exerts adverse cardiovascular effects. PKC activation by hyperglycemia contributes to cardiac fibrosis by stimulating connective tissue growth factor (CTGF) expression as shown in transgenic PKC-β2 mice29. Consistently, the overexpression of PKC-β2 isoform in the heart of transgenic mice resulted in LV hypertrophy, fibrosis and decreased LV ejection fraction (LV EF), similar to that of DCM30. PKC inhibition attenuated diastolic dysfunction, myocyte hypertrophy and collagen deposition, and preserved cardiac contractility in a rodent model of diabetic diastolic heart failure, suggesting that PKC-inhibition could represent a novel therapeutic strategy for the prevention of diabetes-associated cardiac dysfunction31.
Intracellular Ca2+ is a major regulator of cardiac contractility. Diabetes is associated with the impairment in myocardial performance as a result of abnormalities in contractile and regulatory protein expression, and in cardiomyocyte Ca2+ sensitivity. In a study of human cardiomyocytes from type 2 diabetes patients, Ca2+ dysregulation was indicated by reduced myofilament Ca2+ sensitivity32. Diabetes is associated with the activation of the renin–angiotensin system33 with consequent overproduction of angiotensin II (Ang II), its main physiological effector molecule. Ang II contributes to several alterations in diabetes including fibrosis by stimulating extracellular matrix component synthesis, apoptosis/proliferation, vascular inflammation and oxidative damage34. The oxidative damage includes deoxyribonucleic acid (DNA) damage, which could be related to activation of poly-(ADP-ribose) polymerase (PARP) enzymes35,36.
The endoplasmic reticulum (ER) is an organelle entrusted with lipid synthesis, calcium homeostasis, protein folding and maturation. Perturbation of ER-associated functions results in an evolutionarily conserved cell stress response, the unfolded protein response that is also called ER stress. ER stress is aimed initially at compensating for damage, but can eventually trigger cell death if ER stress is excessive or prolonged. Now ER stress has been associated with numerous diseases. For instance, our recent studies have shown the important role of ER stress in diabetes-induced cardiac cell death. It is known that apoptosis has been considered to play a critical role in diabetic cardiomyopathy. Inhibition of ER stress can result in the prevention of DCM37–39.
Apoptosis has been considered as the outcome of cardiomyocytes in response to diabetic hyperglycemia, hyperlipidemia, inflammation and ER stress, and is also the key initiating factor to stimulating resident cardiomyocyte hypertrophy and fibroblast proliferation, both leading to cardiac remodeling and eventually dysfunction23,40–42. In fact, autophagy, which is a process of self-cannibalization used by mammalian cells for the recycling of cellular contents, alters the heart of diabetic individuals. Like ER stress, autophagy can be both adaptive and maladaptive under specific settings43. In OVE26 type 1 diabetes mice, the adenosine monophosphate (AMP)-activated protein kinase (AMPK) is reduced along with cardiac dysfunction and decreased cardiac autophagy. Genetic inhibition of AMPK in cardiomyocytes attenuates cardiac autophagy, exacerbates cardiac dysfunction and increases mortality in diabetic mice. More importantly, chronic AMPK activation with metformin, one of the most used antidiabetes drugs and a well-characterized AMPK activator, significantly enhances autophagic activity, preserves cardiac function and prevents most of the primary characteristics of DCM in OVE26 mice, but not in dominant negative-AMPK diabetic mice. Therefore, AMPK activation protects cardiac structure and function by increasing cardiac autophagy in the diabetic heart44, whereas ceramide synthase-mediated lipid-induced autophagy and hypertrophy in cardiomyocytes is a cause of DCM45. Therefore, an urgent need exists to clarify the mechanism of pathogenesis of DCM. Emerging evidence shows that diabetes induces cardiomyocyte apoptosis and suppresses or stimulates cardiac autophagy, indicating that the interplay between autophagy and apoptotic cell death pathways is important in the pathogenesis of DCM42.
Oxidative Stress
Although several potential mechanisms underlying DCM have been addressed, all these pathogenic factors either induce or result from oxidative stress. Oxidative stress occurs when the production of ROS and/or RNS exceeds their degradation by anti-oxidant defenses. PARP proteins are overexpressed in diabetes46,47 as ROS- and/or RNS-induced oxidative DNA damage. Hyperglycemia in both type 1 diabetes and type 2 diabetes48,49 is associated with increased ROS and/RNS production in mitochondria, as shown in Figure1.
Figure 1.
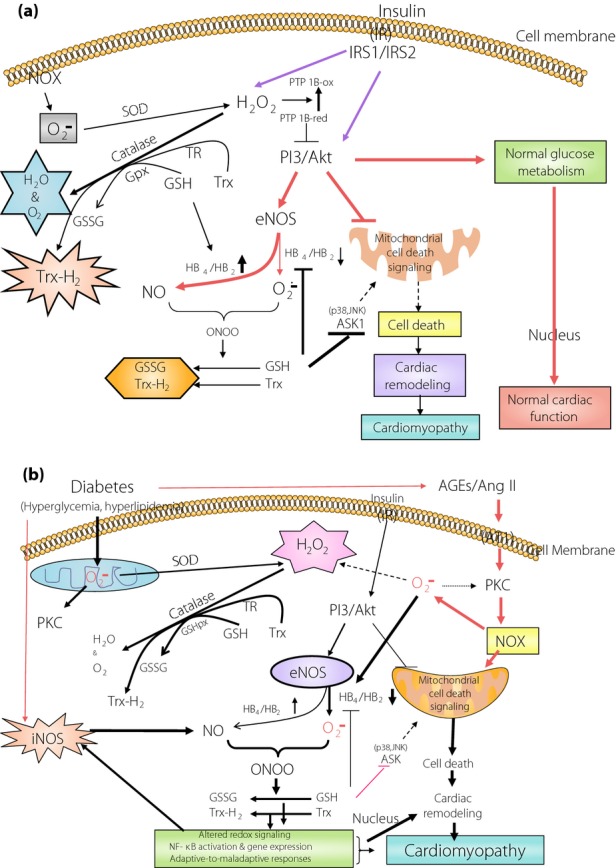
Outline of the possible physiological roles of redox signaling in maintaining the balance between various reactive oxygen species and/or reactive nitrogen species, and anti-oxidants under (a) normal and (b) diabetic conditions. AGE, advanced glycation end-products; Ang II, angiotensin II; ASK1, apoptosis signal-regulating kinase 1; AT1, angiotensin II-1 receptor; eNOS, endothelial nitric oxide synthase; GPx, glutathione peroxidase; GSH, glutathione; GSSG, glutathione disulfide; H2O2, hydrogen peroxide; HB, tetrahydrobiopterin; iNOS, inducible nitric oxide synthase; IR, insulin receptor; IRS1, insulin receptor substrate-1; IRS2, insulin receptor substrate-2; JNK, c-Jun NH2-terminal kinase; NF-κB, nuclear factor-κB; NO, nitric oxide; NOX, nicotinamide adenine dinucleotide phosphate oxidase; ONOO, peroxynitrite; PI3K/AKT, phosphoinositide 3-kinase/Akt; PKC, protein kinase C; PTP-1B, protein tyrosine phosphatase -1B; SOD, super oxide dismutase; TR, thioredoxin reductase; Trx-1, thioredoxin-1.
Mitochondria are a major source of ROS production. Cellular sources of ROS generation within the heart include cardiac myocytes, endothelial cells and neutrophils. The electron transport chain generates superoxide radicals as an inevitable by-product at complex I and complex III in the respiratory chain. ROS leads to cellular damage through several mechanisms (oxidation, interference with nitric oxide [NO] and modulation of detrimental intracellular signaling pathways). Therefore, increased ROS causes cardiac dysfunction by direct damage to proteins and DNA, and by inducing apoptosis.
Elevated free fatty acid (FFA) concentrations in the heart as a result of a lack of insulin-mediated glucose metabolism can increase ROS and/or RNS production from both non-mitochondrial sources, such as activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX), and modulating the mitochondrial electron chain to generate superoxide50. Activated NOX produces superoxide which, in turn, can combine with NO, forming highly reactive and damaging peroxynitrite species. NOXs have emerged as a culprit in this condition51. Cellular sources of ROS and/or RNS generation within the heart include cardiac myocytes, endothelial cells and neutrophils. ROS lead to cellular damage through several mechanisms (oxidation, interference with NO and modulation of detrimental intracellular signaling pathways). Therefore, increased ROS and/or RNS causes cardiac dysfunction by direct damage to proteins and DNA, as well as by inducing apoptosis. Oxidative damage and/or loss of anti-oxidant defense could also stimulate a number of responses associated with ventricular remodeling. These include activation of matrix metalloproteinase to alter the architecture of the extracellular matrix52, modulation of signal transduction pathways that initiate cardiomyocyte hypertrophy53 and apoptosis54, as summarized in Figure1.
Some experiments suggest that cytosolic ROS and/or RNS defense mechanisms might be more significantly impacted by diabetes than mitochondrial defense mechanisms. These include activation of matrix metalloproteinase to alter the architecture of the extracellular matrix52, modulation of signal transduction pathways that initiate cardiomyocyte hypertrophy53 and apoptosis54.
Anti-Oxidants in DCM
Given that oxidative and/or nitrosative stress seems a critical cause for the development of DCM, several anti-oxidant approaches have been investigated for their potential prevention and amelioration of DCM. Although anti-oxidant therapy has been extensively studied for its potential prevention or treatment of various diabetic complications, direct investigations for the prevention by anti-oxidants of DCM remain relatively scarce. Here, we provide some examples first, and then will focus on Nrf2 and MT as the major targets of anti-oxidative therapy for DCM based on our own and other works.
Thioredoxin 1
In recent studies55,56, intramyocardial thioredoxin 1 (Trx1) gene therapy in diabetic rats showed the prevention of angiogenic impairment and cardiac dysfunction severity after cardiac infarction. They found that overexpression of the Trx1 gene could reduce oxidative stress and apoptosis and, thereby, the extent of ventricular remodeling55,56.
Tempol
Super oxide dismutase (SOD) has been found to protect cardiomyocytes from superoxide-induced damage by converting superoxide to hydrogen peroxide57. Tempol, 4-hydroxy-2,2,6,6-tetramethyl piperidinoxyl, is a membrane-permeable SOD mimetic that has been shown to attenuate the effects of peroxynitrite in vascular systems58,59. In the diabetic heart, tempol was also found to significantly reduce diabetes-activated GSK-3β, associated with increases in caspase-3 cleavage and apoptotic cell death at 3 days after streptozotocin (STZ) injection60. The anti-apoptotic effect of tempol was accompanied with attenuation of STZ-induced cardiac hypertrophy and fibrosis, observed at 28 days after STZ injection. Prevention of diabetes-induced cardiac fibrosis was observed at 8 weeks in diabetes patients after 8-week tempol treatment, along with a significant reduction of oxidative stress as evidenced by reducing the expression of p22 phox, ROS production and increasing anti-oxidant enzyme capacity61.
Curcumin
Curcumin is the natural polyphenolic compound that was originally used in traditional Indian medicine over 3,000 years ago, and has shown diverse pharmacological properties including anti-oxidant activity62,63. Soetikno et al.64,65 have shown the prevention of DCM by curcumin in a STZ-induced diabetic rat model, and also found that the prevention of DCM by curcumin is associated with the suppression of NOX activation leading to the overgeneration of ROS and/or RNS, either through inhibition of PKC or activation of Akt pathways. We also reported the prevention of DCM by curcumin analog C66 in a STZ diabetic mouse model through suppression of c-Jun NH2-terminal kinase-related oxidative stress66.
Coenzyme Q10
Coenzyme Q10, a lipophilic cofactor of the mammalian mitochondrial electron transport chain, is not only crucial for mitochondrial energy production (adenosine 5′-triphosphate [ATP]), but also has emerged as an effective anti-oxidant. Coenzyme Q10 has shown some clinical benefits in a number of clinical trials67,68. Huynh et al.69 have treated type 1 diabetic mice at 4 weeks after hyperglycemia that was induced by multiple-low doses of STZ (55 mg/kg/day for 5 days) with coenzyme Q10 or angiotensin-converting-enzyme inhibitor (ACE-I) ramipril for 8 weeks. They found that diabetes upregulated LV Nox2 and superoxide production along with LV oxidative damage along with LV diastolic dysfunction and remodeling. All these were almost completely prevented by coenzyme Q10, which was also compared favorably with improvements observed with ramipril. Therefore, coenzyme Q10 supplementation could thus represent an effective alternative to ACE-Is for the treatment of cardiac complications in type 1 diabetic patients, as both medicines have multiple beneficial effects not only through suppression of oxidative stress.
Resveratrol
Resveratrol (RSV; trans-3,5,4′-trihydroxystilbene), a polyphenolic compound and naturally-occurring phytoalexin present in red wine and vegetable foods, was also used for the potential prevention or therapy for DCM. In experimental models of diabetes, RSV treatment was found to reduce the generation of ROS and the incidence of cardiomyocyte death, along with improvement of cardiac function70–74.
In summary, the aforementioned summarized anti-oxidants have been extensively applied in several other diseases, such as cancer and cardiovascular diseases, but whether these anti-oxidants can also be used for diabetic patients for the prevention of their DCM remains the subject of further systemic investigation, although the preliminary studies discussed here are promising.
Metallothionein
Metallothionein is a cysteine-containing (1/3 of 61 amino acids) and metal-binding protein. Physiological functions of MT include the maintenance of the homeostasis of essential metals and detoxification of heavy metals. MT maintains the redox status by binding and releasing metals (mainly zinc [Zn] under physiological conditions). Although four isoforms of MTs have been characterized, MT-I and MT-II exist as the major isoforms in various human and animal organs including the heart75,76. The physical, chemical and biological properties of MT have been summarized by other reviews77–79. Cardiac protection of MT against various oxidative damage induced by various oxidative stresses has been reviewed by others75,80–83. To address whether MT can protect the heart from diabetes, we have extensively explored the potential protection of MT from DCM since 200184,85. Type 1 diabetes was induced by a single injection of STZ in both cardiac-specific MT transgenic mice (MT-TG) and wild-type (WT) mice. No differences in hyperglycemia, bodyweight gain, and serum lipid peroxide levels were found between the MT-TG and the WT diabetic mice. However, serum creatine phosphokinase activity as a biomarker of cardiac injury was markedly increased in the WT diabetic mice. Significantly, cardiac injury was also found in the WT diabetic mice as evidenced by morphological changes. Therefore, these results provided the first evidence showing the preventive role of MT in the heart against diabetes-induced injury84,85. Following these early studies, several investigations from the authors' own laboratory and other laboratories86–90 in STZ-induced and transgenic OVE26 type 1 diabetic mouse models, and a high-fat diet-induced type 2 diabetic mouse model, further supported the significant protection of cardiac MT overexpression against diabetes-induced morphological and functional changes. Detailed review of the prevention of DCM by MT from several aspects, including experimental evidence, potential mechanisms and clinical implication, has been provided41,91,92. In the following section, we provide some new insights based on the most recent studies.
We have shown the important role of diabetes-induced nitrosative damage in the development of DCM41,87,92, and MT can efficiently prevent DCM development through suppression of nitrosative damage. Recently, we found that diabetes can specifically induce ATPase and succinyl-CoA:3-ketoacid CoA transferase-1 (SCOT; Figure2) and ATP synthase (Figure3)93,94. We have identified a nitrated protein of 58 kDa as SCOT by mass spectrometry in the heart of diabetic mice. We found that although total SCOT expression was not significantly different between MT-TG and WT diabetic mice, the diabetic WT hearts showed significantly increased nitration content and dramatically decreased catalyzing activity of SCOT (Figure2). Although SCOT nitration sites were identified at Tyr76, Tyr117, Tyr135, Tyr226, Tyr368 and Trp374, only Tyr76 and Trp374 were found to be located in the active site by 3-D structure modeling. However, only Trp374 showed a significantly different nitration level between the WT and MT-TG diabetic hearts. These results suggest that MT prevention of diabetes-induced pathological changes in cardiac tissues is most likely mediated by suppression of SCOT nitration at Trp37494; In another study, using LC/MS analysis we showed that diabetes increased tyrosine nitration of the ATP synthase α subunit at Tyr271, Tyr311 and Tyr476, and the β subunit at Tyr269 and Tyr508, and also significantly reduced ATP synthase activity by ∼32% (Figure3). These changes were not observed in MT-TG diabetic mice93. These results suggest that MT can preserve SCOT and ATP synthase activity in STZ-induced diabetes, probably through the inhibition of SCOT and ATP synthase nitration.
Figure 2.
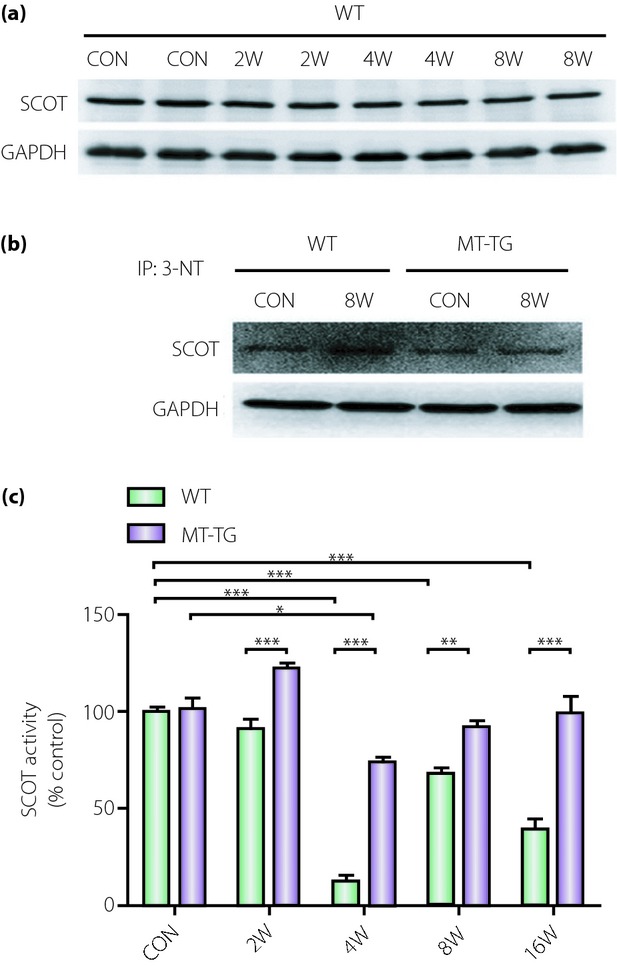
3-Ketoacid CoA transferase-1 (SCOT) protein expression, nitration, and activity in the hearts of diabetic wild-type (WT) and cardiac-specific metallothionein-overexpressing transgenic (MT-TG) mice. Type 1 diabetes was induced with a single dose of streptozotocin (STZ) in Friend virus B-type (wild-type) and cardiac-specific overexpression of MT-TG mice used for the study94. (a) Cardiac tissues collected at the indicated post-induction times were examined by western blot to show changes in SCOT expression. (b) Cardiac tissues from non-diabetic and diabetic WT and MT-TG mice at week 8 post-induction were immunoprecipitated with 3-nitrotyrosine (3-NT) antibody and western blotted with antibodies against succinyl-CoA:3 SCOT. Comparison of SCOT catalytic activity between non-diabetic and diabetic WT and MT-TG mice at the indicated post-induction times using spectrophotometric monitoring of acetoacetyl-CoA formation. (c) SCOT catalytic activity was decreased in diabetic WT mice at weeks 4, 8 and 16 post-induction, but MT prevented this effect. *P < 0.05, **P < 0.01, ***P < 0.001. This work by the authors has been published94. CON, control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Figure 3.
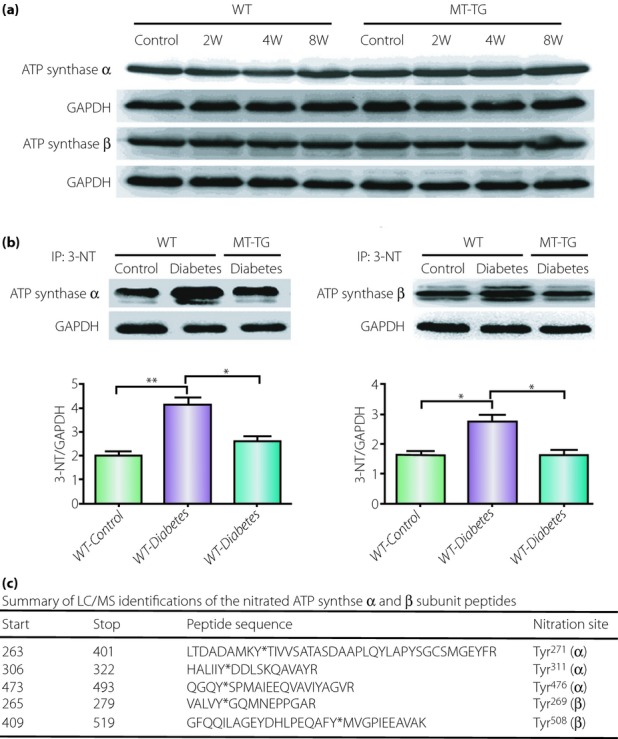
Adenosine 5′-triphosphate (ATP) synthase α and β subunit expression and nitration. Type 1 diabetes was induced with a single dose of STZ in Friend virus B-type (wild-type) and cardiac-specific overexpression of metallothionein transgenic (MT-TG) mice used for the study93. (a) Total expressions of ATP synthase α and β subunits in wild-type (WT) and MT-TG mice from control or diabetes at 2, 4 and 8 weeks after diabetes onset were unchanged (30 μg total proteins/lane). (b) To define the nitration of ATP synthase α and β subunits, cardiac tissues (2 mg total proteins/lane) from WT control and diabetic mice along with MT-TG diabetic mice were immunoprecipitated with 3-nitrotyrosine (3-NT) antibody, followed by western blot for ATP synthase α and β subunits with anti-ATP synthase α and anti-ATP synthase β antibody, respectively. Diabetes-induced increased 3-NT in the heart was prevented in MT-TG mice. *P < 0.05; **P < 0.01. (c) The summary of LC/MS identifications of the nitrated sites for ATP synthase α and β subunits peptides. Bold letters in the table are LC/MS identifications for the nitrated sites of ATP synthase α and β subunit peptides, which are the nitrated sites at Tyr271, Tyr311, Tyr476, Tyr269 and Tyr508. This work by the authors has been published93. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Velic et al.90 have utilized light microscopy (LM) and transmission electron microscopy (TEM) to show cardiomyocyte morphology and myocardial capillary basement membrane (CBM) thickness in the LV. Although the molecular mechanisms regulating CBM width remain elusive, it seems possible that despite a significant hyperglycemic environment, MT anti-oxidant activity could mitigate local oxidative stress and reduce downstream excess microvascular extracellular matrix (ECM) formation. In addition, the reduction of intra- and perivascular ROS and/or RNS might protect against incipient endothelial damage and the CBM thickening that results from such injury.
In summary, MT has shown strong cardioprotection from diabetes under different conditions: type 1 diabetes (STZ-induced and OVE26 transgenic diabetic mouse models), obesity-related diabetes in mice and even in humans with type 2 diabetes95,96. As Zn is an important trace element that is required for more than 300 enzymes and transcription factors, and can induce cardiac MT97,98, and MT plays an important role in intracellular Zn, Zn supplementation could be an alternative approach to intervention of DCM in clinics in the future.
Nrf2
Nrf2, a transcription factor belonging to the Cap'n'co'l'r/basic region leucine zipper (CNC-bZIP) family of transcription factors, plays the role as a chief controller of intracellular redox status and the detoxification process99,100. Nrf2 is bound to Kelch-like ECH-associated protein 1 (Keap1) in the cytoplasm, which facilitates its proteasomal degradation101. Nrf2 can promote its target genes, such as NADPH quinone oxidoreductase (NQO1), glutathione S-transferase, heme oxygenase-1 (HO1) and γ-glutamylcysteine synthetase, to play a role in the process of detoxification, anti-oxidative stress and anti-inflammation102. As Nrf2 is ubiquitously expressed, the implication of its roles in a variety of pathologies has been described mainly through the use of Nrf2 knock-out mice (Nrf2-KO) in different disease models.
The potential therapeutic action of Nrf2 in the treatment of several diseases including DCM has been suggested99. In order to answer whether Nrf2 can provide protection against high glucose-induced oxidative damage, we carried out the first study to compare oxidative stress, apoptosis and the contractile activities in both Nrf2 WT cardiomyocytes and Nrf2-KO cardiomyocytes in vitro103. Treated with different concentrations of glucose and 3-nitropropionic acid (inhibition of the mitochondrial respiratory complex II) caused increases of these effects in both kinds of cells in a concentration-dependent manner. Furthermore, the lever of oxidative damage was more severe, and contractile activities were worse in Nrf2-KO cells than WT cells. It means that Nrf2 is critical in protecting cardiomyocytes from high glucose- and mitochondrial damage-induced oxidative lesions in cardiomyocytes103. Following this early study, we and others have extensively investigated the potential protection of Nrf2 against diabetic complications including DCM. An early review regarding this topic is available100,104,105. In the following section we will provide some new information regarding the expression of Nrf2 in the heart under diabetic conditions, and how to protect the heart from diabetes by manipulating Nrf2 expression.
Nrf2 as an Adaptive Response Protein is Upregulated at the Early Stage of Diabetes, but Downregulated at the Late Stage
It is known that Nrf2 expression and function in cells in vitro and tissues in vivo are increased in response to oxidative stress. Several studies have shown the induction of ROS and/or RNS by high levels of glucose (HG) in cultured cardiovascular cells, and whether HG could elevate the Nrf2 expression and activation and its downstream gene expression in these cells has been investigated103. Treatment with glucose at 20 and 40 mmol/L for 24 h increased the expression of Nrf2 messenger ribonucleic acid in primary cardiomyocytes or the H9C2 cardiac cell line. Nqo1, a prototype of Nrf2-regulated chemical-detoxification gene, was induced to be overexpressed in cardiomyocytes by such HG exposure too. Immunoblotting confirmed the protein expression and induction of Nrf2 and HO1 in cardiomyocytes. Immunofluorescent confocal microscopic examination of the cells showed that HG treatment significantly increased the nuclear and total cell staining of Nrf2 in comparison with the control cells, indicating that glucose indeed increased the protein level and nuclear accumulation of Nrf2103.
Upregulation of Nrf2 and/or its downstream anti-oxidant genes in response to hyperglycemia were found not only in the cultured cells, but also observed in the heart of diabetic mice. We have used C57BL/6 mice to induce type 1 diabetes with a single dose of STZ. At 2 weeks after hyperglycemia onset, we found a significant upregulation of Nrf2 downstream genes NQO1 and HO1 messenger ribonucleic acid expression103. Tan et al.106 have shown a different finding in terms of Nrf2 expression in the cardiac tissue from diabetic patients and controls. Tissue sections of left ventricles were obtained from autopsy heart specimens of humans with or without diabetes (all diabetic males had histories of hypertension and cardiac dysfunction). Nrf2 expression in the nuclei was significantly downregulated compared with the control hearts.
In support of the aforementioned point, our recent finding showed that Nrf2 protein expression was slightly increased in the heart of mice with hyperglycemia for 3 months, but significantly decreased in the heart of mice with hyperglycemia for 6 months107. Combined with our early study103 in which Nrf2 downstream genes were increased in the heart of diabetic mice at 2 weeks after STZ-induced hyperglycemia, we assume that Nrf2 is adaptively trying to remain functional to overcome diabetic damage at the early stage of diabetes. At the late stage of diabetes, however, cardiac anti-oxidant function is further impaired, leading to a decrease in cardiac Nrf2 expression. Therefore, these aforementioned studies imply the preventive function of Nrf2 against diabetes-induced oxidative damage.
Upregulation of Nrf2 Protects the Heart from Diabetes
MG132 is a specific, potent, reversible and cell-permeable proteasome inhibitor, and thus is able to reduce the degradation of ubiquitin-conjugated proteins in mammalian cells. MG-132 as a proteasome inhibitor can upregulate Nrf2-mediated anti-oxidative function and downregulate nuclear factor-kappa-light-chain-enhancer of activated B cells (NF-κB)-mediated inflammation; therefore, it has been used to prevent diabetic renal damage108,109. We have investigated too whether through the aforementioned two mechanisms MG-132 could provide a therapeutic effect on DCM in the OVE26 type 1 diabetes mouse model. OVE26 mice develop hyperglycemia at 2–3 weeks after birth, and show albuminuria and cardiac dysfunction at 3 months-of-age. Therefore, 3-month-old OVE26 diabetic and age-matched control mice were intraperitoneally treated with MG-132 daily for 3 months. Diabetic mice showed significant cardiac dysfunction, including increased LV systolic diameter and wall thickness, and decreased LV EF with an increase of the heart weight-to-tibia length ratio. Diabetic hearts showed structural derangement and remodeling (fibrosis and hypertrophy). In diabetic mice, there was also increased systemic and cardiac oxidative damage and inflammation. All of these pathogenic changes were reversed by MG-132 treatment. MG-132 treatment significantly increased the cardiac expression of Nrf2 and its downstream anti-oxidant genes with a significant increase of total anti-oxidant capacity, and also significantly decreased the expression of NF-κB inhibitor alpha and the nuclear accumulation and DNA-binding activity of NF-κB in the heart. These results suggest that MG-132 has a therapeutic effect on diabetic cardiomyopathy in OVE26 diabetic mice, possibly through the upregulation of Nrf2-dependent anti-oxidative function and downregulation of NF-κB-mediated inflammation110.
Sulforaphane (SFN) is a molecule containing an isothiocyanate functional group, which is obtained from cruciferous vegetables, such as broccoli, Brussels sprouts or cabbages, and shows anticancer and antidiabetic properties in experimental models111. We have investigated whether the chronic use of SFN can prevent the development of DCM in a type 1 diabetes mouse model that was induced with multiple low-dose STZ (MLD-STZ). Diabetic and control mice were treated with SFN for 3 months and then kept for another 3 months without SFN treatment. SFN could almost completely prevent the development of DCM along with an upregulation of Nrf2 expression and transcription function in the heart. The data showed that the exposure of the cultured cardiac H9c2 cells to HG increased the fibrotic effect, which could be completely prevented by pretreatment with SFN, which also significantly increased Nrf2 expression and transcription. Silencing Nrf2 expression completely abolished the prevention by SFN of a HG-induced fibrotic effect, suggesting the important role of Nrf2 in the cardiac protection by SFN against HG in vitro or diabetes in vivo107.
Similarly, diallyl trisulfide (DATS) is a most powerful anti-oxidant among the sulfur-containing compounds in garlic oil. It has been reported that treatment of H9c2 cells with HG resulted in an increase in intracellular ROS level and caspase-3 activity, which were markedly reduced by the administration of DATS. DATS treatment significantly increased Nrf2 protein stability and nuclear translocation, upregulated downstream gene HO-1 and suppressed its repressor, Keap1. However, apoptosis was not inhibited by DATS in cells transfected with Nrf2-specific small interfering ribonucleic acid. Similar results were also observed in high glucose-exposed neonatal primary cardiomyocytes and streptozotocin-treated diabetic rats fed DATS at a dose of 40 mg/kg bodyweight. The findings show that DATS protects against hyperglycemia-induced ROS-mediated apoptosis by upregulating the Nrf2 pathway to further activate Nrf2-regulated anti-oxidant enzymes in cardiomyocytes exposed to HG112.
Therefore, Nrf2 as a new target of anti-oxidant therapy could have a great potential for clinical application, as these Nrf2 activators have a less toxic effect. In terms of the mechanism by which SFN prevents the heart from diabetes, we assumed that it is mediated by upregulation of Nrf2 because of the direct role of Nrf2 in the prevention of the HG-induced fibrotic effect in cultured H9c2 cardiac cells; however, whether the protection by SFN from DCM in vivo is also completely mediated by the upregulation of cardiac Nrf2 expression and function remains to be further examine, as well as whether SFN treatment is able to protect the heart from diabetes in the Nrf2-KO mouse model.
Conclusions
Diabetes mellitus has been recognized as a major cause of morbidity and mortality for decades. There is a large body of evidence to suggest that diabetic patients are prone to significant perturbations at the cellular and molecular level causing structural and functional abnormalities in the myocardium and vasculature, leading to DCM. As already described, some extra research steps are required so as to pinpoint the molecular mechanisms underlying the DCM before proceeding to the proposition of relevant clinical trials. New insights into the mechanisms that increase oxidative stress in diabetes might lead to novel treatment strategies. Clinically relevant translational studies in this direction might help to further unravel the underlying mechanisms of DCM.
Acknowledgments
The work cited from the authors' group was supported in part by the grants from the American Diabetes Association (05-07-CD-02, 1-11-BA-17 to LC) and Development Program of Jilin Province Science and Technology Agency (20130206048SF to QL).
References
- Grundy SM, Benjamin IJ, Burke GL, et al. Diabetes and cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation. 1999;100:1134–1146. doi: 10.1161/01.cir.100.10.1134. [DOI] [PubMed] [Google Scholar]
- Francis GS. Diabetic cardiomyopathy: fact or fiction? Heart. 2001;85:247–248. doi: 10.1136/heart.85.3.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowers JR, Epstein M, Frohlich ED. Diabetes, hypertension, and cardiovascular disease: an update. Hypertension. 2001;37:1053–1059. doi: 10.1161/01.hyp.37.4.1053. [DOI] [PubMed] [Google Scholar]
- Rubler S, Dlugash J, Yuceoglu YZ, et al. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30:595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes. 1999;48:1–9. doi: 10.2337/diabetes.48.1.1. [DOI] [PubMed] [Google Scholar]
- Koufen P, Ruck A, Brdiczka D, et al. Free radical-induced inactivation of creatine kinase: influence on the octameric and dimeric states of the mitochondrial enzyme (Mib-CK) Biochem J. 1999;344(Pt 2):413–417. [PMC free article] [PubMed] [Google Scholar]
- Kowluru RA, Engerman RL, Kern TS. Diabetes-induced metabolic abnormalities in myocardium: effect of antioxidant therapy. Free Radic Res. 2000;32:67–74. doi: 10.1080/10715760000300071. [DOI] [PubMed] [Google Scholar]
- Ustinova EE, Barrett CJ, Sun SY, et al. Oxidative stress impairs cardiac chemoreflexes in diabetic rats. Am J Physiol Heart Circ Physiol. 2000;279:H2176–H2187. doi: 10.1152/ajpheart.2000.279.5.H2176. [DOI] [PubMed] [Google Scholar]
- Uemura S, Matsushita H, Li W, et al. Diabetes mellitus enhances vascular matrix metalloproteinase activity: role of oxidative stress. Circ Res. 2001;88:1291–1298. doi: 10.1161/hh1201.092042. [DOI] [PubMed] [Google Scholar]
- Fang ZY, Prins JB, Marwick TH. Diabetic cardiomyopathy: evidence, mechanisms, and therapeutic implications. Endocr Rev. 2004;25:543–567. doi: 10.1210/er.2003-0012. [DOI] [PubMed] [Google Scholar]
- Cai L, Kang YJ. Oxidative stress and diabetic cardiomyopathy: a brief review. Cardiovasc Toxicol. 2001;1:181–193. doi: 10.1385/ct:1:3:181. [DOI] [PubMed] [Google Scholar]
- Pang Y, Hunton DL, Bounelis P, et al. Hyperglycemia inhibits capacitative calcium entry and hypertrophy in neonatal cardiomyocytes. Diabetes. 2002;51:3461–3467. doi: 10.2337/diabetes.51.12.3461. [DOI] [PubMed] [Google Scholar]
- Ligeti L, Szenczi O, Prestia CM, et al. Altered calcium handling is an early sign of streptozotocin-induced diabetic cardiomyopathy. Int J Mol Med. 2006;17:1035–1043. [PubMed] [Google Scholar]
- Finck BN, Han X, Courtois M, et al. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci USA. 2003;100:1226–1231. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwan A, Tran T, Misra A, et al. Inflammatory mediators and the failing heart: a translational approach. Curr Mol Med. 2003;3:161–182. doi: 10.2174/1566524033361537. [DOI] [PubMed] [Google Scholar]
- Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- Bell DS. Diabetic cardiomyopathy. A unique entity or a complication of coronary artery disease? Diabetes Care. 1995;18:708–714. doi: 10.2337/diacare.18.5.708. [DOI] [PubMed] [Google Scholar]
- Rodrigues B, McNeill JH. The diabetic heart: metabolic causes for the development of a cardiomyopathy. Cardiovasc Res. 1992;26:913–922. doi: 10.1093/cvr/26.10.913. [DOI] [PubMed] [Google Scholar]
- Spector KS. Diabetic cardiomyopathy. Clin Cardiol. 1998;21:885–887. doi: 10.1002/clc.4960211205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fein FS, Sonnenblick EH. Diabetic cardiomyopathy. Prog Cardiovasc Dis. 1985;27:255–270. doi: 10.1016/0033-0620(85)90009-x. [DOI] [PubMed] [Google Scholar]
- Solang L, Malmberg K, Ryden L. Diabetes mellitus and congestive heart failure. Further knowledge needed. Eur Heart J. 1999;20:789–795. doi: 10.1053/euhj.1998.1472. [DOI] [PubMed] [Google Scholar]
- Bauters C, Lamblin N, Mc Fadden EP, et al. Influence of diabetes mellitus on heart failure risk and outcome. Cardiovasc Diabetol. 2003;2:1. doi: 10.1186/1475-2840-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Kang YJ. Cell death and diabetic cardiomyopathy. Cardiovasc Toxicol. 2003;3:219–228. doi: 10.1385/ct:3:3:219. [DOI] [PubMed] [Google Scholar]
- Li X, Xu Z, Li S, et al. Redox regulation of Ito remodeling in diabetic rat heart. Am J Physiol Heart Circ Physiol. 2005;288:H1417–H1424. doi: 10.1152/ajpheart.00559.2004. [DOI] [PubMed] [Google Scholar]
- Retnakaran R, Zinman B. Type 1 diabetes, hyperglycaemia, and the heart. Lancet. 2008;371:1790–1799. doi: 10.1016/S0140-6736(08)60767-9. [DOI] [PubMed] [Google Scholar]
- Ginsberg BJ, Mazze R. Clinical consequences of the Diabetes Control and Complications Trial. N J Med. 1994;91:221–224. [PubMed] [Google Scholar]
- Wang J, Song Y, Wang Q, et al. Causes and characteristics of diabetic cardiomyopathy. Rev Diabet Stud. 2006;3:108–117. doi: 10.1900/RDS.2006.3.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen S, Hartog JW, Hummel YM, et al. Tissue advanced glycation end products are associated with diastolic function and aerobic exercise capacity in diabetic heart failure patients. Eur J Heart Fail. 2011;13:76–82. doi: 10.1093/eurjhf/hfq168. [DOI] [PubMed] [Google Scholar]
- Way KJ, Isshiki K, Suzuma K, et al. Expression of connective tissue growth factor is increased in injured myocardium associated with protein kinase C beta2 activation and diabetes. Diabetes. 2002;51:2709–2718. doi: 10.2337/diabetes.51.9.2709. [DOI] [PubMed] [Google Scholar]
- Wakasaki H, Koya D, Schoen FJ, et al. Targeted overexpression of protein kinase C beta2 isoform in myocardium causes cardiomyopathy. Proc Natl Acad Sci USA. 1997;94:9320–9325. doi: 10.1073/pnas.94.17.9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly KA, Kelly DJ, Zhang Y, et al. Inhibition of protein kinase C-beta by ruboxistaurin preserves cardiac function and reduces extracellular matrix production in diabetic cardiomyopathy. Circ Heart Fail. 2009;2:129–137. doi: 10.1161/CIRCHEARTFAILURE.108.765750. [DOI] [PubMed] [Google Scholar]
- Jweied EE, McKinney RD, Walker LA, et al. Depressed cardiac myofilament function in human diabetes mellitus. Am J Physiol Heart Circ Physiol. 2005;289:H2478–H2483. doi: 10.1152/ajpheart.00638.2005. [DOI] [PubMed] [Google Scholar]
- Lim HS, MacFadyen RJ, Lip GY. Diabetes mellitus, the renin-angiotensin-aldosterone system, and the heart. Arch Intern Med. 2004;164:1737–1748. doi: 10.1001/archinte.164.16.1737. [DOI] [PubMed] [Google Scholar]
- Zhou G, Li X, Hein DW, et al. Metallothionein suppresses angiotensin II-induced nicotinamide adenine dinucleotide phosphate oxidase activation, nitrosative stress, apoptosis, and pathological remodeling in the diabetic heart. J Am Coll Cardiol. 2008;52:655–666. doi: 10.1016/j.jacc.2008.05.019. [DOI] [PubMed] [Google Scholar]
- Pacher P, Liaudet L, Soriano FG, et al. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51:514–521. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- Chiu J, Farhangkhoee H, Xu BY, et al. PARP mediates structural alterations in diabetic cardiomyopathy. J Mol Cell Cardiol. 2008;45:385–393. doi: 10.1016/j.yjmcc.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Xu J, Zhou Q, Xu W, et al. Endoplasmic reticulum stress and diabetic cardiomyopathy. Exp Diabetes Res. 2012;2012:827971. doi: 10.1155/2012/827971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZW, Zhu HT, Chen KL, et al. Protein kinase RNA-like endoplasmic reticulum kinase (PERK) signaling pathway plays a major role in reactive oxygen species (ROS)-mediated endoplasmic reticulum stress-induced apoptosis in diabetic cardiomyopathy. Cardiovasc Diabetol. 2013;12:158. doi: 10.1186/1475-2840-12-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Zhao Z, Cai T, et al. Liraglutide alleviates diabetic cardiomyopathy by blocking CHOP-triggered apoptosis via the inhibition of the IRE-alpha pathway. Mol Med Rep. 2014;9:1254–1258. doi: 10.3892/mmr.2014.1956. [DOI] [PubMed] [Google Scholar]
- Cai L, Li W, Wang G, et al. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes. 2002;51:1938–1948. doi: 10.2337/diabetes.51.6.1938. [DOI] [PubMed] [Google Scholar]
- Cai L. Suppression of nitrative damage by metallothionein in diabetic heart contributes to the prevention of cardiomyopathy. Free Radic Biol Med. 2006;41:851–861. doi: 10.1016/j.freeradbiomed.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Ouyang C, You J, Xie Z. The interplay between autophagy and apoptosis in the diabetic heart. J Mol Cell Cardiol. 2014;71C:71–80. doi: 10.1016/j.yjmcc.2013.10.014. [DOI] [PubMed] [Google Scholar]
- Mellor KM, Reichelt ME, Delbridge LM. Autophagy anomalies in the diabetic myocardium. Autophagy. 2011;7:1263–1267. doi: 10.4161/auto.7.10.17148. [DOI] [PubMed] [Google Scholar]
- Xie Z, He C, Zou MH. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy. 2011;7:1254–1255. doi: 10.4161/auto.7.10.16740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SB, Baicu CF, Van Laer A, et al. Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J Clin Invest. 2012;122:3919–3930. doi: 10.1172/JCI63888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Zingarelli B, Szabo C. Effect of genetic disruption of poly (ADP-ribose) synthetase on delayed production of inflammatory mediators and delayed necrosis during myocardial ischemia-reperfusion injury. Shock. 2000;13:60–66. doi: 10.1097/00024382-200013010-00011. [DOI] [PubMed] [Google Scholar]
- Szabo C. Roles of poly(ADP-ribose) polymerase activation in the pathogenesis of diabetes mellitus and its complications. Pharmacol Res. 2005;52:60–71. doi: 10.1016/j.phrs.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Wold LE, Ren J. Streptozotocin directly impairs cardiac contractile function in isolated ventricular myocytes via a p38 map kinase-dependent oxidative stress mechanism. Biochem Biophys Res Commun. 2004;318:1066–1071. doi: 10.1016/j.bbrc.2004.04.138. [DOI] [PubMed] [Google Scholar]
- Boudina S, Sena S, O'Neill BT, et al. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- Steinberg HO, Paradisi G, Hook G, et al. Free fatty acid elevation impairs insulin-mediated vasodilation and nitric oxide production. Diabetes. 2000;49:1231–1238. doi: 10.2337/diabetes.49.7.1231. [DOI] [PubMed] [Google Scholar]
- Olukman M, Orhan CE, Celenk FG, et al. Apocynin restores endothelial dysfunction in streptozotocin diabetic rats through regulation of nitric oxide synthase and NADPH oxidase expressions. J Diabetes Complications. 2010;24:415–423. doi: 10.1016/j.jdiacomp.2010.02.001. [DOI] [PubMed] [Google Scholar]
- King MK, Coker ML, Goldberg A, et al. Selective matrix metalloproteinase inhibition with developing heart failure: effects on left ventricular function and structure. Circ Res. 2003;92:177–185. doi: 10.1161/01.res.0000052312.41419.55. [DOI] [PubMed] [Google Scholar]
- Cesselli D, Jakoniuk I, Barlucchi L, et al. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res. 2001;89:279–286. doi: 10.1161/hh1501.094115. [DOI] [PubMed] [Google Scholar]
- Takano H, Zou Y, Hasegawa H, et al. Oxidative stress-induced signal transduction pathways in cardiac myocytes: involvement of ROS in heart diseases. Antioxid Redox Signal. 2003;5:789–794. doi: 10.1089/152308603770380098. [DOI] [PubMed] [Google Scholar]
- Samuel SM, Thirunavukkarasu M, Penumathsa SV, et al. Thioredoxin-1 gene therapy enhances angiogenic signaling and reduces ventricular remodeling in infarcted myocardium of diabetic rats. Circulation. 2010;121:1244–1255. doi: 10.1161/CIRCULATIONAHA.109.872481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adluri RS, Thirunavukkarasu M, Zhan L, et al. Thioredoxin 1 enhances neovascularization and reduces ventricular remodeling during chronic myocardial infarction: a study using thioredoxin 1 transgenic mice. J Mol Cell Cardiol. 2011;50:239–247. doi: 10.1016/j.yjmcc.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- Nassar T, Kadery B, Lotan C, et al. Effects of the superoxide dismutase-mimetic compound tempol on endothelial dysfunction in streptozotocin-induced diabetic rats. Eur J Pharmacol. 2002;436:111–118. doi: 10.1016/s0014-2999(01)01566-7. [DOI] [PubMed] [Google Scholar]
- Jagadeesha DK, Lindley TE, Deleon J, et al. Tempol therapy attenuates medial smooth muscle cell apoptosis and neointima formation after balloon catheter injury in carotid artery of diabetic rats. Am J Physiol Heart Circ Physiol. 2005;289:H1047–H1053. doi: 10.1152/ajpheart.01071.2004. [DOI] [PubMed] [Google Scholar]
- Gurusamy N, Watanabe K, Ma M, et al. Glycogen synthase kinase 3beta together with 14-3-3 protein regulates diabetic cardiomyopathy: effect of losartan and tempol. FEBS Lett. 2006;580:1932–1940. doi: 10.1016/j.febslet.2006.02.056. [DOI] [PubMed] [Google Scholar]
- Taye A, Abouzied MM, Mohafez OM. Tempol ameliorates cardiac fibrosis in streptozotocin-induced diabetic rats: role of oxidative stress in diabetic cardiomyopathy. Naunyn Schmiedebergs Arch Pharmacol. 2013;386:1071–1080. doi: 10.1007/s00210-013-0904-x. [DOI] [PubMed] [Google Scholar]
- Ammon HP, Wahl MA. Pharmacology of Curcuma longa. Planta Med. 1991;57:1–7. doi: 10.1055/s-2006-960004. [DOI] [PubMed] [Google Scholar]
- Bengmark S. Curcumin, an atoxic antioxidant and natural NFkappaB, cyclooxygenase-2, lipooxygenase, and inducible nitric oxide synthase inhibitor: a shield against acute and chronic diseases. JPEN J Parenter Enteral Nutr. 2006;30:45–51. doi: 10.1177/014860710603000145. [DOI] [PubMed] [Google Scholar]
- Soetikno V, Sari FR, Sukumaran V, et al. Curcumin prevents diabetic cardiomyopathy in streptozotocin-induced diabetic rats: possible involvement of PKC-MAPK signaling pathway. Eur J Pharm Sci. 2012;47:604–614. doi: 10.1016/j.ejps.2012.04.018. [DOI] [PubMed] [Google Scholar]
- Yu W, Wu J, Cai F, et al. Curcumin alleviates diabetic cardiomyopathy in experimental diabetic rats. PLoS ONE. 2012;7:e52013. doi: 10.1371/journal.pone.0052013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhou S, Sun W, et al. Inhibition of JNK by novel curcumin analog C66 prevents diabetic cardiomyopathy with a preservation of cardiac metallothionein expression. Am J Physiol Endocrinol Metab. 2014;306:e1239–1247. doi: 10.1152/ajpendo.00629.2013. [DOI] [PubMed] [Google Scholar]
- Kolahdouz MR, Hosseinzadeh-Attar MJ, Eshraghian MR, et al. The effect of coenzyme Q10 supplementation on metabolic status of type 2 diabetic patients. Minerva Gastroenterol Dietol. 2013;59:231–236. [PubMed] [Google Scholar]
- Carrasco J, Anglada FJ, Campos JP, et al. The protective role of coenzyme Q10 in renal injury associated with extracorporeal shockwave lithotripsy: a randomised, placebo-controlled clinical trial. BJU Int. 2014;113:942–950. doi: 10.1111/bju.12485. [DOI] [PubMed] [Google Scholar]
- Huynh K, Kiriazis H, Du XJ, et al. Targeting the upregulation of reactive oxygen species subsequent to hyperglycemia prevents type 1 diabetic cardiomyopathy in mice. Free Radic Biol Med. 2013;60:307–317. doi: 10.1016/j.freeradbiomed.2013.02.021. [DOI] [PubMed] [Google Scholar]
- Palsamy P, Subramanian S. Modulatory effects of resveratrol on attenuating the key enzymes activities of carbohydrate metabolism in streptozotocin-nicotinamide-induced diabetic rats. Chem Biol Interact. 2009;179:356–362. doi: 10.1016/j.cbi.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Zhang H, Morgan B, Potter BJ, et al. Resveratrol improves left ventricular diastolic relaxation in type 2 diabetes by inhibiting oxidative/nitrative stress: in vivo demonstration with magnetic resonance imaging. Am J Physiol Heart Circ Physiol. 2010;299:H985–H994. doi: 10.1152/ajpheart.00489.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirunavukkarasu M, Penumathsa SV, Koneru S, et al. Resveratrol alleviates cardiac dysfunction in streptozotocin-induced diabetes: role of nitric oxide, thioredoxin, and heme oxygenase. Free Radic Biol Med. 2007;43:720–729. doi: 10.1016/j.freeradbiomed.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulaiman M, Matta MJ, Sunderesan NR, et al. Resveratrol, an activator of SIRT1, upregulates sarcoplasmic calcium ATPase and improves cardiac function in diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2010;298:H833–H843. doi: 10.1152/ajpheart.00418.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian B, Yang S, Chaudry IH, et al. Resveratrol improves cardiac contractility following trauma-hemorrhage by modulating Sirt1. Mol Med. 2012;18:209–214. doi: 10.2119/molmed.2011.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Satoh M, Tohyama C, et al. Metallothionein in radiation exposure: its induction and protective role. Toxicology. 1999;132:85–98. doi: 10.1016/s0300-483x(98)00150-4. [DOI] [PubMed] [Google Scholar]
- Templeton DM, Cherian MG. Toxicological significance of metallothionein. Methods Enzymol. 1991;205:11–24. doi: 10.1016/0076-6879(91)05079-b. [DOI] [PubMed] [Google Scholar]
- Davis SR, Cousins RJ. Metallothionein expression in animals: a physiological perspective on function. J Nutr. 2000;130:1085–1088. doi: 10.1093/jn/130.5.1085. [DOI] [PubMed] [Google Scholar]
- Vallee BL. The function of metallothionein. Neurochem Int. 1995;27:23–33. doi: 10.1016/0197-0186(94)00165-q. [DOI] [PubMed] [Google Scholar]
- Bremner I, Beattie JH. Metallothionein and the trace minerals. Annu Rev Nutr. 1990;10:63–83. doi: 10.1146/annurev.nu.10.070190.000431. [DOI] [PubMed] [Google Scholar]
- Cherian MG, Nordberg M. Cellular adaptation in metal toxicology and metallothionein. Toxicology. 1983;28:1–15. doi: 10.1016/0300-483x(83)90101-4. [DOI] [PubMed] [Google Scholar]
- Chiaverini N, De Ley M. Protective effect of metallothionein on oxidative stress-induced DNA damage. Free Radic Res. 2010;44:605–613. doi: 10.3109/10715761003692511. [DOI] [PubMed] [Google Scholar]
- Kang YJ. The antioxidant function of metallothionein in the heart. Proc Soc Exp Biol Med. 1999;222:263–273. doi: 10.1046/j.1525-1373.1999.d01-143.x. [DOI] [PubMed] [Google Scholar]
- Nath R, Kumar D, Li T, et al. Metallothioneins, oxidative stress and the cardiovascular system. Toxicology. 2000;155:17–26. doi: 10.1016/s0300-483x(00)00273-0. [DOI] [PubMed] [Google Scholar]
- Cai L, Kelein JB, Kang YJ. Metallothionein prevents diabetic cardiomyopathy. Toxicol Sci. 2001;60:13. [Google Scholar]
- Kang YJ, Cai L. Metallothionein suppression of diabetic cardiomyopathy by inhibition of hyperglycemia-induced oxidative stress. Free Radical Biol Med. 2001;31:74. [Google Scholar]
- Liang Q, Carlson EC, Donthi RV, et al. Overexpression of metallothionein reduces diabetic cardiomyopathy. Diabetes. 2002;51:174–181. doi: 10.2337/diabetes.51.1.174. [DOI] [PubMed] [Google Scholar]
- Cai L, Wang J, Li Y, et al. Inhibition of superoxide generation and associated nitrosative damage is involved in metallothionein prevention of diabetic cardiomyopathy. Diabetes. 2005;54:1829–1837. doi: 10.2337/diabetes.54.6.1829. [DOI] [PubMed] [Google Scholar]
- Cai L, Wang Y, Zhou G, et al. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J Am Coll Cardiol. 2006;48:1688–1697. doi: 10.1016/j.jacc.2006.07.022. [DOI] [PubMed] [Google Scholar]
- Wold LE, Ceylan-Isik AF, Fang CX, et al. Metallothionein alleviates cardiac dysfunction in streptozotocin-induced diabetes: role of Ca2+ cycling proteins, NADPH oxidase, poly(ADP-Ribose) polymerase and myosin heavy chain isozyme. Free Radic Biol Med. 2006;40:1419–1429. doi: 10.1016/j.freeradbiomed.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Velic A, Laturnus D, Chhoun J, et al. Diabetic basement membrane thickening does not occur in myocardial capillaries of transgenic mice when metallothionein is overexpressed in cardiac myocytes. Anat Rec (Hoboken) 2013;296:480–487. doi: 10.1002/ar.22646. [DOI] [PubMed] [Google Scholar]
- Cai L. Metallothionein as an adaptive protein prevents diabetes and its toxicity. Nonlinearity Biol Toxicol Med. 2004;2:89–103. doi: 10.1080/15401420490464367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L. Diabetic cardiomyopathy and its prevention by metallothionein: experimental evidence, possible mechanisms and clinical implications. Curr Med Chem. 2007;14:2193–2203. doi: 10.2174/092986707781389646. [DOI] [PubMed] [Google Scholar]
- Cong W, Zhao T, Zhu Z, et al. Metallothionein prevents cardiac pathological changes in diabetes by modulating nitration and inactivation of cardiac ATP synthase. J Nutr Biochem. 2014;25:463–474. doi: 10.1016/j.jnutbio.2013.12.007. [DOI] [PubMed] [Google Scholar]
- Cong W, Ma W, Zhao T, et al. Metallothionein prevents diabetes-induced cardiac pathological changes, likely via the inhibition of succinyl-CoA:3-ketoacid coenzyme A transferase-1 nitration at Trp(374) Am J Physiol Endocrinol Metab. 2013;304:E826–E835. doi: 10.1152/ajpendo.00570.2012. [DOI] [PubMed] [Google Scholar]
- Giacconi R, Bonfigli AR, Testa R, et al. +647 A/C and +1245 MT1A polymorphisms in the susceptibility of diabetes mellitus and cardiovascular complications. Mol Genet Metab. 2008;94:98–104. doi: 10.1016/j.ymgme.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Yang L, Li H, Yu T, et al. Polymorphisms in metallothionein-1 and -2 genes associated with the risk of type 2 diabetes mellitus and its complications. Am J Physiol Endocrinol Metab. 2008;294:E987–E992. doi: 10.1152/ajpendo.90234.2008. [DOI] [PubMed] [Google Scholar]
- Miao X, Sun W, Fu Y, et al. Zinc homeostasis in the metabolic syndrome and diabetes. Front Med. 2013;7:31–52. doi: 10.1007/s11684-013-0251-9. [DOI] [PubMed] [Google Scholar]
- Li B, Tan Y, Sun W, et al. The role of zinc in the prevention of diabetic cardiomyopathy and nephropathy. Toxicol Mech Methods. 2013;23:27–33. doi: 10.3109/15376516.2012.735277. [DOI] [PubMed] [Google Scholar]
- Cho HY, Reddy SP, Kleeberger SR. Nrf2 defends the lung from oxidative stress. Antioxid Redox Signal. 2006;8:76–87. doi: 10.1089/ars.2006.8.76. [DOI] [PubMed] [Google Scholar]
- Li B, Liu S, Miao L, et al. Prevention of diabetic complications by activation of Nrf2: diabetic cardiomyopathy and nephropathy. Exp Diabetes Res. 2012;2012:216512. doi: 10.1155/2012/216512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- de Haan JB. Nrf2 activators as attractive therapeutics for diabetic nephropathy. Diabetes. 2011;60:2683–2684. doi: 10.2337/db11-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Kan H, Cai L, et al. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J Mol Cell Cardiol. 2009;46:47–58. doi: 10.1016/j.yjmcc.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Ramprasath T, Selvam GS. Potential impact of genetic variants in Nrf2 regulated antioxidant genes and risk prediction of diabetes and associated cardiac complications. Curr Med Chem. 2013;20:4680–4693. doi: 10.2174/09298673113209990154. [DOI] [PubMed] [Google Scholar]
- Chartoumpekis DV, Kensler TW. New player on an old field; the keap1/Nrf2 pathway as a target for treatment of type 2 diabetes and metabolic syndrome. Curr Diabetes Rev. 2013;9:137–145. doi: 10.2174/1573399811309020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Ichikawa T, Li J, et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes. 2011;60:625–633. doi: 10.2337/db10-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Cui W, Xin Y, et al. Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of Nrf2 expression and transcription activation. J Mol Cell Cardiol. 2013;57:82–95. doi: 10.1016/j.yjmcc.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Luo ZF, Qi W, Feng B, et al. Prevention of diabetic nephropathy in rats through enhanced renal antioxidative capacity by inhibition of the proteasome. Life Sci. 2011;88:512–520. doi: 10.1016/j.lfs.2010.12.023. [DOI] [PubMed] [Google Scholar]
- Cui W, Li B, Bai Y, et al. Potential role for Nrf2 activation in the therapeutic effect of MG132 on diabetic nephropathy in OVE26 diabetic mice. Am J Physiol Endocrinol Metab. 2013;304:E87–E99. doi: 10.1152/ajpendo.00430.2012. [DOI] [PubMed] [Google Scholar]
- Wang Y, Sun W, Du B, et al. Therapeutic effect of MG-132 on diabetic cardiomyopathy is associated with its suppression of proteasomal activities: roles of Nrf2 and NF-kappaB. Am J Physiol Heart Circ Physiol. 2013;304:H567–H578. doi: 10.1152/ajpheart.00650.2012. [DOI] [PubMed] [Google Scholar]
- Fahey JW, Talalay P. Antioxidant functions of sulforaphane: a potent inducer of Phase II detoxication enzymes. Food Chem Toxicol. 1999;37:973–979. doi: 10.1016/s0278-6915(99)00082-4. [DOI] [PubMed] [Google Scholar]
- Tsai CY, Wang CC, Lai TY, et al. Antioxidant effects of diallyl trisulfide on high glucose-induced apoptosis are mediated by the PI3K/Akt-dependent activation of Nrf2 in cardiomyocytes. Int J Cardiol. 2013;168:1286–1297. doi: 10.1016/j.ijcard.2012.12.004. [DOI] [PubMed] [Google Scholar]