

Abstract
Paenibacillus larvae is the etiological agent of American Foulbrood (AFB) a world-wide distributed devastating disease of the honey bee brood. Previous comparative genome analysis and more recently, the elucidation of the bacterial genome, provided evidence that this bacterium harbors putative functional nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs) and therefore, might produce nonribosomal peptides (NRPs) and polyketides (PKs). Such biosynthesis products have been shown to display a wide-range of biological activities such as antibacterial, antifungal or cytotoxic activity. Herein we present an in silico analysis of the first NRPS/PKS hybrid of P. larvae and we show the involvement of this cluster in the production of a compound named paenilamicin (Pam). For the characterization of its in vitro and in vivo bioactivity, a knock-out mutant strain lacking the production of Pam was constructed and subsequently compared to wild-type species. This led to the identification of Pam by mass spectrometry. Purified Pam-fractions showed not only antibacterial but also antifungal and cytotoxic activities. The latter suggested a direct effect of Pam on honey bee larval death which could, however, not be corroborated in laboratory infection assays. Bee larvae infected with the non-producing Pam strain showed no decrease in larval mortality, but a delay in the onset of larval death. We propose that Pam, although not essential for larval mortality, is a virulence factor of P. larvae influencing the time course of disease. These findings are not only of significance in elucidating and understanding host–pathogen interactions but also within the context of the quest for new compounds with antibiotic activity for drug development.
Keywords: Antibacterial activity, antifungal activity, cytotoxic activity, nonribosomal peptide-polyketide hybrid, Paenibacillus larvae, paenilamicin
Introduction
Insect pathogenic bacteria have only recently been recognized as a rich but poorly explored source of structurally diverse secondary metabolites. Nonribosomal peptides (NRPs) and polyketides (PKs) produced by entomopathogens have been shown to have important biological roles during pathogenesis such as antagonism of microbial competitors, motility, and contribution to virulence. In addition, some of them might even have a future as pharmaceuticals (reviewed in Bode 2009). The emergence of whole genome-sequencing projects and improved genome mining techniques paved the way for the identification of new NRP synthetase (NRPS) and PK synthase (PKS) gene clusters. To date, known bacterial entomopathogens producing secondary metabolites are for instance Bacillus thuringiensis (Broderick et al. 2000), Pseudomonas entomophila (Vallet-Gely et al. 2010), Serratia marcescens (Li et al. 2005), and Xenorhabdus ssp. (Reimer et al. 2009). Recently, Paenibacillus larvae has been added to the list of secondary metabolite producing bacterial entomopathogens (Fünfhaus et al. 2009). Comparative genomics and whole genome comparison within the species P. larvae revealed the existence of several NRPS and NRPS/PKS gene clusters (Fünfhaus et al. 2009; Djukic et al. 2014; Schild et al. 2014) including an NRPS gene cluster leading to the production of a novel nonribosomal peptide antibiotic, sevadicin (Garcia-Gonzalez et al. 2014).
Paenibacillus larvae is the etiological agent of AFB, a serious disease of the European honey bee (Apis mellifera) larvae (Genersch et al. 2006). AFB is the most contagious and destructive bacterial disease affecting honey bees. Although only larvae are susceptible to and inevitably die from the disease, AFB is able to kill entire colonies. Hence, AFB is a notifiable epizootic in most countries and in the case of an outbreak, burning of diseased colonies and contaminated hive material is usually considered the only workable control measure. As a result, AFB leads to considerable economic losses in apiculture. In addition, colony losses due to AFB also compromise agricultural and natural ecosystems because of the indispensable role of honey bees as pollinators of many fruit, crops, and wild flowers.
Despite the enormous impact of this disease, molecular mechanisms of infection are not fully understood. To date, we know that four different genotypes within the species P. larvae exist named ERIC I to ERIC IV based on the primers used for differentiation (Genersch et al. 2006). These genotypes not only differ in virulence (Genersch et al. 2005; Rauch et al. 2009) but also in their genomic makeup (Fünfhaus et al. 2009; Djukic et al. 2014). All genotypes share the general steps in pathogenesis: Honey bee larvae become infected by ingesting spores, which germinate in the midgut; vegetative P. larvae then massively proliferate in the midgut, breach the peritrophic membrane and the epithelium, and finally decompose the larva to a ropy mass (Yue et al. 2008; Garcia-Gonzalez and Genersch 2013). Recently, the first virulence factors of P. larvae have been elucidated and were proved to be genotype specific: P. larvae ERIC I expresses two AB toxins, Plx1 and Plx2, missing in P. larvae ERIC II (Fünfhaus et al. 2013) whereas the S-layer protein SplA, involved in bacterial adhesion to the midgut epithelium, was shown to be expressed only in P. larvae ERIC II (Fünfhaus and Genersch 2012; Poppinga et al. 2012). The relevance of secondary metabolites like sevadicin (Garcia-Gonzalez et al. 2014) during pathogenesis of P. larvae infections still remains elusive.
NRPs and PKs are secondary metabolites synthesized by dedicated complex multimodular biosynthetic machineries termed nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs). Each module consists of at least three domains responsible for a specific catalytic reaction in the incorporation of an amino acid (NRPS) or malonyl coenzyme A (PKS). These modular enzyme machineries sequentially activate and condense specific amino acids or acyl units. The modules of the megaenzymes of the NRPS-type (reviewed in Finking and Marahiel 2004) commonly consist of the following domains: The adenylation (A) domain of an NRPS specifically binds and activates an amino acid by transforming it into an amino acyl adenylate. The thiolation (T) domain covalently binds the activated amino acid to the module via a phosphopantetheinyl arm. The condensation (C) domain catalyzes the formation of a peptide bond between two amino acids of neighbored modules. The terminal module consists of an additional thioesterase (TE) domain which releases the peptide from the NRPS. It may occur that NRPSs contain additional domains for further peptide modification. An important representative of such a domain is the epimerization (E) domain which catalyzes the inversion of the stereocenter of the activated l-amino acid into its d-amino acid enantiomer.
Likewise the modules of type I PKSs consist of at least three core domains (reviewed in Hill 2006): the acyltransferase (AT) domain binds an appropriate acyl unit and transfers it into the acyl carrier protein (ACP) domain. The elongation step is performed by the ketosynthetase (KS) domain which catalyzes the formation of a C–C bond via Claisen condensation with the acyl unit of the next module. Optionally, the present domains for further tailoring of the substrates are ketoreductase (KR) domains that catalyse the reduction of the β-keto group to a β-hydroxy group. A fully saturated acyl backbone additionally requires the reductive action of dehydratase (DH), and enoyl reductase (ER) domains.
Here, we report on the identification, isolation, and biological characterization of a novel NRP/PK hybrid, named paenilamicin (Pam), produced by P. larvae. By means of construction of a gene-inactivation mutant followed by mass spectrometric analysis, we were able to link functionality of the pam gene cluster responsible for Pam production and antibiotic activity. Further in vitro and in vivo assays corroborated the antibacterial, antifungal and cytotoxic activities of this metabolite. Moreover, in vivo functional analyses suggested that Pam might act as a putative virulence factor in AFB pathogenesis. The study of antibiotic substances produced by the entomopathogen P. larvae is of outstanding interest because of both, the identification of a novel bioactive NRPS/PKS hybrid and the characterization of the biological function of this substance.
Materials and Methods
Bacterial strains and culture conditions
Wild-type P. larvae bacteria (DSM25430; ERIC II (Genersch et al. 2005)) were cultivated either in MYPGP liquid broth (Dingman and Stahly 1983) or on Columbia sheep blood agar (CSA) plates at 37°C as previously described (Genersch and Otten 2003; Neuendorf et al. 2004). Gene inactivation mutants of P. larvae were cultivated on MYPGP-agar plates supplemented with 5 μg/mL chloramphenicol for selection and incubated at 37°C for 2–3 days as previously described (Poppinga et al. 2012).
Paenibacillus alvei, Bacillus subtilis, Bacillus licheniformis, Bacillus megaterium, and Saccharomyces cerevisiae were obtained from the DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Braunschweig, Germany). Pichia pastoris yDT39 was a generous gift from Prof. Budisa (Technische Universität, Berlin, Germany) and Fusarium oxysporum ETH1536/9 was obtained from the Centraalbureau voor Schimmelcultures (Utrecht, the Netherlands). These strains were used for testing the antibacterial activity of P. larvae culture supernatants or purified Pam. All strains were grown in LB (Luria Bertani), MYPGP, YPD or FCM media (broth or agar plates).
Paenibacillus larvae spore suspensions were generated by resuspending about 100 colonies in 300 μL brain heart infusion (BHI) broth to inoculate the liquid part of CSA slants. These were incubated at 37°C for 10 days. Spore concentrations were determined by cultivating serial dilutions as already described (Genersch and Otten 2003; Neuendorf et al. 2004).
Escherichia coli DH5α cells (Invitrogen, Karlsruhe, Germany) transformed with the plasmid pTT_pamA (see below) were cultivated in selective LB media (agar and broth) supplemented with 30 μg/mL chloramphenicol. Plasmid DNA was prepared following the manufacturer's protocols (QIAprep Spin Miniprep kit; Qiagen, Hilden, Germany). Concentration and purity of the plasmid preparations were analyzed by photometric analysis (Nanodrop; Peqlab, Erlangen, Germany) and agarose gel electrophoresis.
Paenibacillus larvae cultivation for secondary metabolite production
Paenibacillus larvae was cultivated as follows: bacteria were grown on CSA plates at 37°C for 2–3 days. A preculture of 10 mL MYPGP medium was inoculated with a single colony and grown overnight. A 50 mL of MYPGP broth was inoculated with the preculture to reach a final (calculated) OD600 of 0.001. This main culture was incubated at 30°C with gentle shaking (80 rpm, Thermoshake; Gerhardt, Bonn, Germany) for 72 h. Cultures were centrifuged for 10 min at 4°C with 3200 g and supernatants were stored at −20°C until further use.
Agar diffusion assay for antibacterial activity
To test the antibiotic activity of P. larvae supernatants, P. alvei, B. subtilis, B. licheniformis and B. megaterium were used as bacterial indicator strains and S. cerevisiae, Pichia pastoris, and Fusarium oxysporum as fungal indicator strains. The activity against P. alvei, B. subtilis, B. licheniformis, B. megaterium, and S. cerevisiae was tested as follows. A preculture was grown in 5 mL of the correspondent broth (LB or MYPGP) and cultured overnight at 37°C with gentle shaking. A 10 mL of the correspondent broth was inoculated with 500 μL of this preculture and cultivated at 37°C with gentle shaking for 5 h. Subsequently, the OD600 of the cultures (typically between 0.5 and 0.7) was determined. A 40 mL of molten, 37°C warm LB or MYPGP agar was inoculated to reach a final (calculated) OD600 of 0.05 and poured on Petri plates, while 20 μL of the test substrate (P. larvae supernatants or purified Pam) was dispensed on a filter disk and dried at RT. Subsequently, the disks were carefully placed on the agar. Alternatively, holes of 6 mm in diameter were made into the agar and filled with 40 μL of the tested substance. The plates were incubated at 37°C overnight. Clear zones around the filter disks or the holes were considered indicative of growth inhibition. P. pastoris yDT39 was grown in 5 mL YPD medium at 30°C and 200 rpm until reaching OD600 of around 0.8–0.9. 200 μL of this culture were plated on YPD agar plates (10 mL per plate). Plates were incubated at 30°C overnight. F. oxysporum ETH 1536/9 was grown in 5 mL FCM medium at 27°C and 115 rpm for 48 h. FCM agar plates for bioactivity tests were made the same way as described for P. pastoris above. The agar plates were incubated at 27°C for 2–3 days.
Sequence analysis
The genome sequence of Paenibacillus larvae (P. larvae DSM25430 (Djukic et al. 2014), NCBI Reference Sequence NC_023134.1) was screened for polyketide synthase (PKS) – and nonribosomal peptide synthetase (NRPS) – coding sequences. This analysis revealed the occurrence of nine genes consisting of five NRPSs, two PKSs, and two NRPS/PKS hybrid genes (NC_023134.1, position 1729923–1788675). Sequence similarity searches were performed using BLASTX and BLASTP at the NCBI website (Altschul et al. 1997). Domain organization, extraction of binding pocket signatures and substrate specificity prediction were analyzed by using in silico tools: NRPSpredictor2 (Rausch et al. 2005; Röttig et al. 2011), PKS/NRPS analysis (Bachmann and Ravel 2009), and SBSPKS (Anand et al. 2010).
Construction of a P. larvae gene inactivation mutant (knock-out strain)
The gene pamA (Gene ID: 18058116) from the pam gene cluster was disrupted in the genome of P. larvae DSM25430 via a recently described strategy (Zarschler et al. 2009). Vector pTT_wsfA243 (Zarschler et al. 2010) was used for constructing a targetron vector for targeted group II intron insertion at position 1080 from the start codon of pamA of the pam gene cluster. Retargeting of the LI.LtrB targetron of vector pTT_wsfA243 prior to transformation into P. larvae was performed following the manufacturer's protocol and essentially as already described for disruption of the P. larvae S-layer gene splA (Poppinga et al. 2012). In brief, a computer algorithm provided by the manufacturer (http://www.sigma-genosys.com/targetron) identified potential insertion sites for the intron into pamA open reading frames and designed suitable PCR primers (Table1) for the modification of the intron RNA. The algorithm predicted a high insertion efficiency for the position 1080 in the gene and designed corresponding PCR primers. These primers were used for modification of the LI.LtrB targetron to generate the new vector pTT_pamA, respectively, which were subsequently transformed into E. coli DH5α cells for plasmid replication and preparation. The targeted sequence for intron insertion was unique inside the DSM25430 genome, assuring the specificity of gene inactivation.
Table 1.
Primers used in this study.
| Primer name | Primer sequences (5′–3′) | Used for |
|---|---|---|
| IBS_cltIII_pksA | AAAAAAGCTTATAATTATCCTTATCCGGCGTCGCAGTGCGCCCAGATAGGGTG | creation of pamA knock-out |
| EBS1d_cltIII_pksA | CAGATTGTACAAATGTGGTGATAACAGATAAGTCGTCGCAGGTAACTTACCTTTCTTTGT | |
| EBS2_cltIII_pksA | TGAACGCAAGTTTCTAATTTCGGTTCCGGATCGATAGAGGAAAGTGTCT | |
| PKSA_F | GGCACGAGCATGGGTGATCCG | sequencing; verification of pamA knock-out |
| PKSA_R | CGTGGACATTTGTCCCGCCCA |
For generation of a P. larvae gene inactivation mutant, electrocompetent P. larvae DSM25430 cells (ERIC II) were prepared as described (Murray and Aronstein 2008) and 1 μg of plasmid pTT_pamA was transformed by electroporation as recently established (Poppinga and Genersch 2012). Cells were regenerated in MYPGP broth for 16 h, plated on MYPGP-agar containing 5 μg/mL chloramphenicol and incubated for 3 days. Positive clones were identified by PCR analysis using primers flanking the intron insertion positions of the targeted ORF: 948 and 1276 (Table1, Fig.1A), leading to the construction of the strain DSM25430 ΔpamA. Germination, growth in liquid broth, and sporulation of P. larvae DSM25430 ΔpamA was tested and did not significantly differ from the parent wild-type strain (Fig.1B; two-way ANOVA; P = 0.607) thus ruling out that the outcome of the exposure bioassays is influenced by unknown side effects of the knock-out strategy on relevant features of the bacteria as also already described for other P. larvae knock-out mutants generated with the targetron strategy (Poppinga et al. 2012; Fünfhaus et al. 2013).
Figure 1.
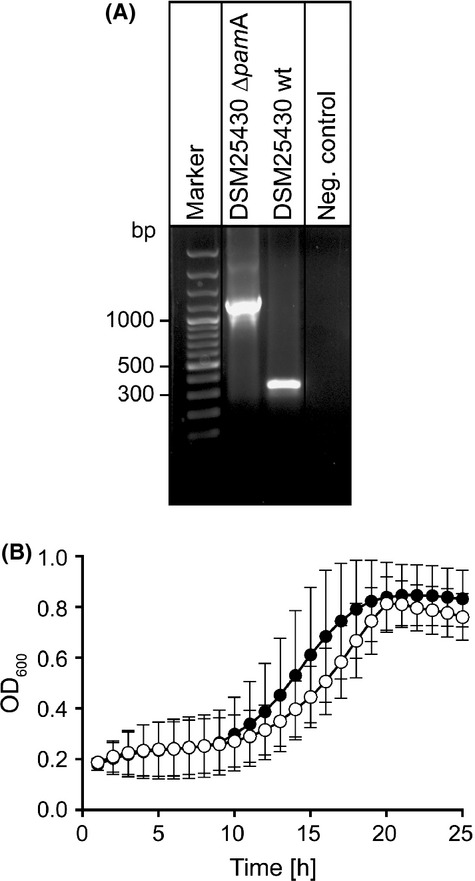
Determination of successful knock-out construction and growth analysis of knockout mutant. (A) PCR analysis of the constructed knock-out mutant DSM25430 ΔpamA, revealed successful intron insertion. Primers flanking the insertion sites in the pamA gene in the wild-type strain DSM25430 wt, amplified a fragment that was 915 bp smaller than the fragment amplified from DSM25430ΔpamA because of intron insertion. (B) Growth of DSM25430 wt and DSM25430 ΔpamA was analyzed with three biological and three technical replicates each. No significant difference in growth could be observed (two-way ANOVA, P = 0.607). PCR, polymerase chain reaction.
Liquid chromatography-electrospray ionization-mass spectrometry analytics
Polypropylene columns (1 mL) (Qiagen) were filled with Amberlite XAD-16 adsorption beads (Sigma, St. Louis, MO) (1.5 g), and used for subsequent chromatographic fractionation. Thus, prepared columns were then equilibrated by flushing with three column volumes (CVs) of distilled water. Frozen supernatants of P. larvae cultures were thawed, and the column was loaded with supernatant. A step gradient was applied using 1 CV of distilled water followed by aqueous methanol of decreasing polarity (1 CV each in 10% steps: 10%, 20%, 30%; etc. 100%). Eluates were tested for bioactivity against B. megaterium. Chromatographic fractions from P. larvae wild-type showing bioactivity and the corresponding fractions from P. larvae gene inactivation mutant were pooled and dried in vacuo. For LC-ESI-MS analytics, the extracts were resolved in 5 mL 10% aqueous acetonitrile + 0.1% formic acid and a portion of 2 μL was injected into the HPLC (high-pressure liquid chromatography) column.
LC-ESI-MS analytics were performed using an Agilent 6410 Triple Quadrupole LC/MS system in MS2-scan mode coupled to an UHPLC 1290 Infinity-Series (Agilent Technologies, Waldbronn, Germany). As a column, an Eclipse Plus C18 B 1.5 μm, 2.1 × 50 mm (Grace; Grace GmbH & Co KG, Worms, Germany) was used for separation. Samples of the same concentration were analyzed by linear gradient elution using H2O + 0.1% formic acid as solvent A and acetonitrile + 0.1% formic acid as solvent B. The gradient was from 5% to 100% solvent B in 6 min with a 2-min isocratic elution at 100% for solvent B.
Bioassay-guided isolation of Pam for bioactivity assays
Small-scale purification was performed by a step-wise fractionation protocol. Bioactive fractions against B. megaterium XAD-16 fractions were submitted to Sephadex® LH-20 column (25 × 1000 mm, filled with 492 g Sephadex® LH-20 media, GE Healthcare, Uppsala, Sweden). The injection volume was 5 mL with a flow rate of 0.5 mL/min. 1.2 CVs were fractionated isocratically with 100% H2O with a fraction size of 2 mL. Antibiotically active fractions were pooled and dried in vacuo and purified subsequently by using a Reveleris® Flash Cartridge (Grace; Grace GmbH & Co KG, Worms, Germany) filled with 4 g C18 reversed phase resin coupled to a Reveleris® X2 Flash System (Grace; Grace GmbH & Co KG). The separation was accomplished by linear gradient elution using H2O + 0.1% formic acid as solvent A and methanol + 0.1% formic acid as solvent B. The gradient was from 3% to 15% solvent B in 20 min, followed by a 5-min gradient to 100% B ending at 100% B isocratic for 5 min at a flow rate of 18 mL/min.
Bioactive fractions were pooled, concentrated and purified subsequently by using a Grace HPLC column (250 × 20 mm) filled with 10 μm GROM-Sil 120 ODS-5 ST coupled to an Agilent 1100 HPLC system (Agilent Technologies) with a multi wavelength ultraviolet detector (MWD). The separation was accomplished by linear gradient elution using H2O + 0.1% formic acid as solvent A and methanol + 0.1% formic acid as solvent B. The gradient was from 3% to 15% solvent B in 45 min, followed by a 5-min gradient to 100% B ending at 100% B isocratic for 5 min at a flow rate of 10 mL/min.
Subsequently, bioactive fractions were loaded onto a HPLC column (150 × 4.0 mm) filled with 5 μm, 12 nm YMC-Pack C-8 (Octyl) (YMC Europe GmbH, Dinslaken, Germany) coupled to an Agilent 1100 HPLC system (Agilent Technologies) with a DAD UV detector. The samples were separated by linear gradient elution using H2O + 0.1% formic acid as solvent A and acetonitrile + 0.1% formic acid as solvent B at a flow rate of 1.5 mL/min. The gradient was from 3% to 20% for solvent B in 8 min with a 2-min hold at 20% for solvent B followed by a 2-min gradient to 100% B ending at 100% B isocratic for 5 min. Pam-containing fractions (mass spectrometric control) were used for bioactivity assays.
High-resolution-LC-ESI-MS analytics
For the analysis of pure Pam an LTQ Orbitrap XL mass spectrometer (Thermo Scientific, Bremen, Germany) coupled to an Agilent 1260 HPLC system (Agilent Technologies) was used. A Grom-Sil ODS-4 HR 3 μm, 50 × 2.0 mm column (Grace; Grace GmbH & Co KG) was used for separation. The sample was analyzed by linear gradient elution using H2O + 0.1% formic acid as solvent A and acetonitrile + 0.1% formic acid as solvent B. The elution started with an initial hold at 5% B for 1.6 min followed by an increase of solvent B to 100% in 7.9 min with a hold at 100% for 2.5 min.
Cell toxicity assays
Cytotoxicity in vitro tests were performed using the commercial available cell line: BTI-Tn-5B1-4 cells (generous gift from Dr. Holger Hübner from the Friedrich-Alexander-Universität Erlangen-Nürnberg, Germany). The cells were maintained in 75 cm2 cell culture flasks (Roth) filled with 20 mL of serum-free Sf 900II medium (Lonza, Walkersville, MD, USA) and cultivated at 27°C in a cooling incubator. Cells were passaged once a week adjusting its initial concentration to 2E + 05 cells per mL using a counting chamber.
2E + 04 cells (100 μL) were seeded in each well of a 96-well microtiter plate and incubated at 27°C for 24 h to allow cell adherence. Microtiter plates were centrifuged with 220g for 15 min and medium was removed and replaced with 100 μL medium plus the different samples: Pam isolated from the supernatant of DSM25430 wt was resuspended (1:100) in 100 μL SF 900 II medium with a 250 μg mL−1 penicillin/streptomycin premix. Controls were performed as follows: SF 900 II medium served as a blank, HPLC solvent (1:100 diluted 5% acetonitrile + 0.1% formic acid) served as a negative control, and 10 μL of lysis buffer (99.4 mL DMSO, 0.6 mL acetic acid and 10% SDS w/v) served as a positive control for cell toxicity. After 40 h of incubation at 33°C, plates were centrifuged (220 g for 15 min) and the medium was discarded. Subsequently, 100 μL of fresh MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid) solution (0.5 mg/mL in SF 900 II medium) was added to the cells and incubated at 27°C for 3 h. Microtiter plates were centrifuged again at 220g for 10 min and the supernatant was carefully aspirated without damaging the cell pellet. To disrupt the cells and liberate formazan, 100 μL lysis buffer (99.4% DMSO mL, 0.6% acetic acid, 10% SDS w/v) were added and the plate was incubated for 5 min at 300 rpm on a vibrate platform shaker (Titramax 1000; Heidolph, Schwabach, Germany) at room temperature. The OD corresponding to cell-absorbed formazan, and thus to cell vitality after the treatment was analyzed with the SynergyHT ELISA reader (BioTek, Bad Friedrichshall, Germany) using 595 nm excitation wavelength.
Infection assays
To analyze the functional role of Pam during pathogenesis, an exposure bioassay was performed as described by Genersch et al. (2005 and) Genersch et al. (2006). Preparation of spore suspension for infection assays was performed as previously described (Genersch et al. 2005, 2006; Rauch et al. 2009; Poppinga et al. 2012; Fünfhaus et al. 2013). Spore concentrations were calculated by plating out serial dilutions on CSA agar plates and counting colony forming units (CFUs). Spore suspensions with equal concentrations of CFUs were prepared for both wild-type and knock-out. Larvae of first instar were infected with P. larvae ERIC II wild-type (DSM25430) and the Pam deficient knock-out mutant (DSM25430 ΔpamA) by adding corresponding spores in a final concentration of 500 cfu/mL (about LC100) to the larval diet to ensure that infection of larvae was initiated by the same number of vegetative P. larvae for wild-type and knock-out bacteria. Control larvae were fed with normal larval diet, consisting of 66% royal jelly (v/v), 3.3% glucose (w/v) and 3.3% fructose (w/v). After 24 h, larvae were transferred to fresh food daily and monitored for 15 days. Dead larvae were removed, streaked out on CSA, incubated at 37°C for 2–3 days, and checked for AFB phenotypically and by PCR (Kilwinski et al. 2004). Only larvae that died of AFB infected by P. larvae ERIC II (DSM25430 wt) or by the Pam-deficient knock-out (DSM25430 ΔpamA) mutant, respectively, were considered for statistical calculations. Knock-out stability was proved by PCR with the gene-specific primer pair (Table1) PKSA_F and PKSA_R, flanking the intron insertion site 1080. PCR amplicons were analyzed by gel electrophoresis on a 1% agarose gel, stained with ethidium bromide and visualized by UV light.
Total mortality was depicted as mean values ± SD and statistically analyzed by Student's t-test. Survival curves were plotted and two-way ANOVA was used to calculate significant differences. Infection assays for DSM25430 wt, DSM25430 ΔpamA mutant and control larvae were carried out three times with 30 replicates each, statistical analysis was performed using GraphPad Prism 6 software.
Results and Discussion
Genomic organization of the Pam NRPS/PKS hybrid gene cluster
The complete genome of Paenibacillus larvae DSM25430 (ERIC II) has been sequenced (NCBI Reference Sequence NC_023134.1) and it has been shown to harbor several NRPS and NRPS/PKS hybrid clusters (Djukic et al. 2014). One of these clusters codes for a trimodular NRPS synthesising the tripeptide sevacidin (Garcia-Gonzalez et al. 2014). Further analysis now revealed a gene cluster coding for a mixed NRPS/PKS hybrid gene cluster termed pam cluster, with the compound Pam as its correspondent biosynthesis product. The genomic organization of the pam cluster revealed a size of ∼60 kb comprising five NRPS genes, two PKS genes, and two NRPS/PKS hybrid genes (gene locus tags ERIC2_c18040 – ERIC2 _c180170) (Fig.2, Table S1). The hybrid NRPS/PKS coding genes were assigned as pamA and pamB. Each of these hybrids contains an A domain (A1 and A2) for activation of corresponding amino acid substrates. The genes pamC, pamD, pamE, pamH, and pamN code for NRPSs whereas pamF and pamG code for PKSs.
Figure 2.
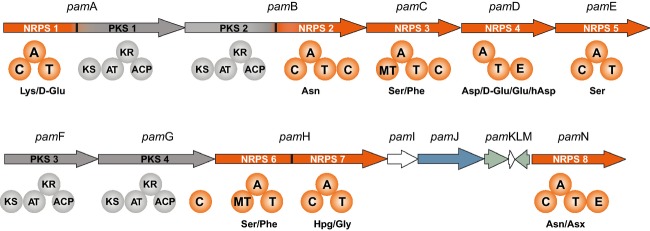
Domain organization derived from the pam secondary metabolite gene cluster in the genome of Paenibacillus larvae DSM25430. In the genome of P. larvae DSM25430 (NCBI Reference Sequence NC_023134.1) an NRPS/PKS hybrid gene cluster could be identified. The gene cluster of the putative NRP/PK hybrid paenilamicin (pam cluster) consisted of five NRPS, two PKS, and two NRPS/PKS hybrid genes. A, adenylation domain; C, condensation domain; T, thiolation domain; E, epimerization domain; MT, N-methyl transferase; KS, ketosynthetase; AT, acetyltransferase; KR, ketoreductase, and ACP, acyl carrier protein. Domain prediction was performed using SBSPKS (Anand et al. 2010). NRPSs, nonribosomal peptide synthetases; PKSs, polyketide synthases.
BLAST analysis (Altschul et al. 1990, 1997) did not reveal any similarity to known NRPS/PKS gene clusters. In order to gain insight into the cognate amino acid substrates, we used several software tools and databases which allow predicting of substrate specificity of amino acid-activating adenylation domains (A domains) of NRPSs. NRPSpredictor2 predicts A-domain substrate specificity based on sequence and structural information about the active site of the domain (http://nrps.informatik.uni-tuebingen.de/) (Rausch et al. 2005; Röttig et al. 2011). This program recognized the A2, A5, and A8 domains as well conserved and predicted the activation of Asn (90%), Ser (100%), and Asn (100%), respectively (Table2). The other A domains of the pam cluster were less well conserved and, hence, provided only poor predictions: Lys (60%), Phe (50%), Asp (70%), Phe (50%), and Gly (70%) were predicted to be recruited by the A1, A3, A4, A6, and A7 domains, respectively (Table2). Modules NRPS4 and NRPS8 contain an additional epimerization domain for the stereochemical conversion of the activated l-amino acid into its d-amino acid enantiomer, suggesting d-Asp and d-Asn as constituents of Pam, respectively. Closer inspection of the four PKS modules revealed, next to a standard set of ketosynthase (KS), AT and ACP, that they each contains a KR domain, indicating the transformation of a ß-carbonyl into a ß-hydroxy group. The pamG gene codes for a PKS with a C-terminal C domain. While the predictor software was unable to predict the function of the second domain, a BLAST comparison of the domain sequence suggested an acyltransferase domain. We next used the NRPS-PKS tool of the SBSPKS software (http://www.nii.ac.in/∼pksdb/sbspks/master.html) which allows identifying various catalytic domains in the sequence of Type I PKS proteins and comparing them with experimentally characterized PKS/NRPS clusters cataloged in the backend databases of SBSPKS (Anand et al. 2010). Analysis of the pam gene cluster using this software predicted two additional domains in NRPS module 3 and NRPS module 6, suggesting an N-methylation of the incorporated amino acids, presumably Phe (Table2). Finally, we used NRPS/PKS analysis software (http://nrps.igs.umaryland.edu/nrps/2metdb), which has been designed for the prediction of microbial secondary metabolic pathways from DNA sequence data of gene clusters leading to the production of modular PKs and nonribosomally encoded peptides (Bachmann and Ravel 2009). Comparison of the results obtained by all three software tools (SBSPKS, NRPSpredictor2 and NRPS/PKS analysis) for the A domain specificity verified recruitment of Asn by A2 (NRPSpredictor 2 and SBSPKS), Ser by A5 (NRPSpredictor, SBSPKS, NRPS/PKS analysis), 4-hydroxyphenylglycine (Hpg) by A7 (PKS/NRPS analysis, SBSPKS), and Asn (SBSPKS, NRPSpredictor2) or Asx (PKS/NRPS analysis) by A8 (Table2). Between pamH and pamN, five genes (pamI, pamJ, pamK, pamL, pamM) are located (Fig.2) that were analyzed by BLAST. PamI and pamL could not be identified (hypothetical proteins) whereas pamJ, pamK, and pamM were identified and annotated as cyclic peptide transporter, 3-hydroxy-CoA dehydrogenase, and transcription antiterminator-like protein NusG, respectively (Djukic et al. 2014).
Table 2.
In silico prediction of A-domain specificity.
| A domain | NRPSpredictor2 | SBSPKS | PKS/NRPS analysis |
|---|---|---|---|
| 1 | Lys | d-Glu | NO HIT |
| 2 | Asn | Asn | NO HIT |
| 3 | Phe | Ser | NO HIT |
| 4 | Asp | d-Glu | Asp/hAsp/Glu |
| 5 | Ser | Ser | Ser |
| 6 | Phe | Ser | NO HIT |
| 7 | Gly | Hpg | Hpg |
| 8 | Asn | Asn | Asx |
PKS, polyketide synthases; NRPS, nonribosomal peptide synthetases.
Recently, it was noted that the here described 3′-end of the Pam biosynthesis gene cluster comprising pamH to pamN and the xenocoumacin (XCN) biosynthesis gene cluster identified in Xenorhabdus nematophila contain homologous genes (Reimer et al. 2011). The P. larvae NRPS7 (PamH) was reported to recruit Hpg confirming our prediction (see above) and the NRPS PamN was shown to be homologous to the starting module XcnA of the XCN biosynthesis gene cluster (Reimer et al. 2011) as were corresponding modules in the biosynthesis gene clusters for amicoumacin (B. pumilus), zwittermicin (B. thuringiensis), colibactin (E. coli), and other similar NRPS/PKS clusters with unknown products (Reimer et al. 2011). All of these starting modules are predicted to be specific to Asn or Asx (Reimer et al. 2011). The XCN biosynthesis gene cluster also harbors the gene xcnG which was originally described as encoding a peptide transporter involved in the resistance mechanism (Reimer et al. 2009). However, most recent studies revealed that XcnG has a peptidase function which is necessary for prodrug activation (Reimer et al. 2011). The X. nematophila peptidase XcnG was shown to be a homolog of PamJ (Reimer et al. 2011) suggesting that pamJ might be a peptidase gene rather than a peptide transporter gene. However, peptidase function of PamJ needs to be confirmed experimentally especially since considerable structural differences between XcnG and PamJ exist. Reimer and co-workers proposed two different domain architecture types for these prodrug-activating peptidases. Type I with XcnG as the model peptidase comprises a signal peptide followed by a peptidase domain and three C-terminal transmembrane helices. In contrast, P. larvae PamJ has type II domain architecture with nine transmembrane helices followed by an ABC transporter domain presumably located in the cytoplasm (Reimer et al. 2011). Therefore, an additional transport function of PamJ is likely but needs to be confirmed experimentally.
Xenocoumacins are produced by bacteria of the genus Xenorhabdus. These bacteria live in symbiosis with nematodes of the genus Steinernema which infect and kill insects. The proposed role of XCN in this symbiotic entomopathogenic complex is either to help killing the insect or to help killing the bacteria living inside the insect gut and competing with Xenorhabdus for food (Reimer et al. 2011). Interestingly, the same roles can be discussed for Pam. It might either act as an antibiotic killing bacterial competitors of P. larvae in the honey bee larval gut or it might act as a toxin helping P. larvae to kill the honey bee larvae. To address these questions, we functionally analyzed Pam.
Paenilamicin is a nonribosomal peptide antibiotic
Paenibacillus larvae have been reported to produce antibacterial activity against different Gram-negative and Gram-positive bacteria including B. subtilis and P. alvei (Holst 1945). Nonribosomally synthesized peptide-polyketide hybrids like Pam often have antibiotic activity. In order to elucidate the contribution of the functional pam gene cluster and hence its biosynthetic product to the antibiotic activity of P. larvae, a knock-out mutant of P. larvae (DSM25430 ΔpamA) was generated, which was interrupted in the NRPS/PKS gene pamA. Subsequently, culture supernatants of wild-type P. larvae (DSM25430 wt) and mutant P. larvae lacking Pam production (DSM25430 ΔpamA) were tested for absence or presence of inhibitory activity against various bacteria (P. alvei, B. subtilis, B. licheniformis, and B. megaterium) and fungi (Sacchoaromyces cerevisiae, Pichia pastoris, and Fusarium oxysporum) selected because of their potential association with honey bees (Bailey 1963; Gilliam et al. 1974; Gilliam and Prest 1987; Snowdon and Cliver 1996; Gilliam 1997; Yoder et al. 2013). Culture supernatants of the wild-type P. larvae strain inhibited growth of all tested microorganisms although differences in inhibitory potential were evident (Fig.3). In contrast, culture supernatants of the DSM25430 ΔpamA mutant did not show any inhibitory activity. These results clearly linked integrity and thus functionality of the pam gene cluster to antibacterial/antifungal activity of P. larvae culture supernatants. So far we only tested some of the microorganisms P. larvae might encounter when infecting honey bee larvae. Once the larval microbiome is fully analyzed and, hence the potential competitors of P. larvae in the larval gut and the dead larvae are known, it will be of special interest to identify the target organism(s) of Pam. It will also be interesting to explore the potential of Pam in anti-infective therapy by elucidating the spectrum of medically relevant microorganisms inhibited by this antibiotic.
Figure 3.

Contribution of paenilamicin to the antibacterial and antifungal activity of P. larvae culture supernatants. P. larvae ERIC II wild-type strain DSM25430 and mutant strain DSM25430 ΔpamA lacking paenilamicin production were grown in MYPGP medium at 30°C. Activity of culture supernatants against different bacteria (Paenibacillus alvei, Bacillus subtilis, B. licheniformis, and B. megaterium) and fungi (Saccharomyces cerevisiae, Pichia pastoris, and Fusarium oxysporum) was tested by agar diffusion assays. Neg. control: MYPGP medium.
Identification and purification of Pam
In order to further characterize the putative biosynthetic product of the pam gene cluster, a comparative metabolic profiling using HPLC mass spectrometry was employed. Direct HPLC-ESI-MS analysis of culture filtrates of P. larvae wild-type and P. larvae ΔpamA did not render any mass peaks which could be assigned to Pam or derivatives (Fig. S1). This finding could be attributed to signal suppression in mass spectrometry by high matrix background of the culture filtrate. Therefore, we performed a polarity-based fractionation of whole P. larvae DSM25430 ΔpamA supernatants and of the corresponding parent wild-type strain P. larvae DSM25430 by XAD-16 chromatography. Briefly, 50 mL of whole supernatants of the wild-type and the pamA knock-out were incubated with XAD-16 resins. Fractionation of the supernatants was accomplished with increasing concentrations of methanol (0–100%) as eluent. Subsequently, antibacterial activity of the eluted fractions was tested against B. megaterium. The DSM25430 wt 90% methanol fraction, that was highly active against B. megaterium, and the corresponding DSM25430 ΔpamA 90% fraction that showed no antibiotic activity were chosen for comparative metabolic profiling using reversed-phase HPLC electrospray ionization mass spectrometry (RP-LC-ESI-MS). Careful comparison of the obtained spectra indicated a compound eluting at Rt = 0.6 min that was absent in the corresponding fraction of the mutant strain (Fig.4A). By HPLC-ESI mass spectrometry two singly-charged ion peaks with molecular masses of [M+H]+ = 1009.6 and 1037.6 Da could be identified in the 90% methanol fractions of the P. larvae wild-type strain that were absent in the 90% methanol fractions of the DSM25430 ΔpamA strain (Fig.4B). Due to the gene inactivation experiments of the pam gene cluster we could unambiguously assign these peaks as products of the NRPS/PKS machinery that is encoded by the pam gene cluster.
Figure 4.
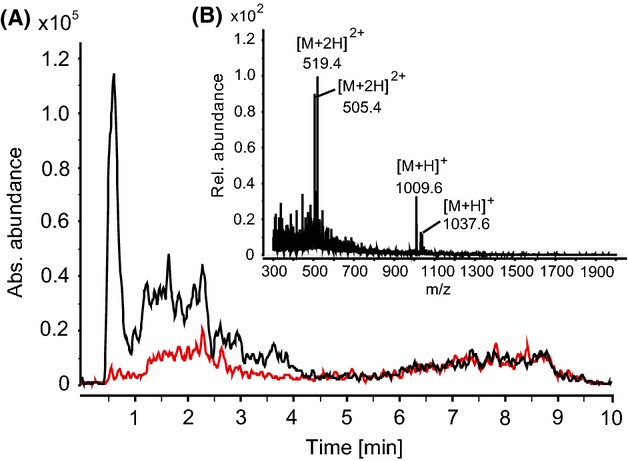
Identification of paenilamicin by HPLC-ESI-triple quadrupole MS from P. larvae secretome after sample workup. (A) Comparison of extracted ion chromatograms (EIC, m/z 519.0–519.5 corresponding to twofold charge state (z = 2) of paenilamicins) of a 90% XAD-16 fraction from wild-type strain DSM25430 (black) with mutant strain DSM25430 ΔpamA (red) (cultivation time 72 h). (B) Mass spectrum of a fraction (90% XAD-16) of P. larvae DSM25430 wild-type. Molecular masses of paenilamicins: [M+H]+ = 1009.6 Da and 1037.6 Da (elution time 0.397–0.843 min). No corresponding peaks were found for the mutant strain (data not shown). HPLC, high-pressure liquid chromatography; MS, mass spectrometry.
To verify our assumption that the identified peaks are responsible for the antibiotic activity, we performed a bioactivity-guided small-scale purification from 1 L culture of P. larvae DSM25430 wt by a stepwise fractionation protocol with B. megaterium as the indicator strain (see Materials and Methods). Fractions containing Pam were collected from the semi-preparative C8 HPLC separation and pooled. High sample purity was confirmed by a single peak in the chromatogram (MS detection) and high-quality MS data with little background (Fig.5A and B). Subsequent high-resolving HPLC-ESI Orbitrap-MS of the pooled fractions showed that Pam consists of two components ([M+H]+ = 1009.6718 and 1037.6780 Da; Fig.5B). Thus, the purified Pam mixture (∼1.2 mg) was used for further functional assays to characterizing bioactivities.
Figure 5.

HR-MS analysis of purified paenilamicin. (A) Total ion chromatogram (TIC) of purified paenilamicin. (B) Corresponding HR mass spectrum (1.26–2.37 min) showing both paenilamicin derivatives with different charge states. m/z 391.2838 is a compound also present in blank measurements. HR, high resolution; MS, mass spectrometry.
Purified Pam has antibacterial, antifungal, and cytotoxic activity
To further confirm the results obtained with P. larvae culture supernatants, we used purified Pam in agar diffusion assays performed with B. megaterium, B. licheniformis, P. alvei, P. pastoris, and F. oxysporum as bacterial and fungal indicator strains, respectively. Halos around the disk impregnated with 20 μL Pam (B. megaterium and B. licheniformis) or the hole containing 40 μL Pam (P. alvei, P. pastoris, and F. oxysporum) confirmed the results obtained with culture supernatants and further substantiated that Pam has antibacterial and antifungal activity (Fig.6A). This activity was concentration dependent as revealed by an increased inhibition of B. megaterium and F. oxysporum when increasing concentrations of purified Pam (0.3–20.0 and 5.0–40.0 μg, respectively) were tested (Fig.6B). Some secondary peptide metabolites also have cytotoxic activity. In order to further converge to the in vivo function of Pam we, therefore, investigated its putative cytotoxicity on insect cells by using the insect cell line Tn5 derived from Trichoplusia ni (Lepidotera). Cell vitality was determined via MTT test for Tn5 cells incubated in cell culture medium alone (SF 900 II medium) or in cell culture medium supplemented with HPLC solvent (1:100 diluted 5% acetonitrile + 0.1% formic acid) as negative control, with cell lysis buffer as positive control or with Pam (Fig.6C). While cell vitality was unaltered in the presence of HPLC solvent (100 ± 10.51% vital cells; P = 0.89), incubation with Pam led to a significant decrease in cell vitality (12.86 ± 11.91% vital cells; P = 0.0024, Student's t-test) after 40 h comparable to the effect of cell lysis buffer (4.01 ± 0.22% vital cells; P = 0.0008, Student's t-test). These results suggested that in addition to its antibacterial and antifungal activity, Pam also is cytotoxic to insect cells.
Figure 6.
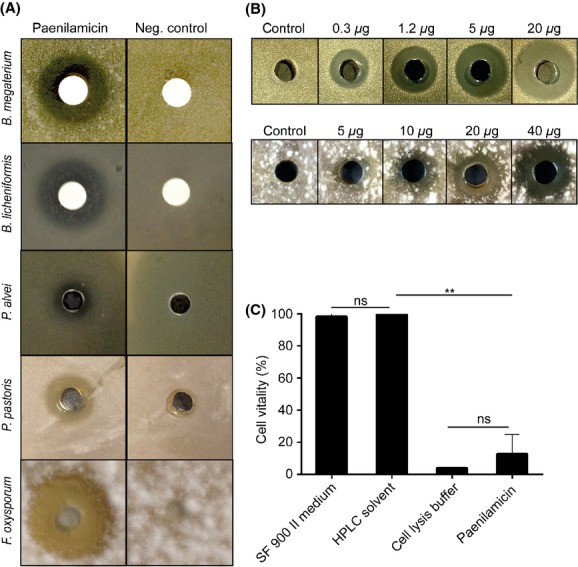
Antibacterial, antifungal, and cytotoxic activity of purified paenilamicin. (A) Isolated paenilamicin (left column) showed antibiotic activity against B. megaterium, B. licheniformis, P. alvei, P. pastoris, and F. oxyporum. HPLC solvent served as negative control. (B) Increasing concentrations of purified paenilamicin were tested in agar diffusion assays against B. megaterium (0.3–20.0 μg Pam) and F. oxysporum (5.0–40.0 μg Pam) as bacterial and fungal indicator strain, respectively. Water was used as a control. (C) Tn5 cells were incubated for 40 h in cell culture medium (SF900 II) or in cell culture medium supplemented with HPLC solvent as negative control, cell lysis buffer as positive control, and paenilamicin isolated from DSM25430 wt. Quantitative measure of cell vitality obtained by the MTT test is shown in the bar graph. Cells incubated with paenilamicin showed a significantly decreased vitality compared to the two negative controls. Bars represent mean values ± SD of three replicates, analyzed by Student's t-test (**P < 0.01). HPLC, high-pressure liquid chromatography.
Biosynthesis gene clusters containing genes coding for homologs of XcnA and XcnG are described to be evolutionary conserved (Reimer et al. 2011). Among these clusters is the biosynthesis gene cluster leading to the production of colibactin (Homburg et al. 2007; Reimer et al. 2011). The polyketide-peptide hybrid colibactin, produced by strains of Escherichia coli harboring a genomic island called “pks,” has been described as cytotoxin or, more precisely, as genotoxin (Nougayrede et al. 2006; Cuevas-Ramosa et al. 2010). However, insect cells intoxicated by Pam did not show the cytopathic effect described for colibactin, i.e., megalocytosis, characterized by a progressive enlargement of the cell body and nucleus and the absence of mitosis, indicating that cytotoxicity of Pam is different from colibactin's effect on eukaryotic cells.
Paenilamicin is involved in the time course of infection
Based on the in vitro cytotoxic activity of purified Pam against insect cells we hypothesized that Pam might act as toxin during P. larvae infection. To test this assumption we performed exposure bioassays with wild-type P. larvae (DSM25430 wt) and the mutant strain lacking Pam production (DSM25430 ΔpamA). Surprisingly, total mortality did not differ significantly between the groups infected with wild-type or mutant bacteria (Fig.7A). This result did not corroborate a function of Pam as toxin during infection despite its cytotoxicity in vitro and exemplified that in the P. larvae/honey bee larvae system it is obviously not always possible to infer from in vitro data on in vivo function.
Figure 7.
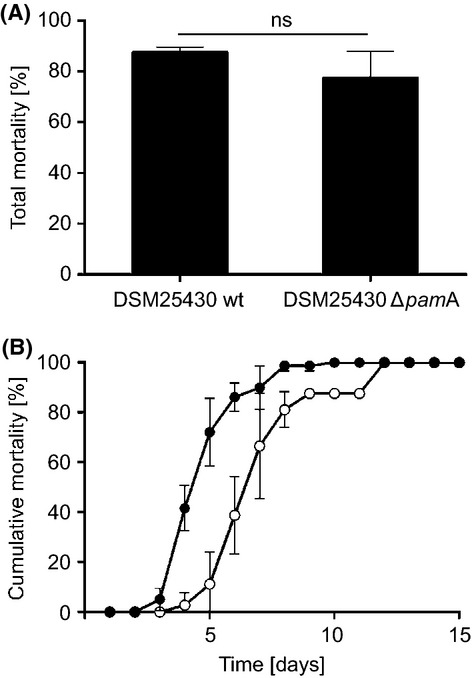
Role of paenilamicin during infection of honey bee larvae. (A) First instar honey bee larvae were infected with P. larvae DSM25430 wt and DSM25430 ΔpamA. Daily mortality due to P. larvae infection was recorded and total mortality was calculated. Bars represent the percentage of exposed larvae that died from American Foulbrood after 15 days. Bars represent mean values ± SD of three independent exposure bioassays each with three groups (control, wt and mutant bacteria) and 30 larvae per group. Data were analyzed by Student′s t-test. No significant difference in total mortality was observed. (B) Daily mortality due to P. larvae infection was recorded and cumulative mortality was calculated. Larvae infected with DSM25430 ΔpamA died as fast as larvae that had been infected with wild-type bacteria, however, onset of larval dying was delayed resulting in a significant parallel shift of the cumulative mortality curve to the right. Curves represent mean cumulative mortality ± SD for each day of three replicas with 30 larvae each. Data were analyzed by two-way ANOVA (P = 0.028).
Analyzing the time course of infection revealed that the curve for cumulative mortality was shifted to the right for the groups infected with the mutant strain (Fig.7B) indicating that in these groups the onset of larval dying was significantly delayed in comparison to the groups infected with wild-type bacteria (P = 0.028; two-way ANOVA). Accordingly, the LT100 also significantly differed between both strains (Student's t-test, P = 0.0142). The wild-type strain DSM25430 needed 8.33 ± 1.52 days to kill all infected larvae, whereas it took the mutant strain DSM25430 ΔpamA 12.0 ± 0 days to accomplish killing all infected animals. These results suggested that Pam although not influencing total mortality influenced the time course of disease and, hence, can be considered a virulence factor.
These assays were all performed with P. larvae DSM25430, a strain belonging to the genotype ERIC II, and a mutant thereof. So far, no toxin genes could be identified in this genotype (Fünfhaus et al. 2009; Djukic et al. 2014) while two functional AB toxins, Plx1 and Plx2, have been characterized in P. larvae ERIC I quite recently (Fünfhaus et al. 2013). Gene disruption in these toxin genes led to a significant reduction in mortality indicating that they are virulence factors and play an important role during pathogenesis (Fünfhaus et al. 2013). If the function of Pam were to act as cytotoxin during infection and to help breaching the epithelial cell layer (Yue et al. 2008) we would have expected a similar significant reduction in total mortality in the absence of this toxin, especially because so far no toxin gene has been identified in this strain (Djukic et al. 2014). Instead, absence or presence of Pam production did not make a difference in total mortality but instead a delay in the onset of larval dying was observed in those larvae that were infected with the mutant bacteria. This suggested that Pam might play a role in the very early phase of infection when P. larvae germinates and starts to colonize the larval midgut. However, the observed effect caused by the absence of Pam cannot simply be explained by differences in germination efficiency between the wild-type and the mutant strains. Both, germination and growth of the mutant strain were not negatively influenced in cultured P. larvae when compared to the wild-type strain (Fig. 1) and all larvae were infected with the same concentration of CFUs, hence, with the same concentration of germinating spores. Therefore, more experimental work is necessary to unravel the exact function of Pam in pathogenesis of P. larvae infections and to explain its influence on virulence of P. larvae.
In summary, we here presented the identification, isolation, and biological characterization of Pam, a peptide-polyketide hybrid produced by the bacterial honey bee pathogen P. larvae. We were able to prove that Pam which consists of two components with molecular masses of [M+H]+ = 1009.6718 and 1037.6780 Da has antibacterial and antifungal activity. In addition, we demonstrated that paenilamcin has cytotoxic activity against insect cells, although a function as toxin during P. larvae infection of honey bee larvae could not be confirmed. However, in larvae infected by P. larvae lacking Pam production a delayed onset of larval mortality by unaltered total mortality could be observed in comparison to larvae infected by wild-type bacteria. These results suggested that Pam is involved in determining the time course of disease and, hence, can be considered a virulence factor of P. larvae. Further experimental work is necessary to elucidate the chemical structures of Pam as well as mechanistic details of Pam activity during AFB pathogenesis which will finally unravel the full potential of this secondary metabolite.
Acknowledgments
This work was supported by grants from the Ministries for Agriculture from Brandenburg and Sachsen-Anhalt, Germany, and through the German Research Foundation (Graduate School 1121) as well as by the Cluster of Excellence “Unifying Concepts in Catalysis” (UniCat) coordinated by TU Berlin.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. of LC-ESI-Orbitrap-MS profiling of metabolites present in the crude supernatant. (A) Total ion chromatogram (TIC) of P. larvae DSM25430 wt and (B) total ion chromatogram (TIC) of P. larvae DSM25430 ΔpamA. Paenilamicin could not be detected directly from crude supernatants due to early elution in the dead volume and ion suppression. The secondary metabolite sevadicin is indicated with a red arrow.
Figure S2. MS/MS fragmentation studies on paenilamicin by HPLC-ESI-triple quadrupole MS from P. larvae secretome after sample workup. (A) MS/MS fragmentation spectrum with [M + 2H]2+ = 505.4 as precursor ion (♦). (B) MS/MS fragmentation spectrum with [M + 2H]2+ = 519.4 as precursor ion (♦).
Figure S3. HPLC UV analytics of pure paenilamicin. (A) Chromatogram of pure paenilamicin monitored at λ = 214 nm. (B) Chromatogram of a blank measurement (H2O) monitored at λ = 214 nm. Paenilamicin shows practically no retention at the C18 column.
Table S1. Homology analysis of the pam gene cluster products.
References
- Altschul SF, Gish W, Miller W, Meyers EW. Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–33402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand S, Prasad MVR, Yadav G, Kumar N, Shehara J, Ansari MZ, et al. SBSPKS: structure based sequence analysis of polyketide synthases. Nucleic Acids Res. 2010;38:W487–W496. doi: 10.1093/nar/gkq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann BO. Ravel J. In silico prediction of microbial secondary metabolic pathways from DNA sequence data. Methods Enzymol. 2009;458:181–217. doi: 10.1016/S0076-6879(09)04808-3. [DOI] [PubMed] [Google Scholar]
- Bailey L. Pathogenicity for honeybee larvae of microorganisms associated with European Foulbrood. J. Insect Pathol. 1963;5:198–205. [Google Scholar]
- Bode HB. Entomopathogenic bacteria as a source of secondary metabolites. Curr. Opin. Chem. Biol. 2009;13:224–230. doi: 10.1016/j.cbpa.2009.02.037. [DOI] [PubMed] [Google Scholar]
- Broderick NA, Goodman RM, Raffa KF. Handelsman J. Synergy between zwittermicin A and Bacillus thuringiensis subsp. kurstaki against gypsy moth (Lepidoptera: Lymantriidae) Environ. Entomol. 2000;29:101–107. [Google Scholar]
- Cuevas-Ramosa G, Petita CR, Marcqa I, Bourya M, Oswalda E. Nougayrèdea J-P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. USA. 2010;107:11537–11542. doi: 10.1073/pnas.1001261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingman DW. Stahly DP. Medium promoting sporulation of Bacillus larvae and metabolism of medium components. Appl. Environ. Microbiol. 1983;46:860–869. doi: 10.1128/aem.46.4.860-869.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djukic M, Brzuszkiewicz E, Fünfhaus A, Voss J, Gollnow K, Poppinga L, et al. How to kill the honey bee larva: genomic potential and virulence mechanisms of Paenibacillus larvae. PLoS One. 2014;9:e90914. doi: 10.1371/journal.pone.0090914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finking R. Marahiel MA. Biosynthesis of nonribosomal peptides. Ann. Rev. Microbiol. 2004;58:453–488. doi: 10.1146/annurev.micro.58.030603.123615. [DOI] [PubMed] [Google Scholar]
- Fünfhaus A. Genersch E. Proteome analysis of Paenibacillus larvae reveals the existence of a putative S-layer protein. Environ. Microbiol. Rep. 2012;4:194–202. doi: 10.1111/j.1758-2229.2011.00320.x. [DOI] [PubMed] [Google Scholar]
- Fünfhaus A, Ashiralieva A, Borriss R. Genersch E. Use of suppression subtractive hybridization to identify genetic differences between differentially virulent genotypes of Paenibacillus larvae, the etiological agent of American Foulbrood of honeybees. Environ. Microbiol. Rep. 2009;1:240–250. doi: 10.1111/j.1758-2229.2009.00039.x. [DOI] [PubMed] [Google Scholar]
- Fünfhaus A, Poppinga L. Genersch E. Identification and characterization of two novel toxins expressed by the lethal honey bee pathogen Paenibacillus larvae, the causative agent of American foulbrood. Environ. Microbiol. 2013;15:2951–2965. doi: 10.1111/1462-2920.12229. [DOI] [PubMed] [Google Scholar]
- Garcia-Gonzalez E. Genersch E. Honey bee larval peritrophic matrix degradation during infection with Paenibacillus larvae, the aetiological agent of American foulbrood of honey bees, is a key step in pathogenesis. Environ. Microbiol. 2013;15:2894–2901. doi: 10.1111/1462-2920.12167. [DOI] [PubMed] [Google Scholar]
- Garcia-Gonzalez E, Müller S, Ensle P, Süssmuth RD. Genersch E. Elucidation of sevadicin, a novel nonribosomal peptide secondary metabolite produced by the honey bee pathogenic bacterium Paenibacillus larvae. Environ. Microbiol. 2014;16:1297–1309. [PubMed] [Google Scholar]
- Genersch E. Otten C. The use of repetitive element PCR fingerprinting (rep-PCR) for genetic subtyping of German field isolates of Paenibacillus larvae subsp. larvae. Apidologie. 2003;34:195–206. [Google Scholar]
- Genersch E, Ashiralieva A. Fries I. Strain- and genotype-specific differences in virulence of Paenibacillus larvae subsp. larvae, the causative agent of American foulbrood disease in honey bees. Appl. Environ. Microbiol. 2005;71:7551–7555. doi: 10.1128/AEM.71.11.7551-7555.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genersch E, Forsgren E, Pentikäinen J, Ashiralieva A, Rauch S, Kilwinski J, et al. Reclassification of Paenibacillus larvae subsp. pulvifaciens and Paenibacillus larvae subsp. larvae as Paenibacillus larvae without subspecies differentiation. Int. J. Syst. Evol. Microbiol. 2006;56:501–511. doi: 10.1099/ijs.0.63928-0. [DOI] [PubMed] [Google Scholar]
- Gilliam M. Identification and roles of non-pathogenic microflora associated with honey bees. FEMS Microbiol. Lett. 1997;155:1–10. [Google Scholar]
- Gilliam M. Prest DB. Microbiology of feces of the larval honey bee, Apis mellifera. J. Invertebr. Pathol. 1987;49:70–75. [Google Scholar]
- Gilliam M, Wickerham LJ, Morton HL. Martin RD. Yeasts isolated from honey bees, Apis mellifera, fed 2,4-D and antibiotics. J. Invertebr. Pathol. 1974;24:349–356. doi: 10.1016/0022-2011(74)90143-8. [DOI] [PubMed] [Google Scholar]
- Hill AM. The biosynthesis, molecular genetics and enzymology of the polyketide-derived metabolites. Nat. Prod. Rep. 2006;23:256–320. doi: 10.1039/b301028g. [DOI] [PubMed] [Google Scholar]
- Holst EC. An antibiotic from a bee pathogen. Science. 1945;102:593–594. doi: 10.1126/science.102.2658.593. [DOI] [PubMed] [Google Scholar]
- Homburg S, Oswald E, Hacker J. Dobrindt U. Expression analysis of the colibactin gene cluster coding for a novel polyketide in Escherichia coli. FEMS Microbiol. Lett. 2007;275:255–262. doi: 10.1111/j.1574-6968.2007.00889.x. [DOI] [PubMed] [Google Scholar]
- Kilwinski J, Peters M, Ashiralieva A. Genersch E. Proposal to reclassify Paenibacillus larvae subsp. pulvifaciens DSM 3615 (ATCC 49843) as Paenibacillus larvae subsp. larvae. Results of a comparative biochemical and genetic study. Vet. Microbiol. 2004;104:31–42. doi: 10.1016/j.vetmic.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Li H, Tanikawa T, Sato Y, Nakagawa Y. Matsuyama T. Serratia marcescens gene required for surfactant serrawettin W1 production encodes putative aminolipid synthetase belonging to nonribosomal peptide synthetase family. Microbiol. Immunol. 2005;49:303–310. doi: 10.1111/j.1348-0421.2005.tb03734.x. [DOI] [PubMed] [Google Scholar]
- Murray KD. Aronstein KA. Transformation of the Gram-positive honey bee pathogen, Paenibacillus larvae, by electroporation. J. Microbiol. Methods. 2008;75:325–328. doi: 10.1016/j.mimet.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Neuendorf S, Hedtke K, Tangen G. Genersch E. Biochemical characterization of different genotypes of Paenibacillus larvae subsp. larvae, a honey bee bacterial pathogen. Microbiology. 2004;150:2381–2390. doi: 10.1099/mic.0.27125-0. [DOI] [PubMed] [Google Scholar]
- Nougayrede JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313:848–851. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- Poppinga L. Genersch E. Heterologous expression of green fluorescent protein in Paenibacillus larvae, the causative agent of American Foulbrood of honey bees. J. Appl. Microbiol. 2012;112:430–435. doi: 10.1111/j.1365-2672.2011.05214.x. [DOI] [PubMed] [Google Scholar]
- Poppinga L, Janesch B, Fünfhaus A, Sekot G, Garcia-Gonzalez E, Hertlein G, et al. Identification and functional analysis of the S-layer protein SplA of Paenibacillus larvae, the causative agent of American Foulbrood of honey bees. PLoS Pathog. 2012;8:e1002716. doi: 10.1371/journal.ppat.1002716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch S, Ashiralieva A, Hedtke K. Genersch E. Negative correlation between individual-insect-level virulence and colony-level virulence of Paenibacillus larvae, the etiological agent of American foulbrood of honeybees. Appl. Environ. Microbiol. 2009;75:3344–3347. doi: 10.1128/AEM.02839-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch C, Weber T, Kohlbacher O, Wohlleben W. Huson DH. Specificity prediction of adenylation domains in nonribosomal peptide synthetases (NRPS) using transductive support vector machines (TSVMs) Nucleic Acids Res. 2005;33:5799–5808. doi: 10.1093/nar/gki885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer D, Luxenburger E, Brachmann AO. Bode HB. A new type of pyrrolidine biosynthesis is involved in the late steps of xenocoumacin production in Xenorhabdus nematophila. ChemBioChem. 2009;10:1997–2001. doi: 10.1002/cbic.200900187. [DOI] [PubMed] [Google Scholar]
- Reimer D, Pos KM, Thines M, Grün P. Bode HB. A natural prodrug activation mechanism in nonribosomal peptide synthesis. Nat. Chem. Biol. 2011;7:888–890. doi: 10.1038/nchembio.688. [DOI] [PubMed] [Google Scholar]
- Röttig M, Medema MH, Blin K, Weber T, Rausch C. Kohlbacher O. NRPSpredictor2 – a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 2011;39:W362–W367. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild H-A, Fuchs SW, Bode HB. Grünewald B. Low-molecular-weight metabolites secreted by Paenibacillus larvae as potential virulence factors of American Foulbrood. Appl. Environ. Microbiol. 2014;80:2484–2492. doi: 10.1128/AEM.04049-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snowdon JA. Cliver DO. Microorganisms in honey. Int. J. Food Microbiol. 1996;31:1–26. doi: 10.1016/0168-1605(96)00970-1. [DOI] [PubMed] [Google Scholar]
- Vallet-Gely I, Opota O, Boniface A, Novikov A. Lemaitre B. A secondary metabolite acting as a signalling molecule controls Pseudomonas entomophila virulence. Cell. Microbiol. 2010;12:1666–1679. doi: 10.1111/j.1462-5822.2010.01501.x. [DOI] [PubMed] [Google Scholar]
- Yoder JA, Jajack AJ, Rosselot AE, Smith TJ, Yerke MC. Sammataro D. Fungicide contamination reduces beneficial fungi in bee bread based on an area-wide field study in honey bee, Apis mellifera, colonies. J. Toxicol. Environ. Health A. 2013;76:587–600. doi: 10.1080/15287394.2013.798846. [DOI] [PubMed] [Google Scholar]
- Yue D, Nordhoff M, Wieler LH. Genersch E. Fluorescence in situ-hybridization (FISH) analysis of the interactions between honeybee larvae and Paenibacillus larvae, the causative agent of American foulbrood of honeybees (Apis mellifera. Environ. Microbiol. 2008;10:1612–1620. doi: 10.1111/j.1462-2920.2008.01579.x. [DOI] [PubMed] [Google Scholar]
- Zarschler K, Janesch B, Zayni S, Schäffer C. Messner P. Construction of a gene knockout system for application in Paenibacillus alvei CCM 2051T, exemplified by the S-layer glycan biosynthesis initiation enzyme WsfP. Appl. Environ. Microbiol. 2009;75:3077–3085. doi: 10.1128/AEM.00087-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarschler K, Janesch B, Pabst M, Altmann F, Messner P. Schäffer C. Protein tyrosine O-glycosylation – A rather unexplored prokaryotic glycosylation system. Glycobiology. 2010;20:787–798. doi: 10.1093/glycob/cwq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. of LC-ESI-Orbitrap-MS profiling of metabolites present in the crude supernatant. (A) Total ion chromatogram (TIC) of P. larvae DSM25430 wt and (B) total ion chromatogram (TIC) of P. larvae DSM25430 ΔpamA. Paenilamicin could not be detected directly from crude supernatants due to early elution in the dead volume and ion suppression. The secondary metabolite sevadicin is indicated with a red arrow.
Figure S2. MS/MS fragmentation studies on paenilamicin by HPLC-ESI-triple quadrupole MS from P. larvae secretome after sample workup. (A) MS/MS fragmentation spectrum with [M + 2H]2+ = 505.4 as precursor ion (♦). (B) MS/MS fragmentation spectrum with [M + 2H]2+ = 519.4 as precursor ion (♦).
Figure S3. HPLC UV analytics of pure paenilamicin. (A) Chromatogram of pure paenilamicin monitored at λ = 214 nm. (B) Chromatogram of a blank measurement (H2O) monitored at λ = 214 nm. Paenilamicin shows practically no retention at the C18 column.
Table S1. Homology analysis of the pam gene cluster products.