

Abstract
Background
It has been suggested that glutamatergic system hyperactivity may be related to the pathogenesis of Parkinson’s disease (PD). Vesicular glutamate transporters (VGLUT1-3) import glutamate into synaptic vesicles and are key anatomical and functional markers of glutamatergic excitatory transmission. Both VGLUT1 and VGLUT2 have been identified as definitive markers of glutamatergic neurons, but VGLUT 3 is also expressed by non glutamatergic neurons. VGLUT1 and VGLUT2 are thought to be expressed in a complementary manner in the cortex and the thalamus (VL/VM), in glutamatergic neurons involved in different physiological functions. Chronic high-frequency stimulation (HFS) of the subthalamic nucleus (STN) is the neurosurgical therapy of choice for the management of motor deficits in patients with advanced PD. STN-HFS is highly effective, but its mechanisms of action remain unclear. This study examines the effect of STN-HFS on VGLUT1-3 expression in different brain nuclei involved in motor circuits, namely the basal ganglia (BG) network, in normal and 6-hydroxydopamine (6-OHDA) lesioned rats.
Results
Here we report that: 1) Dopamine(DA)-depletion did not affect VGLUT1 and VGLUT3 expression but significantly decreased that of VGLUT2 in almost all BG structures studied; 2) STN-HFS did not change VGLUT1-3 expression in the different brain areas of normal rats while, on the contrary, it systematically induced a significant increase of their expression in DA-depleted rats and 3) STN-HFS reversed the decrease in VGLUT2 expression induced by the DA-depletion.
Conclusions
These results show for the first time a comparative analysis of changes of expression for the three VGLUTs induced by STN-HFS in the BG network of normal and hemiparkinsonian rats. They provide evidence for the involvement of VGLUT2 in the modulation of BG cicuits and in particular that of thalamostriatal and thalamocortical pathways suggesting their key role in its therapeutic effects for alleviating PD motor symptoms.
Keywords: High frequency stimulation, Subthalamic nucleus, Parkinson’s disease, Basal Ganglia, 6-OHDA-lesion, Rat, Glutamate, Vesicular glutamate transporters
Background
It is long recognized that the degeneration of dopaminergic neurons induces an abnormal activation of glutamate systems in the basal ganglia (BG) that is central to the pathophysiology of Parkinson’s disease (PD) [1-4]. Glutamate mediated mechanisms are also thought to play a role in the development of dyskinesias with long-term administration of L-3,4-dihydroxyphenylalanine (L-DOPA), the most efficient treatment for PD. Many experimental studies also evidence that dopamine denervation induces an increase in corticostriatal glutamate [5-11] and that L-DOPA-induced dyskinesia (LID) are linked to BG network glutamate transmission abnormalities [12,13]. Microdialysis studies have suggested that dopamine lesion may also increase glutamate transmission in the BG output structures, substantia nigra pars reticulata (SNr) [5,14-16] and entopeduncular nucleus [6], presumably as a result of the abnormal activation of the subthalamic nucleus (STN) [17].
Three subtypes of vesicular glutamate transporters have been identified: VGLUT1, 2 and 3 [18]. These transporters mediate glutamate uptake inside presynaptic vesicles and are anatomical and functional markers of glutamatergic excitatory transmission [19-25]. VGLUT1-3 are very similar in structure and function, but are used by different neuronal populations. VGLUT1 and VGLUT2 are expressed by the cortical and subcortical neurons respectively. VGLUT3 is expressed by nonglutamatergic neurons, such as cholinergic striatal interneurons, a GABAergic interneuron subpopulation from the cortex and hippocampus and serotoninergic neurons from the dorsal and medial raphe nuclei [22,26].
Since the 1990s, High Frequency Stimulation (HFS) of the STN has become an effective surgical treatment of late-stage Parkinson’s disease (PD), improving all motor symptoms in PD patients, particularly in those who experience motor fluctuations [27-29]. However, the mechanisms underlying the improvement in symptoms remain unclear [30-32]. Beyond its local effect on STN activity, we know that, by activating axons, STN-HFS may generate widespread and heterogeneous distal effects throughout the BG network [32,33]. Indeed, we have already reported in previous studies that in intact or 6-OHDA (6-hydroxydopamine)-lesioned rats, STN-HFS increases extracellular glutamate in the striatum, the globus pallidus and the SNr [14-16,34].
The present study analyzed the effects of DA depletion and for the first time those of STN-HFS on VGLUT1-3 expression in several BG nuclei, by using immunoradioautography with affinity-purified rabbit VGLUT1, VGLUT2 or VGLUT3 antiserum.
We found that DA-depletion did not affect VGLUT1 and VGLUT3 expression in almost all BG structures studied while that of VGLUT2 significantly decreased. Interestingly, STN-HFS did not affect VGLUT1-3 expression in normal rats, but systematically increased their expression in most of the BG nuclei studied in DA-depleted animals.
According to the changes of VGLUT1-3 expression observed and to their known anatomical localization, we suggest that STN-HFS may achieve its therapeutic effect, at least in part, through normalization of the thalamostriatal and thalamocortical pathways.
Methods
Animals
Adult (5 to 7 weeks old) male Sprague–Dawley rats (Janvier, Le Genest St Isle, France), weighing 180 to 270 g, were housed in an animal room on a 12-hour light/dark cycle, with food and water supplied ad libitum. This study was carried out in strict accordance with the recommendations of the European Community Council Directive of 24 November 1986 (86/609/EEC) concerning the care of laboratory animals, French Ministry of Agriculture regulations (Direction Départementale de la Protection des Populations, Préfecture de l’Isère, France, Grenoble Institute of Neuroscience, agreement number: A 38-516-10-008; Marc Savasta, permit number 38-10-08, Carole Carcenac permit number 38-10-23) and French guidelines for the use of live animals in scientific investigations. The protocol was approved by the Committee on the Ethics of Animal Experiments of the “Grenoble Institute of Neuroscience ethical committee” agreement number 04. All surgery was performed under a mixture of xylazine and ketamaine and all efforts were made to minimize the number of animal used and their suffering. All operated rats were intraperitoneally treated with Rimadyl (1 ml.kg-1) to prevent post-surgery suffering.
Lesion procedure
Forty rats (n = 40) were anesthetized with a mixture of xylazine (10 mg.kg-1, intraperitoneal) and ketamine (100 mg.kg-1, intraperitoneal) and secured in a Kopf stereotaxic apparatus (Phymep, Paris, France). All animals received desipramine (25 mg/kg s.c.) pretreatment, to protect noradrenergic neurons. Lesioned animals (n = 20) received a unilateral injection of 9 μg of 6-hydroxydopamine (6-OHDA) (Sigma, St. Quentin-Fallavier, France) dissolved in 3 μl of 0.9% sterile NaCl supplemented with 0.2% ascorbic acid, administered at a flow rate of 0.5 μl · min-1 to the left SNc. An identical procedure was used for controls (n = 20) but with the injection of NaCl 0.9%. The stereotaxic coordinates for the injection site relative to the bregma were as follows: anteroposterior (AP), -5.3 mm; lateral (L), +2.35 mm; dorsoventral (DV), -7.5 mm, with the incisor bar at 3.3 mm below the interaural plane, according to the stereotaxic atlas of Paxinos and Watson [35]. After injections, animals were kept warm and allowed to recover from the anesthetic before being returned to the animal house for three weeks until the stimulation experiments. This time interval was left to allow the DA system degeneration induced by the neurotoxin to stabilize.
Implantation of the stimulation electrode
Rats from the two experimental groups (sham-operated controls, n = 20, and 6-OHDA lesioned, n = 20) were first anesthetized by the inhalation (1 l.min-1) of a mixture of 3% isoflurane in air (the air used being composed of 22% O2, 78% N2) and mounted in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). The dorsal skull was exposed and holes were drilled for the implantation of the stimulation electrode into the left STN. During the implantation and stimulation procedure, anesthesia was maintained with an inhaled mixture of 1% isoflurane in air (1 l.min-1) and body temperature was maintained at 37°C with a feedback-controlled heating pad (Harvard Apparatus, Edenbridge, UK). Stereotaxic coordinates were chosen according to the atlas of Paxinos and Watson [35] and were as follows relative to the bregma: AP, -3.7 mm; L, +2.4 mm; and DV, -7.8 mm as previously described [14-16,34,36].
Electrical stimulation
For electrical stimulation, we used a concentric stimulating bipolar electrode (SNEX 100, Rhodes Medical Instruments, Woodland Hills, CA), with an outer diameter of 250 μm and a distance between the poles of 1 mm. Stimuli were delivered under anesthesia during 4 hours with a World Precision Instrument (Stevenage, UK) acupulser and stimulus isolation units giving a rectangular pulse. This duration of stimulation (> 1 h) was chosen to be sure that the proteic expression of VGLUTs can be detected and stabilized and almost corresponds to that used in previous studies analyzing mRNA levels of different target proteins of basal ganglia circuits [37]. As previously reported, the stimulation parameters (130 Hz, 60 μs, 200 μA) matched those routinely used in Parkinsonian patients [14,34,36]. At the end of each experiment, an electrical lesion was created in the STN so that the position of the electrode could be checked post-mortem. In control rats (sham-operated and 6-OHDA-lesioned) the stimulation was never switched “on”.
Histology
At the end of the electrical stimulation, all animals were perfused transcardially with 0.9% saline, under chloral hydrate anesthesia. Brains were rapidly removed and frozen in cooled (−40°C) isopentane, then stored at −20°C. Serial frontal sections (14-μm thick) were cut with a cryostat (Microm HM 500, Microm, Francheville, France), collected on microscopic slides and stored at −20°C. Tissue sections from different BG nuclei and related structures (striatum (caudate-putamen), nucleus accumbens, motor and somatosensory cortices, thalamus (VL/VM), subthalamic nucleus, globus pallidus and substantia nigra pars reticulata (SNr)) were selected to analyze changes in VGLUT expression.
The correct location of the stimulation electrode was checked by collecting several subthalamic tissue sections (n = 12 sections per stimulated rat) (14 μm thick from AP, -3,6 to −4,3 mm relative to the bregma, Paxinos and Watson, [35]) and counterstaining with cresyl violet. The tip of the electrode was systematically implanted directly in the STN at the top of its dorsal part. These histological controls were systematically carried out for all the animals in each experimental group. All animals with incorrectly positioned stimulation electrodes were excluded (controls, n = 3 and 6-OHDA lesioned, n = 4).
TH-immunohistochemistry
We assessed the extent of the dopaminergic denervation induced by nigral 6-OHDA injection by TH immunostaining on striatal and nigral sections from the fixed brains of lesioned animals. TH immunostaining was carried out as previously described [14]. Briefly, striatal and nigral tissue sections from 6-OHDA-lesioned rats were mounted on silane-coated microscope slides. Tissue sections were postfixed in 4% paraformaldehyde, thoroughly washed with Tris buffered-saline (TBS, 0.1 M, pH 7.4) and incubated for 1 hour in 0.3% Triton X-100 in TBS (TBST) and 3% normal goat serum (NGS, Sigma-Aldrich, St Quentin Fallavier, France). They were then incubated with primary antisera diluted in TBST supplemented with 1% normal goat serum (NGS) for 24 h, at 4°C. The antiserum was diluted 1:500 for TH staining (mouse monoclonal antibody; Chemicon, Temecula, CA). Antibody binding was detected with avidin-biotin-peroxidase conjugate (Vectastain ABC Elite, Vector Laboratories, Burlingame, CA), with 3, 3’-diaminobenzidine as the chromagen. The detection reaction was allowed to proceed for one to three minutes, as previously described. Sections were dehydrated in a series of graded ethanol solutions, cleared in xylene, mounted in DPX (DBH Laboratories Supplies, Poole, UK) and covered with a coverslip for microscopy.
VGLUT 1–3 immunoradioautography
Tissue sections were air-dried, post-fixed by immersion in fixative (4% PFA), and then washed in PBS. Nonspecific binding sites were saturated by incubation with 3% bovine serum albumin (BSA) in PBS, 1% NGS and 2 mM NaI (buffer A). Sections were incubated overnight at 4°C in buffer A supplemented with affinity-purified rabbit VGLUT1, VGLUT2 or VGLUT3 antiserum (dilution 1/10000 for VGLUT1 and VGLUT2, 1/5000 for VGLUT3, from Dr Salah El Mestikawy), and then for 1 hour with an affinity-purified goat anti-rabbit [125I] IgG (0.25 μCi/ml, Perkin Elmer, Paris, France) in buffer A supplemented with 0.02% sodium azide. The sections were rinsed in water, dried and placed against X-ray films (Biomax MR, Kodak) for 9 to 11 days.
The specificity of all antisera used in this study have been previously validated by our group (Gras et al. [22], [38]; Herzog et al. [23], [26]). For each labeled section, a background value was estimated by measuring optical density in the corpus callosum, since this structure is devoided of specific staining for VGLUT 1–3 antibodies. This background value was then systematically subtracted from the optical density values obtained for each corresponding section.
Quantification and statistical analysis
For the evaluation of the extent of DA-denervation, striatal and nigral TH immunostained sections were directly processed by using the Calopix software of the computerized image analysis system (TRIBVN, 2.9.2 version, Châtillon, France). Six TH-immunostained sections from each structure (striatum and SNc) and for each rat were used for quantification. The loss of TH immunostaining in the SNc or in the striatum was evaluated by comparing the total surface of both structures, as revealed by the TH immunolabelling, in normal and lesioned animals.
For quantification of VGLUT1-3 contents, four AP levels (+1, -0.92, -3.8 and −5.5 mm relative to bregma (Paxinos et Watson, [35])) were choosen. For each rat, three stained sections of the same AP level were used for quantification and the triplicate OD values obtained for each structure analyzed were averaged. Immunoradioautograms obtained from X-ray films were analyzed with Autoradio V4.03 software (SAMBA Technologies, Meylan, France). Values of optical densities measured from each structure analyzed are expressed as a mean ± standard error (SEM) in Table 1. Histograms presented in figures show the mean ± standard error of the mean (SEM) of optical densities expressed as a percentage of control values. Data were analyzed for each brain structure by Kruskal-Wallis tests with SigmaStat 3.1 software. Post-hoc analyses were carried out with the Dunn’s method.
Table 1.
Effect of 6-OHDA-lesion and STN-HFS on bilateral changes of optical density measurements of immunoreactive signals for VGLUT1-3
| VGLUT1 | VGLUT2 | VGLUT3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Ipsilateral side |
Controlateral side |
Ipsilateral side |
Controlateral side |
Ipsilateral side |
Controlateral side |
||||||
|
A |
Controls |
6-OHDA |
Controls |
6-OHDA |
Controls |
6-OHDA |
Controls |
6-OHDA |
Controls |
6-OHDA |
Controls |
6-OHDA |
| CPu |
51,35 (±3,2) |
53,33 (±1,9) |
48,78 (±3,4) |
52,59(±1,7) |
20,15 (±2,38) |
12,64*(±1,5) -37% |
19,76 (±2,3) |
13,38*(±0,6) -33% |
16, 65(±0,8) |
18,15 (±1,7) |
15,37 (±0,8) |
16, 33 (±3,1) |
| PM Cx |
55,54 (±3,3) |
48,3 (±2,1) |
55,81 (±2,9) |
48,07 (±2,8) |
18,56 (±1,7) |
10,93*(±0,6) -42% |
18,5 (±2,1) |
11,12*(±0,6) -40% |
14,34 (±0,9) |
13,09 (±2,4) |
14,39 (±0,9) |
13, 02 (±2,5) |
| SS Cx |
45,93 (±3) |
35,37 (±1,6) |
45,93 (±1,6) |
38,93 (±1,6) |
15,79 (±1,81) |
9,22*(±0,4) -42% |
16,44 (±1,8) |
10,57*(±0,6) -36% |
12,01 (±0,6) |
10,28 (±2,1) |
12,89 (±0,7) |
11,63 (±2,4) |
| Acb |
54,15 (±2,8) |
63,43 (±2,8) |
51,52 (±3,6) |
59,65 (±3,1) |
22,55 (±2,7) |
13,56*(±2,1) -40% |
22,14 (±2,6) |
13,91*(±2,1) -47% |
18,84 (±0,9) |
23,33 (±1,1) |
19,04 (±1,1) |
23 (±1,2) |
| Thalamus |
41,18 (±3,4) |
34,15 (±1,7) |
41,86 (±3,3) |
32,47 (±1,1) |
16,83 (±2,3) |
8,85*(±0,4) -48% |
17,49 (±2,4) |
9,05*(±0,7) -48% |
11,38 (±0,5) |
9,85 (±1,3) |
11,43 (±0,5) |
9,79 (±1,1) |
| STN |
21,13 (±2,7) |
23, 56 (±4,6) |
19,25 (±2,5) |
20,65 (±4,2) |
16,83 (±2,1) |
7,66*(±1,3) -55% |
15,53 (±2,3) |
9,32*(±0,5) -40% |
ND |
ND |
ND |
ND |
| GP |
ND |
ND |
ND |
ND |
12,78 (±1,9) |
7,97(±1) |
13,39 (±1,7) |
8,83 (±1,1) |
ND |
ND |
ND |
ND |
| SNr |
ND |
ND |
ND |
ND |
51,1 (±2,4) |
11,26*(±0,3) -25% |
15,76 (±2,2) |
11,39*(±0,5) -28% |
ND |
ND |
ND |
ND |
| |
Ipsilateral side |
Controlateral side |
Ipsilateral side |
Controlateral side |
Ipsilateral side |
Controlateral side |
||||||
|
B |
6-OHDA |
6-OHDA + STN-HFS |
6-OHDA |
6-OHDA + STN-HFS |
6-OHDA |
6-OHDA + STN-HFS |
6-OHDA |
6-OHDA + STN-HFS |
6-OHDA |
6-OHDA + STN-HFS |
6-OHDA |
6-OHDA + STN-HFS |
| CPu |
53,33 (±1,8) |
65,07*(±3,4) +22% |
52,59 (±1,7) |
64,43(±3,2) +23% |
12,64 (±1,5) |
17,81*(±1,4) +41% |
13,38 (±0,6) |
17,61*(±1,8) +32% |
18,15 (±1,7) |
26,13*(±2,9) +44% |
16,33 (±3,1) |
24,88*(±1,8) +53% |
| PM Cx |
48,3 (±2,2) |
63,08*(±3,3) +31% |
48,07 (±2,8) |
64, 79*(±3,5) +35% |
10,93 (±0,6) |
16,93*(±1,6) +55% |
11,12 (±0,6) |
16,86*(±1,8) +52% |
13,09 (±2,4) |
20*(±1,9) +53% |
13,02 (±2,5) |
16,62*(±1,7) +51% |
| SS Cx |
35,75 (±1) |
54,25*(±3,8) +52% |
38,93 (±1,6) |
55,08*(±3,8) +41% |
9,22 (±0,4) |
14,03*(±1,4) +52% |
10,57 (±0,6) |
13,38*(±1,6) +27% |
10,28 (±2,1) |
20,44*(±2,1) +99% |
11,63 (±2,4) |
16,78*(±1,4) +44% |
| Acb |
63,43 (±2,8) |
65,39 (±3,9) |
59,65 (±4,1) |
65,06 (±4,1) |
13,56 (±2,1) |
20,25*(±1,3) +49% |
13,91 (±2,1) |
18,78*(±1,2) +35% |
23,23 (±1,1) |
31,7* (1,2) +36% |
23 (±1,2) |
31,89*(±1,2) +38% |
| Thalamus |
34,15 (±1,7) |
48,1*(±3,8) +41% |
32,47(±1,1) |
46,37 (±3,6) +43% |
8,85 (±0,4) |
14,63*(±1,6) +65% |
9,05 (±0,7) |
13,5*(±1,5) +449% |
9,85 (±1,3) |
20,3*(±1,6) +106% |
9,79 (±1,1) |
19,38*(±2,5) +98% |
| STN |
23,56 (±4,6) |
17,78 (±2,6) |
20,65 (±4,2) |
19,34 (±3) |
7,66 (±1,3) |
14,79*(±1,5) +93% |
9,32 (±0,5) |
15,94*(±1,9) +71% |
ND |
ND |
ND |
ND |
| GP |
ND |
ND |
ND |
ND |
7.97 (±1) |
10,18 (±0,9) |
8,83 (±1,1) |
9,61 (±1) |
ND |
ND |
ND |
ND |
| SNr | ND | ND | ND | ND | 11,26 (±0,3) | 15,84*(±1,3) +41% | 11,39 (±0,5) | 16,64*(±1,4) +46% | ND | ND | ND | ND |
A, Modifications of VGLUT1-3 expression induced by unilateral 6-OHDA-lesion of SNc (control rats, n = 9; 6-OHDA rats, n = 6). *p<0.05, vs control group.
B, Modifications of VGLUT1-3 expression induced by unilateral STN-HFS in 6-OHDA lesioned rats (6-OHDA rats, n = 6; 6-OHDA rats + STN-HFS, n = 10). *p<0.05, vs non stimulated 6-OHDA-lesioned group. Values of optical densities measurements are expressed as a mean ± standard error (SEM). Data were analyzed for each brain structure by Kruskal-Wallis tests with SigmaStat 3.1 software. Post-hoc analyses were carried out with the Dunn’s method. Bold numbers correspond to significant differences. Ipsilateral side: related to the lesion and/or stimulation side; Controlateral side: related to the lesion and/or stimulation side; Acb, Accumbens nucleus; CPu, Caudate Putamen (striatum); GP, Globus Pallidus; PM Cx, Premotor Cortex; SNr, Substantia nigra pars reticulata; SS Cx, Somatosensory Cortex; STN, Subthalamic nucleus; Thalamus (VL/VM).
Results and discussion
Histological controls of the extent of the dopamine lesion and of electrode location
Three weeks after the unilateral injection of 6-OHDA, all lesioned animals presented a substantial loss of TH immunostaining in the ipsilateral SNc and the striatum (caudate-putamen nucleus), as shown by comparison with the contralateral side (Figure 1A, B) or with control animals. An analysis of densitometric measurements of TH immunostaining showed an absence of statistical difference between the two lesioned groups (non stimulated and stimulated).
Figure 1.
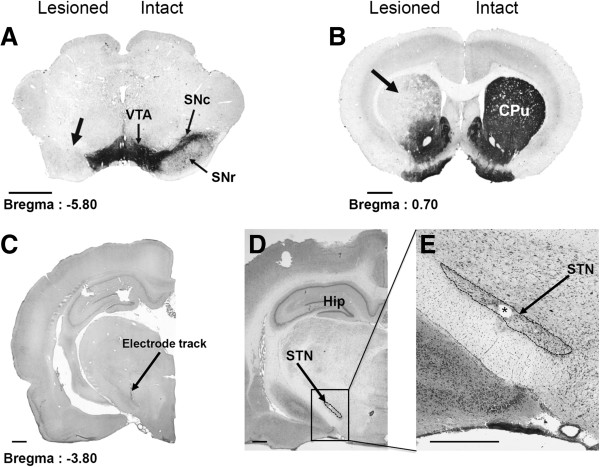
Photographs of TH-immunostained coronal rat-brain sections at the nigral (A) and striatal (B) levels and of cresyl violet-stained coronal rat-brain sections at subthalamic (C, D and E) levels in 6-OHDA-lesioned rats. Note, on the lesioned side (left), the loss of dopaminergic cells in the SNc (A) and the loss of dopaminergic terminals in the striatum (B). Note also the correct implantation of the stimulation electrode within the STN (C, D, E). C, The arrow indicates the electrode track. E, The asterisk indicates the point of stimulation. CPu: Caudate Putamen; Hip, Hippocampus; SNc, Substantia nigra pars compacta; SNr, Substantia nigra pars reticulata; STN, Subthalamic nucleus; VTA, Ventral Tegmental Area. Scale bar, 0.75 mm.
In DA-depleted animals, the loss of SNc TH + neurons was evaluated by comparing the total SNc surface on the intact side with the homologous area on the lesioned side. A loss of 92 ± 5% (p < 0.001) of TH immunolabeled surface was measured. In the striatum of the same rats, the loss of DA nerve terminals, as revealed by TH immunostaining mainly affected the dorsal part of the striatum (Figure 1B). This loss affected around 83 ± 4% of the striatal surface as compared to the total striatal surface of the control side. In this denervated striatal area, TH immunolabeling, as evaluated by a mean of densitometric values, was decreased by 85 ± 5% (p < 0.001) when compared to the controlateral intact side.
The correct implantation of the stimulation electrode in the STN is illustrated in Figure 1C-E. Figure 1E shows, at a higher magnification, the small electrical lesion (asterisk) created at the end of the experiment, indicating the point stimulated.
Regional distribution of VGLUT1-3 in control rats (without lesioning and stimulation)
VGLUT1-3 expression was qualitatively analyzed in control rats that had been neither lesioned nor stimulated, to ensure the validity and specificity of the immunoradioautographical staining. Immunoradioautograms from the different sections showed a distribution of VGLUT1-3 similar to that previously reported [26,39], confirming the validity of our VGLUT1-3 staining procedure and the lack of cross-reactivity between the antibodies used.
VGLUT1 immunostaining was dense in almost all the structures studied, including, especially, the striatum, nucleus accumbens, cortex, the motor part of the thalamus (VL/VM) and hippocampus. By contrast, no VGLUT1 labeling was found in the globus pallidus, the substantia nigra and in most of the brainstem (Figure 2 E-H).
Figure 2.

Regional distribution of VGLUT1-3 proteins in control (without lesion and stimulation) rats. A-D, Schematic diagrams adapted from the stereotaxic atlas of Paxinos and Watson [1982]. E-P, Photographs of immunoradioautograms obtained by incubating coronal rat-brain sections of control rats (non lesioned and non stimulated) with affinity-purified anti-VGLUT1 (E-H), anti-VGLUT2 (I-L) and anti-VGLUT3 (M-P) antisera and then with anti-rabbit [125I] IgG. Note the different distributions of the three VGLUTs in the brain structures studied. Acb, Accumbens nucleus; Amy, Amygdaloid nucleus; CPu, Caudate Putamen; Cg, Cingulate cortex; GP, Globus Pallidus; Hip, Hippocampus; PM Cx, Premotor Cortex; SNr, Substantia nigra pars reticulata; SS Cx, Somatosensory Cortex; STN, Subthalamic nucleus; Tha, Thalamus (VL/VM). Scale bar, 0.4 mm.
VGLUT2 proteins were detected in almost the same set of structures as VGLUT1 although the density of VGLUT2 immunostaining was slightly lower than that for VGLUT1 in striatal, cortical and thalamic areas, whereas the opposite was observed in many sub-cortical structures. These data are consistent with the well-described complementary pattern of expression of VGLUT1 and VGLUT2 in the rat brain. VGLUT2 staining, unlike that for VGLUT1, was detectable in the substantia nigra pars reticulata, hypothalamic nuclei and midbrain, which displayed widespread staining. Different, complementary patterns of immunostaining for VGLUT1 and VGLUT2 were observed in the hippocampus. The density of VGLUT2 proteins was highest in layers IV and VI of the cortex and in the hypothalamus, the central gray matter and the superior colliculus (Figure 2 I-L).
VGLUT3 staining was weaker than that for VGLUT1 and VGLUT2, but was also observed in many different areas. VGLUT3 levels were moderate in the striatum, but high in the hippocampus, with a complementary distribution for VGLUT1 and VGLUT2 (Figure 2M-P).
Effects of 6-OHDA-lesion and/or STN-HFS on VGLUT1-3 expression
DA lesion and STN stimulation were unilaterally performed in this study. However, we found similar changes of VGLUTs expression on both sides, as revealed by the optical density measurements of immunoreactive signals for VGLUT1-3 in all structures examined (see Table 1). In order to simplify the presentation of our data, we decided to only show the results obtained from the ipsilateral side (the lesioned and/or stimulated side) on Figures 3 and 4. As precised in materials and methods, changes in VGLUTs expression induced by 6-OHDA lesion and STN stimulation were first analyzed by Kruskal-Wallis tests with SigmaStat 3.1 software. Results of these tests for each brain structure are presented in legends of Figures 3 and 4. Post-hoc analyses were then carried out with the Dunn’s method.
Figure 3.
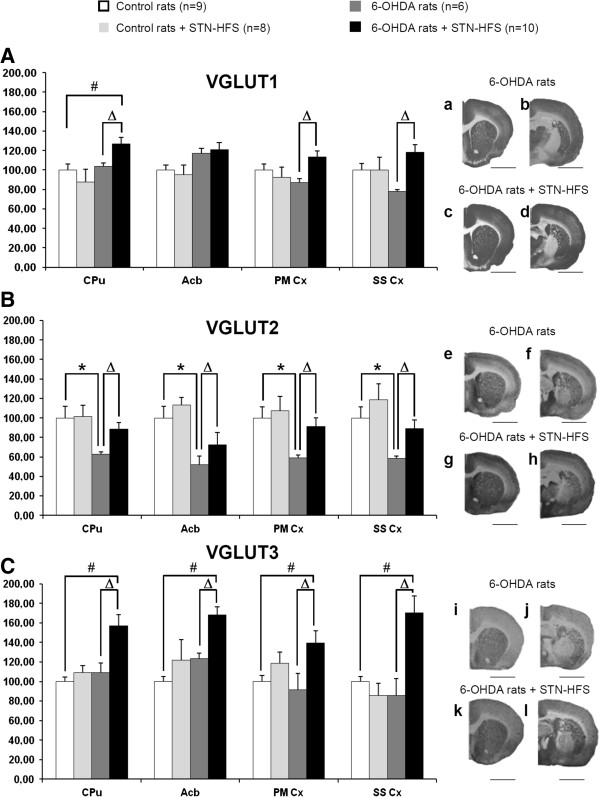
Effects of 6-OHDA-lesion and STN-HFS on striatal and cortical VGLUT1-3 expression. A-C, Histograms show the mean ± standard error of the mean (SEM) of optical density values expressed as a percentage of values of control rats (non lesioned and non stimulated). Data were analyzed for each brain structure by Kruskal-Wallis tests with SigmaStat 3.1 software. Post-hoc analyses were carried out with the Dunn’s method. Kruskal-Wallis tests (VGLUT1, CPu, p = 0.024; Acb, p = 0.05; PMCx, p = 0.049; SSCx, p = 0.02), (VGLUT2, CPu, p = 0.009; Acb, p = 0.002; PMCx, p = 0.012; SSCx, p = 0.002), (VGLUT3, CPu, p = 0.003; Acb, p<0.001; PMCx, p = 0.043; SSCx, p = 0.002). a-l, Photographs of immunoradioautograms obtained by incubating coronal rat-brain sections with affinity-purified anti-VGLUT1 (a-d), anti-VGLUT2 (e-h) and anti-VGLUT3 (i-l) antisera and then with 125I-labeled anti-rabbit IgG. Acb, Accumbens nucleus; CPu, Caudate putamen; PM Cx, Premotor cortex; SS Cx, Somatosensory Cortex. *, controls vs 6-OHDA rats; #, controls vs 6-OHDA + STN-HFS rats; Δ, 6-OHDA rats vs 6-OHDA + STN-HFS rats: p < 0.05. Scale bar, 0.4 mm.
Figure 4.

Effects of 6-OHDA-lesion and STN-HFS on thalamic, pallidal and nigral VGLUT1-3 expression. A-C, Histograms show the mean ± standard error of the mean (SEM) of optical density values expressed as a percentage of values of control rats (non lesioned and non stimulated). Data were analyzed for each brain structure by Kruskal-Wallis tests with SigmaStat 3.1 software. Post-hoc analyses were carried out with the Dunn’s method. Kruskal-Wallis tests: (VGLUT1, Thalamus VL/VM, p = 0.036; STN, p = 0.536), (VGLUT2, Thalamus (VL/VM); p = 0.036; STN, p = 0.015; SNr, p = 0.049; GP, p = 0.048), (VGLUT3, Thalamus (VL/VM), p<0.001. a-l, Photographs of immunoradioautograms obtained by incubating coronal rat-brain sections with affinity-purified anti-VGLUT1 (a-d), anti-VGLUT2 (e-h) and anti-VGLUT3 (i-l) antisera and then 125I-labeled anti-rabbit IgG. GP, Globus Pallidus; SNr, Substantia nigra pars reticulata; STN, Subthalamic nucleus; Tha, Thalamus (VL/VM). *, controls vs 6-OHDA rats; #, controls vs 6-OHDA + STN-HFS rats; Δ, 6-OHDA rats vs 6-OHDA + STN-HFS rats: p < 0.05. Scale bar, 0.4 mm.
Effect of the 6-OHDA-lesion on VGLUT1-3 expression
DA depletion did not affect VGLUT1 and VGLUT3 expression whatever the brain structure analyzed. Slight changes were observed in the striatum, nucleus accumbens, somatosensory cortex and thalamus (VL/VM), but they were not statistically significant (Table 1, Figures 3 and 4, white versus dark grey histograms).
By contrast, 6-OHDA lesioning induced a significant decrease in VGLUT2 expression of nearly 50% with respect to that in sham-operated rats (Figures 3 and 4). This decrease was particularly strong in the STN, thalamus (VL/VM) (Figure 4B), and cortical areas (Figure 3B) which displayed decreases of 55%, 48% and 42% with respect to control (non-lesioned) rats, respectively (p < 0.05, n = 6).
Effect of STN-HFS on VGLUT1-3 expression in sham-operated control (non-lesioned) rats and in 6-OHDA-lesioned animals
No significant change in VGLUT1-3 expression was detected after four hours of STN-HFS in sham-operated control rats (non-lesioned) rats. Levels of VGLUT1-3 expression were similar between the two experimental groups (non-lesioned rats with and without STN stimulation) in all different structures studied, as show in Figures 3 and 4.
On the contrary, STN-HFS induced a marked increase in VGLUT1-3 expression in 6-OHDA-lesioned rats. This increase affected almost all the brain structures studied (Table 1, Figures 3 and 4). VGLUT1 levels were always higher for all brain areas studied in stimulated 6-OHDA rats when compared to non-stimulated 6-OHDA-rats except for the nucleus accumbens (Acb) and STN (Figure 3A, a-d, for examples of autoradiographs and Figure 4A, a-d). The largest differences were found in the somatosensory cortex (+52%) and the motor part of the thalamus (VL/VM) (+41%) (p < 0.05, n = 10). For many brain structures analyzed, VGLUT1 levels measured in stimulated 6-OHDA-rats were comparable to those detected in control (without lesion and stimulation) rats. Interestingly, for the striatum and the thalamus (VL/VM), VGLUT1 levels remained moderately overexpressed versus controls (+27%, p < 0.05 and +17%, p < 0.05, respectively, n = 10) (Figures 3A, a-d, and 4A, a-d).
STN-HFS induced a significant increase in VGLUT2 expression in all structures studied in 6-OHDA-lesioned rats when compared to non-stimulated 6-OHDA-rats. However this increase did not affect the globus pallidus. (Figures 3B, e-h and 4B, e-h). Thus, STN-HFS more or less completely reversed the decrease in VGLUT2 expression induced by the DA-depletion in all structures analyzed.
STN-HFS induced a strong upregulation of VGLUT3 expression in 6-OHDA-lesioned rats (Figures 3C, i-l and 4C, i-l). Interestingly, this effect was particularly marked in the thalamus (VL/VM) (+106% versus non-stimulated 6-OHDA-lesioned rats, p < 0.05, n = 10) and the somatosensorial cortex (+99% versus non-stimulated 6-OHDA-lesioned rats, p < 0.05, n = 10).
Discussion
The key findings of this study were: 1) DA depletion decreased VGLUT2 (−40 to -50%) in all brain structures studied; 2) STN-HFS did not affect VGLUT1-3 expression in control (sham-operated) rats whatever the brain structure analyzed; 3) STN-HFS increased VGLUT1-3 expression in 6-OHDA-lesioned rats in almost all structures analyzed. Thereby STN-HFS: i) normalized VGLUT2 levels after the decrease induced by DA depletion, and ii) significantly increased VGLUT3 levels above those detected in control animals. This was also true for VGLUT1 levels but only for the striatum.
This remodeling suggests that the mode of action of STN-HFS results from a global effect on basal ganglia network and related structures and that its therapeutic efficacy may to be linked, at least in part, to the normalization of thalamostriatal and thalamocortical neurotransmissions.
Bilateral effects of unilateral DA lesioning and STN-HFS on VGLUT expression
As stated above, unilateral DA lesioning and STN-HFS caused similar changes in VGLUT expression on both sides of the brain. Bilateral effects of unilateral DA lesioning have already been reported in the striatum for tissue concentrations of glutamate [9], extracellular glutamate content assessed by microdialysis or voltammetry [34,40], glutamate receptor mRNA [41] and the glial glutamate transporter GLT-1 [42]. Similarly, unilateral STN-HFS has been reported to induce bilateral increases in striatal and nigral glutamate content [14,15,34]. These bilateral effects may result from crossed glutamatergic projections from the cortex or the thalamus (VL/VM) innervating the BG on the contralateral side, consistent with cross-talk in cortico-BG-cortical loops [42].
Effect of 6-OHDA-SNc-lesioning on VGLUT expression
We found here that 6-OHDA-lesions had no effect on VGLUT1 or VGLUT3 levels in any of the structures studied. By contrast, VGLUT2 levels decreased significantly three weeks after lesioning. At first glance, our observations contrast with previous reported data showing that dopamine depletion is associated with an increase in synaptic glutamate release [43-45] and with high striatal extracellular glutamate levels and glutamatergic activity [9,11,34,44,46-48] and greater thalamostriatal activity [48,49], two to four weeks after lesioning of nigral dopaminergic neurons. However, other studies have reported an absence of change in glutamate levels [50]. These differences may be accounted for by differences in the extent of the dopamine lesion, lesion sites, methodologies and time courses. We cannot exclude the possibility that different cellular mechanisms underlie presynaptic glutamate processes and extracellular glutamate release after lesioning. Indeed, striatal extracellular glutamate levels have been reported to depend on a complex balance between vesicular release and non vesicular release via glutamate transporters on both neurons and glia and the cysteine-glutamine antiporter [50,51]. Dopamine depletion leads to complex, biphasic changes in striatal glutamatergic transmission over the first few weeks, possibly stabilizing over three months. Contradictory data have been reported, for the cysteine-glutamate antiporter [52] and glial transporters [42,53,54] for example.
Furthermore, the changes in VGLUTs expression induced by 6-OHDA lesions are also complex. VGLUT1 levels increase in the three weeks following the injection but then decrease, whereas VGLUT2 levels decrease and then normalize [42,55-57]. In monkeys, MPTP treatment increases VGLUT1 expression but does not affect VGLUT2 levels [53,58]. In postmortem samples of Parkinsonian patients VGLUT1 and VGLUT2 levels are increased in the putamen while VGLUT1 levels is lowered in the prefrontal and temporal cortex [56].
However, the decrease in VGLUT2 levels observed here in all the brain structures of 6-OHDA rats closely parallels the thalamic hypoactivity induced by the strengthening of GABAergic inputs from the SNr and EP/GPi by the STN overactivity observed in DA-depleted BG networks [59]. Furthermore, neuronal degeneration has been observed postmortem in the thalamic nuclei of PD patients [60] and in the parafascicular nucleus in 6-OHDA-lesioned rats [49]. VGLUT2 is massively expressed by thalamic nuclei [61] and have be postulated as selective marker of thalamo-striatal activity [62]. This observation support the notion of a potential decreased glutamatergic afferences from the thalamus (VL/VM).
Effect of STN-HFS on VGLUT1-3 expression in control and 6-OHDA-lesioned rats
STN-HFS had no effect on VGLUT1-3 expression in any of the brain structures studied in control rats. This suggests that in the absence of dopamine depletion, STN-HFS did not affect VGLUT1-3 expression. These data are rather surprising since we reported in previous microdialysis study that in intact rats STN-HFS increases extracellular glutamate in the striatum, the globus pallidus and the SNr [14-16,34]. However, the duration of stimulation used here was longer than that used in our previous studies. Thus, we can speculate that increase of extracellular glutamate levels induced by STN-HFS in physiological conditions mainly involves non vesicular release. By contrast, after 6-OHDA lesions, STN-HFS induced an increase in VGLUT1-3 expression in almost all the structures analyzed. In these DA depleted conditions, we cannot exclude the possibility that the balance between non vesicular and vesicular release of glutamate is disturbed, involving more glutamate transporters on both neurons and glia [50,51]. Indeed, it is well documented that following DA nigrostriatal lesion, there is an increase in the number of glial cells, including astrocytes and microglia. Therefore, the non vesicular release may be due to an increase in membrane transporters, such as glial glutamate transporters (GLT1 and GLAST) and the neuronal glutamate transporter EEAC1 [55,63,64]. Thus, our previous findings concerning increased extracellular glutamate levels in the striatum, globus pallidus and SNr of 6-OHDA-lesioned rats [15,16,34] are consistent and confirm that STN-HFS affects not only its direct targets, but also more distant structures of the BG network [32,33]. The mechanisms underlying the therapeutic effects of STN-HFS are not fully elucidated. STN neuron inhibition by HFS, with loss of the drive of the internal part of the globus pallidus and disinhibition of the thalamus (VL/VM), would be consistent with the classical BG model [59,65]. However, far more complex effects and circuitry are probably involved. For example, the direct activation of nearby thalamostriatal and pallidonigral fibres [16,66] or direct or antidromic cortex activation [67,68]. These mechanisms might lead to corticostriatal fiber activation and the observed increase in VGLUT1 levels.
Similarly, the increase in VGLUT2 levels induced here by STN-HFS may reflect a release of the classical thalamic inhibition induced by dopamine lesions. As VGLUT2 is found mostly in the thalamostriatal neurons specifically affected by DA depletion, these data strongly suggest a major role for this pathway in the therapeutic effects of STN-HFS in PD [69,70]. However, the normalization of VGLUT2 levels we observed in the somatosensory and premotor cortices may also play a non negligeable role in these effects of STN-HFS.
Interestingly, VGLUT2 was also overexpressed in the SNr following STN-HFS in 6-OHDA-lesioned rats. As VGLUT2 is the only VGLUT expressed by STN glutamatergic projections to the SNr [71], this confirms our hypothesis that information transmission via the transsubthalamic pathway is not completely blocked during STN-HFS [15,16,31,32,67,68] although the effect on VGLUT2 expression in the SNr might also be mediated by the modulation of thalamic afferents during STN-HFS.
VGLUT3 is expressed in striatal cholinergic interneurons linked to the dopaminergic, GABAergic and glutamatergic systems in the cortico-BG-cortical loop [38]. It is therefore unsurprising that STN-HFS, which affects the whole BG network, affects VGLUT3 expression in the striatum and related structures, such as the cortex, hippocampus, nucleus accumbens, thalamus (VL/VM) and amygdala. Indeed, STN-HFS has been reported to affect not only dopamine, glutamate and GABA [14-16,34,36,68] but also serotonin and cholinergic transmissions [72-74]. Consequently, it is difficult to explain the role of VGLUT3 in STN-HFS mechanisms. However, VGLUT3 in cholinergic interneurons has been shown to increase acetylcholine tone and to release both glutamate and acetylcholine [18]. This mechanism might explain the efficacy of STN-HFS, via complex modulation of the BG network [32,33].
Conclusion
In conclusion, this is the first study of the expression of the three VGLUT subtypes in various brain structures in control and 6-OHDA-lesioned rats subjected to STN-HFS. We know that all the three types of VGLUTs are expected to be found in axon terminals and that immunoradioautography cannot really disclose between neuronal vs. axonal vs. glial expression of VGLUTs. In situ hybridization looking for different VGLUT transcripts will probably provide some added morphological value to our present study. These experiments are planned in our lab. However, our present data can suggest that STN-HFS may achieve its therapeutic effect, at least in part, through normalization of the thalamostriatal and the thalamocortical pathways.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed the experiments: MF, SB, SEM, MS. Performed the experiments: MF, GD, CC. Analyzed the data: MF, CC, MS. Contributed reagents/materials/analysis tools: SEM, MS. Wrote the paper: MF, SB, SEM, MS. All authors read and approved the final manuscript.
Contributor Information
Mathieu Favier, Email: mathieu.h.favier@wanadoo.fr.
Carole Carcenac, Email: carole.carcenac@ujf-grenoble.fr.
Guillaume Drui, Email: guillaume.drui@gmail.com.
Sabrina Boulet, Email: sabrina.boulet@ujf-grenoble.fr.
Salah El Mestikawy, Email: salah.elmestikawy@mcgill.ca.
Marc Savasta, Email: marc.savasta@ujf-grenoble.fr.
Acknowledgments
This work was supported by the Institut National de la Santé et de la Recherche Médicale, Université Joseph Fourier and Région Rhône-Alpes (ARC 2). We thank, Dr Emilie Lacombe for technical assistance and Dr Sebastien Carnicella for critical reading of this manuscript.
References
- Carlsson M, Carlsson A. Interactions between glutamatergic and monoaminergic systems within the basal ganglia–implications for schizophrenia and Parkinson’s disease. Trends Neurosci. 1990;13:272–276. doi: 10.1016/0166-2236(90)90108-M. [DOI] [PubMed] [Google Scholar]
- Lang AE, Lozano AM. Parkinson’s disease. First of two parts. N Engl J Med. 1998;339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- Lang AE, Lozano AM. Parkinson’s disease. Second of two parts. N Engl J Med. 1998;339:1130–1143. doi: 10.1056/NEJM199810153391607. [DOI] [PubMed] [Google Scholar]
- Lange KW, Kornhuber J, Riederer P. Dopamine/glutamate interactions in Parkinson’s disease. Neurosci Biobehav Rev. 1997;21:393–400. doi: 10.1016/S0149-7634(96)00043-7. [DOI] [PubMed] [Google Scholar]
- Abarca J, Gysling K, Roth RH, Bustos G. Changes in extracellular levels of glutamate and aspartate in rat substantia nigra induced by dopamine receptor ligands: in vivo microdialysis studies. Neurochem Res. 1995;20:159–169. doi: 10.1007/BF00970540. [DOI] [PubMed] [Google Scholar]
- Biggs CS, Starr MS. Dopamine and glutamate control each other’s release in the basal ganglia: a microdialysis study of the entopeduncular nucleus and substantia nigra. Neurosci Biobehav Rev. 1997;21:497–504. doi: 10.1016/S0149-7634(96)00032-2. [DOI] [PubMed] [Google Scholar]
- Blandini F, Porter RH, Greenamyre JT. Glutamate and Parkinson’s disease. Mol Neurobiol. 1996;12:73–94. doi: 10.1007/BF02740748. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT. Glutamate-dopamine interactions in the basal ganglia: relationship to Parkinson’s disease. J Neural Transm Gen Sect. 1993;91:255–269. doi: 10.1007/BF01245235. [DOI] [PubMed] [Google Scholar]
- Lindefors N, Ungerstedt U. Bilateral regulation of glutamate tissue and extracellular levels in caudate-putamen by midbrain dopamine neurons. Neurosci Lett. 1990;115:248–252. doi: 10.1016/0304-3940(90)90463-J. [DOI] [PubMed] [Google Scholar]
- Walker RH, Koch RJ, Sweeney JE, Moore C, Meshul CK. Effects of subthalamic nucleus lesions and stimulation upon glutamate levels in the dopamine-depleted rat striatum. NeuroReport. 2009;20:770–775. doi: 10.1097/WNR.0b013e32832ad556. [DOI] [PubMed] [Google Scholar]
- Walker RH, Moore C, Davies G, Dirling LB, Koch RJ, Meshul CK. Effects of subthalamic nucleus lesions and stimulation upon corticostriatal afferents in the 6-hydroxydopamine-lesioned rat. PLoS One. 2012;7:e32919. doi: 10.1371/journal.pone.0032919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana A, Sgambato-Faure V, Savasta M. Effects of L-DOPA and STN-HFS dyskinesiogenic treatments on NR2B regulation in basal ganglia in the rat model of Parkinson’s disease. Neurobiol Dis. 2012;48:379–390. doi: 10.1016/j.nbd.2012.06.009. [DOI] [PubMed] [Google Scholar]
- Sgambato-Faure V, Cenci MA. Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog Neurobiol. 2012;96:69–86. doi: 10.1016/j.pneurobio.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Boulet S, Lacombe E, Carcenac C, Feuerstein C, Sgambato-Faure V, Poupard A, Savasta M. Subthalamic stimulation-induced forelimb dyskinesias are linked to an increase in glutamate levels in the substantia nigra pars reticulata. J Neurosci. 2006;26:10768–10776. doi: 10.1523/JNEUROSCI.3065-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windels F, Bruet N, Poupard A, Urbain N, Chouvet G, Feuerstein C, Savasta M. Effects of high frequency stimulation of subthalamic nucleus on extracellular glutamate and GABA in substantia nigra and globus pallidus in the normal rat. Eur J Neurosci. 2000;12:4141–4146. doi: 10.1046/j.1460-9568.2000.00296.x. [DOI] [PubMed] [Google Scholar]
- Windels F, Carcenac C, Poupard A, Savasta M. Pallidal origin of GABA release within the substantia nigra pars reticulata during high-frequency stimulation of the subthalamic nucleus. J Neurosci. 2005;25:5079–5086. doi: 10.1523/JNEUROSCI.0360-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassani OK, Mouroux M, Feger J. Increased subthalamic neuronal activity after nigral dopaminergic lesion independent of disinhibition via the globus pallidus. Neuroscience. 1996;72:105–115. doi: 10.1016/0306-4522(95)00535-8. [DOI] [PubMed] [Google Scholar]
- El Mestikawy S, Wallén-Mackenzie A, Fortin GM, Descarries L, Trudeau LE. From glutamate co-release to vesicular synergy: vesicular glutamate transporters. Nat Rev Neurosci. 2011;12:204–216. doi: 10.1038/nrn2969. [DOI] [PubMed] [Google Scholar]
- Aihara Y, Mashima H, Onda H, Hisano S, Kasuya H, Hori T, Yamada S, Tomura H, Yamada Y, Inoue I, Kojima I, Takeda J. Molecular cloning of a novel brain-type Na(+)-dependent inorganic phosphate cotransporter. J Neurochem. 2000;74:2622–2625. doi: 10.1046/j.1471-4159.2000.0742622.x. [DOI] [PubMed] [Google Scholar]
- Bellocchio EE, Reimer RJ, Fremeau RT, Edwards RH. Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science. 2000;289:957–960. doi: 10.1126/science.289.5481.957. [DOI] [PubMed] [Google Scholar]
- Fremeau RT Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science. 2004;304:1815–1819. doi: 10.1126/science.1097468. [DOI] [PubMed] [Google Scholar]
- Gras C, Herzog E, Bellenchi GC, Bernard V, Ravassard P, Pohl M, Gasnier B, Giros B, El Mestikawy S. A third vesicular glutamate transporter expressed by cholinergic and serotoninergic neurons. J Neurosci. 2002;22:5442–5451. doi: 10.1523/JNEUROSCI.22-13-05442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog E, Bellenchi GC, Gras C, Bernard V, Ravassard P, Bedet C, Gasnier B, Giros B, El Mestikawy S. The existence of a second vesicular glutamate transporter specifies subpopulations of glutamatergic neurons. J Neurosci. 2001;21:RC181. doi: 10.1523/JNEUROSCI.21-22-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Rhee JS, Rosenmund C, Jahn R. Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature. 2000;407:189–194. doi: 10.1038/35025070. [DOI] [PubMed] [Google Scholar]
- Takamori S, Rhee JS, Rosenmund C, Jahn R. Identification of differentiation-associated brain-specific phosphate transporter as a second vesicular glutamate transporter (VGLUT2) J Neurosci. 2001;21:RC182. doi: 10.1523/JNEUROSCI.21-22-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog E, Gilchrist J, Gras C, Muzerelle A, Ravassard P, Giros B, Gaspar P, El Mestikawy S. Localization of VGLUT3, the vesicular glutamate transporter type 3, in the rat brain. Neuroscience. 2004;123:983–1002. doi: 10.1016/j.neuroscience.2003.10.039. [DOI] [PubMed] [Google Scholar]
- Benabid AL, Pollak P, Gross C, Hoffmann D, Benazzouz A, Gao DM, Laurent A, Gentil M, Perret J. Acute and long-term effects of subthalamic nucleus stimulation in Parkinson’s disease. Stereotact Funct Neurosurg. 1994;62:76–84. doi: 10.1159/000098600. [DOI] [PubMed] [Google Scholar]
- Krack P, Batir A, Van Blercom N, Chabardes S, Fraix V, Ardouin C, Koudsie A, Limousin PD, Benazzouz A, LeBas JF, Benabid AL, Pollak P. Five-year follow-up of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med. 2003;349:1925–1934. doi: 10.1056/NEJMoa035275. [DOI] [PubMed] [Google Scholar]
- Limousin P, Pollak P, Benazzouz A, Hoffman D, Broussolle E, Perret JE, Benabid AL. Bilateral subthalamic nucleus stimulation for severe Parkinson’s disease. Mov Disord. 1995;10:672–674. doi: 10.1002/mds.870100523. [DOI] [PubMed] [Google Scholar]
- Dostrovsky JO, Lozano AM. Mechanisms of deep brain stimulation. Mov Disord. 2002;17(Suppl 3):S63–S68. doi: 10.1002/mds.10143. [DOI] [PubMed] [Google Scholar]
- McIntyre CC, Savasta M, Kerkerian-Le Goff L, Vitek JL. Uncovering the mechanism(s) of action of deep brain stimulation: activation, inhibition, or both. Clin Neurophysiol. 2004;115:1239–1248. doi: 10.1016/j.clinph.2003.12.024. [DOI] [PubMed] [Google Scholar]
- Savasta M, Carcenac C, Boulet S. In: Diagnosis and Treatment of Parkinson’s Disease. Rana AQ, editor. Rijeka-Croatia: In Tech; 2011. Mechanisms of High Frequency Stimulation of the Subthalamic Nucleus in Parkinson’s Disease: From Local to Distal Effects on the Basal Ganglia Network; pp. 211–232. [Google Scholar]
- Deniau JM, Degos B, Bosch C, Maurice N. Deep brain stimulation mechanisms: beyond the concept of local functional inhibition. Eur J Neurosci. 2010;32:1080–1091. doi: 10.1111/j.1460-9568.2010.07413.x. [DOI] [PubMed] [Google Scholar]
- Bruet N, Windels F, Carcenac C, Feuerstein C, Bertrand A, Poupard A, Savasta M. Neurochemical mechanisms induced by high frequency stimulation of the subthalamic nucleus: increase of extracellular striatal glutamate and GABA in normal and hemiparkinsonian rats. J Neuropathol Exp Neurol. 2003;62:1228–1240. doi: 10.1093/jnen/62.12.1228. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. Stereotaxic coordinates. 4. San Diego: Academic Press; 1982. The rat brain. [Google Scholar]
- Bruet N, Windels F, Bertrand A, Feuerstein C, Poupard A, Savasta M. High frequency stimulation of the subthalamic nucleus increases the extracellular contents of striatal dopamine in normal and partially dopaminergic denervated rats. J Neuropathol Exp Neurol. 2001;60:15–24. doi: 10.1093/jnen/60.1.15. [DOI] [PubMed] [Google Scholar]
- Salin P, Manrique C, Forni C, Kerkerian-Le Goff L. High-frequency stimulation of the subthalamic nucleus selectively reverses dopamine denervation-induced cellular defects in the output structures of the basal ganglia in the rat. J Neurosci. 2002;22:5137–5148. doi: 10.1523/JNEUROSCI.22-12-05137.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gras C, Amilhon B, Lepicard EM, Poirel O, Vinatier J, Herbin M, Dumas S, Tzavara ET, Wade MR, Nomikos GG, Hanoun N, Saurini F, Kemel ML, Gasnier B, Giros B, El Mestikawy S. The vesicular glutamate transporter VGLUT3 synergizes striatal acetylcholine tone. Nat Neurosci. 2008;11:292–300. doi: 10.1038/nn2052. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Fujiyama F, Hioki H. Immunohistochemical localization of candidates for vesicular glutamate transporters in the rat brain. J Comp Neurol. 2002;444:39–62. doi: 10.1002/cne.10129. [DOI] [PubMed] [Google Scholar]
- Dzahini K, Dentresangle C, Le Cavorsin M, Bertrand A, Detraz I, Savasta M, Leviel V. Pre-synaptic glutamate-induced activation of DA release in the striatum after partial nigral lesion. J Neurochem. 2010;113:1459–1470. doi: 10.1111/j.1471-4159.2010.06682.x. [DOI] [PubMed] [Google Scholar]
- Fan XD, Li XM, Ashe PC, Juorio AV. Lesion of the substantia nigra pars compacta downregulates striatal glutamate receptor subunit mRNA expression. Brain Res. 1999;850:79–86. doi: 10.1016/S0006-8993(99)02106-X. [DOI] [PubMed] [Google Scholar]
- Massie A, Schallier A, Vermoesen K, Arckens L, Michotte Y. Biphasic and bilateral changes in striatal VGLUT1 and 2 protein expression in hemi-Parkinson rats. Neurochem Int. 2010;57:111–118. doi: 10.1016/j.neuint.2010.04.019. [DOI] [PubMed] [Google Scholar]
- Meshul CK, Cogen JP, Cheng HW, Moore C, Krentz L, McNeill TH. Alterations in rat striatal glutamate synapses following a lesion of the cortico- and/or nigrostriatal pathway. Exp Neurol. 2000;165:191–206. doi: 10.1006/exnr.2000.7467. [DOI] [PubMed] [Google Scholar]
- Meshul CK, Emre N, Nakamura CM, Allen C, Donohue MK, Buckman JF. Time-dependent changes in striatal glutamate synapses following a 6-hydroxydopamine lesion. Neuroscience. 1999;88:1–16. doi: 10.1016/S0306-4522(98)00189-4. [DOI] [PubMed] [Google Scholar]
- Robinson S, Freeman P, Moore C, Touchon JC, Krentz L, Meshul CK. Acute and subchronic MPTP administration differentially affects striatal glutamate synaptic function. Exp Neurol. 2003;180:74–87. doi: 10.1016/S0014-4886(02)00050-X. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Mercuri NB, Sancesario G, Bernardi G. Electrophysiology of dopamine-denervated striatal neurons. Implications for Parkinson’s disease. Brain. 1993;116:433–452. doi: 10.1093/brain/116.2.433. [DOI] [PubMed] [Google Scholar]
- Gubellini P, Eusebio A, Oueslati A, Melon C, Kerkerian-Le Goff L, Salin P. Chronic high-frequency stimulation of the subthalamic nucleus and L-DOPA treatment in experimental parkinsonism: effects on motor behaviour and striatal glutamate transmission. Eur J Neurosci. 2006;24:1802–1814. doi: 10.1111/j.1460-9568.2006.05047.x. [DOI] [PubMed] [Google Scholar]
- Villalba RM, Smith Y. Differential structural plasticity of corticostriatal and thalamostriatal axo-spinous synapses in MPTP-treated Parkinsonian monkeys. J Comp Neurol. 2011;519:989–1005. doi: 10.1002/cne.22563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymerich MS, Barroso-Chinea P, Pérez-Manso M, Muñoz-Patiño AM, Moreno-Igoa M, González-Hernández T, Lanciego JL. Consequences of unilateral nigrostriatal denervation on the thalamostriatal pathway in rats. Eur J Neurosci. 2006;23:2099–2108. doi: 10.1111/j.1460-9568.2006.04741.x. [DOI] [PubMed] [Google Scholar]
- Herrera-Marschitz M, You ZB, Goiny M, Meana JJ, Silveira R, Godukhin OV, Chen Y, Espinoza S, Pettersson E, Loidl F, Lubec G, Andersson K, Nylander I, Terenius L, Ungerstedt U. On the origin of extracellular glutamate levels monitored in the basal ganglia of the rat by in vivo microdialysis. J Neurochem. 1996;66:1726–1735. doi: 10.1046/j.1471-4159.1996.66041726.x. [DOI] [PubMed] [Google Scholar]
- Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22:9134–9141. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/S0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Chung EK, Chen LW, Chan YS, Yung KK. Downregulation of glial glutamate transporters after dopamine denervation in the striatum of 6-hydroxydopamine-lesioned rats. J Comp Neurol. 2008;511:421–437. doi: 10.1002/cne.21852. [DOI] [PubMed] [Google Scholar]
- Lievens JC, Salin P, Nieoullon A, Kerkerian-Le Goff L. Nigrostriatal denervation does not affect glutamate transporter mRNA expression but subsequent levodopa treatment selectively increases GLT1 mRNA and protein expression in the rat striatum. J Neurochem. 2001;79:893–902. doi: 10.1046/j.1471-4159.2001.00644.x. [DOI] [PubMed] [Google Scholar]
- Chung EK, Chen LW, Chan YS, Yung KK. Up-regulation in expression of vesicular glutamate transporter 3 in substantia nigra but not in striatum of 6-hydroxydopamine-lesioned rats. Neurosignals. 2006;15:238–248. doi: 10.1159/000101704. [DOI] [PubMed] [Google Scholar]
- Kashani A, Betancur C, Giros B, Hirsch E, El Mestikawy S. Altered expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in Parkinson disease. Neurobiol Aging. 2007;28:568–578. doi: 10.1016/j.neurobiolaging.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robelet S, Melon C, Guillet B, Salin P, Kerkerian-Le Goff L. Chronic L-DOPA treatment increases extracellular glutamate levels and GLT1 expression in the basal ganglia in a rat model of Parkinson’s disease. Eur J Neurosci. 2004;20:1255–1266. doi: 10.1111/j.1460-9568.2004.03591.x. [DOI] [PubMed] [Google Scholar]
- Raju DV, Ahern TH, Shah DJ, Wright TM, Standaert DG, Hall RA, Smith Y. Differential synaptic plasticity of the corticostriatal and thalamostriatal systems in an MPTP-treated monkey model of parkinsonism. Eur J Neurosci. 2008;27:1647–1658. doi: 10.1111/j.1460-9568.2008.06136.x. [DOI] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-X. [DOI] [PubMed] [Google Scholar]
- Henderson JM, Carpenter K, Cartwright H, Halliday GM. Degeneration of the centre median-parafascicular complex in Parkinson’s disease. Ann Neurol. 2000;47:345–352. doi: 10.1002/1531-8249(200003)47:3<345::AID-ANA10>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Smith Y, Raju D, Nanda B, Pare JF, Galvan A, Wichmann T. The thalamostriatal systems: anatomical and functional organization in normal and parkinsonian states. Brain Res Bull. 2009;78:60–68. doi: 10.1016/j.brainresbull.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey CJ, Boyes J, Gerlach O, Chen L, Magill PJ, Bolam JP. GABA(B) receptors at glutamatergic synapses in the rat striatum. Neuroscience. 2005;136:1083–1095. doi: 10.1016/j.neuroscience.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Gomide VC, Silveira GA, Chadi G. Transient and widespread astroglial activation in the brain afetr a striatal 6-OHDA-induced-partial lesion of the nigrostriatal system. Intern J Neurosci. 2005;115:99–117. doi: 10.1080/00207450490512696. [DOI] [PubMed] [Google Scholar]
- Henning J, Strauss U, Wree A, Gimsa J, Rolfs A, Benecke R, Gimsa U. Differential astroglia activation in 6-hydroxydopaminemodels of parkinson’s disease. Neurosci Res. 2008;62:246–253. doi: 10.1016/j.neures.2008.09.001. [DOI] [PubMed] [Google Scholar]
- DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281–285. doi: 10.1016/0166-2236(90)90110-V. [DOI] [PubMed] [Google Scholar]
- Hahn PJ, McIntyre CC. Modeling shifts in the rate and pattern of subthalamopallidal network activity during deep brain stimulation. J Comput Neurosci. 2010;28:425–441. doi: 10.1007/s10827-010-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Arbuthnott GW, Jutras MJ, Goldberg JA, Jaeger D. Resonant antidromic cortical circuit activation as a consequence of high-frequency subthalamic deep-brain stimulation. J Neurophysiol. 2007;98:3525–3537. doi: 10.1152/jn.00808.2007. [DOI] [PubMed] [Google Scholar]
- Maurice N, Thierry AM, Glowinski J, Deniau JM. Spontaneous and evoked activity of substantia nigra pars reticulata neurons during high-frequency stimulation of the subthalamic nucleus. J Neurosci. 2003;23:9929–9936. doi: 10.1523/JNEUROSCI.23-30-09929.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouve L, Salin P, Melon C, Kerkerian-Le Goff L. Deep brain stimulation of the center median-parafascicular complex of the thalamus has efficient anti-parkinsonian action associated with widespread cellular responses in the basal ganglia network in a rat model of Parkinson’s disease. J Neurosci. 2010;30:9919–9928. doi: 10.1523/JNEUROSCI.1404-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkerian-Le Goff L, Bacci JJ, Jouve L, Melon C, Salin P. Impact of surgery targeting the caudal intralaminar thalamic nuclei on the pathophysiological functioning of basal ganglia in a rat model of Parkinson’s disease. Brain Res Bull. 2009;78:80–84. doi: 10.1016/j.brainresbull.2008.08.010. [DOI] [PubMed] [Google Scholar]
- Varoqui H, Schäfer MK, Zhu H, Weihe E, Erickson JD. Identification of the differentiation-associated Na+/PI transporter as a novel vesicular glutamate transporter expressed in a distinct set of glutamatergic synapses. J Neurosci. 2002;22:142–155. doi: 10.1523/JNEUROSCI.22-01-00142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Cui WY, Liu CG. Modulation of apomorphine-induced rotations in unilaterally 6-hydroxydopamine lesioned rats by cholinergic agonists and antagonists. Life Sci. 1997;60:PL 317–PL 323. doi: 10.1016/s0024-3205(97)00168-9. [DOI] [PubMed] [Google Scholar]
- Navailles S, Benazzouz A, Bioulac B, Gross C, De Deurwaerdère P. High-frequency stimulation of the subthalamic nucleus and L-3,4-dihydroxyphenylalanine inhibit in vivo serotonin release in the prefrontal cortex and hippocampus in a rat model of Parkinson’s disease. J Neurosci. 2010;30:2356–2364. doi: 10.1523/JNEUROSCI.5031-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temel Y, Boothman LJ, Blokland A, Magill PJ, Steinbusch HW, Visser-Vandewalle V, Sharp T. Inhibition of 5-HT neuron activity and induction of depressive-like behavior by high-frequency stimulation of the subthalamic nucleus. Proc Natl Acad Sci U S A. 2007;104:17087–17092. doi: 10.1073/pnas.0704144104. [DOI] [PMC free article] [PubMed] [Google Scholar]