

Abstract
Background
Tuberculosis (TB) poses a worldwide threat due to advancing multidrug-resistant strains and deadly co-infections with Human immunodeficiency virus. Today large amounts of Mycobacterium tuberculosis whole genome sequencing data are being assessed broadly and yet there exists no comprehensive online resource that connects M. tuberculosis genome variants with geographic origin, with drug resistance or with clinical outcome.
Description
Here we describe a broadly inclusive unifying Genome-wide Mycobacterium tuberculosis Variation (GMTV) database, (http://mtb.dobzhanskycenter.org) that catalogues genome variations of M. tuberculosis strains collected across Russia. GMTV contains a broad spectrum of data derived from different sources and related to M. tuberculosis molecular biology, epidemiology, TB clinical outcome, year and place of isolation, drug resistance profiles and displays the variants across the genome using a dedicated genome browser. GMTV database, which includes 1084 genomes and over 69,000 SNP or Indel variants, can be queried about M. tuberculosis genome variation and putative associations with drug resistance, geographical origin, and clinical stages and outcomes.
Conclusions
Implementation of GMTV tracks the pattern of changes of M. tuberculosis strains in different geographical areas, facilitates disease gene discoveries associated with drug resistance or different clinical sequelae, and automates comparative genomic analyses among M. tuberculosis strains.
Keywords: Mycobacterium tuberculosis, Genome variations, Mutation, Genetic diversity, Whole genome sequencing, Database
Background
Tuberculosis (TB) remains an ongoing threat to worldwide public health, which in 2011 caused some 8.7 new cases and killed 1.4 million people, including 430,000 co-infected with Human immunodeficiency virus (HIV) [1]. The incidence of multidrug-resistant strains is rising in spite of increasing financial resources being released to stem the epidemic. With globalization, improvement of the health care and epidemic control systems in one country may not guarantee prevention of this airborne disease in others. Russia reported 180,000 TB cases and 20,000 TB deaths in 2011 and shows the highest incidence of new multidrug-resistant strains developed largely due to noncompliant drug regimens [1,2].
Molecular genetic studies of Mycobacterium tuberculosis strains using various genotyping technologies offer an approach to monitor strain dispersal and evolutionary adaptations, important to stem bacterial and disease spread. Genetic markers that track TB transmission include IS6110, polymorphic GC-reach repetitive sequences, direct repeat regions and mycobacterial interspersed repetitive units [3-7]. Recently, it was shown that bacterial whole genome sequencing (WGS) provides greater discriminative power [8-11]. WGS of multiple isolates may address a broad range of topics – from questions on the transmission of clinical strains to how M. tuberculosis evolves over long and short time scales. Rapid analysis of WGS data allows to detect bacterial genetic variants based on single nucleotide polymorphisms (SNPs) and insertion/deletions (Indels), including mutations associated with drug resistance or genetic lineage.
Increasingly large quantities of genome sequence data are becoming available from different types of M. tuberculosis studies [12-16]. Numerous M. tuberculosis WGS studies have been used for phylogenetic analyses and to identify genetic factors involved in TB drug resistance [15,17,18]. Several databases were developed to systematize and compare genomic data. TubercuList (http://tuberculist.epfl.ch) database provides gene-based information of M. tuberculosis H37Rv genome [19]. Tuberculosis Database (TBDB http://www.tbdb.org) is an integrated database providing access to TB genomic sequence data and resources from Mycobacterium species and M. tuberculosis strains, relevant to the discovery and development of TB drugs, vaccines and biomarkers. Currently TBDB contains information about 21 mycobacterial species whole genome sequences, nine of which belong to M. tuberculosis complex [20]. Mycobacterial Genome Divergence Database (MGDD), allows to find genetic differences between two strains or species of M. tuberculosis complex [21]. A web-based comprehensive information system Pathosystems Resource Integration Center (PATRIC, http://patricbrc.org) provides comparative analysis for genomes of different bacterial pathogens, one of which is M. tuberculosis[22]. To date PATRIC contains 201 mycobacterial isolates whole genome sequences, 68 of which are M. tuberculosis genomes.
Although these TB information databases have been established, there is no comprehensive online resource that brings together detailed information on M. tuberculosis genome variations associated with phylogeographic distribution, drug resistance and clinical outcome of TB. Here we describe and release a broadly inclusive unifying database – Genome-wide Mycobacterium tuberculosis Variation (GMTV) – that catalogues genome variations of Russian M. tuberculosis strains combined with available clinical data. GMTV helps to discover genomic variants of M. tuberculosis strains from different geographical areas and lists genetic markers associated with drug resistance and different clinical TB signs. GMTV allows association analysis between molecular variation and clinical consequences as well as facilitating epidemiological surveillance of TB and HIV/TB co-infection. Our hope is that the database will allow to find efficacious strategies to control TB infection and spread.
Construction and content
Database construction
GMTV database contains a broad spectrum of data derived from different sources and relates to M. tuberculosis molecular biology, epidemiology, TB clinical outcome, year and place of isolation, and drug resistance profiles. Access to GMTV database is distributed through web application with Python backend that connected to our MySQL database. Every record in the database is identified by the unique sample ID. Each sample ID corresponds to the set of SNPs and Indels and other information (e.g. medical, geographical and drug resistance data). The database includes information from following databases: NCBI, KEGG metabolic pathways [23,24] and TubercuList [19], the web interface provides access to the corresponding websites through hyperlinks. The GMTV genome browser is an essential tool for genome variations visualization that could be used as an analytical tool to compare nucleotide variations. The core of our MySQL database is manually curated and contain M. tuberculosis genome sequences assessed at Theodosius Dobzhansky Center for Genome Bioinformatics (St. Petersburg), Research Institute of Physical-Chemical Medicine (Moscow) and publicly available data of sequenced M. tuberculosis strains obtained in Russia.
Mycobacterium tuberculosis H37Rv reference genome (NC_000962.3) was used for SNP and Indel calling. For reference assisted assembly sequence reads were aligned on reference genome (H37Rv) using bowtie2 program [25] with standard parameters (bowtie2 -x H37Rv - p30 -U raw_reads.fq -S aligned_reads.sam). For SNP calling and VCF file processing a combination of samtools and vcftools was used [26,27].
A web genome browser, GMTVB, based on JBrowse platform [28,29] is an essential component of GMTV. GMTVB allows one to compare distinct regions or genes among M. tuberculosis strains, including an option to select a particular reference sequence, e.g. H37Rv. GMTVB implements an AJAX paradigm, which increases reaction time. Distinctive regions, genes, genomic features are displayed in tracks. Some tracks are permanent for the reference (e.g. genes, CDS, repeats, etc.). The interface allows one to add, delete and substitute tracks as well as to change reference sequences. GMTV clinical and genetic data combined with graphical tools incorporated in GMTVB makes the database an effective instrument for TB analysis (available at http://mtb.dobzhanskycenter.org).
M. tuberculosis isolates and genome sequence
Whole genome sequences from 1084 M. tuberculosis isolates with various medical datasets from different regions of the Russian Federation comprise the present database. The database contains information on 73 isolates sequenced by our research group and 1011 publicly available genome sequences.
Sequence data for 73 M. tuberculosis strains were provided by Theodosius Dobzhansky Center for Genome Bioinformatics (St. Petersburg State University) and Research Institute of Physical-Chemical Medicine. These M. tuberculosis strains were collected in St. Petersburg (n = 47), Leningrad Oblast (n = 7), Moscow (n = 7), Volgograd (n = 2), Kalmykiya (n = 2), Buryatya (n = 1), Arkhangelsk Oblast (n = 1), Chelyabinsk Oblast (n = 1), Kaliningrad Oblast (n = 1), Novgorod Oblast (n = 1), Nizhniy Novgorod Oblast (n = 1), Ulyanovsk Oblast (n = 1) and Zabaykalsky Krai (n = 1) (Figure 1). Bacterial isolates were provided by Saint-Petersburg Research Institute of Phthisiopulmonology (M. tuberculosis All-Russian Collection) and Moscow Scientific-Practical Center of Treatment of Tuberculosis of Moscow Healthcare. Genomic DNA of 73 M. tuberculosis strains was isolated using standard extraction method [4]. DNA samples were used for library preparation and sequenced using Illumina MiSeq Sequencing Platform and Roche 454 Life Sciences Genome Sequencer FLX following the manufacturer’s instructions. Sequencing data for M. tuberculosis sequenced genomes were deposited in the NCBI Sequence Read Archive [30] under accession numbers PRJNA218508 and PRJNA181180. The accuracy of these data stored at GMTV database is guaranteed by institutions that revealed the disease, performed microbiological tests and genomic data analysis (spoligotyping and WGS).
Figure 1.
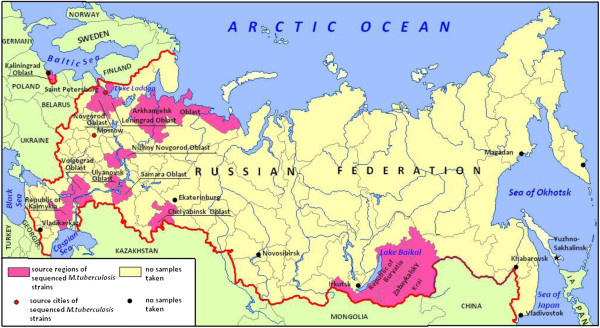
Regions of the Russian Federation, from which M. tuberculosis isolates were obtained. Source regions of M. tuberculosis strains genomes included into the database are highlighted with pink color.
Whole genome sequence reads of other 1011 Russian M. tuberculosis isolates obtained in Samara region (Russia) were downloaded from the European Nucleotide Archive (http://www.ebi.ac.uk/ena/) submitted under accession no. ERP000192 [31,32]. The region of M. tuberculosis strains isolation and available drug susceptibility tests results provided by the authors were deposited to GMTV database.
Information about microbiological drug susceptibility tests is available for the majority of sequenced bacterial strains. Medical data were not available for all samples, however it will be updated as far as possible. To date GMTV database contains information on eleven isolates obtained from HIV-infected patients, 19 from HIV-negative, the rest of isolates were collected from people with unknown HIV status. Classical spoligotyping method was performed for 61 sequenced bacterial isolates [35]. Following spoligotype families were identified according to SpolDB4 database: Beijing (n = 38), H4 (n = 12), LAM9 (n = 9), T1 (n = 2) (Figure 2). Based on SNP analysis described earlier [16] we identified following genetic lineages among 1084 M. tuberculosis genomes: Beijing (n = 685), New/Uganda/X-type/not defined (n = 135), Ural (n = 111), LAM (n = 96), Haarlem (n = 40), S-type (n = 4), Dehli/CAS (n = 3), Not defined (n = 10) (Figure 2). Spoligotype International Type (SIT) number could be used to generate query for SNPs/Indels search. The clinical outcome of the TB was known for 30 isolates: 12 isolates were collected from patients with extrapulmonary TB, 15 from patients with pulmonary TB and 3 from patients with both pulmonary and extrapulmonary localizations. Four hundred eighty-seven isolates had proved multiple drug resistance (MDR) and 60 Extensive Drug Resistance (XDR). Limiting clinical data represented in the database without patient identifiers protects patients’ privacy.
Figure 2.
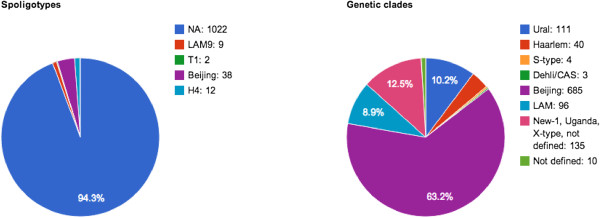
Genotypes of M. tuberculosis isolates included into GMTV database. Spoligotypes were identified using conventional spacer oligonucleotide typing technique and genetic clades were identified using SNP analysis.
Utility and discussion
The current version of GMTV database has a web interface for the retrieval of genomic diversity, geography, drug resistance and clinical information. A derived genome variation table contains several types of information:
1. Sample: Sample ID, HIV status of the patient, patients’ gender, ear of strain isolation, spoligotype family name based on SpolDB4 [33], genetic clades based on SNP analysis [16], and geographical region.
2. Genes: Gene ID on NCBI database, gene name, locus tag, coordinates of gene start and end.
3. Variations (SNPs and Indels): SNP/Indel coordinates, Nucleic acid variations, Amino acid substitutions, Effect of the nucleic acid substitution (synonymous or nonsynonymous), various VCF file statistics, The SNP/Indels sections allows to download VCF file and easily get appropriate annotation of genome variations results. SNP, Indel or SNP/Indel options could be selected.
4. Databases: Information about protein function and protein functional category, according to TubercuList database [19] and Metabolic pathways, according to the KEGG database [23,24].
The input file for the database is a VCF file and a FASTQ file of the assembled genome, which can be downloaded from the website. Presently the GMTV database contains 1084 genomes and over 45,000 SNPs and 23,000 Indel variants across whole genomes with Quality (Q) score 30 threshold (Table 1). The Q score is one of the VCF file statistics associated with the probability of each substitution. The Q30 threshold means that the probability of incorrect base substitution is 1 in 1000. More than half of SNPs in coding regions (64%) are nonsynonymous. Analysis of Indels size, derived from VCF files, showed that one- and nine-nucleotide Indels are the most common, but other Indels are also widely spread (Figure 3).
Table 1.
Genome variants (SNPs and Indels) in GMTV database filtered by Q30
| Genome variation | SNPs | Indels |
|---|---|---|
| Overall quantity |
45655 |
23975 |
| In CDS |
39808 |
18537 |
| Nonsynonymous mutations in CDS |
24124 |
- |
| Synonymous mutations in CDS |
13392 |
- |
| Variations in STOP-codons in CDS |
684 |
- |
| Frameshift mutations in CDS | - | 10993 |
Figure 3.
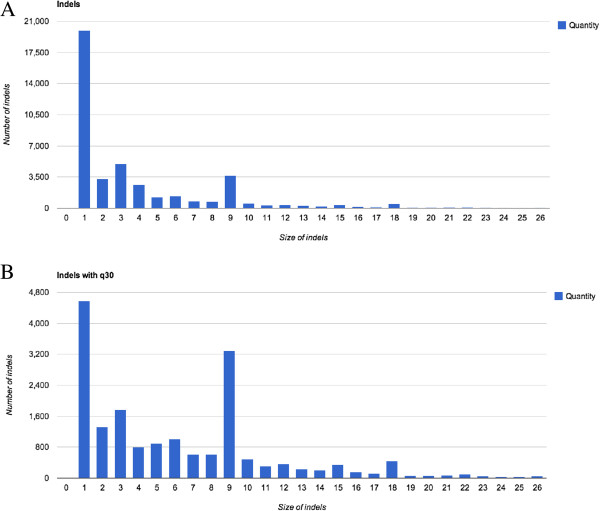
Size distribution of Indels in M. tuberculosis genome. (A) Indels distribution without Quality threshold, (B) Indels distribution with Quality threshold 30. One- and nine-nucleotide size Indels are the most common among M. tuberculosis isolates in GMTV database.
The GMTV web interface contains detailed information about genomic variations revealed from WGS data and provides for convenient analysis of genomic variation to facilitate searches of disease associations. Currently GMTV database allows:
• Review SNPs and Indels of M. tuberculosis isolates filtered by quality score and sequence coverage selected by the user;
• Identify functions and related metabolic pathways of genes where genome variations were identified using the links to KEGG and TubercuList databases;
• Compare SNPs between several isolates selected by drug resistance, clinical outcome, geographical distribution, genetic lineage and other characteristics. It is possible to select all, common or unique genome variations, synonymous and nonsynonymous mutations are highlighted;
• Annotate genome variations using integrated online-tool, download a table with annotated genome variations in CSV (Comma Separated Values) format for further research, and visualize results with the genome browser.
• Download VCF files with nucleotide genome variations, FQ files with reference-assisted assemblies of MTB genomes and FASTA files with sequences of selected genes.
The work with the database starts from “Genome variations” page. To get required information the user can select a sample or some features (e.g. drug resistance, medical or genetic features) and click “Table” on the left bottom of the page. After generating the result, the user selects the type of information to be displayed in the top section of the page (Figure 4). There are five types of information which could be selected by the user: “sample info” provides medical, genetic and geographical information; “gene info” provides information about genes; “SNP/Indel info” provides statistics from VCF files; “Tuberculist database” provides information about genes functions; “Metabolic Pathways Info” provides available information from KEGG database. Information represented in the generated table could be filtered by sample ID, nucleotide position, gene name, geographical region etc. SNPs and Indels are analyzed in separate tables; the type of nucleotide variations could be selected in the top left section. It is possible to compare genome variations of bacterial isolates by downloading tables with mutations found in each group of genomes. The user may compare mutations in selected genes by listing several genes separated with “;” on the “Select genome region” section. This feature allows detecting mutations associated with drug resistance as well as finding compensatory mutations in other genes.
Figure 4.
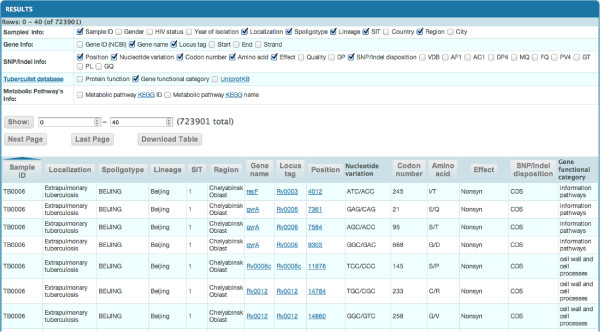
An output of a query (SNP). Query was based on SNPs in M. tuberculosis isolates with streptomycin and isoniazid resistance in coding regions with Quality threshold 30.
The “Download page” allows one to select some characteristics of M. tuberculosis isolates or ID of the interested isolates and to generate a table with information about genotype, geographic region and drug resistance. Each genome could be downloaded as a VCF file representing genome variations, or as an FQ file representing reference-assisted assembly of the genome. It is also possible to download FASTA files with a specific gene or genes, for this purpose the user have to select a special point “Gene” at the “Select genome region” section on the left bar region of the page and list one or several genes separated with ‘;’. This function is useful for comparative studies, for example, it allows analyzing selected protein-coding genes without intergenic regions.
The “Comparison samples” page is developed to compare single nucleotide genome variations. It is possible to browse variations in the whole genome sequence or gene-by-gene in selected M. tuberculosis isolates. For comparative analysis the user selects the sample ID in the left bar (the number is not limited) or to select interested features (medical, geographical, genotype or drug resistance). User may set Q score and coverage for SNPs. Generated table displays nucleotide variations and their position. It is possible to download the whole table or to browse mutations at the genome browser.
The combination of the database with a genome browser makes the GMTV an effective tool for interactive analysis and research. The browser illustrates a scalable map feature of the genome in tracks representing a site’s position, strand, value and supplement notes. Permanent tracks are preloaded into the browser (e.g. original DNA sequence, genes, repeats, etc. as well as defined SNPs, Indels) and listed on the left side of the screen. Tracks may be hidden or shown and there is an option to form tracks ad hoc based on SQL request. Such ad hoc tracks are created temporarily to provide opportunities to analyze the combination of data in visual form. Entire tracks can be downloaded in FASTA format. Scalable views of the genome elements inside the GMTVB let a user look at genome picture both with bird’s eye and as a detailed representation on the DNA level. GMTVB allows one to compare genomic features or to download selected feature for further analysis.
GMTV database is designed to assist in identification of genetic variants associated with drug resistance, clinical outcome or geographic distribution of the pathogen. It allows comparing nucleotide variations based on WGS data in different groups of M. tuberculosis isolates. Bacterial isolates could be divided into categories based on their geographic origin, drug resistance pattern, genetic clade or medical data. GMTV database functions allow using for phylogeographic, epidemiological and evolutionary studies.
GMTV is the first M. tuberculosis database to integrate clinical, epidemiological and microbiological description with genome variations based on whole genome sequencing data, a part of the large epidemiological database established at St. Petersburg Research Institute of Phthisiopulmonology. The development of a M. tuberculosis genome variations database will allow empirical exploring of influences of SNP and Indels around clinical outcomes. GMTV will facilitate the epidemiological surveillance of TB and HIV/TB co-infection and will help to develop effective strategies to control these infections in the population.
Conclusions
GMTV allows association analysis between molecular variation and clinical consequences as well as facilitates epidemiological surveillance of TB and HIV/TB co-infection. Our hope is to inform efficacious strategies for TB control.
Availability and requirements
The web server can be accessed at http://mtb.dobzhanskycenter.org.
Competing interests
The authors declare no competing financial interests.
Authors’ contributions
EC, PD, MR, SS developed the database construction. SS, MR performed web-based interface development, on-line annotation tool creation and JBrowse integration. PD, EC, ES and DI developed the pipeline and performed sequence reads analysis (M. tuberculosis annotations and reference-assisted genome assemblies). IK, EK and EC performed WGS of 73 M. tuberculosis strains. VZ, OM, IM, ON, AV, YI and EN provided M. tuberculosis samples, associated drug resistance, spoligotyping, epidemiological and clinical data on 73 M. tuberculosis isolates included in the GMTV. AL, EC, EI, VG, MS, PY and SJO designed the database. EC, ES, AL, MS and SJO wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Ekaterina N Chernyaeva, Email: echernya@gmail.com.
Marina V Shulgina, Email: mshulgina@spbniif.ru.
Mikhail S Rotkevich, Email: rotke@yandex.ru.
Pavel V Dobrynin, Email: pdobrynin@gmail.com.
Serguei A Simonov, Email: sergueisimonov@gmail.com.
Egor A Shitikov, Email: egorshtkv@gmail.com.
Dmitry S Ischenko, Email: ischenko.dmitry@gmail.com.
Irina Y Karpova, Email: irysya87@mail.ru.
Elena S Kostryukova, Email: el-es@yandex.ru.
Elena N Ilina, Email: ilinaen@gmail.com.
Vadim M Govorun, Email: govorun_vadim@mail.ru.
Vyacheslav Y Zhuravlev, Email: zhuravlev.viacheslav@gmail.com.
Olga A Manicheva, Email: olgamanicheva@rambler.ru.
Peter K Yablonsky, Email: spbniif_all@mail.ru.
Yulia D Isaeva, Email: isaeva1980@mail.ru.
Elena Y Nosova, Email: Nosova_EYu@mail.ru.
Igor V Mokrousov, Email: imokrousov@mail.ru.
Anna A Vyazovaya, Email: elmtree2001@mail.ru.
Olga V Narvskaya, Email: onarvskaya@gmail.com.
Alla L Lapidus, Email: piterlabs@gmail.com.
Stephen J O’Brien, Email: lgdchief@gmail.com.
Acknowledgements
Research was supported in part by the Russian Ministry of Education and Science, Mega-grant no. 11.G34.31.0068 and grant no. 16.522.11.2003.
References
- World Health Organization. Global Tuberculosis Report 2012. France: WHO; 2012. Available: http://who.int/tb/publications/global_report/gtbr12_main.pdf. Accessed 15 April 2014. [Google Scholar]
- Phillips L. Infectious disease: TB’s revenge. Nature. 2013;493(7430):14–16. doi: 10.1038/493014a. doi:10.1038/493014a. [DOI] [PubMed] [Google Scholar]
- Ross BC, Raios K, Jackson K, Dwyer B. Molecular cloning of a highly repeated DNA element from Mycobacterium tuberculosis and its use as an epidemiological tool. J Clin Microbiol. 1992;30(4):942–946. doi: 10.1128/jcm.30.4.942-946.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Embden JD, Cave MD, Crawford JT, Dale JW, Eisenach KD, Gicquel B, Hermans P, Martin C, McAdam R, Shinnick TM, Small PM. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: recommendations for a standardized methodology. J Clin Microbiol. 1993;31(2):406–409. doi: 10.1128/jcm.31.2.406-409.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Bunschoten A, Molhuizen H, Shaw R, Goyal M, van Embden J. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35(4):907–914. doi: 10.1128/jcm.35.4.907-914.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frothingham R, Meeker-O’Connell WA. Genetic diversity in the Mycobacterium tuberculosis complex based on variable numbers of tandem DNA repeats. Microbiology. 1998;144(Pt 5):1189–1196. doi: 10.1099/00221287-144-5-1189. [DOI] [PubMed] [Google Scholar]
- Mazars E, Lesjean S, Banuls AL, Gilbert M, Vincent V, Gicquel B, Tibayrenc M, Locht C, Supply P. High-resolution minisatellite-based typing as a portable approach to global analysis of Mycobacterium tuberculosis molecular epidemiology. Proc Natl Acad Sci U S A. 2001;98(4):1901–1906. doi: 10.1073/pnas.98.4.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardy JL, Johnston JC, Ho Sui SJ, Cook VJ, Shah L, Brodkin E, Rempel S, Moore R, Zhao Y, Holt R, Varhol R, Birol I, Lem M, Sharma MK, Elwood K, Jones SJ, Brinkman FS, Brunham RC, Tang P. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. 2011;364(8):730–739. doi: 10.1056/NEJMoa1003176. [DOI] [PubMed] [Google Scholar]
- Walker TM, Monk P, Grace Smith E, Peto TE. Contact investigations for outbreaks of Mycobacterium tuberculosis: advances through whole genome sequencing. Clin Microbiol Infect. 2013. doi:10.1111/1469-0691.12183. Epub ahead of print. [DOI] [PubMed]
- Roetzer A, Diel R, Kohl TA, Rückert C, Nübel U, Blom J, Wirth T, Jaenicke S, Schuback S, Rüsch-Gerdes S, Supply P, Kalinowski J, Niemann S. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. PLoS Med. 2013;10(2):e1001387. doi: 10.1371/journal.pmed.1001387. doi:10.1371/journal.pmed.1001387. Epub 2013 Feb 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Roychowdhury T, Kumar P, Kumar A, Kalra P, Singh J, Singh S, Prasad HK, Bhattacharya A. Genetic heterogeneity revealed by sequence analysis of Mycobacterium tuberculosis isolates from extra-pulmonary tuberculosis patients. BMC Genomics. 2013;14:404. doi: 10.1186/1471-2164-14-404. doi:10.1186/1471-2164-14-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreevatsan S, Pan X, Stockbauer KE, Connell ND, Kreiswirth BN, Whittam TS, Musser JM. Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc Natl Acad Sci U S A. 1997;94(18):9869–9874. doi: 10.1073/pnas.94.18.9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filliol I, Motiwala AS, Cavatore M, Qi W, Hazbón MH, Bobadilla del Valle M, Fyfe J, García-García L, Rastogi N, Sola C, Zozio T, Guerrero MI, León CI, Crabtree J, Angiuoli S, Eisenach KD, Durmaz R, Joloba ML, Rendón A, Sifuentes-Osornio J, Ponce de León A, Cave MD, Fleischmann R, Whittam TS, Alland D. Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: insights into tuberculosis evolution, phylogenetic accuracy of other DNA fingerprinting systems, and recommendations for a minimal standard SNP set. J Bacteriol. 2006;188(2):759–772. doi: 10.1128/JB.188.2.759-772.2006. [Erratum in: J Bacteriol 2006, 188(8):3162–3163] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux S, DeRiemer K, Van T, Kato-Maeda M, De Jong BC, Narayanan S, Nicol M, Niemann S, Kremer K, Gutierrez MC, Hilty M, Hopewell PC, Small PM. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2006;103(8):2869–2873. doi: 10.1073/pnas.0511240103. Epub 2006 Feb 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, Ernst JD, Gagneux S. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet. 2010;42(6):498–503. doi: 10.1038/ng.590. doi:10.1038/ng.590. Epub 2010 May 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homolka S, Projahn M, Feuerriegel S, Ubben T, Diel R, Nübel U, Niemann S. High resolution discrimination of clinical Mycobacterium tuberculosis complex strains based on single nucleotide polymorphisms. PLoS One. 2012;7(7):e39855. doi: 10.1371/journal.pone.0039855. doi:10.1371/journal.pone.0039855. Epub 2012 Jul 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioerger TR, Koo S, No EG, Chen X, Larsen MH, Jacobs WR Jr, Pillay M, Sturm AW, Sacchettini JC. Genome analysis of multi- and extensively-drug-resistant tuberculosis from KwaZulu-Natal, South Africa. PLoS One. 2009;4(11):e7778. doi: 10.1371/journal.pone.0007778. doi:10.1371/journal.pone.0007778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilina EN, Shitikov EA, Ikryannikova LN, Alekseev DG, Kamashev DE, Malakhova MV, Parfenova TV, Afanas’ev MV, Ischenko DS, Bazaleev NA, Smirnova TG, Larionova EE, Chernousova LN, Beletsky AV, Mardanov AV, Ravin NV, Skryabin KG, Govorun VM. Comparative genomic analysis of Mycobacterium tuberculosis drug resistant strains from Russia. PLoS One. 2013;8(2):e56577. doi: 10.1371/journal.pone.0056577. doi:10.1371/journal.pone.0056577. Epub 2013 Feb 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew JM, Kapopoulou A, Jones LM, Cole ST. TubercuList–10 years after. Tuberculosis (Edinb) 2011;91(1):1–7. doi: 10.1016/j.tube.2010.09.008. doi:10.1016/j.tube.2010.09.008. Epub 2010 Oct 25. PubMed PMID: 20980199. [DOI] [PubMed] [Google Scholar]
- Reddy TB, Riley R, Wymore F, Montgomery P, DeCaprio D, Engels R, Gellesch M, Hubble J, Jen D, Jin H, Koehrsen M, Larson L, Mao M, Nitzberg M, Sisk P, Stolte C, Weiner B, White J, Zachariah ZK, Sherlock G, Galagan JE, Ball CA, Schoolnik GK. TB database: an integrated platform for tuberculosis research. Nucleic Acids Res. 2009;37(Database issue):D499–D508. doi: 10.1093/nar/gkn652. doi:10.1093/nar/gkn652. Epub 2008 Oct3. PubMed PMID: 18835847; PubMed Central PMCID: PMC2686437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnoi A, Srivastava A, Roy R, Bhattacharya A. MGDD: Mycobacterium tuberculosis genome divergence database. BMC Genomics. 2008;9:373. doi: 10.1186/1471-2164-9-373. doi:10.1186/1471-2164-9-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie JJ, Wattam AR, Cammer SA, Gabbard JL, Shukla MP, Dalay O, Driscoll T, Hix D, Mane SP, Mao C, Nordberg EK, Scott M, Schulman JR, Snyder EE, Sullivan DE, Wang C, Warren A, Williams KP, Xue T, Yoo HS, Zhang C, Zhang Y, Will R, Kenyon RW, Sobral BW. PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect Immun. 2011;79(11):4286–4298. doi: 10.1128/IAI.00207-11. doi:10.1128/IAI.00207-11. Epub 2011 Sep 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40(Database issue):D109–D114. doi: 10.1093/nar/gkr988. doi:10.1093/nar/gkr988. Epub 2011 Nov 10. PubMed PMID: 22080510; PubMed Central PMCID: PMC3245020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. PubMed PMID: 10592173; PubMed Central PMCID: PMC102409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. doi:10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 1000 Genome Project Data Processing Subgroup. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers C, Banks E, DePristo M, Handsaker R, Lunter G, Marth G, Sherry S, McVean G, Durbin R. 1000 Genomes Project Analysis Group. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–2158. doi: 10.1093/bioinformatics/btr330. doi:10.1093/bioinformatics/btr330. Epub 2011 Jun 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner ME, Uzilov AV, Stein LD, Mungall CJ, Holmes IH. JBrowse: a next-generation genome browser. Genome Res. 2009;19(9):1630–1638. doi: 10.1101/gr.094607.109. doi:10.1101/gr.094607.109. Epub 2009 Jul 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westesson O, Skinner M, Holmes I. Visualizing next-generation sequencing data with JBrowse. Brief Bioinform. 2013;14(2):172–177. doi: 10.1093/bib/bbr078. doi:10.1093/bib/bbr078. Epub 2012 Mar 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI Sequence Read Archive. [ http://www.ncbi.nlm.nih.gov/Traces/sra/]
- Casali N, Nikolayevskyy V, Balabanova Y, Ignatyeva O, Kontsevaya I, Harris SR, Bentley SD, Parkhill J, Nejentsev S, Hoffner SE, Horstmann RD, Brown T, Drobniewski F. Microevolution of extensively drug-resistant tuberculosis in Russia. Genome Res. 2012;22(4):735–745. doi: 10.1101/gr.128678.111. doi:10.1101/gr.128678.111. Epub 2012 Jan 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casali N, Nikolayevskyy V, Balabanova Y, Harris SR, Ignatyeva O, Kontsevaya I, Corander J, Bryant J, Parkhill J, Nejentsev S, Horstmann RD, Brown T, Drobniewski F. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat Genet. 2014;46(3):279–286. doi: 10.1038/ng.2878. doi:10.1038/ng.2878. Epub 2014 Jan. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudey K, Driscoll JR, Rigouts L, Prodinger WM, Gori A, Al-Hajoj SA, Allix C, Aristimuño L, Arora J, Baumanis V, Binder L, Cafrune P, Cataldi A, Cheong S, Diel R, Ellermeier C, Evans JT, Fauville-Dufaux M, Ferdinand S, Garcia de Viedma D, Garzelli C, Gazzola L, Gomes HM, Guttierez MC, Hawkey PM, van Helden PD, Kadival GV, Kreiswirth BN, Kremer K, Kubin M. et al. Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 2006;6:23. doi: 10.1186/1471-2180-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]