

Abstract
BET bromodomain inhibition has contributed new insights into gene regulation and emerged as a promising therapeutic strategy in cancer. Structural analogy of early methyl-triazolo BET inhibitors has prompted a need for structurally dissimilar ligands as probes of bromodomain function. Using fluorous-tagged multicomponent reactions, we developed a focused chemical library of bromodomain inhibitors around a 3,5-dimethylisoxazole biasing element with micromolar biochemical IC50. Iterative synthesis and biochemical assessment allowed optimization of novel BET bromodomain inhibitors based on an imidazo[1,2-a]pyrazine scaffold. Lead compound 32 (UMB-32) binds BRD4 with a Kd of 550 nM and 724 nM cellular potency in BRD4-dependent lines. Additionally, compound 32 shows potency against TAF1, a bromodomain-containing transcription factor previously unapproached by discovery chemistry. Compound 32 was cocrystallized with BRD4, yielding a 1.56 Å resolution crystal structure. This research showcases new applications of fluorous and multicomponent chemical synthesis for the development of novel epigenetic inhibitors.
Introduction
Cellular identity is imprinted on the genome and epigenome via covalent chemical modification. Post-translational modification of the DNA and histone proteins around which the genome is structured is dynamically maintained by chromatin-modifying enzymes, which “write” or “erase” an expansive and diverse set of marks.1−4 Some of these marks are retained through cell division, contributing to epigenetic mitotic memory. Others mediate open (actively transcribed) or closed (actively repressed) conformations of chromatin, enforcing gene expression programs that govern specialized cellular functions. Interpretation of epigenomic marks is accomplished by families of “reader” proteins which are recruited to sites of context-specific modification where they nucleate higher-ordered assemblies that influence gene expression and chromatin condensation.2
As end-effectors of signaling pathways, regulators of transcription, and emerging unrecognized cancer dependencies, epigenetic reader proteins are attractive therapeutic targets in cancer.5 However, the function of these agents via protein–protein interaction poses a challenge to the discipline of ligand discovery.6 Indeed, few high-quality chemical probes exist for transcriptional proteins, perhaps owing to perceptions regarding the difficulty of disrupting protein–protein interactions.7 Recent work from our laboratory established the feasibility of targeting the acetyl-lysine (Kac) recognition module, or bromodomain.8 A bromodomain is a conserved structural module comprised of four antiparallel α-helices which creates a Kac-recognition pocket between two interhelical loops.9−13 Molecular recognition of Kac is often facilitated by a conserved asparagine residue and a network of structured water molecules.
In 2010, we reported a first, methyl-triazolo bromodomain inhibitor, compound 1 (JQ1), which selectively displaces the BET (bromodomain and extra-terminal domain) subfamily of human bromodomains from chromatin.8 The basic research guiding our understanding of BET bromodomains has been empowered by the availability of this chemical tool. We now appreciate that the BET family (BRD2, BRD3, BRD4, and BRDT) functions as transcriptional coactivator proteins, which relay signals from master regulatory transcription factors, such as MYC in cancer and NFkB in inflammation, to RNA polymerase II (RNA Pol II).14−16 BET proteins function to promote release of RNA Pol II from promoters, leading to effective transcriptional elongation.17−19 Beyond the central role in chromatin-dependent transcriptional signaling, BET bromodomains are also important mediators of cell cycle progression13 and facilitate developmental transitions such as spermiogenesis.20
Deregulation of BET bromodomain function is observed in numerous malignancies, and two BET bromodomains (BRD3 and BRD4) are proto-oncogenes in aggressive epithelial cancers.13,21 BET-rearranged lung and head and neck cancer demonstrates in an in-frame fusion to the NUT gene, resulting in aberrant expression of a chimeric oncoprotein that retains the tandem N-terminal bromodomains.21 Exposure of patient-derived cell lines from BET-rearranged lung cancers to compound 1 results in immediate squamous differentiation and subsequent cell death, establishing a compelling rationale for the development of BET-targeted therapy in this disease.8 Beyond these solid tumors, compound 1 has demonstrated potent antiproliferative efficacy in models of multiple myeloma, acute lymphoid leukemia, and acute myeloid leukemia.8,14,22,23 Informed by this research, first-generation methyl-triazolo BET inhibitors analogous to compound 1 have already been translated to human clinical investigation by at least four pharmaceutical companies.24
Beyond BETs, there are 38 additional bromodomain-containing proteins for which high-quality small-molecule probes are urgently needed. Transcription initiation factor TFIID subunits 1 (TAF1) and 1L (TAF1L) are two such proteins. As components of the STAGA complex, which contains TRRAP, GCN5, TFIID, CBP/P300, mediator,25 and Sp1,26 TAF1 is susceptible to oncogenic activation by MYC. Moreover, TAF1 has been shown to block p53 activity,27 and inactivation of TAF1 triggers a DNA damage response.28 In addition, the TFIID complex, of which TAF1 is a significant member, is vital to stem cell reprogramming.29 Inhibitors of TAF1 may help further elucidate its biological role and potentially function to inhibit cancer cell growth and survival.
Toward the development of a next-generation of bromodomain inhibitors, we have endeavored to build focused libraries of novel small molecules possessing one of several biasing elements with structural or electronic analogy to the methyl-triazolo warhead of compound 1. Iterative synthesis and biochemical testing is employed to efficiently compare chemical cores and to explore appending groups. Complex, nonscalable, and wasteful reactions can significantly impede iterative screening efforts. Techniques involving the use of fluourous reagents have shown great versatility, high-yield, rapid deployment, and are relatively eco-friendly. Complex molecules may be synthesized in multicomponent reactions (MCRs)30 with perfluoroalkyl “phase tags” which can be used to facilitate purification by fluorous solid-phase extraction (F-SPE).31 Subsequent Suzuki-type reactions may replace the fluorous tag to form a biaryl compound.32 Benefits of such reactions include high yielding reactivity with facile purification. Reactions have proven viable to create substituted proline analogues,33 imidazo[1,2-a]-pyridines,34 diazepines,35 and others.36,37 Such heterocyclic compounds form easily substitutable, drug-like scaffolds for the discovery of probe compounds.
Structurally dissimilar chemical tools are valuable reagents for biological research when utilized in parallel, allowing observed biological phenotypes to be unambiguously linked to common target function.7 Today, coordinated efforts in ligand discovery are working to develop additional BET bromodomain inhibitors within our group, and beyond.38−40 Drawing upon robust fluorous chemical synthesis and previously developed screening capabilities, we have rapidly diversified and optimized a set of potent and highly specific inhibitors. Building from the dimethylisoxazole biasing element, we sought to determine preferred regiochemistry, optimal scaffold (accessible via MCR), and favorable appending groups. We report here a novel class of bromodomain inhibiting compounds using the recently identified 3,5-dimethylisoxazole biasing element.38,39,41,42 In addition to demonstrating a new approach to targeting BET bromodomains, these compounds and near derivatives could create opportunities to drug other bromodomain-containing proteins.
Results and Discussion
Analysis of existing small-molecule bromodomain inhibitors, shown in Figure 1A, has defined a structure–function model in which the competitive small molecule binds through two chemical features: an acetyl-lysine mimetic warhead (red) and a core region with appending surface-recognition features (black). Nonobvious Kac mimetics in existing BET inhibitors, such as the triazole ring in 1(8) and 2 (I-BET),40 the 3,5-dimethylisoxazole ring in 3 (iBET-151),43 and the quinazolinone in 4 (PFI-1),44 form a highly specific hydrogen bond to the conserved asparagine in bromodomains. The core of the molecule establishes shape complementarity with the contour of the binding pocket to increase binding affinity primarily through hydrophobic interactions. Appending groups provide an opportunity to enhance potency and selectivity via surface and loop region interactions. The 3,5-dimethylisoxazole headgroup of 3 was identified as a promising and chemically accessible biasing moiety for further development as a chemical probe. Our screening strategy created an extensive series of diverse chemical features attached to the 4-position of the 3,5-dimethylisoxazole (Figure 1B). As a starting point, we focused on small, ligand efficient compounds that could serve as leads for further derivatization.
Figure 1.

Chemical strategies to inhibit BET bromodomain proteins. (A) Existing BET bromodomain inhibitors with biasing moiety in red. (B) Synthetic strategy for the creation of small fragments based on dimethyl isoxazole based BET bromodomain biasing moiety (red). X signifies varied heteroatoms and R signifies varied substituents. (C) Perfluoroalkyl synthetic strategy with reaction conditions for small fragments shown. (D) Small fragment dimethylisoxazole inhibitors with potency values indicated. (E) Representative inhibitory curves for small fragment inhibitors. Error is shown based on duplicate technical replicates. (F) Docking of compound 9 into BRD4 crystal structure (PDB: 3MXF). The conserved asparagine interaction is indicated. (G) Ligand interaction diagram of compound 9.
This approach to library development was empowered by recent advances in use of perfluoroalkyl substituents to enable efficient Suzuki-type coupling reactions, shown in Figure 1C. Purification of reaction intermediates is greatly facilitated by F-SPE. In this case, aryl perfluorooctylsulfonates (ArOSO2(CF2)7CF3) were reacted with 3,5-dimethylisoxazole-4-boronic acid pinacol esters to produce a low-molecular-weight chemical series. The resulting compounds were purified using F-SPE. Initial compounds included positional isomers of carbonyl functionalities around the phenyl ring system (Figure 1D). Compound potency was initially assessed by relative average IC50 determined by an AlphaScreen competition assay (representative assay shown in Figure 1E). IC50 values were found to be highly dependent on the steric arrangement of functional groups around the phenyl ring. ortho- and meta-Aldehyde substitutions were poorly tolerated, while a para-aldehyde (compound 8) resulted in potency of 1.70 μM with a ligand efficiency (as defined by Hopkins et al.)45 of 0.54. Substitution with the more chemically stable para-acetyl group (compound 9) did not affect potency. Previous study of similar compounds had only explored variation at meta ring positions.38
We performed computational modeling to establish hypotheses regarding the mode of molecular recognition and to guide further medicinal chemistry (Figure 1F). Using previously published crystal structures of both compounds 1 and 3, we were able to dock fragments from our emerging chemical library into the Kac binding site of BRD4 domain 1 (BRD4(1)).8,43 As is the case with 3, compound 9 is thought to bind through a conserved hydrogen bond with asparagine-140 through the ring oxygen, while the ring nitrogen coordinates through a structured water that interacts with the hydroxyl group of tyrosine-97 (Figure 1G). On the basis of this interaction model, we proceeded to build off the 4-position of the phenyl ring to improve potency by establishing protein surface interactions near the BC loop region.
We sought to explore a variety of chemical scaffolds while maintaining the dimethyl isoxazole biasing moiety. A diverse set of molecules was synthesized using reactions compatible with fluorous-tagged multicomponent synthesis (Table 1). These reactions create structural diversity by changing the substituent groups on each fractional component, allowing generation of diverse small-molecule libraries around a biasing element. The perfluoroalkyl tags can substituted with a binding motif of choice via Suzuki coupling (see Figure 1C).32 Synthesized compounds included tertiary amines, pyrimidines, and fused heterocyclic ring systems in order to explore the optimal shape for exploiting protein–inhibitor contour interactions. In addition to biochemical IC50, these compounds were selectively evaluated in a BRD4-dependent cell viability study. Effects on cell proliferation (EC50) were assessed using a surrogate measure of cellular viability (ATP content; PerkinElmer ATPlite) in the BET-rearranged 797 cell line and reported as an average value of multiple experiments. The use of a fluorous-tagged Groebke–Blackburn–Bienayme34,46 multicomponent reaction was used to develop the para-imidazo[1,2-a]pyridine scaffold (compound 11), which was found to be a promising lead with biochemical and cellular inhibitory values of 11.0 and 14.1 μM, respectively. In addition, the compound 11 scaffold is accessible at a variety of positions for diversification to drive potency and develop a further understanding of structure–activity relationships (SAR).
Table 1. Exploration of Compound Scaffold Region.
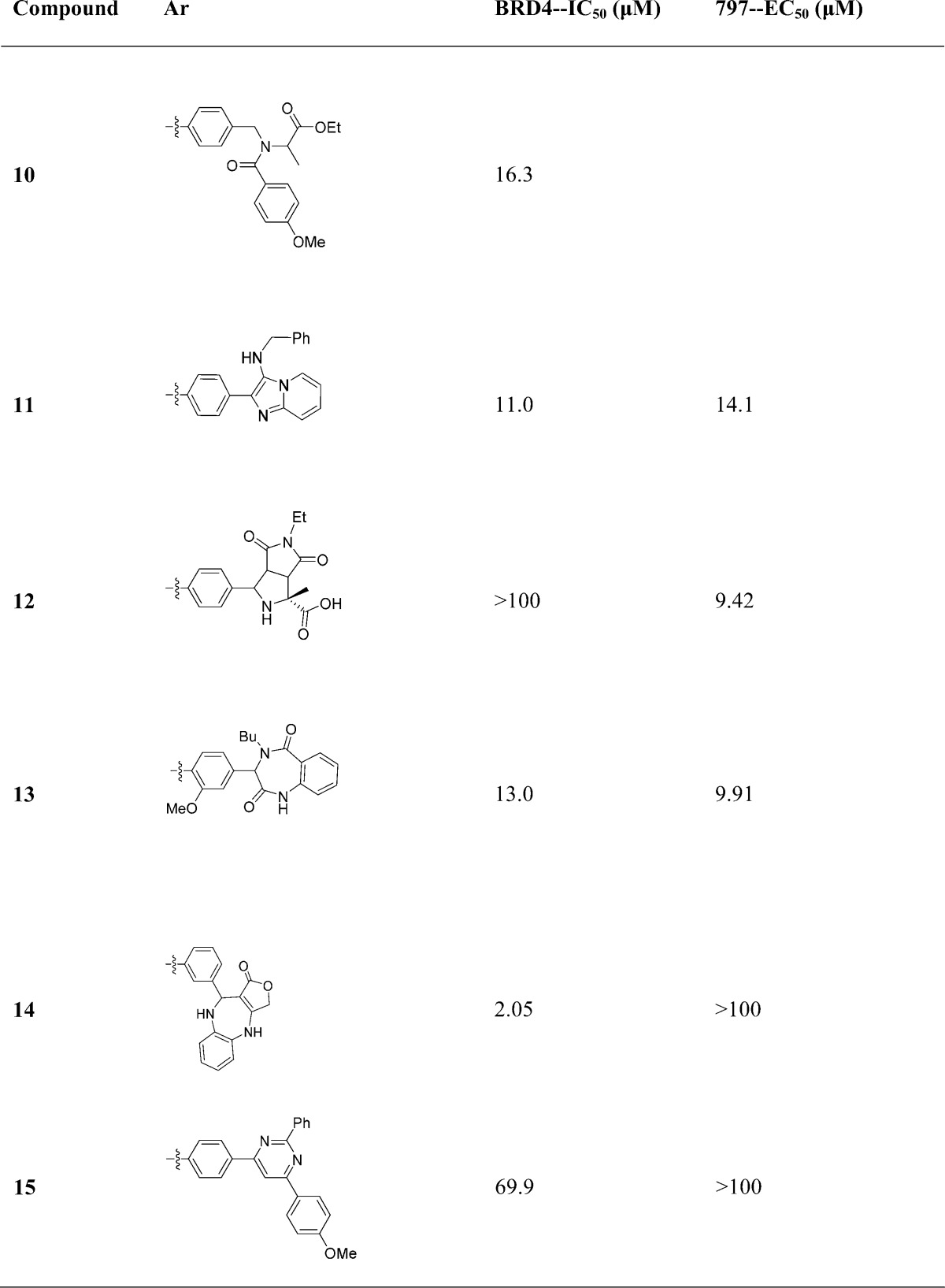
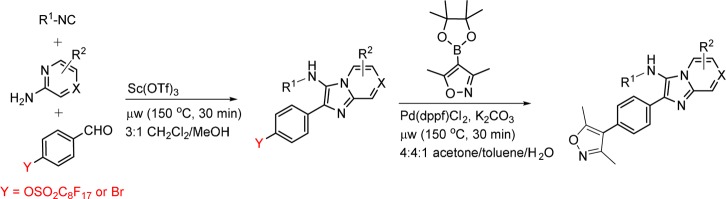
Having selected a core scaffold in compound 11, we assembled a focused library of compounds to optimize placement of heteroatoms and appending groups. Synthesis of the eventual lead compound 32 (UMB-32) is shown in Scheme 1 as an exemplar of the route used to assemble the focused SAR series. Bromobenzaldehyde reagents were substituted for fluorosulfonylbenzaldehydes in the subsequent MCRs. The compound was synthesized in a two-step reaction. The first step included combination of t-butylisocyanide, 2-aminopyrazine, and 4-bromobenzaldehyde. The reaction is catalyzed by scandium triflate and microwaved at 150 °C for 30 min in 3:1 CH2Cl2/MeOH. Flash chromatography yielded the pure product 80%. This intermediate was then coupled to 3,5-dimethylisoxazole-4-boronic acid pinacol ester in a Suzuki coupling. The resulting crude mixture was purified by flash chromatography with yield 57% and purity of >95% as determined by liquid chromatography.
Scheme 1. Synthesis of Compound 32 and Close Analogues.
Analogues were synthesized similarly to explore the functional consequence of steric and electronic modification to the fused bicyclic scaffold, shown in Table 2. Variations of R1 from the compound 11 benzyl group to bulky, nonaromatic groups like cyclohexyl (compound 20) and t-Bu (compound 29) improved potency. An acid at the R1 position (compound 26) completely inactivated the ligand. Appending R2 groups at the 6-, 7-, and 8-positions of the fused ring system universally decreased potency compared to parent compounds. Ring-nitrogens at the 7- and 8-positions (compound 32 and 31, respectively) had little effect on biochemical potency. A 6-position ring-nitrogen reduced compound potency from 479 nM (compound 29) to 20 μM (compound 30). Cellular potency, however, modestly improved between compound 29 and 32 upon substitution of a 7-N. For this reason, compound 32 emerged as a lead compound.
Table 2. Elaboration of Imidazopyridine Scaffold.
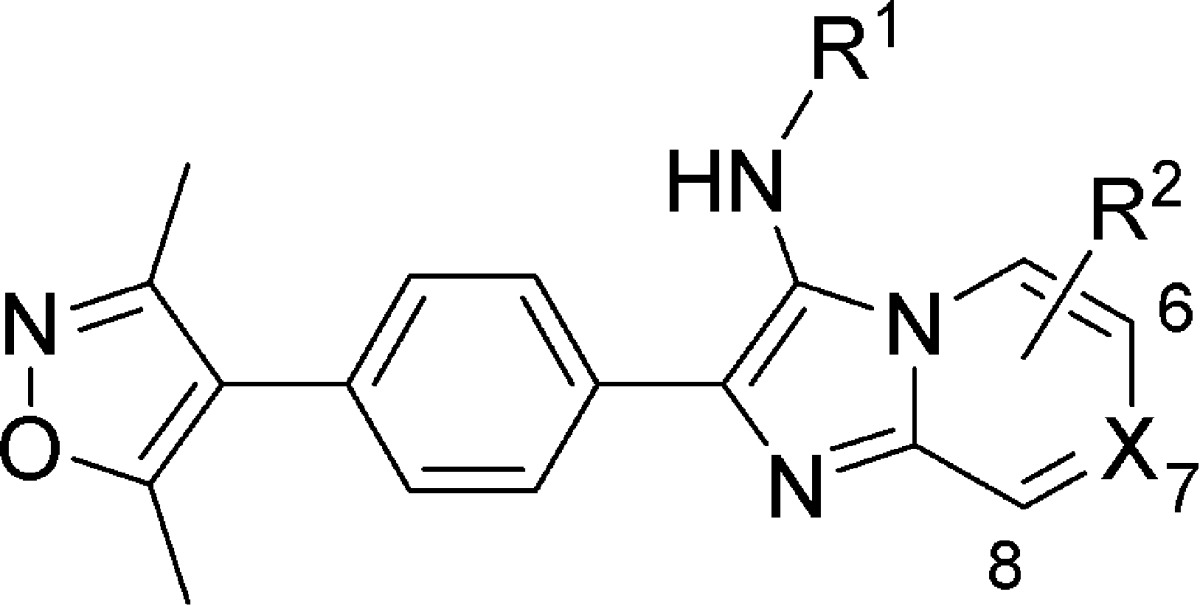
| compd | R1 | R2 | X | BRD4 IC50 (μM) | 797 EC50 (μM) |
|---|---|---|---|---|---|
| 16 | H | H | CH | 3.89 | |
| 17 | Bn | 6-Me | CH | >100 | |
| 18 | Bn | 6-Cl | CH | 11.8 | 18.4 |
| 19 | Bn | H | 7-N | 18.8 | 49.1 |
| 20 | cyclohexyl | H | CH | 0.904 | 5.07 |
| 21 | 4-(OMe)Ph | H | CH | 2.76 | 7.36 |
| 22 | 4-(OMe)Ph | 6-Cl | CH | 31.5 | 20.7 |
| 23 | 4-(OMe)Ph | 7-OMe | 6-N | 2.89 | 11.8 |
| 24 | 4-(OMe)Ph | H | 7-N | 7.86 | 32.8 |
| 25 | (S)-1-PhEt | H | 7-N | 2.86 | |
| 26 | –CH2CO2H | H | 7-N | >100 | |
| 27 | –CH2CO2Et | H | 7-N | 4.53 | |
| 28 | –CH2CO2tBu | H | 7-N | 4.96 | |
| 29 | t-Bu | H | CH | 0.479 | 2.04 |
| 30 | t-Bu | H | 6-N | 20.7 | 2.06 |
| 31 | t-Bu | H | 8-N | 0.860 | |
| 32 | t-Bu | H | 7-N | 0.637 | 0.724 |
| 33 | t-Bu | 6-Cl | CH | 1.17 | 2.19 |
| 34 | t-Bu | 6-Me | CH | 3.17 | |
| 35 | t-Bu | 8-CF3 | CH | 11.9 | |
| 36 | t-Bu | 6-CO2CH2 | CH | 1.62 | |
| 37 | t-Bu | 6-COOH | CH | 0.968 | |
| 38 | i-Pr | H | 7-N | 0.807 | 0.494 |
| 39 | n-Bu | H | 7-N | 1.66 |
A limited structural study around the linking phenyl ring is shown in Table 3. Substitution of the phenyl ring for pyridine resulted in decreased potency (compound 40 and 41), and the addition of electron-withdrawing fluorine (compound 42) was detrimental as well. Electron-donating methoxy substituents appear to be mildly beneficial in biochemical assays. Additional work regarding this linking region of the molecule may be a focus of ongoing medicinal chemistry.
Table 3. Variations to UMB-32 Linker.
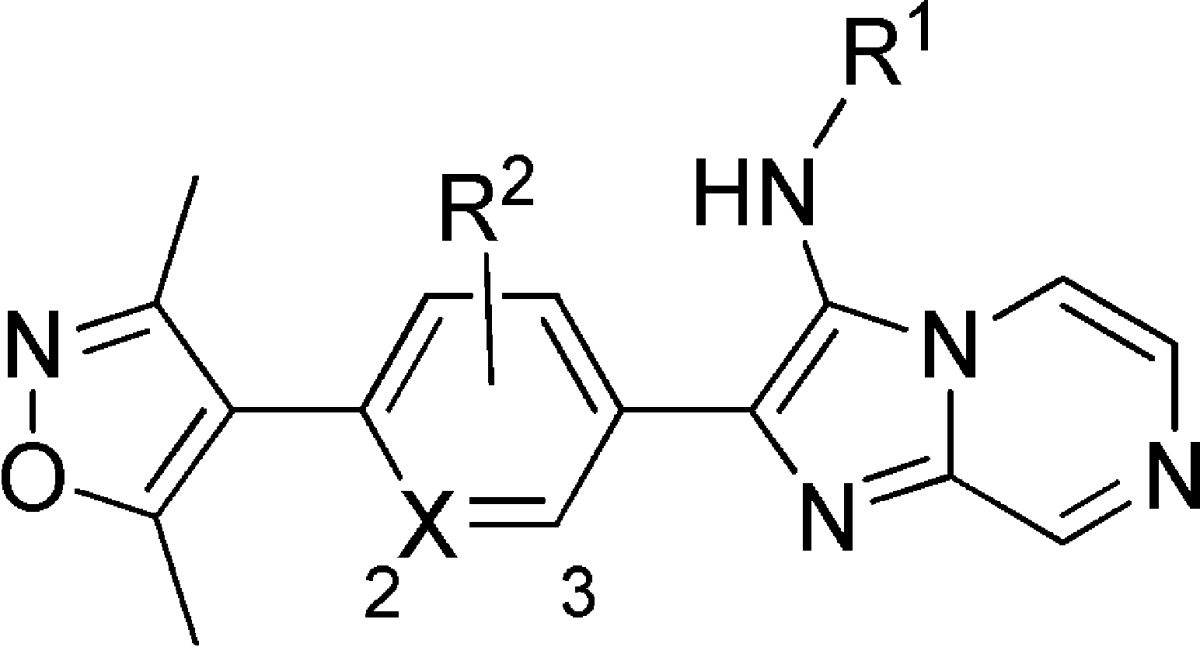
| compd | R1 | R2 | X | BRD4 IC50 (μM) | 797 EC50 (μM) |
|---|---|---|---|---|---|
| 40 | t-Bu | H | 2-N | 51.9 | >100 |
| 41 | t-Bu | H | 3-N | 12.9 | 8.29 |
| 42 | t-Bu | 2-F | CH | 13.0 | 12.8 |
| 43 | t-Bu | 3-OMe | CH | 0.562 | |
| 44 | i-Pr | 3-OMe | CH | 0.474 |
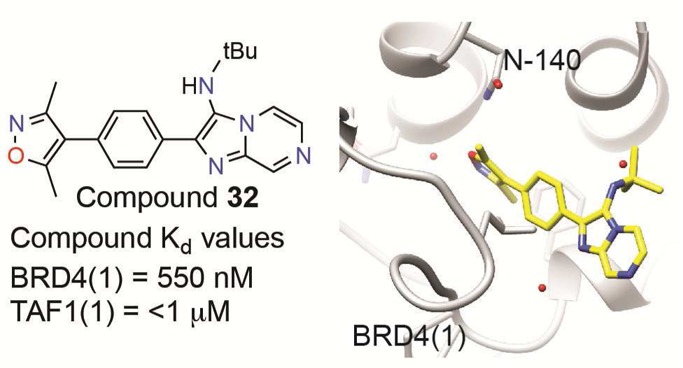
The potency of 32 was accomplished by two key changes from compound 11 (Figure 2A). Compound 32 has substituted t-Bu for benzyl and inserted a 7-N. Comparison of compound 11, 19, and 32 show the critical substitutions in the development of 32, which led to an improved biochemical IC50 (Figure 2B, top). Biochemical potency was supported by cellular screening of compounds again in the BET-rearranged 797 cell line (Figure 2B, bottom). The addition of 7-N and the benzyl to t-butyl substitution clearly establish 32 as a front-runner with dramatically improved biological activity. While there is often a drop in potency when compounds shift from biochemical to cellular assays, we observed similar potency for compound 32 in both experiments. This likely reflects high target availability of free drug, intrinsic cell permeability of this chemical series, and high sensitivity of the genetically leveraged BRD4-NUT cells (797). Notably, we previously observed parity between biochemical and cellular potency during the initial assessment of compound 1,8 which were confirmed in the present study (IC50 of 81.5 nM and EC50 of 71.7 nM, data not shown). Characterization of compound 32 binding kinetics to BRD4(1) by isothermal titration calorimetry (ITC) further supported the improved biochemical affinity (Supporting Information, Figure S1). The resultant Kd was shown to be 550 nM, in good agreement with the biochemical measurements. The binding process is shown to be exothermic and positively entropic with a 1:1 binding ratio.
Figure 2.

Compound 32 is a potent, selective inhibitor of BRD4. (A) Selected compounds leading to compound 32. (B) Representative biochemical inhibitory curves (top, duplicate technical replicates) and 797 cellular viability (bottom, quadruplicate technical replicates). (C) Evaluation of compound 32 selectivity against a panel of bromodomains. (D) Crystal structure of 32 bound to BRD4(1). Blue is oxygen and red is nitrogen (PDB: 4WIV). (E) Ligand interaction diagram of 32 from the cocrystal structure with BRD4. (F) Crystal structure of 32 overlaid on 1 (left) and rotated 90° (right).
Lead compounds 11 (Supporting Information, Table S1) and 32 (Figure 2C) were analyzed for bromodomain selectivity using a commercially available, phage-based, multiplexed bromodomain displacement assay (BromoScan; DiscovRx). We have noted somewhat increased sensitivity of the BromoScan assay compared to AlphaScreen. Compound 11 demonstrates broad activity against the tested bromodomains with some selectivity for the BET family. In contrast, 32 showed markedly increased potency for the BET proteins associated with a dramatic improvement in selectivity. Interestingly, 32 revealed potent binding to the TAF1 (560 nM) and TAF1L (1.3 μM) bromodomains. Albeit a significantly weaker interaction, this observation suggests a starting point for development of inhibitors of the previously undrugged TAF1 bromodomain.
To understand the mode of molecular recognition, cocrystallization of 32 and BRD4(1) was pursued, yielding a rather high-resolution structure of the cocomplex (PDB: 4WIV; 1.56 Å; Figure 2D,E). Compound 32 interacts with BRD4(1) via the expected hydrogen bond to the conserved asparagine. Hydrogen bonding to structural waters and π-stacking with tryptophan-81 illustrate the contributions to binding of the 32 region farthest from the warhead over other similar compounds in the SAR series. The additional heteroatom at 7-N is solvent exposed. In addition, the t-Bu group rests in the groove between the WPF shelf and the BC loop similar to the chlorophenyl in compound 1 and other diazepine bromodomain inhibitors. An analysis the crystal structure of 32 overlaid on 1 (PDB: 3MXF; Figure 2F) underscores this feature. The central phenyl ring in 32 occupies the same region as the fused thienodiazepine ring system of 1. As previously mentioned, the t-Bu group of 32 takes the place of chlorophenyl group of 1. In contrast, 32 has no bulky appending group analogous to the t-Bu in 1, suggesting a means of differential binding of 32 to TAF1.
Conclusion
Bromodomain proteins have emerged as master regulators of development and disease. Numerous functional connections to cancer and inflammation pathophysiology has established a fervent interest in the development of direct-acting inhibitors. Here we report a new functional class of bromodomain inhibitors that potently inhibits BET bromodomains and TAF1/TAF1L. Further optimization to develop a selective probe to the bromodomain of TAF1 is ongoing in our laboratories. In sum, we illustrate a facile synthetic strategy and biochemical platform capable of efficiently optimizing selective inhibitors from small organic biasing features. Compound 32 emerges from this research as a chemical probe available to the scientific community for mechanistic and translational research.
Experimental Section
AlphaScreen BRD Binding Assay
Assays were performed with minor modifications from the manufacturer’s protocol (PerkinElmer, USA). All reagents were diluted in 50 mM HEPES, 150 mM NaCl, 0.1% w/v BSA, 0.01% w/v Tween20, pH 7.5, and allowed to equilibrate to room temperature prior to addition to plates. After addition of Alpha beads to master solutions, all subsequent steps were performed in low light conditions. A 2× solution of components with final concentrations of BRD (see Supporting Information) at 40 nM, Ni-coated acceptor bead at 25 μg/mL, and 20 nM biotinylated-JQ1 (Supporting Information, Figure S2, synthesized as previously described)47 was added in 10 μL to 384-well plates (AlphaPlate-384, PerkinElmer, USA). After a 1 min 1000 rpm spin-down, 100 nL of compounds in DMSO from stock plates were arrayed in duplicate dose response by pin transfer using a Janus Workstation (PerkinElmer, USA). The streptavidin-coated donor beads (25 μg/mL final) were added as with previous solution in a 2×, 10 μL volume. Following this addition, the plates were sealed with foil to block light exposure and to prevent evaporation. The plates were spun down again at 1000 rpm for 1 min. Next, the plates were incubated in the room with the plate reader (for temperature equilibration) for 1 h prior to reading the assay. Signal is stable for up to 3 h after donor bead addition. AlphaScreen measurements were performed on an Envision 2104 (PerkinElmer, USA) utilizing the manufacturer’s protocol. Reported IC50 values are based on averages of multiple experiments, except where noted.
Cell Viability Assay
Assays were performed with endogenous BRD4-NUT-expressing midline carcinoma cell lines, 797.48 Cells were counted and adjusted to 60000 cells/mL. Using a Biotek EL406, 50 μL of cells are media were distributed into 384-well white plates from Thermo. Immediately after plating, compound in DMSO was distributed to plates. For large plate sets, cells were returned to 37 °C incubator while not in use. Compounds were added to plates using a 100 nL 384-well pin transfer manifold on a Janus workstation. Stocks were arrayed in 10 point quadruplicate dose response in DMSO stock in 384-well Greiner compound plates. After addition of compound, plates were incubated for 3 days in a 37 °C incubator. Cell viability was read out using ATPlite from PerkinElmer. Plates were removed from the incubator and brought to room temperature prior to use. Lyophilized powder was resuspended in lysis buffer and diluted 1:2 with DI water. Then 25 μL of this solution was added to each well using the Biotek liquid handler. Plates were sealed with adherent aluminum seals prior to vortexing and spinning down at 1000g for 1 min. Plates were incubated for 15 min at room temperature before a signal was read on an Envision plate reader. Reported EC50 values are based on averages of multiple experiments, except where noted.
Computational Methods
All computational work was performed in Schrodinger Suite (Schrodinger, LLC). Conformational analysis of lead compounds was performed using Schrodinger’s Conformational Search function. Possible poses were prepared for docking by Ligprep. In both cases, default settings were used (OPLS2005 force field, water solvent). Docking was conducted using Glide. The cocrystal of BRD4 and compound 1 (PDB: 3MXF) was used to define the ligand receptor grid. Water molecules outside the binding pocket were excluded, and hydrogen bonding interactions were optimized prior to docking.
General Synthetic Information
All chemicals and solvents were purchased from commercial suppliers and used as received. All biologically evaluated compounds were found to be >95% pure as determined by NMR and LCMS. 1H NMR (300 or 400 MHz) and 13C NMR (75 MHz) spectra were recorded on a Varian NMR spectrometer. CDCl3 was used as the solvent unless otherwise specified. LC-MS were performed on an Agilent 2100 system with a C18 (5.0 μm, 6.0 mm × 50 mm) LC column. The mobile phase is MeOH and water both containing 0.01% trifluoroacetic acid. A linear gradient was started from 75:25 MeOH/H2O to 100% MeOH in 5.0 min at a flow rate of 0.7 mL/min. The chromatograms were recorded at UV 210, 254, and 365 nm and subsequently used to determine compound purity. Low resolution mass spectra were recorded in APCI (atmospheric pressure chemical ionization). Flash chromatography separation was performed on YAMAZEN AI-580 system with Agela silica gel (12 or 20 g, 230–400 μm mesh) cartridges. The microwave reactions were performed on a Biotage Initiator 8 system.
General Procedures for the Synthesis of Compounds 11, 16–25, and 28–39
The synthesis of these compounds was accomplished by a reported two-step synthesis shown in Scheme 1.34 The three-component reaction (Groebke–Blackburn–Bienayme reaction) was followed by the Suzuki coupling.
Representative Procedure for the Three-Component Reaction: Synthesis of 2-(4-Bromophenyl)-N-(t-butyl)imidazo[1,2-a]pyrazin-3-amine (X = N, R1 = t-Bu, R2 = H).34
A sealed 10 mL microwave tube charged with t-butylisocyanide (0.043 g, 0.52 mmol), 2-aminopyrazine (0.05 g, 0.52 mmol), 4-bromobenzaldehyde (0.074 g, 0.40 mmol), and Sc(OTf)3 (0.010 g, 0.02 mmol) in 2 mL of 3:1 CH2Cl2/MeOH was heated under microwave at 150 °C for 30 min. The mixture was filtered and washed with EtOAc (4 mL). Concentration of the organic phase gave a crude product which was purified by flash chromatography eluted with 2:8 EtOAc/hexane to provide product as a yellow solid (110 mg, 80%).
Representative Procedure for the Suzuki-Coupling Reaction: Synthesis of N-(tert-Butyl)-2-(4-(3,5-dimethylisoxazol-4-yl)phenyl)imidazo[1,2-α]pyrazin-3-amine (32).34
A sealed 10 mL microwave tube charged with 2-(4-bromophenyl)-N-(t-butyl)imidazo[1,2-a]pyrazin-3-amine (X = N, R1 = t-Bu, R2 = H, 0.110 g, 0.32 mmol), 3,5-dimethylisoxazole-4-boronic acid pinacol ester (0.093 g, 0.42 mmol), Pd(dppf)Cl2·CH2Cl2 (0.021 g, 0.026 mmol, 8% mol), and K2CO3 (0.088 g, 0.64 mmol) in 2 mL of 4:4:1 acetone/toluene/H2O was heated under microwave at 130 °C for 40 min. The mixture was filtered and washed with EtOAc (4 mL). Concentration of the organic phase gave a crude product which was purified by flash chromatography eluting with 3:7 EtOAc/hexane to give 32 as brownish solid (>95% purity, 66 mg, 57%). 1H NMR (300 MHz, CDCl3) δ 9.01 (s, 1H), 8.16 (d, J = 4.8 Hz, 1H), 8.05 (d, J = 6.6 Hz, 2H), 7.88 (d, J = 4.8 Hz, 1H), 7.38 (d, J = 6.6 Hz, 2H), 3.18 (s, 1H, NH), 2.46 (s, 3H), 2.33 (s, 3H), 1.11 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 165.0, 158.34, 143.0, 141.2, 137.1, 133.2, 129.8, 128.7, 128.6, 128.3, 124.9, 116.1, 116.1, 56.7, 30.2, 11.4, 10.6. MS (APCI) m/z 362.2 (M+ + 1).
Acknowledgments
We acknowledge undergraduate students Yuan Xia and Shiva Dastjerdi for their assistance. The work in this study was supported by National Institutes of Health U54 grant CA156732 (J.E.B. and W.Z.), NIH U01 grant HD076508 (J.E.B.), the Damon Runyon Cancer Research Foundation (J.E.B.), and The William Lawrence and Blanche Hughes Foundation (J.E.B.). D.L.B. is a Merck Fellow of the Damon Runyon Cancer Research Foundation (DRG-2196-14).
Glossary
Abbreviations Used
- Kac
acetyl lysine
- BET
bromodomain and extraterminal domain
- BRD
bromodomain containing protein
- BRD4(1)
first bromodomain of bromodomain containing protein 4
- TAF
transcription initiation factor TFIID subunits
- F-SPE
fluorous solid phase extraction
Supporting Information Available
Synthesis and characterization of all compounds and further experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
4WIV: co-crystal of compound 32 and BRD4(1).
Author Contributions
Authors M.R.M., D.L.S., H.F., and S.L. contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sanchez R.; Zhou M. M. The role of human bromodomains in chromatin biology and gene transcription. Curr. Opin. Drug Discovery Dev. 2009, 12, 659–665. [PMC free article] [PubMed] [Google Scholar]
- Schreiber S. L.; Bernstein B. E. Signaling network model of chromatin. Cell 2002, 111, 771–778. [DOI] [PubMed] [Google Scholar]
- Frank S. R.; Parisi T.; Taubert S.; Fernandez P.; Fuchs M.; Chan H. M.; Livingston D. M.; Amati B. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 2003, 4, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervoorts J.; Luscher-Firzlaff J. M.; Rottmann S.; Lilischkis R.; Walsemann G.; Dohmann K.; Austen M.; Luscher B. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep. 2003, 4, 484–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J. S.; Jones P. A. Cancer genetics and epigenetics: two sides of the same coin?. Cancer Cell 2012, 22, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg T. Inhibition of transcription factors with small organic molecules. Curr. Opin. Chem. Biol. 2008, 12, 464–471. [DOI] [PubMed] [Google Scholar]
- Frye S. V. The art of the chemical probe. Nature Chem. Biol. 2010, 6, 159–161. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinjha R. K.; Witherington J.; Lee K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol. Sci. 2012, 33, 146–153. [DOI] [PubMed] [Google Scholar]
- Mujtaba S.; He Y.; Zeng L.; Yan S.; Plotnikova O.; Sachchidanand; Sanchez R.; Zeleznik-Le N. J.; Ronai Z.; Zhou M. M. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol. Cell 2004, 13, 251–263. [DOI] [PubMed] [Google Scholar]
- Mujtaba S.; Zeng L.; Zhou M. M. Structure and acetyl-lysine recognition of the bromodomain. Oncogene 2007, 26, 5521–5527. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J. P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Muller S.; Pawson T.; Gingras A. C.; Arrowsmith C. H.; Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkina A. C.; Denis G. V. BET domain co-regulators in obesity, inflammation and cancer. Nature Rev. Cancer 2012, 12, 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore J. E.; Issa G. C.; Lemieux M. E.; Rahl P. B.; Shi J.; Jacobs H. M.; Kastritis E.; Gilpatrick T.; Paranal R. M.; Qi J.; Chesi M.; Schinzel A. C.; McKeown M. R.; Heffernan T. P.; Vakoc C. R.; Bergsagel P. L.; Ghobrial I. M.; Richardson P. G.; Young R. A.; Hahn W. C.; Anderson K. C.; Kung A. L.; Bradner J. E.; Mitsiades C. S. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z.; Huang B.; Wu X.; Zhang H.; Qi J.; Bradner J.; Nair S.; Chen L. Brd4 maintains constitutively active NF-κB in cancer cells by binding to acetylated RelA. Oncogene 2014, 33, 2395–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B.; McKeown M. R.; Lin C. Y.; Monti S.; Roemer M. G.; Qi J.; Rahl P. B.; Sun H. H.; Yeda K. T.; Doench J. G.; Reichert E.; Kung A. L.; Rodig S. J.; Young R. A.; Shipp M. A.; Bradner J. E. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 2013, 24, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder S.; Cho S.; Zeng L.; Zhang Q.; Kaehlcke K.; Mak L.; Lau J.; Bisgrove D.; Schnolzer M.; Verdin E.; Zhou M. M.; Ott M. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J. Biol. Chem. 2012, 287, 1090–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S.; Sowa M. E.; Ottinger M.; Smith J. A.; Shi Y.; Harper J. W.; Howley P. M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol. 2011, 31, 2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A.; Chitsaz F.; Abbasi A.; Misteli T.; Ozato K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 8758–8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzuk M. M.; McKeown M. R.; Filippakopoulos P.; Li Q.; Ma L.; Agno J. E.; Lemieux M. E.; Picaud S.; Yu R. N.; Qi J.; Knapp S.; Bradner J. E. Small-molecule inhibition of BRDT for male contraception. Cell 2012, 150, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French C. A. Pathogenesis of NUT midline carcinoma. Annu. Rev. Pathol.: Mech. Dis. 2012, 7, 247–265. [DOI] [PubMed] [Google Scholar]
- Zuber J.; Shi J.; Wang E.; Rappaport A. R.; Herrmann H.; Sison E. A.; Magoon D.; Qi J.; Blatt K.; Wunderlich M.; Taylor M. J.; Johns C.; Chicas A.; Mulloy J. C.; Kogan S. C.; Brown P.; Valent P.; Bradner J. E.; Lowe S. W.; Vakoc C. R. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott C. J.; Kopp N.; Bird L.; Paranal R. M.; Qi J.; Bowman T.; Rodig S. J.; Kung A. L.; Bradner J. E.; Weinstock D. M. BET bromodomain inhibition targets both c-MYC and IL7R in high-risk acute lymphoblastic leukemia. Blood 2012, 120, 2843–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ClinicalTrials.gov; U.S. National Library of Medicine: Bethesda, MD, 2014; www.ClinicalTrials.gov.
- Liu X.; Vorontchikhina M.; Wang Y. L.; Faiola F.; Martinez E. STAGA recruits Mediator to the MYC oncoprotein to stimulate transcription and cell proliferation. Mol. Cell. Biol. 2008, 28, 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi F.; Wirapati P.; Naef F. Identifying synergistic regulation involving c-Myc and sp1 in human tissues. Nucleic Acids Res. 2007, 35, 1098–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. H.; Li A. G.; Sheppard H. M.; Liu X. Phosphorylation on Thr-55 by TAF1 mediates degradation of p53: a role for TAF1 in cell G1 progression. Mol. Cell 2004, 13, 867–878. [DOI] [PubMed] [Google Scholar]
- Buchmann A. M.; Skaar J. R.; DeCaprio J. A. Activation of a DNA damage checkpoint response in a TAF1-defective cell line. Mol. Cell. Biol. 2004, 24, 5332–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijnappel W. W.; Esch D.; Baltissen M. P.; Wu G.; Mischerikow N.; Bergsma A. J.; van der Wal E.; Han D. W.; Bruch H.; Moritz S.; Lijnzaad P.; Altelaar A. F.; Sameith K.; Zaehres H.; Heck A. J.; Holstege F. C.; Scholer H. R.; Timmers H. T. A central role for TFIID in the pluripotent transcription circuitry. Nature 2013, 495, 516–519. [DOI] [PubMed] [Google Scholar]
- Zhang W. Fluorous linker-facilitated chemical synthesis. Chem. Rev. 2009, 109, 749–795. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Curran D. P. Synthetic applications of fluorous solid-phase extraction (F-SPE). Tetrahedron 2006, 62, 11837–11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Chen C. H.-T.; Lu Y.; Nagashima T. A highly efficient microwave-assisted Suzuki coupling reaction of aryl perfluorooctylsulfonates with boronic acids. Org. Lett. 2004, 6, 1473–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Chen C. H.-T. Fluorous synthesis of biaryl-substituted proline analogs by 1,3-dipolar cycloaddition and Suzuki coupling reactions. Tetrahedron Lett. 2005, 46, 1807–1810. [Google Scholar]
- Lu Y.; Zhang W. Microwave-assisted synthesis of a 3-aminoimidazo[1,2-a]-pyridine/pyrazine library by fluorous multicomponent reactions and subsequent cross-coupling reactions. QSAR Comb. Sci. 2004, 23, 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.; Zhang W.; Yan B. Use of cyclohexylisocyanide and methyl 2-isocyanoacetate as convertible isocyanides for microwave-assisted fluorous synthesis of 1,4-benzodiazepine-2, 5-dione library. J. Comb. Chem. 2009, 12, 206–214. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Nagashima T. Palladium-catalyzed Buchwald–Hartwig type amination of fluorous arylsulfonates. J. Fluorine Chem. 2006, 127, 588–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.-H.; Lü H.-Y.; Yang S.-H.; Gao J.-W. Synthesis of 2,3-dihydroquinazolin-4(1H)-ones by three-component coupling of isatoic anhydride, amines, and aldehydes catalyzed by magnetic Fe3O4 nanoparticles in water. J. Comb. Chem. 2010, 12, 643–646. [DOI] [PubMed] [Google Scholar]
- Hewings D. S.; Wang M.; Philpott M.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Vuppusetty C.; Marsden B.; Knapp S.; Conway S. J.; Heightman T. D. 3,5-Dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. J. Med. Chem. 2011, 54, 6761–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewings D. S.; Fedorov O.; Filippakopoulos P.; Martin S.; Picaud S.; Tumber A.; Wells C.; Olcina M. M.; Freeman K.; Gill A.; Ritchie A. J.; Sheppard D. W.; Russel A. J.; Hammond E. M.; Knapp S.; Brennan P. E.; Conway S. J. Optimization of 3,5-dimethylisoxazole derivatives as potent bromodomain ligands. J. Med. Chem. 2013, 56, 3217–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E.; Jeffrey K. L.; Schaefer U.; Beinke S.; Dewell S.; Chung C.-w.; Chandwani R.; Marazzi I.; Wilson P.; Coste H.; White J.; Kirilovsky J.; Rice C. M.; Lora J. M.; Prinjha R. K.; Lee K.; Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamborough P.; Diallo H.; Goodacre J. D.; Gordon L.; Lewis A.; Seal J. T.; Wilson D. M.; Woodrow M. D.; Chung C.-w. Fragment-based discovery of bromodomain inhibitors part 2: optimization of phenylisoxazole sulfonamides. J. Med. Chem. 2012, 55, 587–596. [DOI] [PubMed] [Google Scholar]
- Gehling V. S.; Hewitt M. C.; Vaswani R. G.; Leblanc Y.; Côté A.; Nasveschuk C. G.; Taylor A. M.; Harmange J.-C.; Audia J. E.; Pardo E.; Joshi S.; Sandy P.; Mertz J. A.; Sims R. J.; Bergeron L.; Bryant B. M.; Bellon S.; Poy F.; Jayaram H.; Sankaranarayanan R.; Yellapantula S.; Srinivasamurthy N. B.; Birudukota S.; Albrecht B. K. Discovery, design, and optimization of isoxazole azepine BET inhibitors. ACS Med. Chem. Lett. 2013, 4, 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M. A.; Prinjha R. K.; Dittmann A.; Giotopoulos G.; Bantscheff M.; Chan W. I.; Robson S. C.; Chung C. W.; Hopf C.; Savitski M. M.; Huthmacher C.; Gudgin E.; Lugo D.; Beinke S.; Chapman T. D.; Roberts E. J.; Soden P. E.; Auger K. R.; Mirguet O.; Doehner K.; Delwel R.; Burnett A. K.; Jeffrey P.; Drewes G.; Lee K.; Huntly B. J. P.; Kouzarides T. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picaud S.; Da Costa D.; Thanasopoulou A.; Filippakopoulos P.; Fish P. V.; Philpott M.; Fedorov O.; Brennan P.; Bunnage M. E.; Owen D. R.; Bradner J. E.; Taniere P.; O’Sullivan B.; Muller S.; Schwaller J.; Stankovic T.; Knapp S. PFI-1, a highly selective protein interaction inhibitor, targeting BET bromodomains. Cancer Res. 2013, 73, 3336–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Bienayme H.; Bouzid K. A new heterocyclic multicomponent reaction for the combinatorial synthesis of fused 3-aminoimidazoles. Angew. Chem., Int. Ed 1998, 37, 2234–2237. [DOI] [PubMed] [Google Scholar]
- Anders L.; Guenther M. G.; Qi J.; Fan Z. P.; Marineau J. J.; Rahl P. B.; Lovén J.; Sigova A. A.; Smith W. B.; Lee T. I.; Bradner J. E.; Young R. A. Genome-wide localization of small molecules. Nature Biotechnol. 2013, 32, 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toretsky J. A.; Jenson J.; Sun C. C.; Eskenazi A. E.; Campbell A.; Hunger S. P.; Caires A.; Frantz C.; Hill J. L.; Stamberg J. Translocation (11;15;19): a highly specific chromosome rearrangement associated with poorly differentiated thymic carcinoma in young patients. Am. J. Clin. Oncol. 2003, 26, 300–306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.