

Abstract
Background
Endometrial cancers (ECs) are the most common form of gynecologic malignancy. Recent studies have reported that ECs reveal distinct markers for molecular pathogenesis, which in turn is linked to the various histological types of ECs. To understand further the molecular events contributing to ECs and endometrial tumorigenesis in general, a more precise identification of cancer-associated molecules and signaling networks would be useful for the detection and monitoring of malignancy, improving clinical cancer therapy, and personalization of treatments.
Results
ECs-specific gene co-expression networks were constructed by differential expression analysis and weighted gene co-expression network analysis (WGCNA). Important pathways and putative cancer hub genes contribution to tumorigenesis of ECs were identified. An elastic-net regularized classification model was built using the cancer hub gene signatures to predict the phenotypic characteristics of ECs. The 19 cancer hub gene signatures had high predictive power to distinguish among three key principal features of ECs: grade, type, and stage. Intriguingly, these hub gene networks seem to contribute to ECs progression and malignancy via cell-cycle regulation, antigen processing and the citric acid (TCA) cycle.
Conclusions
The results of this study provide a powerful biomarker discovery platform to better understand the progression of ECs and to uncover potential therapeutic targets in the treatment of ECs. This information might lead to improved monitoring of ECs and resulting improvement of treatment of ECs, the 4th most common of cancer in women.
Keywords: Endometrial cancer; WGCNA, Network analysis; Hub gene, TCA cycle
Background
Endometrial cancers (ECs) are the most common type of uterine cancer. With more than 280,000 cases occurring annually worldwide, it has become the fourth most common cancer in women worldwide [1]. The incidence rate of ECs is higher than uterine cervix cancer and ovarian cancer in the United State [2]. These tumors have been broadly classified into two major subtypes I and II, based on the clinic-pathological characteristics such as prognosis and aggressiveness, as well as, molecular alterations that impact tumor response to therapies [3]. Type I tumors are the most frequent subtype which have been linked to obesity, estrogen excess, and low-grade (differentiated) inflammation. Type II endometrial tumors are serous carcinoma that have been associated with older, non-obese, post-menopausal women, high-grade (undifferentiated), and also with worst outcomes. It has been suggested that the molecular-genetic alterations may be responsible for the distinct morphology and biologic behavior of the different subtypes of human ECs [4]. For example, low-grade or early-stage Type I tumors may progress to high-grade or late-stage; nonetheless Type I and II cancers appear to be separate entities in most cases, and different molecular abnormalities would result in unique cellular functions and distinctive tumor morphology [5]. Therefore, it is crucial to discover with more accuracy the putative molecular signatures of ECs, which should allow for improved detection and monitoring of endometrial tumorigenesis, since such knowledge could be beneficial for early diagnostic, enhanced prognostic, and more effective therapeutic strategies.
ECs are characterized by a variety of genetic alterations and significant gene expression modifications. Over the last decade, multiple studies have identified aberrant gene expression of several important genes in ECs, with the mutation frequency varying according to the histological classification [6]. Type I tumors are frequently characterized by the loss or altered expression of phosphatase and tensin homolog (PTEN). PTEN modulates cell survival and proliferation through its effects on downstream factors, mainly phospholipid phosphatidylinositol (3, 4, 5)-triphosphate (PIP3) and protein kinase B (PKB, Akt). PTEN inactivation leads to a decrease of lipid and protein phosphatase activity and promotes cell cycle progression to the G1/S phase [7]. Other genes are linked to abnormalities in Type I tumors includingβ-catenin, K-ras and DNA-mismatch repair genes [7-10]. In comparison, Type II tumors have been reported to be associated with abnormalities in TP53 and Her2/neu[6]. The gene TP53 encodes a tumor suppressor p53, the most frequently mutated protein in cancer. P53 prevents cell cycle progression after DNA damage, inducing cell arrest and apoptosis through several regulator proteins such as p21, Cyclin D1, and RB1. TP53 mutations occur as an early event in Type II tumorigenesis and may occur as manifestations of late-stage molecular changes in Type I lesions. Overexpression of Her-2/neu observed in Type II carcinomas has been linked to coding alterations for a transmembrane receptor tyrosine kinase involved in cell signaling [11]. Although these studies provide important insights into the molecular basis of endometrial cancers, a limited set of well-known cancer genes was obtained from these studies. In fact, until now, a large-scale screen of the gene expression analyses incorporating systematic methods to discover cancer subtypes and their molecular alterations in ECs has not been globally conducted and explored.
Recent advances in constructing genetic network approaches have enabled the unprecedented characterization of studying a variety of somatic alterations and gene expression in cancer genomes. Therefore, these advances allow connecting the existent gap of understanding the association of individual genes to complex diseases such as cancer by the systematic investigation of the observed relationship between gene products and tumorigenesis. A weighted gene co-expression network approach (WGCNA) has been proposed to reconstruct gene co-expression networks (modules) in terms of large-scale gene expression profiles and as well as for the distinction of centrally located genes (hub genes) driving key cellular signaling pathways [12,13]. The WGCNA approach provides a functional interpretation in Systems Biology and leads to new insights into cancer pathophysiology [14-17].
Here, we aimed to establish a systematic framework for constructing for the first time, the ECs-associated gene co-expression networks and pin-pointing cancer hub genes contributing to endometrial tumorigenesis and progression. This study provides a novel and broad application platform for the identification of cancer gene signatures of ECs tumorigensis and for the discovery of potential new molecular targets for the development of more effective therapies for the treatment of ECs.
Results
Systematic framework for identifying cancer hub genes in ECs
In this study, a novel systematic analysis was developed to integrate WGCNA and elastic-net analysis to identify cancer hub genes in ECs (Figure 1). This method is divided into three main parts. First, we performed a very large human microarray-based ECs meta-analysis by merging multiple platforms to reveal the differentially expressed genes on 273 EC samples properly matched with normal samples. Second, we conducted the WGCNA analysis to reconstruct EC-associated gene co-expression networks (modules) and discover the cancer hub genes. Third, we developed a cancer hub genes-based classifier model to distinguish the phenotypic characteristics of ECs (i.e., grade, type and stage). Finally, we used the hub genes as the gene signatures to validate its biological and phenotypic characteristics relevance using 10-fold cross-validation and independent data set validation. Figure 1 presents an overview of approach in this study, and the detailed methods are described in Supporting Information (Additional file 1: SI Materials and Methods).
Figure 1.
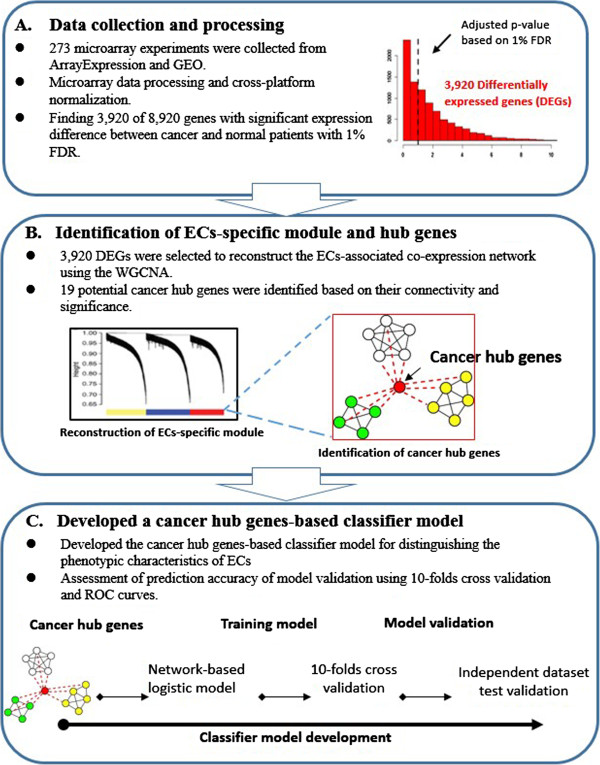
Framework for deriving the cancer hub genes and validating its phenotypic relevance in ECs: (A) Data collection and processing for 273 microarray experiments to find 3,920 DEGs, (B) Identification of ECs-specific module and 19 cancer hub genes, and (C) Developed a cancer hub genes-based classifier model using 10-fold cross validation and ROC curves to assess the prediction accuracy of model.
Reconstruction of EC-specific gene co-expression network
In the initial analysis, we identified 3,920 genes having significant expression difference between subjects with cancer and subjects without cancer by applying a 1% FDR (Additional file 2: Table S1). These endometrial cancer-related genes were used to reconstruct the EC-associated co-expression network (module) and identify a number of modules of high co-expression genes. As shown in Figure 2, these modules are significantly enriched for biologically important processes that are relevant to cancer, including cell-cycle regulation, antigen processing, immune response, and cell adhesion (Table 1). Among the modules, yellow-colored module (Myellow) that specifically corresponds to clinical information of ECs, showed a high Pearson correlation with phenotypic characteristics of ECs including grade (r = 0.44, Bonferroni-adjusted p-value = 1.2E−16), type (r = 0.34, Bonferroni-adjusted p-value = 6.3E−9) and stage (r = 0.31, Bonferroni-adjusted p-value = 2.1E−7) in ECs. The blue-colored module (Mblue) was only significantly correlated with the stage of ECs (r = 0.42, Bonferroni-adjusted p-value = 6.0E−19). By contrast, other modules showed a much lower correlation with the phenotypic characteristics of ECs. Interestingly, the Myellow module was significantly enriched for cell-cycle regulation (Bonferroni-adjusted p-value = 1.2E−31). Conversely, Mblue gene ontology categories included antigen processing (Bonferroni-adjusted p-value = 8.7E−12) and the citric acid (tricarboxylic acid; TCA) cycle (Bonferroni-adjusted p-value = 4.5E−12).
Figure 2.
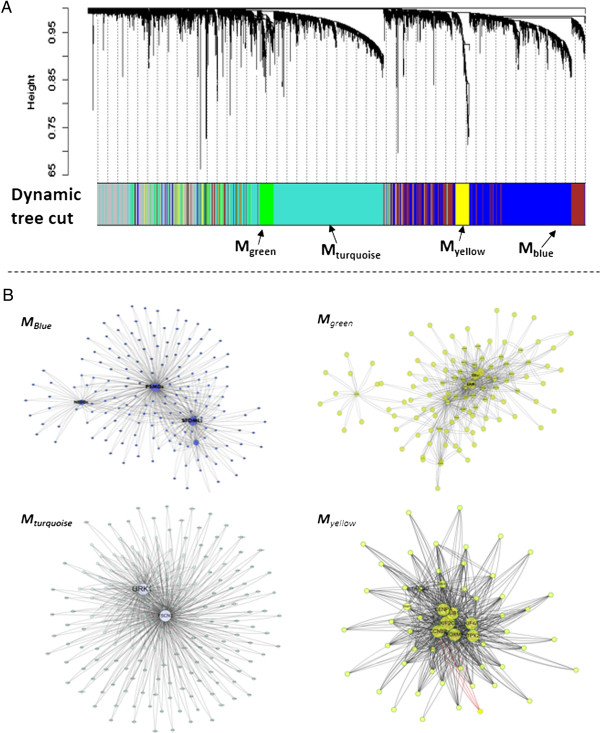
WGCNA analysis on the large-scale microarray datasets. (A) Dendrogram showing relationship for the topological overlap of genes and their relationship to modules, which are color-coded. (B) Graphic depiction of the blue-color module (Mblue), green-color module (Mgreen), turquoise-color module (Mturquoise) and yellow-color module (Myellow) using Cytoscape. For each viewing module, pairs of genes with the highest intramodular topological overlap are illustrated, with each link corresponding to a topology overlap measure (TOM) between the connected nodes.
Table 1.
Module significance in ECs and GO analysis
|
Module |
Correlation
a
with phenotypic characteristics of ECs (p-value) |
Gene ontology enrichment analysis |
||||
|---|---|---|---|---|---|---|
| Grade | Type | Stage | Term | p-value b | FDR | |
| Blue |
−0.002 (9.0E−1) |
0.08 (2.2E−1) |
0.42 (6.0E−19) |
Antigen processing |
8.7E−12 |
1.7E−12 |
| |
|
|
|
TCA cycle |
4.5E−12 |
2.1E−12 |
| Green |
0.006 (9.1E−1) |
−0.02 (6.1E−5) |
0.01 (9.3E−1) |
Immune response |
3.7E−43 |
6.3E−40 |
| Turquoise |
−0.17 (6.4E−3) |
−0.19 (2.4E−3) |
0.17 (6.2E−3) |
Cell adhesion |
2.9E−29 |
1.1E−29 |
| Yellow | 0.44 (1.2E−16) | 0.34 (6.3E−9) | 0.31 (2.1E−7) | Cell-cycle regulation | 4.9E−35 | 8.6E−32 |
aThe correlation coefficient was calculated from the module eigengenes (i.e., first principal component of the expression values across subjects) and phenotypic characteristics of ECs using Pearson correlation.
bp-value used the Bonferroni-adjusted p-value.
Identification of cancer hub genes
Genes with the highest degree of connectivity within a module (centrally located genes of co-expressed genes) are termed hub genes and are expected to be drivers required for signaling pathways of essential cellular function. To identify the cancer hub genes in the Myellow and Mblue modules, we estimated the scale connectivity (K) for each gene and for gene significance (GS) based on its Pearson correlation with phenotypic characteristics of ECs (Additional file 3: Table S2) and predicted the frequency (f) by using the elastic net regression model combined with bootstrap approaches in the modules (Figure 3). We set the weighted cutoff value (defined as r > 0.2, K > 0.25 and f > 750) to identify cancer hub genes with strongest connections to other genes and to link to phenotypic characteristics of ECs. As a result, we identified 19 cancer hub genes with at least 50 connections derived from the Myellow and Mblue Modules. These hub genes associated with grade (TP53, BUB1, AURKB and CENPA), type (AURKB, PRC1, CDC6, E2F2, KIF20A) and stage (BUB1, FEN1, KIF23, CDC20 and PRC1) in the Myellow, module. Nonetheless, 5 hub genes associated with stage including IDH3G, NDUFV2, ATP5B, PSMB3 and PSMB7 were identified in the Mblue module (Figure 3A- 3D, Table 2). Figure 4 illustrated the relationship among these genes suggesting a complex regulatory gene network with varying topology.
Figure 3.
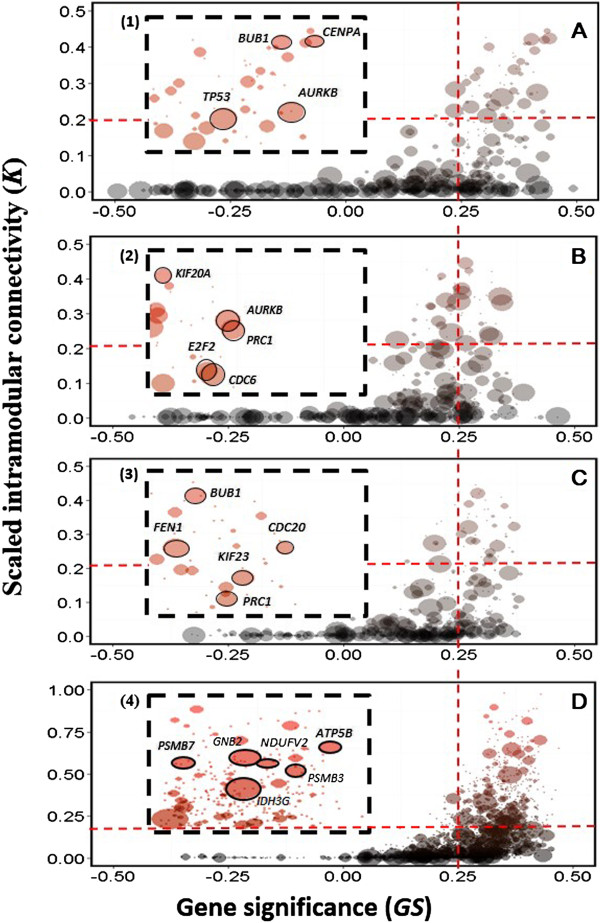
A systematic screen in ECs-associated networks identifies cancer hub genes. A plot representation of the elastic-net regression gene selection results showing the Gene significance (GS, x-axis) and scaled gene connectivity (K, y-axis) of all genes of Myellow network that are associated with (A) Grade, (B) Type, (C) Stage and that of the Mblue network that are associated with Stage (D). Each circle represents a single phenotype-gene interaction and the size is proportional to the frequency (f) calculated from the elastic-net regression analysis. The red dashed line indicates the cancer hub genes selection criteria (GS > 0.25, K > 0.2). Insets (1) - (4) are magnified views of selected cancer hub genes.
Table 2.
List of hub gene associated with grade, type and stage o ECs
|
Symbol |
Gene names |
Hub gene significance
*
|
||
|---|---|---|---|---|
| f | K | GS | ||
|
Hub genes for grade | ||||
| TP53 |
Tumor protein 53 |
954 |
0.40 |
0.31 |
| AURKB |
Aurora kinase B |
885 |
0.41 |
0.37 |
| BUB1 |
Budding uninhibited by benzimidazoles 1 |
768 |
0.43 |
0.42 |
| CENPA |
Centromere protein A |
764 |
0.44 |
0.42 |
|
Hub genes for type | ||||
| AURKB |
Aurora kinase B |
995 |
0.41 |
0.35 |
| PRC1 |
Protein regulator of cytokinesis 1 |
893 |
0.34 |
0.33 |
| CDC6 |
Cell division cycle 6 |
849 |
0.26 |
0.24 |
| E2F2 |
E2F transcription factor 2 |
776 |
0.31 |
0.22 |
| KIF20A |
Kinesin family member 20A |
760 |
0.46 |
0.21 |
|
Hub genes for stage (derived from module yellow) | ||||
| BUB1 |
Budding uninhibited by benzimidazoles 1 |
768 |
0.43 |
0.42 |
| FEN1 |
Flap structure-specific endonuclease 1 |
764 |
0.27 |
0.32 |
| KIF23 |
Kinesin family member 23 |
763 |
0.34 |
0.27 |
| CDC20 |
Cell division cycle 20 |
762 |
0.37 |
0.32 |
| PRC1 |
Protein regulator of cytokinesis 1 |
760 |
0.34 |
0.27 |
|
Hub genes for stage (derived from module blue) | ||||
| IDH3G |
Isocitrate dehydrogenase 3 (NAD+) gamma |
934 |
0.36 |
0.49 |
| NDUFV2 |
NADH dehydrogenase (ubiquinone) flavoprotein 2 |
873 |
0.35 |
0.42 |
| ATP5B |
ATP synthase, H + transporting, mitochondrial F1 complex, beta polypeptide |
769 |
0.52 |
0.49 |
| PSMB3 |
Proteasome (prosome, macropain) subunit, beta type, 3 |
762 |
0.41 |
0.47 |
| PSMB7 | Proteasome (prosome, macropain) subunit, beta type, 7 | 760 | 0.32 | 0.42 |
*This study defined the hub gene significance (HGS) by integrating WGCNA and elastic-net analysis, estimating the gene significance (GS), scaled connectivity (K) and frequency (f). Only genes present in GS > 0.2, K > 0.2 and f > 750 were selected as ECs-specific hub genes.
Figure 4.
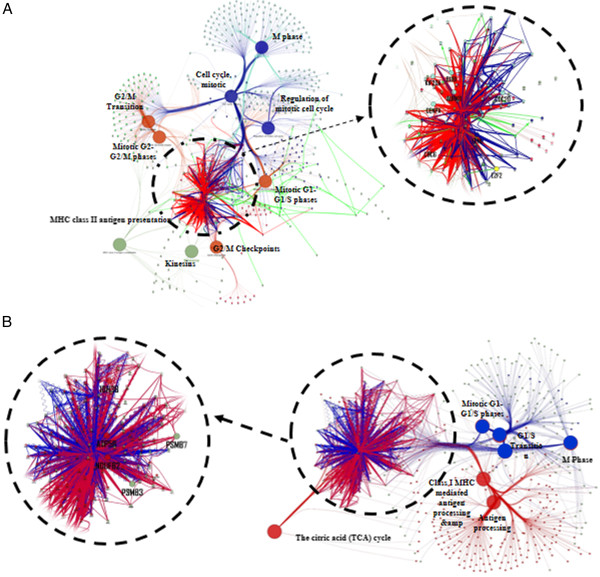
Differentially activated pathways features between Type I and Type II ECs subtypes in (A) the cell‒cycle regulator network and (B) the antigen processing network. Color arrows reflect pathway differences between subtypes (blue: Type I, red: Type II). Green lines represent activation. A zoomed in view of the cancer hub gene signatures are also shown.
Pathway analysis of hub genes
This study reconstructed and identified the gene networks of cancer hub genes to search the potential key regulators of endometrial tumorigenesis and to identify regulatory relationships among cancer hub genes. Using functional enrichment analysis, we identified three major signaling pathways; cell-cycle regulated networks (mitotic, G2/M or G1/S phases), antigen processing (Class I MHC antigen processing) and the TCA cycle, as the main Gene Ontology biological processes overrepresented in the gene networks deriving from the cancer hub genes (Figure 4). In the cell-cycle network, BUB1, AURKB, CENPA, KIF20A, CDC20, CDC6, E2F2 and FEN1 seem to regulate distinct co-expression pathways that contributed to mitotic G2-G2/M phase, G2/M checkpoints, and kinesins in Type II ECs, whereas these genes regulate the M phase and mitotic G1-G1/S phase in Type I ECs (Figure 4A). In addition, the antigen processing and TCA cycle networks are regulated by the hub genes IDH3G, NDUFV2, ATP5B, PSMB3 and PSMB7, contributing to tumorigenesis in Type II ECs (Figure 4B).
Cancer hub genes classification based model
This study performed receiver-operator characteristic curve (ROC) analysis to assess the predictive accuracy of the cancer hub gene signatures. As follows, an AUC (area under curve) value of 0.5 indicated that the predictive performance equals chance, while values greater than 0.5 indicated high predictive capacity. Using a strict 10-fold cross-validation, the classification capacity of cancer hub gene signatures proved to be significantly better than random predictability (Additional file 4: Figure S1A-S1D for second supporting information figure, p <10−6, AUC = 0.72 ~ 0.85). Furthermore, the module showed a substantial capacity to distinguish ECs grade (AUC = 0.91, p < 10−12), type (AUC = 0.98, p < 10−5) and stage (Myellow: AUC = 0.93, p < 10−12; Mblue: AUC = 0.73, p < 10−4) in independent datasets (Figure 5). Strikingly, these hub genes deriving from the co-expression networks of Myellow and Mblue provided significant predictive power in distinguishing the phenotypic characteristics of ECs.
Figure 5.
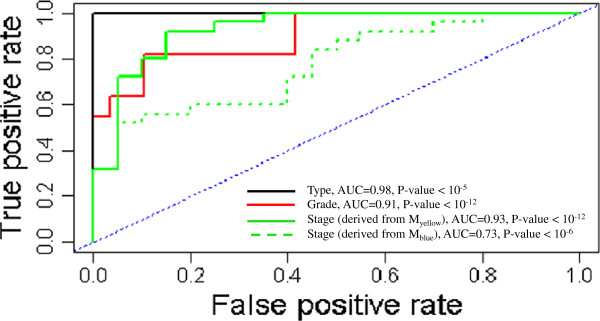
ROC curve to assess the accuracy of the cancer hub genes signature in the independent dataset. True positive rate represent the model sensitivity, whereas false positive rate is one minus the specificity or true negative rate and represents chance.
Discussion
In this study, we integrated a large-scale transcriptional profiling of 273 ECs datasets to identify appropriate cancer subtypes and biomarkers. Our long-term goal is to provide insights into disease biology and diagnostic classification, which may guide early-phase of clinical therapeutic applications. We also determined that co-expression networks reflect causative relationships between different gene-gene interactions. First, this study found that an EC-specific co-expression network majorly regulated the tumorigenesis of ECs. This network not only enriched genes known to play critical roles in cell cycle regulation but also showed significant correlation with grade, type, and stage of ECs. Second, 19 highly connected hub genes were defined into two EC-associated co-expression networks. Detailed and systematic network analysis revealed that these hub genes played roles as major regulators in cell-cycle regulation, kinase modulation, and the development of various tumors. Finally, for independent analysis of microarray data as validated data sets, these hub genes can provide excellent diagnostic power in distinguishing grade, type, and stage of ECs. Altogether these findings strongly suggest that the role of these hub genes in the EC-associated networks may provide new insights into the underlying biological mechanisms driving the tumorigenesis of ECs.
Tumor cell progression is typically associated with aberrant cell-cycle regulation, and this study identified several unique hub genes in the networks associated with the grade of ECs including TP53, BUB1, AURKB, CENPA. In agreement with previous reports [17-20], the disruption of these hub genes was observed in a variety of human tumors and often correlates with tumorigenesis. For example, BUB1 has been suggested to play a direct role in the suppression of p53-mediated cell death via physical interaction with p53 at kinetochores in response to mitotic spindle damage [18]. Altered expression of BUB1 is associated with therapy failure and death in patients with multiple types of cancer [19]. Fu et al. (2007) [20] reported a possible mechanism on the roles of aurora kinases in mitosis and tumorigenesis. They found that overexpression of AURKA and AURKB disruption overrides spindle checkpoint, resulting in the aneuploidy or polyploidy that occurs during cell division. When cells lose the normal function of p53, they enter mitosis and become aneuploid, which is considered a cancerous manifestation.
Recently, up-regulation of BUB1 gene has been linked to the inhibition of p53-dependent senescence, hyper-activation of AURKB and phosphorylation of CENP-A, and might be considered as a potential oncogene in driving the aneuploidiation and tumorigenesis [21]. Interestingly, this study found BUB1 not only highly connected with other genes in the co-expression network, but also significantly correlated with the transformation of ECs. Therefore, BUB1 gene seems to modulate the expression of tens or hundreds of genes suggesting that it might allow undifferentiated cancer cells to overcome apoptotic checkpoints favoring aberrant progression through mitosis.
Previous reports suggested that PTEN mutations occur early in the neoplastic process of Type I ECs and co-exist frequently with other mutations in the PI(3)K/AKT pathway [22], yet Type II ECs showed an alteration of the E2F–retinoblastoma protein–p16 pathway by mutations of p53 and p16[23]. In this study, several unique hub genes (AURKB, PRC1, CDC6, E2F2, KIF20A) were identified in the co-expression network associated with the subtype of ECs, which were linked to the PTEN and p16-associated pathway. The cell division gene, CDC6, is required for the cell cycle G1-to-S transition. Wu et al. (2009) [24] have identified that CDC6 is the putative PTEN target and demonstrated a causal linkage between CDC6 and PTEN in metastatic human prostate cancer. They indicated that the regulation of CDC6 expression by PTEN is mediated through the E2F transcriptional factor and the E2F2 protein. Furthermore, overexpression of AURKB has been suggested to compromise the tumor suppressor function of p53[25]. The gene PRC1, encodes one of the polycomb-group proteins, and is involved in cytokinesis. Of note, the PRC1 and PRC2/3/4 proteins can induce the repression of the PTEN transcription via binding to the PTEN promoter region in leukemic cells [26], while KIF20A is known to be controlled by the E2F–retinoblastoma protein–p16 pathway, and is linked to tumor aggressiveness in human hepatocellular carcinomas [27]. Taken together, it appears that this gene-signature is commonly linked to PTEN, PI(3)K/AKT and E2F–retinoblastoma protein–p16 pathway, and may be marginally associated with the subtype of ECs.
Cancer stage is the most import indicator for selecting an appropriate cancer treatment option for a patient. In this study, two co-expression networks were significantly correlated with the stage of ECs. These two network-regulated signaling events contributed to cell-cycle regulation, antigen processing and TCA cycle, respectively. From the cell-cycle regulation network, 5 hub genes were identified including BUB1, FEN1, KIF23, CDC20 and PRC1. BUB1 and CDC20 are involved in the M phase of mitotic cell cycle and DNA replication, and play critical roles in the cell-cycle regulation [28]. Altered expression of cell-cycle associated protein like BUB1 and CDC20 may lead to chromosomal instability. BUB1 and CDC20 are important regulators of the anaphase-promoting complex (APC) [29]. APC’s function is to trigger the transition from metaphase to anaphase by tagging critical cell-cycle proteins for degradation, and maintaining genomic stability in a normal manner. BUB1 can directly phosphorylate the APC’s co-activator CDC20, leading to the decreased activity of APC, which determines the metaphase-to-anaphase transition. The overexpression of BUB1 or CDC20 induces misregulation of APC, and is associated with the chromosomal instability and poor outcomes in breast cancer patients [30,31]. KIF23 belongs to the kinesin family, and it is part of the central spindle in a complex that clusters PRC1 and AURKB together at the spindle midzone to enable anaphase in dividing cells and regulation of central spindle assembly [32]. Abnormal regulation of these genes may lead to the chromosomal instability, thereby promoting tumor development and progression.
The pathway of MHC class I presentation is an important mechanism in determining whether tumors are able to evade immune response. Down-regulation of MHC Class I has been described in ovary and cervix malignancies. Abnormal expression of MHC Class I genes has been linked to the advanced stage of disease and poor survival in ovarian cancer [33]. Recently, a large cohort study of patients with endometrial cancer indicated that down-regulation of MHC Class I expression in endometrial cancer patients are correlated to late-stage ECs [34]. In this study, we identified a group of hub genes associated with the stage of ECs deriving from the MHC Class I co-expression network. These hub genes, PSMB7 and PSMB3, were found to significantly regulate the network of antigen processing and to contribute to Class I MHC mediated processing. Furthermore, we also discovered 3 hub genes (IDH3G, NDUFV2 and ATP5B) associated with the TCA cycle. No other study to date has reported that these hub genes might be associated with cancer, but recent studies have indicated that the alterations in the TCA cycle enzymes may favor tumorigenesis by impacting on cellular redox state and overall cell metabolism [35,36]. Therefore, it is reasonable to hypothesize that these hub genes could be involved with tumor progression.
To identify the signaling pathway through which hub genes regulated the co-expression network in the progression of ECs, we compared the gene co-expression networks between Type I (early-stage, low-grade) and Type II ECs (late-stage, high-grade) of ECs. In the cell-cycle regulation network, Type II ECs displayed a distinct pathway when compared with Type I ECs. However, BUB1, AURKB, CDC6, CENPA and KIF20A seemed to be critical regulators for the co-expression network in Type II compared to Type I ECs. Among these genes, BUB1 may play a key role in regulating these genes to promote tumor formation. The overexpression of BUB1 regulates AURKB, CENPA, CDC6 and KIF20A in a variety of human cancers [37-39]. Furthermore, from the antigen processing and TCA cycle co-expression networks, we found that the network regulated TCA cycle through the co-regulation of IDH3G, NDUFV2 and ATP5B, and linked to the pathway of Cass I MHC antigen processing key players were PSMB3 and PSMB7 in Type II compared to Type I ECs. Expression of these genes in Type II ECs may facilitate the identification of signaling pathways contributing to tumor progression.
Conclusions
This study used a novel systematic framework to identify two co-expression networks associated with ECs tumorigenesis based on large-scale human microarray data. In addition, a number of novel hub genes in these two co-expression networks were identified contributing to three signaling pathways: cell-cycle regulation, antigen processing and TCA cycle, and presented a high predictive power in distinguishing grade, type and stage of ECs. Although Type I and Type II ECs shared similar genetic information, several critical hub genes were identified that may contribute to progression of ECs. Together, these findings provided a clearer and broader picture of the signaling pathways regulated by co-expression networks contributing to ECs. Furthermore, the characterization of these hub genes might infuse novel insights into the identification of novel clinical markers and potential therapeutic targets for ECs.
Methods
Data collection and processing
Microarray data sets were systematically searched from ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) using the keyword “endometrial cancer”. Only the studies that presented the raw microarray expression data in humans (women) were employed in this study. Samples from both patients with and without cancer were requested along with phenotypic characteristics of ECs such as grade, type, and stage. These samples were controlled in a way that none of the samples had been exposed to any specific treatment, not subjected to any stimulus, nor derived from cell lines, thus only from endometrial tissue of women. A total of 273 microarray datasets from multiple platforms including Affymetrix, Agilent and Illumina were merged across platforms as training datasets to uncover the predictive cancer signatures (Additional file 1: SI Materials and Methods). In addition, 65 samples from Illumina and Swegene microarray platforms were utilized as validation datasets. Therefore, a total of 318 microarray datasets were effectively used in these studies. All the information of the datasets are summarized in Table 3 and Additional file 5: Table S3. To merge the microarray datasets measured with multiple platform chips, we selected genes from all platforms based on the NIH Entrez Gene ID and used the Cross-Platform Normalization (XPN) method of Shabalin et al. [40] implemented in the R package: “CONOR” [41]. These normalization procedures led to a total of 8,920 genes that were selected to further analysis after data processing. Detailed descriptions of the data preprocessing and normalization are summarized in Additional file 6: Figure S2 and Additional file 1: SI Materials and Methods.
Table 3.
Characteristics of microarray datasets for platform, sample groups, and grade, type and stage in endometrial cancers
| Characteristics | Training dataset (n = 273) | Validation set A (n = 40) | Validation set B (n = 90) |
|---|---|---|---|
|
Sample |
|
|
|
| Control |
37 (14%) |
20 (50%) |
45 (50%) |
| Case |
236 (86%) |
20 (50%) |
45 (50%) |
|
Grade
a
|
|
|
|
| G1 |
86 (36%) |
|
20 (44%) |
| G2 |
43 (18%) |
- |
12 (27%) |
| G3 |
52 (22%) |
- |
8 (18%) |
| Unknown |
55 (23%) |
- |
5 (11%) |
|
Type
a
|
|
|
|
| T1 |
113 (48%) |
10 (50%) |
- |
| T2 |
84 (36%) |
10 (50%) |
- |
| Unknown |
39 (17%) |
0 |
- |
|
Stage
a
|
|
|
|
| IA |
31 (13%) |
10 (50%) |
10 (22%) |
| IB |
37 (16%) |
- |
10 (22%) |
| IC or late stage |
23 (10%) |
10 (50%) |
19 (42%) |
| Unknown |
82 (61%) |
0 |
6 (14%) |
|
Platform |
|
|
|
| Affymetrix |
140 |
- |
- |
| Illumina |
19 |
20 |
- |
| Agilent |
114 |
- |
- |
| Swegene | - | - | 45 |
aEndometrial cancers are grouped by the grade (degree of differentiation), type (histopathological types) and stage (status of spread).
Initial data analysis
We reviewed the sample profiles in each of the 273 microarray datasets. From these 273 datasets, at least four samples corresponding to both classes of one analysis of interest were selected for additional analyses. Thus, analyses of interest included cancer versus non-cancer patients, cancer grade, which was further divided into high grade (G3, poorly differentiated) versus low grade (G1, well differentiated), cancer types; Type I (estrogen dependent) versus Type II (estrogen independent), and cancer stage (higher than stage 2) versus early stage (lower than stage 2). After the assignment of samples to classes, we assessed the differential expression using Student’s t test to identify the significantly differential expression of gene profiles. False discovery rates (FDR) were used in these analyses for correcting for multiple comparisons [42]. All differentially expressed analysis used the “limma” program in the R-based Bioconductor package to calculate the level of differential expression [43].
Reconstruction of co-expression network
This study performed WGCNA analysis to construct the modules of co-expression gene for the EC-associated networks and their interactions. From the processed expression files, the networks were formed from the weighted correlation matrices following the protocols of WGCNA. Briefly, the WGCNA converts the gene expression profiles into connection weights that can be visualized as topology overlap measures (TOM) (Additional file 7: Figure S3). We chose expression profiles of 4,500 genes in the co-expression network analysis. These genes were either significantly differentially expressed between non-cancer and cancer samples (FDR < 0.05 and fold change > 1.5 between two groups) or showed a large variability in expression. We defined modules using a hierarchical cluster method, and used the topological overlap dissimilarity measure (1-TOM) as the distance measure with a height cutoff value of 0.95 and a minimum size (gene groups) cutoff value of 100 for the resulting dendrogram. All network analyses were implemented in the package WGCNA in the R environment as previously described [44].
Identification of cancer hub genes
We implemented for the first time an unique systematic framework that applies the elastic-net regularization-based approach and WGCNA to take the ECs-specific gene co-expression networks into account in the process of identification of cancer hub genes. Our approach contain two major steps. Firstly, we used WGCNA to identify the cancer hub genes that functionally contribute to the tumorigenesis of ECs. To distinguish centrally located genes (hub genes) of the co-expressed network, we calculated its scaled connectivity (K) and genes significance (GS) using WGCNA. This approach enabled us to determine the hub genes implicated by both the genetic marker and network connectivity information.
Secondly, to link these potential hub genes to the phenotypic characteristics of ECs, the elastic-net analysis was used to select which of these features were significantly associated with phenotypic characteristics of ECs across the gene co-expression networks. This approach is ideal for building the linear models in situations where the number of variables significantly outweighs the number of samples. In fact, this approach has been used as a powerful classification algorithm for large-scale microarray analysis [45,46]. In addition to all these innovative but highly structured and systematized approaches and procedures, we alsoperformed bootstrap analysis, sampling the datasets with replacements 1,000 times, and calculating the frequency (f) of markers (genes) for inclusion in the model for each bootstrap sample. Only genes present in more than the fourth quartile (f > 750) of all bootstrap samples were selected as ECs-specific hub genes. All elastic-net analysis used the R package “glmnet” [47]. Full methods are available in Support information.
Classifier predictive model
Classification performance was assessed with areas under the receiving operating characteristic (AUC) curve. Using the penalized logistic regression via the elastic-net, a classification model was built, and its discriminatory capacity was first estimated with a strict 10-fold cross-validation methodology (as described in Additional file 1: Materials and Methods). The resulting model was next tested on independent datasets using the cancer hub genes as a model input to predict the classes of particular samples relevant to the process of neoplastic transformation and progression in ECs.
Pathway analysis
The network was visualized through Cytoscape Software 3.0.1 [48]. This study used the Cluepedia plug-in in Cytoscape to identify potential association to pathways of cancer hub genes [49]. CluePedia organizes a functionally grouped pathway with cancer hub genes by integrating heterogeneous expression data and functional network information.
Competing interests
All the authors have declared that no competing interests exist.
Authors’ contributions
Conceived and designed the analysis: WCC CYC. Planned statistical design: WCC CYC ALC MB. Analyzed the data: WCC CYC ALC. Wrote the paper: WCC CYC ALC MB. All authors read and approved the final manuscript.
Supplementary Material
SI Materials and Methods.
Genes differentially expressed between tissues of cancer and non-cancer samples subjects.
List of potential hub genes in the WGCNA network, their gene significance and connectivity to each module.
10-fold cross validation to predict the (A) grade, (B) type, (C, D) stage of ECs using grade-, type- and stage-related cancer hub genes. The ROC curve of stage II is predicted from the cancer hub genes derived from Mblue.
{kind=link}
List of microarray experiments used for meta-analysis.
Detailed process of matching probes among different microarray platforms.
{kind=link}
Topological overlap matrix (TOM) plot of the network connections. Genes in the rows and columns are sorted by the clustering tree.
{kind=link}
Contributor Information
Wei-Chun Chou, Email: chouwcjoe@gmail.com.
An-Lin Cheng, Email: chenga@umkc.edu.
Marco Brotto, Email: brottom@umkc.edu.
Chun-Yu Chuang, Email: cychuang@mx.nthu.edu.tw.
Acknowledgments
This work was supported by Taiwan National Science Council NSC102-2918-I-007-011. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Colombo N, Preti E, Landoni F, Carinelli S, Colombo A, Marini C, Sessa C. Group EGW. Endometrial cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2011;22(Suppl 6):vi35–vi39. doi: 10.1093/annonc/mdr374. [DOI] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- Lax SF, Kurman RJ. A dualistic model for endometrial carcinogenesis based on immunohistochemical and molecular genetic analyses. Verh Dtsch Ges Pathol. 1997;81:228–232. [PubMed] [Google Scholar]
- Okuda T, Sekizawa A, Purwosunu Y, Nagatsuka M, Morioka M, Hayashi M, Okai T. Genetics of endometrial cancers. Obstet Gynecol Int. 2010;2010:1–8. doi: 10.1155/2010/984013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lax SF, Kendall B, Tashiro H, Slebos RJC, Ellenson LH. The frequency of p53, K-ras mutations, and microsatellite instability differs in uterine endometrioid and serous carcinoma - Evidence of distinct molecular genetic pathways. Cancer. 2000;88(4):814–824. doi: 10.1002/(SICI)1097-0142(20000215)88:4<814::AID-CNCR12>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Bansal N, Yendluri V, Wenham RM. The molecular biology of endometrial cancers and the implications for pathogenesis, classification, and targeted therapies. Cancer Control. 2009;16(1):8–13. doi: 10.1177/107327480901600102. [DOI] [PubMed] [Google Scholar]
- Mutter GL, Lin MC, Fitzgerald JT, Kum JB, Baak JPA, Lees JA, Weng LP, Eng C. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Natl Cancer I. 2000;92(11):924–931. doi: 10.1093/jnci/92.11.924. [DOI] [PubMed] [Google Scholar]
- Ryan AJ, Susil B, Jobling TW, Oehler MK. Endometrial cancer. Cell Tissue Res. 2005;322(1):53–61. doi: 10.1007/s00441-005-1109-5. [DOI] [PubMed] [Google Scholar]
- Enomoto T, Inoue M, Perantoni AO, Buzard GS, Miki H, Tanizawa O, Rice JM. K-ras activation in premalignant and malignant epithelial lesions of the human uterus. Cancer Res. 1991;51(19):5308–5314. [PubMed] [Google Scholar]
- Risinger JI, Hayes AK, Berchuck A, Barrett JC. PTEN/MMAC1 mutations in endometrial cancers. Cancer Res. 1997;57(21):4736–4738. [PubMed] [Google Scholar]
- Odicino FE, Bignotti E, Rossi E, Pasinetti B, Tassi RA, Donzelli C, Falchetti M, Fontana P, Grigolato PG, Pecorelli S. HER-2/neu overexpression and amplification in uterine serous papillary carcinoma: comparative analysis of immunohistochemistry, real-time reverse transcription-polymerase chain reaction, and fluorescence in situ hybridization. Int J Gynecol Cancer. 2008;18(1):14–21. doi: 10.1111/j.1525-1438.2007.00946.x. [DOI] [PubMed] [Google Scholar]
- Miller JA, Horvath S, Geschwind DH. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc Natl Acad Sci U S A. 2010;107(28):12698–12703. doi: 10.1073/pnas.0914257107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Dong J. Geometric Interpretation of Gene Coexpression Network Analysis. Plos Comput Biol. 2008;4(8):e1000117. doi: 10.1371/journal.pcbi.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi ZA, Derow CK, Zhang B. Co-expression module analysis reveals biological processes, genomic gain, and regulatory mechanisms associated with breast cancer progression. BMC Syst Biol. 2010;4:74. doi: 10.1186/1752-0509-4-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma SG, Huang Y, Huang J, Fang KN. Gene network-based cancer prognosis analysis with sparse boosting. Genet Res. 2012;94(4):205–221. doi: 10.1017/S0016672312000419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Miller JA, Shapshak P, Gelman B, Singer EJ, Hinkin CH, Commins D, Morgello S, Grant I, Horvath S. Systems analysis of human brain gene expression: mechanisms for HIV-associated neurocognitive impairment and common pathways with Alzheimer's disease. BMC Med Genomics. 2013;6:4. doi: 10.1186/1755-8794-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao FM, Ponte JF, Levy M, Papageorgis P, Cook NM, Ozturk S, Lambert AW, Thiagalingam A, Abdolmaleky HM, Sullivan BA, Thiagalingam S. hBub1 negatively regulates p53 mediated early cell death upon mitotic checkpoint activation. Cancer Biol Ther. 2009;8(7):636–644. doi: 10.4161/cbt.8.7.7929. [DOI] [PubMed] [Google Scholar]
- Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest. 2005;115(6):1503–1521. doi: 10.1172/JCI23412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu JY, Bian ML, Jiang Q, Zhang CM. Roles of aurora kinases in mitosis and tumorigenesis. Mol Cancer Res. 2007;5(1):1–10. doi: 10.1158/1541-7786.MCR-06-0208. [DOI] [PubMed] [Google Scholar]
- Ricke RM, Jeganathan KB, van Deursen JM. Bub1 overexpression induces aneuploidy and tumor formation through Aurora B kinase hyperactivation. J Cell Biol. 2011;193(6):1049–1064. doi: 10.1083/jcb.201012035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005;65(23):10669–10673. doi: 10.1158/0008-5472.CAN-05-2620. [DOI] [PubMed] [Google Scholar]
- Semczuk A, Jakowicki JA. Alterations of pRb1-cyclin D1-cdk4/6-p16(INK4A) pathway in endometrial carcinogenesis. Cancer Lett. 2004;203(1):1–12. doi: 10.1016/j.canlet.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Wu Z, Cho H, Hampton GM, Theodorescu D. Cdc6 and Cyclin E2 Are PTEN-Regulated Genes Associated with Human Prostate Cancer Metastasis. Neoplasia. 2009;11(1):66–76. doi: 10.1593/neo.81048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A, Hoellein A, Kremer M, Graf N, Scheerer M, Hall MA, Goga A, von Bubnoff N, Duyster J, Peschel C, Cleveland JL, Nilsson JA, Keller U. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood. 2010;116(9):1498–1505. doi: 10.1182/blood-2009-11-251074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimi A, Goyama S, Watanabe-Okochi N, Yoshiki Y, Nannya Y, Nitta E, Arai S, Sato T, Shimabe M, Nakagawa M, Imai Y, Kitamura T, Kurokawa M. Evi1 represses PTEN expression and activates PI3K/AKT/mTOR via interactions with polycomb proteins. Blood. 2011;117(13):3617–3628. doi: 10.1182/blood-2009-12-261602. [DOI] [PubMed] [Google Scholar]
- Gasnereau I, Boissan M, Margall-Ducos G, Couchy G, Wendum D, Bourgain-Guglielmetti F, Desdouets C, Lacombe ML, Zucman-Rossi J, Sobczak-Thepot J. KIF20A mRNA and Its Product MKlp2 Are Increased During Hepatocyte Proliferation and Hepatocarcinogenesis. Am J Pathol. 2012;180(1):131–140. doi: 10.1016/j.ajpath.2011.09.040. [DOI] [PubMed] [Google Scholar]
- Margolis RL. Bub1, a gatekeeper for Cdc20-dependent mitotic exit. Dev Cell. 2004;7(5):634–635. doi: 10.1016/j.devcel.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Peters JM. The anaphase-promoting complex: Proteolysis in mitosis and beyond. Mol Cell. 2002;9(5):931–943. doi: 10.1016/S1097-2765(02)00540-3. [DOI] [PubMed] [Google Scholar]
- Yuan BB, Xu Y, Woo JH, Wang YY, Bae YK, Yoon DS, Wersto RP, Tully E, Wilsbach K, Gabrielson E. Increased expression of mitotic checkpoint genes in breast cancer cells with chromosomal instability. Clin Cancer Res. 2006;12(2):405–410. doi: 10.1158/1078-0432.CCR-05-0903. [DOI] [PubMed] [Google Scholar]
- Dai HY, van't Veer L, Lamb J, He YD, Mao M, Fine BM, Bernards R, de Vijver MV, Deutsch P, Sachs A, Stoughton R, Friend S. A cell proliferation signature is a marker of extremely poor outcome in a subpopulation of breast cancer patients. Cancer Res. 2005;65(10):4059–4066. doi: 10.1158/0008-5472.CAN-04-3953. [DOI] [PubMed] [Google Scholar]
- Perez De Castro I, De Carcer G, Malumbres M. A census of mitotic cancer genes: new insights into tumor cell biology and cancer therapy. Carcinogenesis. 2007;28(5):899–912. doi: 10.1093/carcin/bgm019. [DOI] [PubMed] [Google Scholar]
- Morse MA, Secord AA, Blackwell K, Hobeika AC, Sinnathamby G, Osada T, Hafner J, Philip M, Clay TM, Lyerly HK, Philip R. MHC Class I-Presented Tumor Antigens Identified in Ovarian Cancer by Immunoproteomic Analysis Are Targets for T-Cell Responses against Breast and Ovarian Cancer. Clin Cancer Res. 2011;17(10):3408–3419. doi: 10.1158/1078-0432.CCR-10-2614. [DOI] [PubMed] [Google Scholar]
- Bijen CBM, Bantema-Loppe EJ, de Jong RA, Leffers N, Mourits MJE, Eggink HF, van der Zee AGJ, Hollema H, de Bock GH, Nijman HW. The prognostic role of classical and nonclassical MHC class I expression in endometrial cancer. Int J Cancer. 2010;126(6):1417–1427. doi: 10.1002/ijc.24852. [DOI] [PubMed] [Google Scholar]
- Chen JQ, Russo J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Bba-Rev Cancer. 2012;1826(2):370–384. doi: 10.1016/j.bbcan.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Raimundo N, Baysal BE, Shadel GS. Revisiting the TCA cycle: signaling to tumor formation. Trends Mol Med. 2011;17(11):641–649. doi: 10.1016/j.molmed.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Sekido Y, Horio Y, Takahashi M, Saito H, Minna JD, Shimokata K, Hasegawa Y. Infrequent mutation of the hBUB1 and hBUBR1 genes in human lung cancer. Jpn J Cancer Res. 2000;91(5):504–509. doi: 10.1111/j.1349-7006.2000.tb00974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern SL, Qi Y, Pusztai L, Symmans WF, Buchholz TA. Centromere protein-A, an essential centromere protein, is a prognostic marker for relapse in estrogen receptor-positive breast cancer. Breast Cancer Res. 2012;14(3):R72. doi: 10.1186/bcr3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita Y, Akira Y, Tsukamoto H, Senju S, Kuroda Y, Hirayama M, Irie A, Kawahara K, Yatsuda J, Hamada A, Jono H, Yoshida K, Tsunoda T, Kohrogi H, Yoshitake Y, Nakamura Y, Shinohara M, Nishimura Y. Identification of promiscuous KIF20A long peptides bearing both CD4+ and CD8+ T-cell epitopes: KIF20A-specific CD4+ T-cell immunity in patients with malignant tumor. Clin Cancer Res. 2013;19(16):4508–4520. doi: 10.1158/1078-0432.CCR-13-0197. [DOI] [PubMed] [Google Scholar]
- Shabalin AA, Tjelmeland H, Fan C, Perou CM, Nobel AB. Merging two gene-expression studies via cross-platform normalization. Bioinformatics. 2008;24(9):1154–1160. doi: 10.1093/bioinformatics/btn083. [DOI] [PubMed] [Google Scholar]
- Rudy J, Valafar F. Empirical comparison of cross-platform normalization methods for gene expression data. BMC Bioinforma. 2011;12:467. doi: 10.1186/1471-2105-12-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diboun I, Wernisch L, Orengo CA, Koltzenburg M. Microarray analysis after RNA amplification can detect pronounced differences in gene expression using limma. BMC Genomics. 2006;7:252. doi: 10.1186/1471-2164-7-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Horvath S. Network neighborhood analysis with the multi-node topological overlap measure. Bioinformatics. 2007;23(2):222–231. doi: 10.1093/bioinformatics/btl581. [DOI] [PubMed] [Google Scholar]
- Zhuang J, Widschwendter M, Teschendorff AE. A comparison of feature selection and classification methods in DNA methylation studies using the Illumina Infinium platform. BMC Bioinforma. 2012;13:59. doi: 10.1186/1471-2105-13-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler D, Li Y. Survival Analysis with High-Dimensional Covariates: an Application in Microarray Studies. Stat Appl Genet Mol. 2009;8(1):1–14. doi: 10.2202/1544-6115.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw. 2010;33(1):1–22. [PMC free article] [PubMed] [Google Scholar]
- Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, Hanspers K, Isserlin R, Kelley R, Killcoyne S, Lotia S, Maere S, Morris J, Ono K, Pavlovic V, Pico AR, Vailaya A, Wang PL, Adler A, Conklin BR, Hood L, Kuiper M, Sander C, Schmulevich I, Schwikowski B, Warner GJ. et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2(10):2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Galon J, Mlecnik B. CluePedia Cytoscape plugin: pathway insights using integrated experimental and in silico data. Bioinformatics. 2013;29(5):661–663. doi: 10.1093/bioinformatics/btt019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SI Materials and Methods.
Genes differentially expressed between tissues of cancer and non-cancer samples subjects.
List of potential hub genes in the WGCNA network, their gene significance and connectivity to each module.
10-fold cross validation to predict the (A) grade, (B) type, (C, D) stage of ECs using grade-, type- and stage-related cancer hub genes. The ROC curve of stage II is predicted from the cancer hub genes derived from Mblue.
List of microarray experiments used for meta-analysis.
Detailed process of matching probes among different microarray platforms.
Topological overlap matrix (TOM) plot of the network connections. Genes in the rows and columns are sorted by the clustering tree.